

Abstract
Shank3, an abundant excitatory postsynaptic scaffolding protein, has been associated with multiple brain disorders, including autism spectrum disorders (ASD) and Phelan-McDermid syndrome (PMS). However, how cell type-specific Shank3 deletion affects disease-related neuronal and brain functions remains largely unclear. Here, we investigated the impacts of Shank3 deletion in glutamatergic neurons on synaptic and behavioral phenotypes in mice and compared results with those previously obtained from mice with global Shank3 mutation and GABAergic neuron-specific Shank3 mutation. Neuronal excitability was abnormally increased in layer 2/3 pyramidal neurons in the medial prefrontal cortex (mPFC) in mice with a glutamatergic Shank3 deletion, similar to results obtained in mice with a global Shank3 deletion. In addition, excitatory synaptic transmission was abnormally increased in layer 2/3 neurons in mice with a global, but not a glutamatergic, Shank3 deletion, suggesting that Shank3 in glutamatergic neurons are important for the increased neuronal excitability, but not for the increased excitatory synaptic transmission. Neither excitatory nor inhibitory synaptic transmission was altered in the dorsal striatum of Shank3-deficient glutamatergic neurons, a finding that contrasts with the decreased excitatory synaptic transmission in global and Shank3-deficient GABAergic neurons. Behaviorally, glutamatergic Shank3-deficient mice displayed abnormally increased direct social interaction and repetitive self-grooming, similar to global and GABAergic Shank3-deficient mice. These results suggest that glutamatergic and GABAergic Shank3 deletions lead to distinct synaptic and neuronal changes in cortical layer 2/3 and dorsal striatal neurons, but cause similar social and repetitive behavioral abnormalities likely through distinct mechanisms.
Keywords: autism, Phelan-McDermid syndrome, Shank3, medial prefrontal cortex, striatum, social interaction, repetitive behavior
Introduction
Shank3 is an abundant scaffolding protein mainly present in the postsynaptic side of excitatory synapses and contributes to excitatory synapse development and function (Boeckers et al., 2002; Sheng and Hoogenraad, 2007; Sala et al., 2015). Mutations in the SHANK3 gene have been associated with various brain disorders, including autism spectrum disorders (ASD), Phelan-McDermid Syndrome (PMS), schizophrenia, intellectual disability, and mania (Wilson et al., 2003; Gauthier et al., 2010; Bonaglia et al., 2011; Hamdan et al., 2011; Phelan and McDermid, 2012; Boccuto et al., 2013; Han et al., 2013; Guilmatre et al., 2014; Leblond et al., 2014; Cochoy et al., 2015). Mechanisms underlying the development of Shank3-related brain dysfunctions have been suggested through studies of a large number of Shank3-mutant mice (Bozdagi et al., 2010; Peca et al., 2011; Wang X. et al., 2011; Yang et al., 2012; Han et al., 2013; Jiang and Ehlers, 2013; Kouser et al., 2013; Duffney et al., 2015; Lee et al., 2015; Sala et al., 2015; Speed et al., 2015; Jaramillo et al., 2016, 2017; Mei et al., 2016; Wang et al., 2016, 2019a,b; Zhou et al., 2016; Monteiro and Feng, 2017; Vicidomini et al., 2017; Bey et al., 2018; Drapeau et al., 2018; Qin et al., 2018; Yoo et al., 2018).
More recently, Shank3 deletions restricted to specific brain regions and cell types in mice have been attempted (Bey et al., 2018; Yoo et al., 2018). For instance, Shank3 (exons 4–22) deletions in glutamatergic and striatal D1 and D2 GABAergic neurons have been generated using cell type-specific Cre mouse lines, including NEX (dorsal telencephalic glutamatergic neurons mainly found in the cortex and hippocampus), Dlx5/6 (striatal GABAergic neurons), and Drd1/2 (dopaminergic D1 or D2 neurons) (Bey et al., 2018). In addition, a Shank3 (exons 14–16) deletion restricted to GABAergic neurons has also been developed (Yoo et al., 2018). These different Shank3 deletions result in shared as well as distinct phenotypes, including striatal synaptic dysfunctions and ASD-related behavioral deficits (Bey et al., 2018; Yoo et al., 2018).
We previously reported a GABAergic Shank3 (exons 14–16) deletion and described its impacts on synaptic and behavioral phenotypes in mice (Yoo et al., 2018); however, Shank3 is expressed in both glutamatergic and GABAergic neurons (Han et al., 2013; Yoo et al., 2018). In addition, although a glutamatergic Shank3 (exons 4–22) deletion has previously been generated using the NEX-Cre driver line (Bey et al., 2018), in our conditional knockout (cKO)-ready Shank3-mutant mice, exons 14–16 rather than exons 4–22 were targeted, affecting different Shank3 splice variants and leading to different phenotypes, based on the complex alternative splicing patterns in the Shank3 gene (Lim et al., 2001; Durand et al., 2007; Wang X. et al., 2011; Jiang and Ehlers, 2013; Wang et al., 2014a, b; Monteiro and Feng, 2017). We thus attempted to use Emx1-Cre that drives gene expression mainly in the cortex and hippocampus derived from the dorsal telencephalon (Gorski et al., 2002).
The striatum has been strongly associated with ASD (Haznedar et al., 2006; Di Martino et al., 2011; Langen et al., 2013; Kohls et al., 2014; Rothwell et al., 2014; Barak and Feng, 2016; Fuccillo, 2016; Schuetze et al., 2016; Rapanelli et al., 2017). In addition, many previous studies have identified dysfunctions in the corticostriatal pathway involving the dorsal striatum in Shank3-mutant mice (Peca et al., 2011; Filice et al., 2016; Fuccillo, 2016; Jaramillo et al., 2016, 2017; Mei et al., 2016; Peixoto et al., 2016; Wang et al., 2016, 2017; Zhou et al., 2016; Lee Y. et al., 2017; Reim et al., 2017; Vicidomini et al., 2017; Bey et al., 2018; Fourie et al., 2018; Yoo et al., 2018). The medial prefrontal cortex (mPFC) has also been strongly implicated in ASD (Ernst et al., 1997; Mundy, 2003; Pierce et al., 2004; Carper and Courchesne, 2005; Amodio and Frith, 2006; Gilbert et al., 2008; Rinaldi et al., 2008; Shalom, 2009; Courchesne et al., 2011; Yizhar et al., 2011; Testa-Silva et al., 2012; Liang et al., 2015; Barak and Feng, 2016; Ko, 2017; Wang et al., 2019c). However, how the striatum and prefrontal cortex differentially contribute to core ASD-related behaviors remains unclear.
Different cortical layers such as layer 2/3 and layer 5 have been suggested to contribute to ASD. Some previous studies have characterized layer 5 cortical pyramidal neurons in the mPFC or somatosensory cortex in Shank3-mutant mice (Peixoto et al., 2016; Qin et al., 2019; Wang et al., 2019d). Layer 2/3 pyramidal neurons also receive diverse inputs from intracortical and subcortical afferents and provide excitatory inputs onto other cortical layers, including layer 5 (Gabbott et al., 2005; Hoover and Vertes, 2007; Xu and Sudhof, 2013; Lee et al., 2014; Virtanen et al., 2018), and have been implicated in cortical neuronal integration, cognitive functions, and brain disorders such as ASD, schizophrenia, and depression (Parikshak et al., 2013; Shrestha et al., 2015; Li et al., 2016; Page et al., 2018). Indeed, previous studies have highlighted the importance of layer 2/3 cortical neurons in ASD, reporting that superficial cortical layers in the human brain are enriched for genes that are coexpressed in ASD (Parikshak et al., 2013), that inhibitory synaptic transmission in layer 2/3 mPFC cortical neurons is reduced in neuroligin-2–mutant mice with cognitive and social dysfunctions (Liang et al., 2015), and that enhanced synapse remodeling in layer 2/3 pyramidal neurons may be a common pathology in two independent mouse models of ASD (Isshiki et al., 2014). A more recent study reported age-dependent changes in excitatory synaptic transmission and spine density in layer 2/3 mPFC pyramidal neurons (Zhou et al., 2016). However, the role of layer 2/3 pyramidal neurons in ASD-related brain dysfunctions remains incompletely studied.
In the present study, we generated a Shank3 (exons 14–16) deletion in mice restricted to glutamatergic neurons and investigated its impact on mPFC layer 2/3 and dorsal striatal neurons and ASD-related behaviors, and compared the results with those obtained in mice with global or GABAergic Shank3 deletions. Our findings indicate that layer 2/3 pyramidal neurons from Emx1-Cre;Shank3fl/fl (Emx1-Cre;Shank3Δ14–16) mice showed increased neuronal excitability, similar to results in global Shank3-deficient (global Shank3Δ14–16) layer 2/3 neurons. In contrast to the decreased excitatory synaptic transmission in global Shank3Δ14–16 and GABAergic Shank3-deficient (Viaat-Cre;Shank3Δ14–16) neurons, Emx1-Cre;Shank3Δ14–16 dorsolateral striatal neurons showed normal synaptic transmission. Behaviorally, Emx1-Cre;Shank3Δ14–16 mice showed abnormal social interaction and self-grooming, similar to global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice. These results suggest that both glutamatergic and GABAergic Shank3 deletions contribute to the social and repetitive behavioral deficits observed in global Shank3Δ14–16 mice, but lead to distinct synaptic and neuronal alterations in layer 2/3 pyramidal neurons and dorsal striatal GABAergic neurons.
Materials and Methods
Animals
Mice carrying a deletion of exons 14–16 of the Shank3 gene flanked by LoxP sites have been described (Yoo et al., 2018). Homozygous Shank3Δ14–16 cKO mice with a gene deletion restricted to dorsal telencephalic excitatory neurons (Emx1-Cre;Shank3fl/fl mice) were produced by crossing homozygous Shank3fl/fl female mice with double-heterozygous Emx1-Cre;Shank3fl/+ mice. Cre-negative Shank3fl/fl littermates, referred to as wild-type (WT) throughout the manuscript, were used as controls for Emx1-Cre;Shank3fl/fl mice. The Emx1-Cre mouse line, purchased from the Jackson Laboratory (Jackson; #005628) and maintained in a C57BL/6J genetic background for more than five generations, was used for comparisons with all global and cKO mouse lines in the same pure C57BL/6J background. Mice were bred and maintained at the mouse facility of Korea Advanced Institute of Science and Technology (KAIST) according to Animal Research Requirements of KAIST, and all procedures were approved by the Committee of Animal Research at KAIST (KA2016-30). Animals were fed ad libitum and housed under a 12-h light/dark cycle (light phase from 1:00 am to 1:00 pm). Genotypes of Emx1-Cre;Shank3fl/fl mice were determined by polymerase chain reaction (PCR) using the following primer pairs: floxed (478 bp) or WT allele (276 bp), 5′-GGG TTC CTA TGA CAG CCT CA-3′ (forward) and 5′-TTC TGC AGG ATA GCC ACC TT-3′ (reverse); Emx1-Cre (272 bp), 5′-GTG TTG CCG CGC CAT CTG C-3′ (forward) and 5′-CAC CAT TGC CC TGT TTC ACT ATC-3′ (reverse). Only male mice were used for behavioral and electrophysiological experiments, whereas both male and female were used for biochemical experiments.
Brain Lysates
Brains from Emx1-Cre; Shank3fl/fl mice and their Shank3fl/fl littermates (12 weeks; females for Shank3 protein levels, 13 weeks; males for Shank1 and Shank2 protein levels) were extracted and dissected on ice into cortex, thalamus, striatum, and hippocampus, followed by homogenization with ice-cold homogenization buffer (0.32 M sucrose, 10 mM HEPES, pH 7.4, 2 mM EDTA, pH 8.0, 2 mM EGTA, pH 8.0, protease inhibitors, phosphatase inhibitors). Total lysates were prepared by boiling with β-mercaptoethanol directly after homogenization.
Western Blot
Total brain lysates separated in electrophoresis and transferred to a nitrocellulose membrane were incubated with primary antibodies to Shank1 (#2100, guinea pig) (Ha et al., 2016), Shank2 (Synaptic Systems 162 202), Shank3 (#2036 guinea pig polyclonal antibodies raised against aa 1289–1318 of the mouse Shank3 protein) (Lee et al., 2015) and α-tubulin (Sigma T5168) at 4°C overnight. Fluorescent secondary antibody signals were detected using Odyssey® Fc Dual Mode Imaging System.
Behavioral Assays
Male mice (2–8-mo-old) were used for all behavioral assays. Before behavioral experiments, mice were handled for 10 min per day for 3 days. All behavioral assays were initiated after a 30-min habituation in a dark booth. The behavioral tests for Emx1-Cre;Shank3Δ14–16 mice and Emx1-Cre mice were performed in the order indicated in Supplementary Table S1. The order of behavioral tests was designed to minimize stress to the animals.
Three-Chamber Test
Social approach and social novelty recognition were measured using the three-chambered social interaction test (Crawley, 2004; Nadler et al., 2004; Moy et al., 2009; Silverman et al., 2010) under illuminated (70–80 lux) conditions. The 3-chamber test apparatus is a white acrylic box (60 × 40 × 20 cm) divided into three chambers. Both left and right side chambers contained a cage in the upper or lower corner for an object or a stranger mouse. Experimental mice were isolated in a single cage for 3 days prior to the test, whereas unfamiliar stranger mice (129S1/SvlmJ strain) were group-housed (5–7 mice/cage). All stranger mice were age-matched males and were habituated to a corner cage during the previous day (30 min). The test consisted of three phases: empty-empty (habituation), stranger 1-object (S1-O), and stranger 1-stranger 2 (S1-S2). In the first phase (habituation), a test mouse was placed in the center area of the three-chambered apparatus, and allowed to freely explore the whole apparatus for 10 min. The mouse was then gently guided to the center chamber while an inanimate blue cylindrical object (O) and a WT stranger mouse (S1) were placed in the two corner cages. The positions of object (O) and stranger 1 (S1) were alternated between tests to prevent side preference. In the S1-O phase, the test mouse was allowed to explore the stranger mouse or the object freely for 10 min. Before the third phase (S1-S2), the subject mouse was again gently guided to the center chamber while the object was replaced with a new WT stranger mouse (S2). The subject mouse again was allowed to freely explore all three chambers and interact with both stranger mice for 10 min. The duration of sniffing, defined as positioning of the nose of the test mouse within 2.5 cm of a cage, was measured using EthoVision XT10 (Noldus) software.
Direct Social Interaction Test
Direct social interaction tests were performed as described previously (Chung et al., 2015). All mice were isolated for 3 days prior to the day of the experiment. Each individual mouse was habituated to a gray box (30 × 30 × 30 cm; ∼25–30 lux) for two consecutive days (10 min/d). On day 3, pairs of mice of the same genotype (originally housed separately) were placed in the test box for 10 min. Time spent in nose-to-nose interaction, following, and total interaction were measured manually in a blinded manner. Nose-to-nose interaction was defined as sniffing the head part of the other mouse. Following included regular following as well as nose-to-tail sniffing. Total interaction included nose-to-nose interaction, following, body contact, allo-grooming, and mounting.
Tube Test
The tube test assay was performed as described previously (Wang F. et al., 2011). Mice were group-housed (4 in a cage) for 2 weeks before behavioral experiments. We used transparent acryl tubes with 30-cm length and 3-cm inner diameter. During two-day training sessions, each mouse was trained to pass through the tube in either direction for eight times under illuminated (∼30 lux) conditions. As the mice hesitated to move, they were gently pushed by a plastic bar. After this, 3 days of test sessions were proceeded. Animals went through three more training trials before the test. For the test, two different mice were placed into the opposite ends of the test tube and carefully released to meet in the middle of the tube. The mouse that first retreated from the tube was marked as a “loser.” Among six possible pairs between four cage-mates, two pairs were tested per day. Each mouse was ordered by its rank from 1 to 4.
Courtship Ultrasonic Vocalization
Adult subject male mice were isolated in their home cage for 3 days before the test, whereas age-matched intruder female mice were group-housed (6–7 mice/cage). We did not measure female cycles on the assumption that group housing might synchronize cycles. Basal ultrasonic vocalizations (USVs) of an isolated male mouse in its home cage under light conditions of ∼60 lux in a soundproof chamber were recorded for 5 min in the absence of a female intruder. Next, a randomly chosen stranger C57BL/6J female mouse was introduced into the cage, and female-induced courtship USVs were recorded for 5 min during free interaction between the male and female. Avisoft SASLab Pro software was used to automatically analyze the number of USV calls, latency to first call, and total duration of calls from recorded USV files. Signals were filtered from 1 to 100 kHz and digitized with a sampling frequency of 250 kHz, 16 bits per sample (Avisoft UltraSoundGate 116H). Spectrograms were generated using the following parameters: FFT length, 256; frame size, 100; window, FlatTop; overlap, 75%. These parameters yielded a frequency resolution of 977 Hz and a temporal resolution of 0.256 ms. Frequencies lower than 25 kHz were filtered out to reduce white background noise.
Repetitive Behavior and Self-Grooming Test
Each mouse was placed in a lighted (∼60–70 lux), fresh home cage with bedding and recorded for 20 min. Time spent in self-grooming and digging behavior, measured manually, was determined by analyzing the last 10 min. Self-grooming behavior was defined as stroking or scratching of the body or face, or licking body parts. Digging was defined as scattering bedding using the head and forelimbs. Self-grooming behavior was further analyzed by placing mice in an empty home cage without bedding and recording them for 20 min. Time spent in self-grooming behavior was counted manually in a blinded manner during the last 10 min.
Laboras Test
Each mouse was placed in a single cage and recorded for 96 consecutive hours from the start of the night cycle. The illumination condition during light-on periods was ∼60 lux. Basal activities (locomotion, climbing, rearing, grooming, eating, and drinking) were recorded and automatically analyzed using the Laboratory Animal Behavior Observation Registration and Analysis System (LABORAS, Metris). Laboras results were not validated by our own manual analyses, given the availability of previous validation results (Van de Weerd et al., 2001; Quinn et al., 2003, 2006; Dere et al., 2015). Mouse movements during the entire 4-day period were used for quantification of behaviors, except for repetitive behavior, for which analyses were restricted to movements during light-off periods, which yielded clearer results.
Open-Field Test
Mice were placed in the center of an illuminated (90–100 lux) white acrylic box (40 × 40 × 40 cm), and their locomotion was recorded with a video camera for 1 h. The recorded video was analyzed using EthoVision XT10 software (Noldus). The center zone was defined as a 4 × 4-square area at the center of the entire 6 × 6-square region.
Elevated Plus-Maze
The maze consists of two open arms (30 × 6 cm, ∼180 lux) and two closed arms (30 × 6 cm, ∼20 lux) elevated 75 cm from the floor. Mice were introduced into the center of the apparatus with their head oriented toward the open arms and were allowed to freely explore the environment for 8 min. Amounts of time spent in open or closed arms and number of transitions were measured using EthoVision XT10 software (Noldus).
Light-Dark Test
The light-dark (LD) apparatus was divided into light (700 lux; 21 × 29 × 20 cm) and dark (∼5 lux; 21 × 13 × 20 cm) chambers separated by an entrance in the middle wall (5 × 8 cm). Mice were introduced into the light chamber with their head oriented toward the opposite side of the dark chamber and were allowed to freely explore the apparatus for 10 min. Amounts of time spent in light and dark chambers and number of transitions were analyzed using EthoVision XT10 software (Noldus).
Electrophysiology
Male mice at P18–26 (for mPFC measurements) and at P29–41 (for dorsolateral striatum measurements) were anesthetized with isoflurane. Mouse brain sections (300 μm) were sectioned in ice-cold dissection buffer containing (in mM) 212 sucrose, 25 NaHCO3, 10 D-glucose, 2 Na-pyruvate, 1.25 ascorbic acid, 1.25 NaH2PO4, 5 KCl, 3.5 MgSO4, and 0.5 CaCl2 bubbled with 95% O2 and 5% CO2 gases using Leica VT 1200 vibratome. The slices were recovered for 30 min and maintained in artificial cerebrospinal fluid (ACSF) at 32°C (in mM: 124 NaCl, 25 NaHCO3, 10 Glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4 oxygenated with 95% O2 and 5% CO2 gases). All recordings were performed after recovery for additional 30 min at room temperature. During all recordings, brain slices were maintained in a submerge-type recording chamber perfused with 27.5–28.5°C ACSF (2 ml min–1). Recording glass pipettes from borosilicate glass capillaries (Harvard Apparatus) were pulled using an electrode puller (Narishige). All electric responses were amplified and filtered at 2 kHz (Multiclamp 700B, Molecular Devices) and then digitized at 10 kHz (Digidata 1550, Molecular Devices). For whole-cell patch recordings in the mPFC layer 2/3 and dorsolateral striatum, a recording pipette (2.5–3.5 MΩ) was filled with the internal solution (in mM: 100 CsMeSO4, 10 TEA-Cl, 8 NaCl, 10 HEPES, 5 QX-314-Cl, 2 Mg-ATP, 0.3 Na-GTP and 10 EGTA with pH 7.25, 295 mOsm for mEPSCs and sEPSCs; 115 CsCl, 10 EGTA, 8 NaCl, 10 TEACl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 5 QX-314 with pH 7.35, 295 mOsm for mIPSCs and sIPSC; 137 K-gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 10 Na2-phosphocreatine, 4 Mg-ATP, 0.5 Na-GTP with pH 7.2, 280 mOsm for excitability). To measure mEPSCs, mIPSCs, sEPSCs, and sIPSCs, mPFC layer2/3 pyramidal neurons and dorsolateral MSN neurons were voltage-clamped at −70 mV. For mEPSCs and mIPSCs, picrotoxin (60 μM) and NBQX (10 μM) + APV (50 μM) were added to ACSF with TTX (1 μM), respectively. For sEPSCs and sIPSCS, picrotoxin (60 μM) and NBQX (10 μM) + APV (50 μM) without TTX were added, respectively. mE/IPSC and sE/IPSC events were selected based on the properties of the detected currents (rise time < 1 ms, 10 pA < amplitude < 500 pA, and decay half-width > 2 ms). Responses were recorded for 2 min after maintaining stable baseline for 5 min. For neuronal excitability measurement, ACSF contained picrotoxin (60 μM), NBQX (10 μM), and AP5 (50 μM). First minimal currents were introduced to hold the membrane potential around −80 mV in a current clamp mode. To evoke depolarizing voltage sag responses, increasing amounts of depolarizing step currents (by 10 pA, −150–10 pA) were injected. Then, to elicit action potentials, increasing amounts of depolarizing currents (0–330 pA) were injected in a stepwise manner. Input resistance was calculated as the linear slope of current-voltage plots generated from a series of increasing current injection steps.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 software. Details of statistical analyses are presented in Supplementary Table S2. The normality of data distributions was determined using the D’Agostino and Pearson omnibus normality test, followed by Student’s t-test (in the case of a normal distribution) and Mann–Whitney U test (in the case of a non-normal distribution). If samples were dependent on each other, a paired t-test (in the case of a normal distribution) or Wilcoxon signed rank test (in the case of a non-normal distribution) was used. Repeated-measures, two-way analysis of variance (ANOVA) with post hoc Bonferroni test (in the case of significant interactions) was used for time-varying analyses of open-field tests and Laboras tests. In cases where a Grubb’s test showed that a single significant outlier (∗P < 0.05) caused data to be non-normally distributed, the outlier value was removed prior to analysis. A one-sample t-test was used for the analysis of Western blot data. P-values < 0.05 were considered statistically significant; individual P-values are indicated in figures as follows: ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; and ns, not significant.
Results
Generation and Basic Characterization of Emx1-Cre;Shank3Δ14–16 Mice
To analyze the effects of a Shank3 deletion restricted to glutamatergic neurons, we crossed Shank3fl/fl mice (exons 14–16) with an Emx1-Cre mouse line, known to drive gene expression in glutamatergic neurons and glia with a dorsal telencephalic origin (Gorski et al., 2002).
The resulting Emx1-Cre;Shank3Δ14–16 mice, genotyped by PCR (Figure 1A), exhibited strong reductions in the levels of Shank3a and Shank3c/d variants in the hippocampus and cortex (Figures 1B,C), a finding in agreement with previous results on alternative splicing in Shank3 (Lim et al., 1999; Maunakea et al., 2010; Waga et al., 2014; Wang et al., 2014b). In contrast, Shank3 expression was largely unaffected in the thalamus, a brain region that is minimally affected by the Emx1 driver, and the striatum, which is mainly populated by GABAergic inhibitory neurons.
FIGURE 1.

Generation and characterization of Emx1-Cre;Shank3Δ14–16 mice. (A) PCR genotyping of Emx1-Cre;Shank3Δ14–16 mice. Note that the primer set targeting exons 13 and 14 generates a PCR band for the floxed allele (478 bp), and the primer set targeting general Cre generates a PCR band (272 bp) in Emx1-Cre;Shank3Δ14–16 mice, but not in WT mice. (B,C) Reduced levels of Shank3 protein variants in different brain regions of Emx1-Cre;Shank3Δ14–16 mice (12–13 weeks, male and female). Total brain lysates were analyzed by immunoblotting using a Shank3-specific antibody (#2036) (B). Neither the Shank3e isoform in the thalamus nor the Shank3c/d isoform in the striatum was quantified because of their low levels of expression in these regions. Th, thalamus; Str, striatum; Hpc, hippocampus; Ctx, cortex. cKO band signals normalized to α-tubulin are expressed relative to those from WT mice (C). Data are shown as mean ± SEM. n = 5 pairs (WT, cKO), ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, nd, not detectable, ns, not significant, and one sample t-test. (D,E) Normal levels of Shank1 and Shank2 protein variants in different brain regions of Emx1-Cre;Shank3Δ14– 16 mice (12–13 weeks, male and female). Total brain lysates were analyzed by immunoblotting using a Shank1-specific antibody (#2100) and Shank2-specific antibody (162 202, SYSY) (D). cKO band signals normalized to α-tubulin are expressed relative to those from WT mice (E). Data are shown as mean ± SEM. n = 5 pairs (WT, cKO), ns, not significant, and one sample t-test.
Levels of other members of the Shank family of proteins, namely Shank1 (Shank1a variant reported previously) (Lim et al., 1999; Naisbitt et al., 1999) and Shank2 (Shank2a and Shank2b reported previously) (Schmeisser et al., 2012; Won et al., 2012), were unaffected by Shank3 deletion in the tested brain regions (Figures 1D,E), indicative of the lack of compensatory changes.
Increased Excitability in Global Shank3Δ14–16 and Emx1-Cre;Shank3Δ14–16 mPFC Layer 2/3 Pyramidal Neurons
Previous studies have associated Shank3 deletion with altered neuronal excitability in human and rodent neurons (Peixoto et al., 2016; Yi et al., 2016), suggesting the possibility of altered neuronal excitability in Shank3-deficient cortical neurons. To determine whether Shank3 deletion affects intrinsic excitability in layer 2/3 cortical pyramidal neurons in the mPFC, a brain region implicated in ASD, and whether glutamatergic neurons are involved, we measured and compared neuronal excitability in global Shank3Δ14–16 and Emx1-Cre;Shank3Δ14–16 pyramidal neurons in the prelimbic region of the mPFC.
Global Shank3Δ14–16 mice exhibited increased neuronal excitability in layer 2/3 pyramidal neurons, as shown by the current-spike curve and input resistance, two electrophysiolgical parameters that contribute to neuronal excitability in depolarizing and hyperpolarizing ranges of membrane potentials (Figures 2A–C). Emx1-Cre;Shank3Δ14–16 mice also showed similarly increased neuronal excitability in mPFC neurons (Figures 2D–F). These results suggest that glutamatergic neurons contribute to the increased neuronal excitability observed in global Shank3Δ14–16 mice.
FIGURE 2.

Increased excitability in global Shank3Δ14–16 and Emx1-Cre;Shank3Δ14–16 mPFC layer 2/3 pyramidal neurons. (A–C) Enhanced excitability in layer 2/3 pyramidal neurons in the prelimbic region of the mPFC in global Shank3Δ14–16 mice (gKO; P20–25), as shown by current-firing curve (A), current-voltage relationship (B) and input resistance (C). Data are shown as mean ± SEM. n = 15 neurons from 3 mice (WT), 14, 3 (KO), ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, repeated measures two-way ANOVA (for current-firing and current-membrane potential curves), and Student’s t-test (for input resistance). (D–F) Enhanced excitability of layer 2/3 pyramidal neurons in the prelimbic region of the mPFC in Emx1-Cre;Shank3Δ14–16 mice (P20–24), as shown by current-firing curve, current-voltage relationship and input resistance. n = 18 neurons from 4 mice (WT), 17, 4 (cKO), ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, repeated measures two-way ANOVA (for current-firing and current-membrane potential curves), and Student’s t-test (for input resistance).
Altered Excitatory and Inhibitory Spontaneous Synaptic Transmissions in Global Shank3Δ14–16, but Not Emx1-Cre;Shank3Δ14–16, mPFC Layer 2/3 Pyramidal Neurons
Given that neuronal excitability acts together with excitatory and inhibitory synaptic inputs to determine neuronal output function, we next measured excitatory and inhibitory synaptic transmission in Shank3-mutant mPFC neurons.
The frequency, but not amplitude, of miniature excitatory postsynaptic currents (mEPSCs) was increased in the prelimbic region of the mPFC of global Shank3Δ14–16 layer 2/3 neurons compared with WT mice (Figure 3A). In contrast to mEPSCs, miniature inhibitory postsynaptic currents (mIPSCs) were not changed in global Shank3Δ14–16 mice (Figure 3B).
FIGURE 3.

Altered excitatory and inhibitory spontaneous synaptic transmissions in global Shank3Δ14–16, but not Emx1-Cre;Shank3Δ14–16, mPFC layer 2/3 pyramidal neurons. (A,B) Global Shank3Δ14–16 mice (P21–26) show increased frequency, but normal amplitude, of mEPSCs and normal mIPSCs in layer 2/3 pyramidal neurons in the prelimbic region of the mPFC. Data are shown as mean ± SEM. n = 17 neurons from 5 mice (WT), 17, 7 (KO) for mEPSCs, 15, 5 (WT), 16, 6 (KO) for mIPSCs, ∗∗P < 0.01, ns, not significant, Student’s t-test. (C,D) Emx1-Cre;Shank3Δ14–16 mice (P18–22) show normal mEPSCs and mIPSCs in layer 2/3 pyramidal neurons in the prelimbic region of the mPFC. n = 19, 4 (WT), 16, 4 (cKO) for mEPSCs, 14, 4 (WT), 17, 4 (cKO) for mIPSCs, Student’s t-test (amplitude of mEPSC and frequency of mIPSC), and Mann–Whitney U test (frequency of mEPSC and amplitude of mIPSC). (E,F) Global Shank3Δ14–16 mice (P21–24) show normal sEPSCs and increased frequency, but normal amplitude, of sIPSCs in layer 2/3 pyramidal neurons in the prelimbic region of the mPFC. Data are shown as mean ± SEM. n = 17 neurons from 3 mice (WT), 17, 3 (KO) for sEPSCs, 15, 3 (WT), 18, 3 (KO) for sIPSCs, ∗∗P < 0.01, ns, not significant, Student’s t-test. (G,H) Emx1-Cre;Shank3Δ14–16 mice (P20–25) show normal sEPSCs and sIPSCs in layer 2/3 pyramidal neurons in the prelimbic region of the mPFC. n = 15, 3 (WT), 16, 3 (cKO) for sEPSCs,19, 3 (WT), 19, 3 (cKO) for sIPSCs, ns, not significant, Student’s t-test (frequency and amplitude of sEPSC and frequency of sIPSC), and Mann–Whitney U test (amplitude of sIPSC).
In Emx1-Cre;Shank3Δ14–16 layer 2/3 mPFC neurons, both mEPSCs and mIPSCs were normal (Figures 3C,D). This suggests that excitatory neurons are less likely to contribute to the increased mEPSC frequency observed in global Shank3Δ14–16 mPFC neurons.
We also measured excitatory and inhibitory synaptic transmission in the presence of network activity by excluding tetrodotoxin (a blocker of action potential firing) during slice recordings. The frequency and amplitude of spontaneous EPSCs (sEPSCs) were normal in global Shank3Δ14–16 layer 2/3 pyramidal neurons in the prelimbic region of the mPFC compared with those in WT neurons (Figure 3E).
Notably, the frequency, but not amplitude, of spontaneous IPSCs (sIPSCs) was increased in global Shank3Δ14–16 layer 2/3 pyramidal neurons (Figure 3F). In addition, both sEPSCs and sIPSCs were normal in Emx1-Cre;Shank3Δ14–16 mice (Figures 3G,H). These results collectively suggest that global Shank3 deletion leads to increases in mEPSC frequency and sIPSC frequency, whereas glutamatergic Shank3 deletion has no effects on any forms of spontaneous synaptic transmission in layer 2/3 mPFC pyramidal neurons.
Normal Spontaneous Excitatory and Inhibitory Synaptic Transmission in Emx1-Cre;Shank3Δ14–16 Dorsolateral Striatal Neurons
Dysfunctions in striatal regions, where Shank3 is strongly expressed (Peca et al., 2011), have been associated with abnormal behaviors in Shank3-mutant mice (Monteiro and Feng, 2017). Because our previous results revealed decreased excitatory synaptic transmission in dorsolateral striatal neurons in both global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018), we next measured spontaneous excitatory and inhibitory synaptic transmission in dorsolateral striatal neurons.
However, there were no changes in the frequency or amplitude of mEPSCs in Emx1-Cre;Shank3Δ14–16 dorsolateral striatal neurons compared with WT neurons (Figure 4A). In addition, neither the frequency nor amplitude of mIPSCs was altered in Emx1-Cre;Shank3Δ14–16 dorsolateral striatal neurons (Figure 4B). These results contrast with the strongly decreased mEPSC frequency and amplitude in dorsolateral striatal neurons in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018).
FIGURE 4.

Normal spontaneous excitatory and inhibitory synaptic transmission in Emx1-Cre;Shank3Δ14–16 dorsolateral striatal neurons. (A) Emx1-Cre;Shank3Δ14–16 mice (P29–35) show normal frequency and amplitude of mEPSCs in dorsolateral striatal neurons. Data are shown as mean ± SEM. n = 14 neurons from 3 mice (WT), 17, 3 (cKO), ns, not significant, Student’s t-test. (B) Emx1-Cre;Shank3Δ14–16 mice (P34–41) show normal frequency and amplitude of mIPSCs in dorsolateral striatal neurons. n = 15, 3 (WT), 15, 3 (cKO), ns, not significant, Student’s t-test.
Enhanced Direct Social Interaction, but Normal Social Approach and Social Communication, in Emx1-Cre;Shank3Δ14–16 Mice
Given the well-known association between SHANK3 and various neurodevelopmental disorders, including ASD, PMS and schizophrenia (Phelan and McDermid, 2012; Monteiro and Feng, 2017), we first subjected Emx1-Cre; Shank3Δ14–16 mice to behavioral tests in the social domain.
In the three-chambered social interaction test, designed to measure social approach and social novelty recognition (Crawley, 2004; Moy et al., 2004; Silverman et al., 2010), Emx1-Cre;Shank3Δ14–16 mice showed social approach behaviors that are comparable to those of WT mice, as shown by time spent sniffing social and object targets (Figure 5A). In addition, Emx1-Cre;Shank3Δ14–16 mice displayed normal social novelty recognition, as shown by time spent sniffing familiar and novel stranger mice.
FIGURE 5.

Enhanced direct social interaction and repetitive self-grooming in Emx1-Cre;Shank3Δ14–16 mice. (A–D) Emx1-Cre;Shank3Δ14–16 mice (A, 13–20 weeks; B, 15–21 weeks; C, 16–32 weeks; D, 15–21 weeks) show normal social approach in the three-chamber test (A), enhanced direct social interaction (B), normal social dominance in the tube test (C), and normal courtship USVs (D; P = 0.088). Data are shown as mean ± SEM. n = 23 (WT), 20 (cKO) for three-chamber, 10 (WT), 9 (cKO) for direct social interaction, 11 (WT), 9 (cKO) for tube test, and 20 (WT), 20 (cKO) for USV, ∗∗P < 0.01, ∗∗∗P < 0.001, paired t-test (WT S1-O, WT S1-S2, and cKO S1-S2), Wilcoxon signed rank test (cKO S1-O), and Student’s t-test (for the direct social interaction test, and adult USV test). (E–H) Emx1-Cre;Shank3Δ14–16 mice (E, 12–17 weeks; F, 12–18 weeks; G,H, 10–17 weeks) show enhanced self-grooming and normal digging in home cages with bedding (F), but normal self-grooming in a novel home cage without bedding (E) and in Laboras cages (G,H). Data are shown as means ± SEM; data values in panel H represent those from light-off periods (shaded durations). n = 24 (WT), 21 (cKO) for w/o bedding, 16 (WT), 14 (cKO) for w/bedding, and 13 (WT), 14 (cKO) for Laboras, ∗P < 0.05, repeated measures two-way ANOVA (for Laboras, left panel; genotype p-value = 0.3944), Student’s t-test [for digging time of repetitive behavior and Laboras (climbing, rearing, grooming, and eating)], and Mann–Whitney U test [for self-grooming test, self-grooming time of repetitive behavior, and Laboras (drinking)].
Intriguingly, in experiments using genotype- and age-matched mouse pairs, Emx1-Cre;Shank3Δ14–16 mice showed enhanced social interaction in the direct social interaction test, as shown by time spent in nose-to-nose sniffing and total social interaction (Figure 5B). These results indicate that Emx1-Cre;Shank3Δ14–16 mice display normal social approach and social novelty recognition, but abnormally enhanced direct social interaction, similar to the social behaviors of global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice in these tests (Yoo et al., 2018). These changes do not seem to involve altered social dominance, as supported by the lack of genotype difference in the Tube test (Figure 5C). These results suggest that both glutamatergic and GABAergic neurons contribute to the abnormally enhanced direct social interaction in global Shank3Δ14–16 mice.
We next evaluated USVs, which are strongly associated with rodent behaviors and emotional states, including social communication (Knutson et al., 1998, 2002; Portfors, 2007; Scattoni et al., 2009). Adult male Emx1-Cre;Shank3Δ14–16 mice encountering a novel female mouse emitted normal numbers of USVs compared with WT mice (Figure 5D). Notably, this result differs from the suppressed courtship USVs observed in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018), suggesting that GABAergic, but not glutamatergic, neurons strongly contribute to the USV deficits in global Shank3Δ14–16 mice.
Modestly Enhanced Repetitive Self-Grooming, but Normal Digging, in Emx1-Cre;Shank3Δ14–16 Mice
We next evaluated repetitive behaviors, a core component of ASD-related behavior, in Emx1-Cre;Shank3Δ14–16 mice. Emx1-Cre;Shank3Δ14–16 mice displayed enhanced self-grooming in a new home cage with bedding but normal self-grooming in a new home cage without bedding (Figures 5E,F), suggesting that the presence of bedding is required for repetitive behavior in addition to a new cage or environment. This result shows similarities to the strong self-grooming behaviors in global Shank3Δ14–16 mice observed in all three environments (new home cage with bedding, new home cage without bedding, and Laboras cages), but is more comparable to the mildly enhanced self-grooming in Viaat-Cre;Shank3Δ14–16 mice, observed only in a new home cage with bedding (Yoo et al., 2018). Measurements of digging, another method for quantifying repetitive behavior, showed no changes in Emx1-Cre;Shank3Δ14–16 mice compared with WT mice, even in the presence of bedding. This contrasts with the decreased digging observed in both global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018).
Emx1-Cre;Shank3Δ14–16 mice subjected to the Laboras test, designed to measure mouse behaviors for a long period of time (i.e., four consecutive days) in a light/dark-cycling environment with bedding (Quinn et al., 2003, 2006), showed normal levels of self-grooming (Figures 5G,H). The results of these tests, in which mice were fully habituated, especially on days 2–4, suggest that Emx1-Cre;Shank3Δ14–16 mice show enhanced self-grooming only in a particular environment (i.e., novel home cage with bedding). Other behaviors of Emx1-Cre;Shank3Δ14–16 mice, including climbing, rearing, drinking and eating, were unchanged in Laboras cages (Figure 5H).
Normal Locomotor Activity and Enhanced Anxiolytic-Like Behavior in Emx1-Cre;Shank3Δ14–16 Mice
Because hyperactivity and anxiety are observed in ASD, PMS and schizophrenia, we also evaluated locomotor activities of Emx1-Cre;Shank3Δ14–16 mice. In the open-field test, representing a novel environment, Emx1-Cre;Shank3Δ14–16 mice showed normal levels of locomotor activity, as shown by distance moved during 60 min (Figure 6A). In Laboras cages, representing a familiar environment, Emx1-Cre;Shank3Δ14–16 mice showed normal levels of locomotor activities during the last 72 h (Figure 6B). Locomotion in Laboras cages was also unchanged during the first 6 h, similar to the results of the open-field test. These results suggest that glutamatergic Shank3 deletion does not affect locomotor activity, in contrast to the reported hypoactivity of both global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018).
FIGURE 6.

Normal locomotor activity in open-field and Laboras cages, and enhanced anxiety-like behavior in elevated plus-maze (EPM) in Emx1-Cre;Shank3Δ14–16 mice. (A,B) Emx1-Cre;Shank3Δ14–16 mice (A, 11–17 weeks; B, 10–17 weeks) show normal levels of locomotor activity in the open-field test (A) and in Laboras cages (B). Note that these mice spend a normal amount of time in the center region of the open-field arena, indicative of normal anxiety-like behavior in this test. Data are shown as means ± SEM; data values in the bar graph in panel B represent those from light-off periods (shaded durations). n = 23 (WT), 21 (cKO) for open-field and 13 (WT), 14 (cKO) for Laboras, repeated measures of two-way ANOVA (for the left panels in open-field and Laboras; genotype p-values = 0.6889 and 0.3025, respectively), Student’s t-test (for the right panels in open-field and first 6 h of the right panels in Laboras), and Mann–Whitney U test (for the last 72 h of the right panels in Laboras). (C) Emx1-Cre;Shank3Δ14–16 mice (14–19 weeks) spend an increased amount of time in the open arm of the elevated plus-maze (EPM). n = 23 (WT), 20 (cKO), ∗P < 0.05, ∗∗∗P < 0.001, Student’s t-test (right panels of elevated plus maze test) and paired t-test (for left panels of elevated plus maze test). (D) Emx1-Cre;Shank3Δ14–16 mice (14–19 weeks) spend a normal amount of time in the light chamber of the light-dark (LD) apparatus. n = 23 (WT), 21 (cKO), ns, not significant, Student’s t-test.
In anxiety-related behavioral tests, Emx1-Cre;Shank3Δ14–16 mice spent a normal amount of time in the center region of the open-field arena (Figure 6A), but spent an increased amount of time in the open arm of the elevated plus-maze (EPM) (Figure 6C), and a normal amount of time in the light chamber of the LD apparatus (Figure 6D). The normal open-field center time and increased EPM open-arm time in Emx1-Cre;Shank3Δ14–16 mice are similar to behaviors observed in global Shank3Δ14–16 mice, but differ from the decreased open-field center time and normal EPM open-arm time observed in Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018). In addition, the normal light-chamber time in the LD test in Emx1-Cre;Shank3Δ14–16 mice differs from the reduced light-chamber time (anxiety-like behavior) in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018) (summarized in Table 1). Therefore, the two contrasting anxiety-like behaviors in global Shank3Δ14–16 mice—anxiolytic-like behavior in the EPM and anxiety-like behavior in the LD apparatus—seem to more strongly involve glutamatergic and GABAergic neurons, respectively.
TABLE 1.
Comparison of electrophysiological and behavioral phenotypes of global Shank3Δ14–16, Emx1-Cre;Shank3Δ14–16, and Viaat-Cre;Shank3Δ14–16 mice.
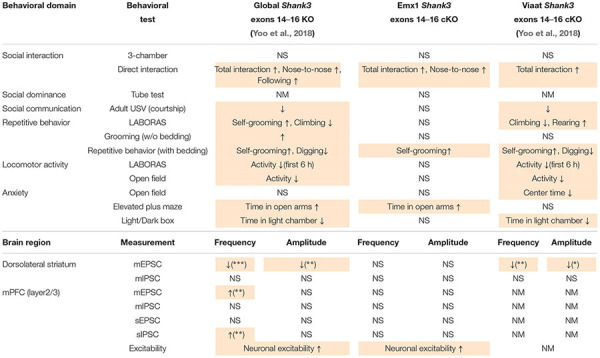 |
Table summarizes increases or decreases in various electrophysiological and behavioral phenotypes in a given mouse line relative to WT/control mice, but is not intended to compare phenotypic severities across different mouse lines. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001; NS, no significant change; NM, not measured; up and down arrows, increases and decreases, respectively. Areas shaded in orange represent similar phenotypes.
Control Emx1-Cre Mice Show Normal Locomotor Activity, Anxiety-Like Behavior, Direct Social Interaction, and Repetitive Behavior
It is conceivable that control mice harboring Emx1-Cre alone might show behavioral abnormalities. To test this, we analyzed the behaviors of Emx1-Cre mice. These mice showed normal behaviors in Laboras cages, including locomotion, climbing, and rearing (Supplementary Figure S1A). In addition, Emx1-Cre mice showed normal levels of locomotor activity in the open-field test and time spent in the center region of the open-field arena (Supplementary Figure S1B). These mice also showed normal levels of time spent in the open arm of the EPM (Supplementary Figure S1C), direct social interaction (Supplementary Figure S1D), and self-grooming and digging in home cages with bedding (Supplementary Figure S1E). These results suggest that control Emx1-Cre mice show normal locomotion, repetitive behavior, and anxiety-related behaviors.
Discussion
In this study, we investigated the impacts of glutamatergic Shank3 (exons 14–16) deletion and compared them with those observed in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice. Our data indicate that the synaptic/neuronal phenotypes of Emx1-Cre;Shank3Δ14–16 mice were similar in part to those observed in global Shank3Δ14–16 mice (summarized in Table 1). Moreover, social and repetitive behavioral deficits were similar between Emx1-Cre;Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice, suggesting that these behaviors involve shared contributions of glutamatergic and GABAergic neurons. However, electrophysiological and behavioral phenotypes of Emx1-Cre;Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice were largely distinct (Table 1).
Electrophysiologically, Emx1-Cre;Shank3Δ14–16 layer 2/3 pyramidal neurons in the prelimbic region of the mPFC showed increased neuronal excitability (Figures 2D–F). The fact that neuronal excitability was similarly increased in global Shank3Δ14–16 layer 2/3 neurons (Figures 2A–C) suggests that glutamatergic neurons strongly contribute to the increased excitability in global Shank3Δ14–16 layer 2/3 neurons. Importantly, a previous study reported that neuronal excitability is increased in human neurons harboring a SHANK3 (exon 13) deletion, in association with increased input resistance and decreased hyperpolarization-activated cation currents (Ih). This latter effect is mediated by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, which directly interact with Shank3 (Yi et al., 2016). In addition, cultured hippocampal neurons from Shank3-deficient mice (exons 13–16) (Peca et al., 2011) display similar increases in neuronal excitability (Yi et al., 2016). These results, together with our demonstration of increased neuronal excitability in global Shank3Δ14–16 and Emx1-Cre;Shank3Δ14–16 mPFC layer 2/3 neurons, collectively suggest that increased neuronal excitability represents a conserved mechanism underlying Shank3 deletion-induced behavioral abnormalities.
In terms of synaptic transmission, neither mEPSCs or mIPSCs in the mPFC were changed in Emx1-Cre;Shank3Δ14–16 layer 2/3 pyramidal neurons (Figures 3C,D). This contrasts with the increased frequency, but not amplitude, of mEPSCs in global Shank3Δ14–16 layer 2/3 pyramidal neurons (Figures 3A,B). Therefore, the increased mEPSC frequency in global Shank3Δ14–16 layer 2/3 pyramidal neurons may not involve changes in glutamatergic neurons, thus implying non-cell-autonomous mechanisms. Indeed, this possibility of non-cell-autonomous mechanisms is in agreement with the known roles of Shank3 as a key component of excitatory postsynaptic compartments (Sheng and Kim, 2000, 2011; Sheng and Sala, 2001; Boeckers et al., 2002; Sheng and Hoogenraad, 2007; Grabrucker et al., 2011; Jiang and Ehlers, 2013; Sala et al., 2015; Monteiro and Feng, 2017; Mossa et al., 2017).
Then how might a Shank3 deletion lead to an increase in mEPSC frequency in global Shank3Δ14–16 layer 2/3 pyramidal neurons? Increased mEPSC frequency may involve increased excitatory synapse number or increased excitatory synaptic transmission through mechanisms, including increased neuronal excitability of presynaptic neurons and increased efficiency of presynaptic release. Therefore, one possibility is that the increased neuronal excitability in global Shank3Δ14–16 mPFC neurons increases the output function of these neurons and activates the intra-cortical network between layer 2/3 neurons, promoting the development of excitatory synapses in target layer 2/3 cortical neurons. Intriguingly, a previous study has shown that Shank3 could be detected in axonal compartments and nerve terminals and negative regulates presynaptic NMDARs (Halbedl et al., 2016), suggesting that the loss of presynaptic Shank3 might contribute to the increased mEPSC frequency. Alternatively, the increased neuronal excitability induced by loss of the interaction between Shank3 and HCN channels (Yi et al., 2016) may promote excitatory synaptic transmission and excitatory synapse development in a cell-autonomous manner; however, this is an unlikely possibility, as noted above.
Our measurements of sEPSCs and sIPSCs provide additional insight into the role of network activity in the context of a Shank3 deletion. Specifically, global Shank3Δ14–16 layer 2/3 pyramidal neurons showed normalized sEPSC frequency and increased sIPSC frequency (Figures 3E,F), findings that contrast with the increased mEPSC frequency and normal mIPSC frequency in the same neurons (Figures 3A,B). These sEPSC/sIPSC phenotypes likely represent compensatory changes that serve to suppress the increased mEPSC frequency as well as the increased neuronal excitability in these neurons and thus normalize the neuronal output. Indeed, the fact that sEPSCs in global Shank3Δ14–16 layer 2/3 pyramidal neurons are normalized suggests that these compensatory changes could actually normalize the neuronal output, at least in the slice preparation, which likely represents baseline conditions. However, the consequence of these compensatory effects seems to be abnormally increased inhibitory synaptic transmission onto pyramidal neurons that still maintain their increased neuronal excitability, as measured in the presence of network activity. Therefore, although neuronal output was apparently normalized in layer 2/3 neurons, the balance between excitatory and inhibitory synaptic transmission, and neuronal activity, might be disrupted. In keeping with this, an imbalance in excitation/inhibition ratio has been implicated in ASD (Rubenstein and Merzenich, 2003; Yizhar et al., 2011; Lee et al., 2015; Nelson and Valakh, 2015; Lee E. et al., 2017; Selimbeyoglu et al., 2017). A disruption in excitation/inhibition balance may also alter network properties such as brain rhythms. Indeed, altered EEG rhythms, including an increase in gamma power, have been observed in Shank3-mutant mice (Han et al., 2013; Wang et al., 2016; Dhamne et al., 2017; Ingiosi et al., 2019; Yoo et al., 2019) as well as in SHANK3-related ASD and PMS (Soorya et al., 2013; Holder and Quach, 2016).
Behaviorally, glutamatergic and GABAergic Shank3 deletions seem to differentially contribute to the abnormal behaviors observed in global Shank3Δ14–16 mice. For instance, increased direct social interaction and normal social approach were observed in all three mouse lines (global, Emx1, and Viaat) (Figures 5A,B). Enhanced self-grooming was also observed in the three mouse lines (Figures 5E–H), although it was stronger in global Shank3Δ14–16 mice. These results suggest that both glutamatergic and GABAergic neurons contribute to the social and repetitive behavioral abnormalities in Emx1-Cre;Shank3Δ14–16 mice. One behavior that deviated from this shared contribution of glutamatergic and GABAergic neurons was courtship USVs, which were normal in Emx1-Cre;Shank3Δ14–16 mice, but suppressed in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Figure 5D). In addition, anxiolytic-like (EPM open-arm time) and anxiety-like (LD light-chamber time) behaviors in global Shank3Δ14–16 mice were observed selectively in Emx1-Cre;Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice, respectively (Figures 6C,D).
Identifying the brain regions and circuit mechanisms underlying this differential recapitulation of behavioral phenotypes of global Shank3Δ14–16 mice in Emx1-Cre;Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice could be a highly speculative undertaking. However, one brain region that is strongly associated with Shank3-dependent social and repetitive behavioral deficits is the striatum (Peca et al., 2011; Filice et al., 2016; Fuccillo, 2016; Jaramillo et al., 2016, 2017; Mei et al., 2016; Peixoto et al., 2016; Wang et al., 2016, 2017; Zhou et al., 2016; Lee Y. et al., 2017; Reim et al., 2017; Vicidomini et al., 2017; Bey et al., 2018; Fourie et al., 2018; Yoo et al., 2018). More recently, the ventral striatum and its upstream regions (ventral tegmental area and dorsal raphe nucleus) have been implicated in the social deficits in Shank3-mutant mice (Bariselli et al., 2016, 2018; Bariselli and Bellone, 2017; Luo et al., 2017). Intriguingly, however, our electrophysiological results indicated that Emx1-Cre;Shank3Δ14–16 mice do not display altered excitatory or inhibitory synaptic transmission in dorsolateral striatal neurons (Figure 4), a finding that strongly contrasts with the reduced excitatory synaptic transmission in dorsolateral striatal neurons in global Shank3Δ14–16 and Viaat-Cre;Shank3Δ14–16 mice (Yoo et al., 2018). Therefore, potential alterations in excitatory synapses in prefrontal cortical neurons in Emx1-Cre;Shank3Δ14–16 mice do not seem to affect excitatory afferents to dorsal striatal neurons. We thus hypothesize that glutamatergic and GABAergic neurons contribute to the social or repetitive behavioral deficits through distinct synapses, circuits and mechanisms, at least in the dorsal striatum.
Notably, a recent study on Shank3Δ4–22 mice carrying deletions in exons 4–22 (not exons 14–16, as in the current study) reported behavioral phenotypes that are surprisingly similar to those observed in our global Shank3Δ14–16 mice (Yoo et al., 2018), including normal social approach, enhanced direct social interaction, suppressed courtship USVs, enhanced self-grooming, open-field hypoactivity, and anxiolytic-like behavior (EPM) (Wang et al., 2016). In addition, a more recent related study investigated the impacts of a Shank3 (exons 4–22) deletion restricted to Nex-positive glutamatergic neurons in the cortex, hippocampus, and amygdala (Nex-Cre;Shank3Δ4–22 mice) (Bey et al., 2018). Intriguingly, Nex-Cre;Shank3Δ4–22 mice recapitulated many behavioral phenotypes of global Shank3Δ4–22 mice, including normal social approach and enhanced self-grooming, similar to the results from global Shank3Δ14–16 and Emx1-Cre;Shank3Δ14–16 mice reported here. In addition, these Nex-Cre;Shank3Δ4–22 mice did not recapitulate the suppressed courtship USV or hypoactivity phenotypes of global Shank3Δ4–22 mice. Again, this is similar to the results from our mice (global and Emx1), which together with our demonstration that Viaat-Cre;Shank3Δ14–16 mice display suppressed courtship USVs suggests (Yoo et al., 2018) that GABAergic neurons may be important for the courtship USV phenotype in Shank3-deficient mice.
However, Nex-Cre;Shank3Δ4–22 mice not only failed to recapitulate the hypoactivity of global Shank3Δ4–22 mice, they actually showed increased locomotor activity in open-field tests (Bey et al., 2018), results in contrast with the normal locomotor activity behavior in our Emx1-Cre;Shank3Δ14–16 mice. Whether these differences involve differentially altered striatal synaptic transmission remains unclear because the previous study on Nex-Cre;Shank3Δ4–22 mice analyzed synaptic transmission only in the hippocampus (Bey et al., 2018). However, these discrepancies could be attributable to differences in the specific exons of Shank3 deleted or specific characteristics of Nex-Cre versus Emx1-Cre mice (Guo et al., 2000; Gorski et al., 2002; Goebbels et al., 2006).
In conclusion, our results suggest that glutamatergic Shank3 (exons 14–16) deletion increases neuronal excitability in layer 2/3 mPFC cortical neurons, but has no effect on synaptic transmission in dorsal striatal neurons. It also induces social and repetitive behavioral deficits, similar to the effects of global and GABAergic Shank3 deletions.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by the Committee of Animal Research at Korea Advanced Institute of Science and Technology (KAIST) (KA2016-30).
Author Contributions
TY, HC, and JL performed the behavioral experiments. TY performed the immunoblot experiments. TY, HP, and HC performed the electrophysiological experiments. TY and EK designed the experiments and wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Institute for Basic Science (IBS-R002-D1 to EK).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00458/full#supplementary-material
Normal locomotor activity, anxiety-like behavior, direct social interaction, and repetitive behavior in control Emx1-Cre mice. (A) Emx1-Cre mice (11–12 weeks) show normal behaviors in Laboras cages. Data are shown as mean ± SEM. n = 19 (WT), 15 (Emx1-Cre), repeated measures two-way ANOVA (for the left panel; genotype p-values = 0.3600), Student’s t-test (distance moved of last 72 h, climbing, rearing, drinking, and eating), Mann–Whitney U test (distance moved of first 6 h and grooming). (B) Emx1-Cre mice (10–12 weeks) show normal locomotor activity and time spent in the center region in the open-field test. n = 19 mice (WT), 16 (Emx1-Cre), repeated measures two-way ANOVA (for the left panel; genotype p-values = 0.9795), Student’s t-test. (C) Emx1-Cre mice (11–12 weeks) show normal anxiety-like behavior in the elevated plus-maze (EPM). n = 18 (WT), 16 (Emx1-Cre), ∗∗∗P < 0.001, paired t-test (for the left panels), and Student’s t-test (for the right panel). (D) Emx1-Cre mice (21–30 weeks) show normal levels of direct social interaction compared with sex- and age-matched conspecific mice. n = 7 (WT), 5 (Emx1-Cre), Mann–Whitney U test. (E) Emx1-Cre mice (11–16 weeks) show normal self-grooming and digging in home cages with bedding. n = 19 (WT), 16 (Emx1-Cre), Student’s t-test (duration of self-grooming), and Mann–Whitney U test (duration of digging).
The order of behavior experiments.
Statistics table.
References
- Amodio D. M., Frith C. D. (2006). Meeting of minds: the medial frontal cortex and social cognition. Nat. Rev. Neurosci. 7 268–277. 10.1038/nrn1884 [DOI] [PubMed] [Google Scholar]
- Barak B., Feng G. (2016). Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci. 19 647–655. 10.1038/nn.4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariselli S., Bellone C. (2017). VTA DA neuron excitatory synapses in Shank3 Deltaex(4-9) mouse line. Synapse 71:e21955. 10.1002/syn.21955 [DOI] [PubMed] [Google Scholar]
- Bariselli S., Contestabile A., Tzanoulinou S., Musardo S., Bellone C. (2018). SHANK3 downregulation in the ventral tegmental area accelerates the extinction of contextual associations induced by juvenile non-familiar conspecific interaction. Front. Mol. Neurosci. 11:360. 10.3389/fnmol.2018.00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariselli S., Tzanoulinou S., Glangetas C., Prevost-Solie C., Pucci L., Viguie J., et al. (2016). SHANK3 controls maturation of social reward circuits in the VTA. Nat. Neurosci. 19 926–934. 10.1038/nn.4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bey A. L., Wang X., Yan H., Kim N., Passman R. L., Yang Y., et al. (2018). Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl. Psychiatry 8:94. 10.1038/s41398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccuto L., Lauri M., Sarasua S. M., Skinner C. D., Buccella D., Dwivedi A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21 310–316. 10.1038/ejhg.2012.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckers T. M., Bockmann J., Kreutz M. R., Gundelfinger E. D. (2002). ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 81 903–910. 10.1046/j.1471-4159.2002.00931.x [DOI] [PubMed] [Google Scholar]
- Bonaglia M. C., Giorda R., Beri S., De Agostini C., Novara F., Fichera M., et al. (2011). Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet. 7:e1002173. 10.1371/journal.pgen.1002173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O., Sakurai T., Papapetrou D., Wang X., Dickstein D. L., Takahashi N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1:15. 10.1186/2040-2392-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carper R. A., Courchesne E. (2005). Localized enlargement of the frontal cortex in early autism. Biol. Psychiatry 57 126–133. 10.1016/j.biopsych.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Chung W., Choi S. Y., Lee E., Park H., Kang J., Park H., et al. (2015). Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat. Neurosci. 18 435–443. 10.1038/nn.3927 [DOI] [PubMed] [Google Scholar]
- Cochoy D. M., Kolevzon A., Kajiwara Y., Schoen M., Pascual-Lucas M., Lurie S., et al. (2015). Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Mol. Autism 6:23. 10.1186/s13229-015-0020-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E., Mouton P. R., Calhoun M. E., Semendeferi K., Ahrens-Barbeau C., Hallet M. J., et al. (2011). Neuron number and size in prefrontal cortex of children with autism. JAMA 306 2001–2010. 10.1001/jama.2011.1638 [DOI] [PubMed] [Google Scholar]
- Crawley J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment. Retard. Dev. Disabil. Res. Rev. 10 248–258. 10.1002/mrdd.20039 [DOI] [PubMed] [Google Scholar]
- Dere E., Winkler D., Ritter C., Ronnenberg A., Poggi G., Patzig J., et al. (2015). Gpm6b deficiency impairs sensorimotor gating and modulates the behavioral response to a 5-HT2A/C receptor agonist. Behav. Brain Res. 277 254–263. 10.1016/j.bbr.2014.04.021 [DOI] [PubMed] [Google Scholar]
- Dhamne S. C., Silverman J. L., Super C. E., Lammers S. H. T., Hameed M. Q., Modi M. E., et al. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 8:26. 10.1186/s13229-017-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martino A., Kelly C., Grzadzinski R., Zuo X. N., Mennes M., Mairena M. A., et al. (2011). Aberrant striatal functional connectivity in children with autism. Biol. Psychiatry 69 847–856. 10.1016/j.biopsych.2010.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapeau E., Riad M., Kajiwara Y., Buxbaum J. D. (2018). Behavioral phenotyping of an improved mouse model of Phelan-McDermid syndrome with a complete deletion of the Shank3 Gene. eNeuro 5 ENEURO.46-18. 10.1523/ENEURO.0046-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney L. J., Zhong P., Wei J., Matas E., Cheng J., Qin L., et al. (2015). Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11 1400–1413. 10.1016/j.celrep.2015.04.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand C. M., Betancur C., Boeckers T. M., Bockmann J., Chaste P., Fauchereau F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39 25–27. 10.1038/ng1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M., Zametkin A. J., Matochik J. A., Pascualvaca D., Cohen R. M. (1997). Low medial prefrontal dopaminergic activity in autistic children. Lancet 350:638 10.1016/s0140-6736(05)63326-0 [DOI] [PubMed] [Google Scholar]
- Filice F., Vorckel K. J., Sungur A. O., Wohr M., Schwaller B. (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourie C., Vyas Y., Lee K., Jung Y., Garner C. C., Montgomery J. M. (2018). Dietary Zinc supplementation prevents autism related behaviors and striatal synaptic dysfunction in Shank3 exon 13-16 mutant mice. Front. Cell. Neurosci. 12:374. 10.3389/fncel.2018.00374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M. V. (2016). Striatal circuits as a common node for autism pathophysiology. Front. Neurosci. 10:27. 10.3389/fnins.2016.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott P. L., Warner T. A., Jays P. R., Salway P., Busby S. J. (2005). Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol. 492 145–177. 10.1002/cne.20738 [DOI] [PubMed] [Google Scholar]
- Gauthier J., Champagne N., Lafreniere R. G., Xiong L., Spiegelman D., Brustein E., et al. (2010). De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 107 7863–7868. 10.1073/pnas.0906232107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S. J., Bird G., Brindley R., Frith C. D., Burgess P. W. (2008). Atypical recruitment of medial prefrontal cortex in autism spectrum disorders: an fMRI study of two executive function tasks. Neuropsychologia 46 2281–2291. 10.1016/j.neuropsychologia.2008.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S., Bormuth I., Bode U., Hermanson O., Schwab M. H., Nave K. A. (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44 611–621. 10.1002/dvg.20256 [DOI] [PubMed] [Google Scholar]
- Gorski J. A., Talley T., Qiu M., Puelles L., Rubenstein J. L., Jones K. R. (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22 6309–6314. 10.1523/jneurosci.22-15-06309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabrucker A. M., Schmeisser M. J., Schoen M., Boeckers T. M. (2011). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21 594–603. 10.1016/j.tcb.2011.07.003 [DOI] [PubMed] [Google Scholar]
- Guilmatre A., Huguet G., Delorme R., Bourgeron T. (2014). The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 74 113–122. 10.1002/dneu.22128 [DOI] [PubMed] [Google Scholar]
- Guo H., Hong S., Jin X. L., Chen R. S., Avasthi P. P., Tu Y. T., et al. (2000). Specificity and efficiency of Cre-mediated recombination in Emx1-Cre knock-in mice. Biochem. Biophys. Res. Commun. 273 661–665. 10.1006/bbrc.2000.2870 [DOI] [PubMed] [Google Scholar]
- Ha S., Lee D., Cho Y. S., Chung C., Yoo Y. E., Kim J., et al. (2016). Cerebellar Shank2 regulates excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors. J. Neurosci. 36 12129–12143. 10.1523/jneurosci.1849-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbedl S., Schoen M., Feiler M. S., Boeckers T. M., Schmeisser M. J. (2016). Shank3 is localized in axons and presynaptic specializations of developing hippocampal neurons and involved in the modulation of NMDA receptor levels at axon terminals. J. Neurochem. 137 26–32. 10.1111/jnc.13523 [DOI] [PubMed] [Google Scholar]
- Hamdan F. F., Gauthier J., Araki Y., Lin D. T., Yoshizawa Y., Higashi K., et al. (2011). Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 88 306–316. 10.1016/j.ajhg.2011.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K., Holder J. L., Jr., Schaaf C. P., Lu H., Chen H., Kang H., et al. (2013). SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503 72–77. 10.1038/nature12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haznedar M. M., Buchsbaum M. S., Hazlett E. A., Licalzi E. M., Cartwright C., Hollander E. (2006). Volumetric analysis and three-dimensional glucose metabolic mapping of the striatum and thalamus in patients with autism spectrum disorders. Am. J. Psychiatry 163 1252–1263. 10.1176/ajp.2006.163.7.1252 [DOI] [PubMed] [Google Scholar]
- Holder J. L., Jr., Quach M. M. (2016). The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss-of-function mutations. Epilepsia 57 1651–1659. 10.1111/epi.13506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover W. B., Vertes R. P. (2007). Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 212 149–179. 10.1007/s00429-007-0150-4 [DOI] [PubMed] [Google Scholar]
- Ingiosi A. M., Schoch H., Wintler T., Singletary K. G., Righelli D., Roser L. G., et al. (2019). Shank3 modulates sleep and expression of circadian transcription factors. eLife 8:e42819. 10.7554/eLife.42819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki M., Tanaka S., Kuriu T., Tabuchi K., Takumi T., Okabe S. (2014). Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun. 5:4742. 10.1038/ncomms5742 [DOI] [PubMed] [Google Scholar]
- Jaramillo T. C., Speed H. E., Xuan Z., Reimers J. M., Escamilla C. O., Weaver T. P., et al. (2017). Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res. 10 42–65. 10.1002/aur.1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo T. C., Speed H. E., Xuan Z., Reimers J. M., Liu S., Powell C. M. (2016). Altered striatal synaptic function and abnormal behaviour in Shank3 Exon4-9 deletion mouse model of autism. Autism Res. 9 350–375. 10.1002/aur.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. H., Ehlers M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78 8–27. 10.1016/j.neuron.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson B., Burgdorf J., Panksepp J. (1998). Anticipation of play elicits high-frequency ultrasonic vocalizations in young rats. J. Comp. Psychol. 112 65–73. 10.1037//0735-7036.112.1.65 [DOI] [PubMed] [Google Scholar]
- Knutson B., Burgdorf J., Panksepp J. (2002). Ultrasonic vocalizations as indices of affective states in rats. Psychol. Bull. 128 961–977. 10.1037//0033-2909.128.6.961 [DOI] [PubMed] [Google Scholar]
- Ko J. (2017). Neuroanatomical substrates of rodent social behavior: the medial prefrontal cortex and its projection patterns. Front. Neural Circuits 11:41. 10.3389/fncir.2017.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohls G., Yerys B. E., Schultz R. T. (2014). Striatal development in autism: repetitive behaviors and the reward circuitry. Biol. Psychiatry 76 358–359. 10.1016/j.biopsych.2014.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouser M., Speed H. E., Dewey C. M., Reimers J. M., Widman A. J., Gupta N., et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33 18448–18468. 10.1523/JNEUROSCI.3017-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen M., Bos D., Noordermeer S. D., Nederveen H., Van Engeland H., Durston S. (2013). Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76 405–411. 10.1016/j.biopsych.2013.08.013 [DOI] [PubMed] [Google Scholar]
- Leblond C. S., Nava C., Polge A., Gauthier J., Huguet G., Lumbroso S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. 10.1371/journal.pgen.1004580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. T., Gee S. M., Vogt D., Patel T., Rubenstein J. L., Sohal V. S. (2014). Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron 81 61–68. 10.1016/j.neuron.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E., Lee J., Kim E. (2017). Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81 838–847. 10.1016/j.biopsych.2016.05.011 [DOI] [PubMed] [Google Scholar]
- Lee Y., Kim S. G., Lee B., Zhang Y., Kim Y., Kim S., et al. (2017). Striatal transcriptome and interactome analysis of Shank3-overexpressing mice reveals the connectivity between Shank3 and mTORC1 signaling. Front. Mol. Neurosci. 10:201. 10.3389/fnmol.2017.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Chung C., Ha S., Lee D., Kim D. Y., Kim H., et al. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell Neurosci. 9:94. 10.3389/fncel.2015.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Nakajima M., Ibanez-Tallon I., Heintz N. (2016). A cortical circuit for sexually dimorphic Oxytocin-dependent anxiety behaviors. Cell 167 60–72.e11. 10.1016/j.cell.2016.08.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Xu W., Hsu Y. T., Yee A. X., Chen L., Sudhof T. C. (2015). Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol. Psychiatry 20 850–859. 10.1038/mp.2015.31 [DOI] [PubMed] [Google Scholar]
- Lim S., Naisbitt S., Yoon J., Hwang J. I., Suh P. G., Sheng M., et al. (1999). Characterization of the shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274 29510–29518. 10.1074/jbc.274.41.29510 [DOI] [PubMed] [Google Scholar]
- Lim S., Sala C., Yoon J., Park S., Kuroda S., Sheng M., et al. (2001). Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Mol. Cell. Neurosci. 17 385–397. 10.1006/mcne.2000.0940 [DOI] [PubMed] [Google Scholar]
- Luo J., Feng Q., Wei L., Luo M. (2017). Optogenetic activation of dorsal raphe neurons rescues the autistic-like social deficits in Shank3 knockout mice. Cell Res. 27 950–953. 10.1038/cr.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea A. K., Nagarajan R. P., Bilenky M., Ballinger T. J., D’souza C., Fouse S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466 253–257. 10.1038/nature09165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y., Monteiro P., Zhou Y., Kim J. A., Gao X., Fu Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530 481–484. 10.1038/nature16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P., Feng G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18 147–157. 10.1038/nrn.2016.183 [DOI] [PubMed] [Google Scholar]
- Mossa A., Giona F., Pagano J., Sala C., Verpelli C. (2017). SHANK genes in autism: defining therapeutic targets. Prog. Neuropsychopharmacol. Biol. Psychiatry 84(Pt B), 416–423. 10.1016/j.pnpbp.2017.11.019 [DOI] [PubMed] [Google Scholar]
- Moy S. S., Nadler J. J., Perez A., Barbaro R. P., Johns J. M., Magnuson T. R., et al. (2004). Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 3 287–302. 10.1111/j.1601-1848.2004.00076.x [DOI] [PubMed] [Google Scholar]
- Moy S. S., Nadler J. J., Young N. B., Nonneman R. J., Grossman A. W., Murphy D. L., et al. (2009). Social approach in genetically engineered mouse lines relevant to autism. Genes Brain Behav. 8 129–142. 10.1111/j.1601-183X.2008.00452.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy P. (2003). Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J. Child Psychol. Psychiatry 44 793–809. [DOI] [PubMed] [Google Scholar]
- Nadler J. J., Moy S. S., Dold G., Trang D., Simmons N., Perez A., et al. (2004). Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 3 303–314. 10.1111/j.1601-183x.2004.00071.x [DOI] [PubMed] [Google Scholar]
- Naisbitt S., Kim E., Tu J. C., Xiao B., Sala C., Valtschanoff J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23 569–582. 10.1016/s0896-6273(00)80809-0 [DOI] [PubMed] [Google Scholar]
- Nelson S. B., Valakh V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87 684–698. 10.1016/j.neuron.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page S. C., Hamersky G. R., Gallo R. A., Rannals M. D., Calcaterra N. E., Campbell M. N., et al. (2018). The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Mol. Psychiatry 23 304–315. 10.1038/mp.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak N. N., Luo R., Zhang A., Won H., Lowe J. K., Chandran V., et al. (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155 1008–1021. 10.1016/j.cell.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peca J., Feliciano C., Ting J. T., Wang W., Wells M. F., Venkatraman T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472 437–442. 10.1038/nature09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto R. T., Wang W., Croney D. M., Kozorovitskiy Y., Sabatini B. L. (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(-/-) mice. Nat. Neurosci. 19 716–724. 10.1038/nn.4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K., McDermid H. E. (2012). The 22q13.3 deletion syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce K., Haist F., Sedaghat F., Courchesne E. (2004). The brain response to personally familiar faces in autism: findings of fusiform activity and beyond. Brain 127 2703–2716. 10.1093/brain/awh289 [DOI] [PubMed] [Google Scholar]
- Portfors C. V. (2007). Types and functions of ultrasonic vocalizations in laboratory rats and mice. J. Am. Assoc. Lab. Anim. Sci. 46 28–34. [PubMed] [Google Scholar]
- Qin L., Ma K., Yan Z. (2019). Chemogenetic activation of prefrontal cortex in Shank3-deficient mice ameliorates social deficits, NMDAR hypofunction, and Sgk2 downregulation. iScience 17 24–35. 10.1016/j.isci.2019.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Ma K., Wang Z. J., Hu Z., Matas E., Wei J., et al. (2018). Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21 564–575. 10.1038/s41593-018-0110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn L. P., Stean T. O., Chapman H., Brown M., Vidgeon-Hart M., Upton N., et al. (2006). Further validation of LABORAS using various dopaminergic manipulations in mice including MPTP-induced nigro-striatal degeneration. J. Neurosci. Methods 156 218–227. 10.1016/j.jneumeth.2006.03.013 [DOI] [PubMed] [Google Scholar]
- Quinn L. P., Stean T. O., Trail B., Duxon M. S., Stratton S. C., Billinton A., et al. (2003). LABORAS: initial pharmacological validation of a system allowing continuous monitoring of laboratory rodent behaviour. J. Neurosci. Methods 130 83–92. 10.1016/s0165-0270(03)00227-9 [DOI] [PubMed] [Google Scholar]
- Rapanelli M., Frick L. R., Xu M., Groman S. M., Jindachomthong K., Tamamaki N., et al. (2017). Targeted interneuron depletion in the dorsal striatum produces autism-like behavioral abnormalities in male but not female mice. Biol. Psychiatry 82 194–203. 10.1016/j.biopsych.2017.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim D., Distler U., Halbedl S., Verpelli C., Sala C., Bockmann J., et al. (2017). Proteomic analysis of post-synaptic density fractions from shank3 mutant mice reveals brain region specific changes relevant to autism spectrum disorder. Front. Mol. Neurosci. 10:26. 10.3389/fnmol.2017.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi T., Perrodin C., Markram H. (2008). Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic Acid animal model of autism. Front. Neural Circuits 2:4. 10.3389/neuro.04.004.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell P. E., Fuccillo M. V., Maxeiner S., Hayton S. J., Gokce O., Lim B. K., et al. (2014). Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158 198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein J. L., Merzenich M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2 255–267. 10.1034/j.1601-183x.2003.00037.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C., Vicidomini C., Bigi I., Mossa A., Verpelli C. (2015). Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 135 849–858. 10.1111/jnc.13232 [DOI] [PubMed] [Google Scholar]
- Scattoni M. L., Crawley J., Ricceri L. (2009). Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 33 508–515. 10.1016/j.neubiorev.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser M. J., Ey E., Wegener S., Bockmann J., Stempel A. V., Kuebler A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486 256–260. 10.1038/nature11015 [DOI] [PubMed] [Google Scholar]
- Schuetze M., Park M. T., Cho I. Y., Macmaster F. P., Chakravarty M. M., Bray S. L. (2016). Morphological alterations in the thalamus, striatum, and pallidum in autism spectrum disorder. Neuropsychopharmacology 41 2627–2637. 10.1038/npp.2016.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A., Kim C. K., Inoue M., Lee S. Y., Hong A. S. O., Kauvar I., et al. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med. 9:eaah6733. 10.1126/scitranslmed.aah6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalom D. B. (2009). The medial prefrontal cortex and integration in autism. Neuroscientist 15 589–598. 10.1177/1073858409336371 [DOI] [PubMed] [Google Scholar]
- Sheng M., Hoogenraad C. C. (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76 823–847. 10.1146/annurev.biochem.76.060805.160029 [DOI] [PubMed] [Google Scholar]
- Sheng M., Kim E. (2000). The Shank family of scaffold proteins. J. Cell Sci. 113(Pt 11), 1851–1856. [DOI] [PubMed] [Google Scholar]
- Sheng M., Kim E. (2011). The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3:a005678. 10.1101/cshperspect.a005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M., Sala C. (2001). PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24 1–29. 10.1146/annurev.neuro.24.1.1 [DOI] [PubMed] [Google Scholar]
- Shrestha P., Mousa A., Heintz N. (2015). Layer 2/3 pyramidal cells in the medial prefrontal cortex moderate stress induced depressive behaviors. eLife 4:e08752. 10.7554/eLife.08752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J. L., Yang M., Lord C., Crawley J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11 490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soorya L., Kolevzon A., Zweifach J., Lim T., Dobry Y., Schwartz L., et al. (2013). Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 4:18. 10.1186/2040-2392-4-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed H. E., Kouser M., Xuan Z., Reimers J. M., Ochoa C. F., Gupta N., et al. (2015). Autism-associated insertion mutation (InsG) of Shank3 Exon 21 causes impaired synaptic transmission and behavioral deficits. J. Neurosci. 35 9648–9665. 10.1523/JNEUROSCI.3125-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa-Silva G., Loebel A., Giugliano M., De Kock C. P., Mansvelder H. D., Meredith R. M. (2012). Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb. Cortex 22 1333–1342. 10.1093/cercor/bhr224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Weerd H. A., Bulthuis R. J., Bergman A. F., Schlingmann F., Tolboom J., Van Loo P. L., et al. (2001). Validation of a new system for the automatic registration of behaviour in mice and rats. Behav. Process. 53 11–20. 10.1016/s0376-6357(00)00135-2 [DOI] [PubMed] [Google Scholar]
- Vicidomini C., Ponzoni L., Lim D., Schmeisser M. J., Reim D., Morello N., et al. (2017). Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 22 689–702. 10.1038/mp.2016.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen M. A., Lacoh C. M., Fiumelli H., Kosel M., Tyagarajan S., De Roo M., et al. (2018). Development of inhibitory synaptic inputs on layer 2/3 pyramidal neurons in the rat medial prefrontal cortex. Brain Struct. Funct. 223 1999–2012. 10.1007/s00429-017-1602-0 [DOI] [PubMed] [Google Scholar]
- Waga C., Asano H., Sanagi T., Suzuki E., Nakamura Y., Tsuchiya A., et al. (2014). Identification of two novel Shank3 transcripts in the developing mouse neocortex. J. Neurochem. 128 280–293. 10.1111/jnc.12505 [DOI] [PubMed] [Google Scholar]
- Wang F., Zhu J., Zhu H., Zhang Q., Lin Z., Hu H. (2011). Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science 334 693–697. 10.1126/science.1209951 [DOI] [PubMed] [Google Scholar]
- Wang X., Mccoy P. A., Rodriguiz R. M., Pan Y., Je H. S., Roberts A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20 3093–3108. 10.1093/hmg/ddr212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Adamski C. J., Bondar V. V., Craigen E., Collette J. R., Pang K., et al. (2019a). A kinome-wide RNAi screen identifies ERK2 as a druggable regulator of Shank3 stability. Mol. Psychiatry. 10.1038/s41380-018-0325-9 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Pang K., Han K., Adamski C. J., Wang W., He L., et al. (2019b). An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry. 10.1038/s41380-018-0324-x [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ma S., Mao M., Li C., Shen X., Xu S., et al. (2019c). RNA-sequencing and bioinformatics analysis of long noncoding RNAs and mRNAs in the prefrontal cortex of mice following repeated social defeat stress. Biomed Res. Int. 2019:7505260. 10.1155/2019/7505260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. J., Zhong P., Ma K., Seo J. S., Yang F., Hu Z., et al. (2019d). Amelioration of autism-like social deficits by targeting histone methyltransferases EHMT1/2 in Shank3-deficient mice. Mol. Psychiatry. 10.1038/s41380-019-0351-2 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Li C., Chen Q., Van Der Goes M. S., Hawrot J., Yao A. Y., et al. (2017). Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127 1978–1990. 10.1172/JCI87997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Bey A. L., Chung L., Krystal A. D., Jiang Y. H. (2014a). Therapeutic approaches for shankopathies. Dev. Neurobiol. 74 123–135. 10.1002/dneu.22084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Xu Q., Bey A. L., Lee Y., Jiang Y. H. (2014b). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5:30. 10.1186/2040-2392-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Bey A. L., Katz B. M., Badea A., Kim N., David L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. 10.1038/ncomms11459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson H. L., Wong A. C., Shaw S. R., Tse W. Y., Stapleton G. A., Phelan M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40 575–584. 10.1136/jmg.40.8.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H., Lee H. R., Gee H. Y., Mah W., Kim J. I., Lee J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486 261–265. 10.1038/nature11208 [DOI] [PubMed] [Google Scholar]
- Xu W., Sudhof T. C. (2013). A neural circuit for memory specificity and generalization. Science 339 1290–1295. 10.1126/science.1229534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Bozdagi O., Scattoni M. L., Wohr M., Roullet F. I., Katz A. M., et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 32 6525–6541. 10.1523/JNEUROSCI.6107-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F., Danko T., Botelho S. C., Patzke C., Pak C., Wernig M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. 10.1126/science.aaf2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O., Fenno L. E., Prigge M., Schneider F., Davidson T. J., O’shea D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477 171–178. 10.1038/nature10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo T., Cho H., Lee J., Park H., Yoo Y. E., Yang E., et al. (2018). GABA neuronal deletion of Shank3 Exons 14-16 in mice suppresses striatal excitatory synaptic input and induces social and locomotor abnormalities. Front. Cell. Neurosci. 12:341. 10.3389/fncel.2018.00341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo Y. E., Yoo T., Lee S., Lee J., Kim D., Han H. M., et al. (2019). Shank3 mice carrying the human Q321R mutation display enhanced self-grooming, abnormal electroencephalogram patterns, and suppressed neuronal excitability and seizure susceptibility. Front. Mol. Neurosci. 12:155. 10.3389/fnmol.2019.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Kaiser T., Monteiro P., Zhang X., Van Der Goes M. S., Wang D., et al. (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89 147–162. 10.1016/j.neuron.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Normal locomotor activity, anxiety-like behavior, direct social interaction, and repetitive behavior in control Emx1-Cre mice. (A) Emx1-Cre mice (11–12 weeks) show normal behaviors in Laboras cages. Data are shown as mean ± SEM. n = 19 (WT), 15 (Emx1-Cre), repeated measures two-way ANOVA (for the left panel; genotype p-values = 0.3600), Student’s t-test (distance moved of last 72 h, climbing, rearing, drinking, and eating), Mann–Whitney U test (distance moved of first 6 h and grooming). (B) Emx1-Cre mice (10–12 weeks) show normal locomotor activity and time spent in the center region in the open-field test. n = 19 mice (WT), 16 (Emx1-Cre), repeated measures two-way ANOVA (for the left panel; genotype p-values = 0.9795), Student’s t-test. (C) Emx1-Cre mice (11–12 weeks) show normal anxiety-like behavior in the elevated plus-maze (EPM). n = 18 (WT), 16 (Emx1-Cre), ∗∗∗P < 0.001, paired t-test (for the left panels), and Student’s t-test (for the right panel). (D) Emx1-Cre mice (21–30 weeks) show normal levels of direct social interaction compared with sex- and age-matched conspecific mice. n = 7 (WT), 5 (Emx1-Cre), Mann–Whitney U test. (E) Emx1-Cre mice (11–16 weeks) show normal self-grooming and digging in home cages with bedding. n = 19 (WT), 16 (Emx1-Cre), Student’s t-test (duration of self-grooming), and Mann–Whitney U test (duration of digging).
The order of behavior experiments.
Statistics table.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.