

Abstract
Background
The dismal survival of glioblastoma (GBM) patients urgently calls for the development of new treatments. Chimeric antigen receptor T (CAR-T) cells are an attractive strategy, but preclinical and clinical studies in GBM have shown that heterogeneous expression of the antigens targeted so far causes tumor escape, highlighting the need for the identification of new targets. We explored if B7-H3 is a valuable target for CAR-T cells in GBM.
Methods
We compared mRNA expression of antigens in GBM using TCGA data, and validated B7-H3 expression by immunohistochemistry. We then tested the antitumor activity of B7-H3-redirected CAR-T cells against GBM cell lines and patient-derived GBM neurospheres in vitro and in xenograft murine models.
Findings
B7-H3 mRNA and protein are overexpressed in GBM relative to normal brain in all GBM subtypes. Of the 46 specimens analyzed by immunohistochemistry, 76% showed high B7-H3 expression, 22% had detectable, but low B7-H3 expression and 2% were negative, as was normal brain. All 20 patient-derived neurospheres showed ubiquitous B7-H3 expression. B7-H3-redirected CAR-T cells effectively targeted GBM cell lines and neurospheres in vitro and in vivo. No significant differences were found between CD28 and 4-1BB co-stimulation, although CD28-co-stimulated CAR-T cells released more inflammatory cytokines.
Interpretation
We demonstrated that B7-H3 is highly expressed in GBM specimens and neurospheres that contain putative cancer stem cells, and that B7-H3-redirected CAR-T cells can effectively control tumor growth. Therefore, B7-H3 represents a promising target in GBM.
Fund
Alex's Lemonade Stand Foundation; Il Fondo di Gio Onlus; National Cancer Institute; Burroughs Wellcome Fund.
Keywords: Glioblastoma, B7-H3, Chimeric antigen receptor, Immunotherapy, Cancer stem cells
Research in context.
Evidence before this study
Glioblastoma (GBM) is the most common primary adult brain tumor and is characterized by poor outcome. Advancements in cell-based immunotherapies opened the way for targeting GBM with chimeric antigen receptor T (CAR-T) cells. However, the molecular heterogeneity of GBM, highlights the need for the identification of novel targets. Moreover, cancer stem cells have been shown to play a central role in tumor progression, treatment failure, and recurrence due to their proliferative capacity and resistance to therapy, and thus targeting cancer stem cells in GBM is important to eradicate the disease.
Added value of this study
We demonstrated in this report that B7-H3 is an attractive target for CAR-T cells in GBM because it is highly expressed in 76% of GBM specimens. Furthermore, B7-H3 is ubiquitously expressed in patient-derived GBM neurospheres that are enriched in tumor stem cells. B7-H3-specific CAR-T cells controlled tumor growth in multiple in vitro and in vivo models, highlighting the efficacy of the proposed approach.
Implications of all available evidence
With the ability to deliver CAR-T cells intracranially, our approach can potentially reduce tumor burden since B7-H3 is highly expressed both within and across GBM tumors, prevent recurrence due to high B7-H3 expression on cancer stem cells, and thus may extend the survival of patients with GBM.
Alt-text: Unlabelled Box
1. Introduction
Glioblastoma (GBM) is an aggressive, malignant brain tumor with abysmal survivorship [1]. Treatment typically consists of surgical resection followed by radiation therapy. The addition of temozolomide increased the median survival (from 12·1 to 14·6 months) and 2-year survival rate (from 10·4% to 26·5%) [2]. Observations of extensive vascular proliferation in GBM led to the use of the VEGF-A inhibiting monoclonal antibody (bevacizumab) that also improved the progression free survival and quality of life of the patients [3].
The systematic molecular assessment of GBM indicates that receptor tyrosine kinase (RTK) genes and the phosphatidylinositol-3-OH kinase (PI3K), p53 and Rb pathways are dysregulated [4]. The identification of these genetic events led to the development of various targeted therapies, such as EGFR-targeting drugs (afatinib, erlotinib, antibody-drug conjugates), and PI3K inhibitors (buparlisib). However, GBM is characterized by great molecular heterogeneity, and different areas within a single tumor can fall under different classification [5], which partially explains the modest improvement of clinical outcome with targeted therapies [6].
Chimeric antigen receptor (CAR) T cells are T lymphocytes genetically modified to express a synthetic receptor that produces activation of the T cell machinery and co-stimulatory pathways upon ligation with a cell surface antigen expressed by tumor cells [7]. CD19-targeting CAR-T cells are FDA-approved for the treatment of refractory/relapsed B-cell malignancies [8,9]. The activity of CAR-T cells in hematologic malignancies stimulated the development of similar strategies in solid tumors including GBM. CAR-T cells targeting EGFRvIII, HER2, and IL-13Rα2 have shown a favorable safety profile and some clinical benefits in patients with GBM [[10], [11], [12]]. However, tumors recur with evidence of immune escape due, at least in part, to antigen loss [[10], [11], [12]]. New promising antigens characterized by high expression in GBM, such as EphA2 and CSPG4, have been explored in preclinical studies [13,14], but tumor heterogeneity remains a concern highlighting the need for the continuous identification of new targets. Here we report that B7-H3, a member of the B7-family, is highly expressed in over 70% of GBM specimens [15,16], and invariably expressed by patient-derived GBM neurospheres (GBM-NS), while it is not detectable in the normal brain. The expression of B7-H3 in GBM-NS is particularly relevant since these cells not only recapitulate the molecular properties of the primary GBM when expanded in vitro or engrafted in immunodeficient mice [17,18], but are also considered to be enriched in putative cancer stem cells (CSCs) [19]. B7-H3-specific CAR-T cells showed antitumor activity both in vitro and in xenograft murine models with either GBM cell lines or GBM-NS, indicating that targeting B7-H3 allows the elimination of both differentiated tumor cells and CSCs.
2. Materials and methods
2.1. Analysis of the cancer genome atlas (TCGA) database
The PanCan mRNA normalized data (http://api.gdc.cancer.gov/data/3586c0da-64d0-4b74-a449-5ff4d9136611) was downloaded, filtered for primary tumors and log2 transformed. The gene expression for CD276 was then plotted by tumor type. GBM samples (primary tumors, recurrent tumors and normal tissue) were also extracted from the PanCan dataset and CD276, CSPG4, EPHA2, ERBB2, and IL13RA2 were plotted by sample type. All analysis was performed in R.
2.2. GBM specimen, GBM-NS, tissue microarrays (TMAs), and cell lines
Patient GBM specimens were obtained from the Department of Neurosurgery (Istituto Neurologico Carlo Besta, Milan Italy) according to a protocol approved by the local institutional review board and upon patients' informed consent. GBM diagnosis was determined according to the WHO Classification [20]. GBM-NS were generated as previously described [21]. GBM and normal brain formalin-fixed paraffin-embedded (FFPE) TMAs were obtained from US BioMax (TMA #: GL801e, BNC17011b, GLN241, RRID: SCR_004295). U-87 MG cells (RRID: CVCL_0022) were obtained from ATCC, U-138 MG cells (RRID: CVCL_0020) from DSMZ, and HL-60 cells (RRID: CVCL_0002) from UNC's Tissue Culture Facility cell line repository which purchases its lines from ATCC. U-87 MG and U-138 MG cells were grown in Dulbecco Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) while HL-60 cells were grown in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products), 1% penicillin/streptomycin (Thermo Fisher Scientific), and 1% GlutaMAX™ (Thermo Fisher Scientific). Cell lines were phenotyped for B7-H3 expression using the 376.96 monoclonal antibody (mAb) followed by APC-goat anti-mouse (BD Biosciences, RRID: AB_398465), or using the B7-H3-BV421 mAb (Clone 7–517; BD Bioscience, RRID: AB_2739369) [22]. SFG plasmid harboring green fluorescent protein and firefly luciferase genes made in-house (SFG.GFP-FFLuc) was used to generate dual GFP- and FFLuc-expressing cell lines. GBM cell lines and GBM-NS were routinely tested to confirm the absence of mycoplasma (Lonza) and for the expression of the targeted antigen by flow cytometry.
2.3. Immunohistochemistry (IHC)
Carnoy-fixed GBM specimens, and paraffin-embedded xenograft gliomas were processed as previously described [14]. The staining was performed using the anti-B7-H3 mAb (1:200 dilution, AF1027, R&D systems, RRID: AB_354546). Staining was detected using an anti-goat biotin secondary mAb for two hour at room temperature, then a streptavidin-HRP for one hour and the chromogen DAB/substrate reagent for the GBM specimens and VECTASTAIN® ABC HRP Kit for xenograft gliomas. Slides were counterstained with hematoxylin (Sigma Aldrich, Inc), dehydrated and mounted. A semiquantitative analysis of B7-H3 expression was performed on 46 human GBM specimens by an experienced pathologist (BP). The expression levels were scored as <5% positive cells or 5–20% positive cells (− and +/−, respectively; low expression); as 20–50% positive cells and > 50% positive cells (+ and ++, respectively; high expression). GBM and normal brain TMAs were stained using the anti-B7-H3 mAb (AF1027, R&D Systems, RRID: AB_354546) by the Translational Pathology Lab core facility and analyzed using the Aperio software. Total IHC score for each core in the TMAs was calculated using Aperio Color Deconvolution Algorithm v9 as follows: Score = (% area with IHC score 0) x 0 + (% area with IHC score 1+) x 1 + (% area with IHC score 2+) x 2 + (% area with IHC score 3+) x 3. The score of each core was normalized to a positive control to compare stains across slides.
2.4. Coculture experiments
The B7-H3.CAR cassettes were generated using the single chain variable fragment (scFv) from the anti-B7-H3 376.96 mAb [22]. The control CD19.CAR was previously described [23]. CAR-T cells were generated from healthy donor peripheral blood T cells (obtained from buffy coats, Gulf Coast Regional Blood Center) as previously described [24]. B7-H3.CAR expression was assessed by incubating the CAR-T cells with B7-H3-GFP [22]. CD19.CAR expression was assessed by incubation with anti-CD19.CAR mAb [23]. To test the antitumor activity in vitro of CAR-T cells, tumor cells were seeded at 1 × 105 cells/well in tissue culture-treated plates (Corning) at either 1:5 or 1:10 effector-to-target (E:T) ratio with the CAR-T cells. CAR-T cells were plated in cytokine-free media for 24 h before being added to the tumor cells. In each experiment, the percentage of CAR-T cells in each group was normalized to the group with the lowest CAR expression using control T cells. To measure cytokine release, the supernatant was collected 24 h after CAR-T cell seeding. Cells were harvested non-enzymatically, using 480 μM ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific) in phosphate buffered saline (PBS) (Thermo Fisher Scientific), and stained with an anti-CD3 mAb (clone SK7; BD Biosciences) to detect T cells, and with mAbs specific for tumor markers to detect tumor cells, and separately for viability using ZombieAqua viability dye (BioLegend). CAR-T cells and tumor cells were quantified using CountBright™ Absolute Counting Beads (Thermo Fisher Scientific). To identify the tumor cells in coculture, we used an anti-AN2 mAb (Clone 1E6.4; Miltenyi Biotec, RRID: AB_2651232) for U-87 MG and U-138 MG cells, and an anti-CD33 mAb (Clone WM53; BD Biosciences, RRID: AB_2713912) for HL-60 due to their uniform expression in their respective tumor cell lines (Supplementary Fig. S1). In coculture experiments GBM tumor cell lines were identified using the anti-AN2 mAb to avoid the potential masking of the B7-H3 antigen by the interaction with the B7-H3.CAR that may cause inaccurate enumeration of residual tumor cells. For coculture experiments with GBM-NS, cells were plated in tissue culture-treated 24-well plates (Corning) at 1:5 E:T ratio with the CAR-T cells in serum-free DMEM/F12 (Thermo Fisher Scientific) with B27™ supplement (Thermo Fisher Scientific) [14]. The supernatant was collected 24 h after CAR-T cell seeding to measure cytokine release. Cells were harvested after 1, 2, 3, or 5 days and stained with the anti-CD3 (Clone REA613; Miltenyi Biotec, RRID: AB_2657063) and anti-B7-H3 (Clone FM276; Miltenyi Biotec, RRID: AB_2661015) mAbs and analyzed by flow cytometry. Interferon γ (IFNγ) and interleukin-2 (IL-2) levels in 24-h coculture supernatant were measured in duplicates using specific enzyme-linked immunosorbent assay (ELISA) kits (R&D System) following the manufacturer's instructions. GBM-NS coculture experiments were analyzed using MACSQuant® Analyzer (Miltenyi Biotec) and FlowLogic V7.1 Software. All remaining flow cytometry experiments were acquired using FACS Canto II (BD Bioscience), and analyzed using the FlowJo software (Version 10.0). Carboxyfluorescein succinimidyl ester (CFSE) staining was performed as previously described [14].
2.5. Xenograft brain slice co-culture model
Hemicoronal brain slices (250 μm thick) from postnatal day 8 CD Sprague Dawley rats (Charles River) were prepared using a vibratome (Leica) and placed in 6-well plates with filtered transwell inserts (Corning). The slices were maintained with 1 mL culture medium (Neurobasal-A medium supplemented with 10% heat-inactivated porcine serum, 5% heat-inactivated rat serum, 10 mM KCl, 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 100 U/mL Penicillin/Streptomycin, 1 mM Sodium Pyruvate, 1 mM l-Glutamine, and 1 μM MK801) under the filter, incubating at 37 °C and 5% CO2. HF2303 tumor neurospheres were obtained from Dr. Ana deCarvalho and the Hermelin Brain Tumor Bank at Henry Ford Health System, Detroit MI, USA. The neurospheres were infected using LV-GFP (RRID: Addgene_25999) virus at 10 multiplicity of infection (MOI) with 5 μg/mL polybrene overnight and were maintained in NM medium (DMEM/F12 supplemented with, 500 μg/mL bovine serum albusmin (BSA), 12·5 μg/mL gentamycin, antibiotic-antimycotic, N-2 supplement). U87-GFP-FFLuc cells were counted using Nexelcom Cellometer Auto 2000 and prepared at 10,000,000 cells/mL. HF2303-GFP cells counted using the hemacytometer were prepared at 200,000 cells/mL. Each brain slice was plated with 5 μL of respective cell suspension (50,000 U87-GFP-FFLuc cells or 10,000 HF2303-GFP cells). One hundrend μL of media from each well was collected at 24 h after plating for cytokine analysis.
2.6. Xenograft mouse models
The antitumor activity of CAR-T cells was tested in nu/nu mice (Animal Studies Core Facility, UNC) according to an Institutional Animal Care and Use Committee (IACUC) approved protocol. Female nu/nu mice ages 6–10 weeks were injected with 1 × 105 U87-GFP-FFLuc cells in 3 μL PBS intracranially at the following coordinates relative to the bregma, corresponding to the caudate nucleus: 0·7 mm posteriorly, 3 mm laterally to the right, and 3·5 mm deep. A week later, tumor burden was assessed using bioluminescence (BLI) measured by the IVIS Kinetic (Caliper LifeSciences), and 2 × 106 CAR-T cells in 5 μL PBS were injected intratumorally. Tumor BLI was quantified weekly thereafter. Mice were monitored every 2–3 days, and culled upon reaching humane end points in accordance with institutional guidelines. Overall survival was assessed using Kaplan-Meier analysis. Explanted tumors were dissociated using Human Tumor Dissociation Kit (Miltenyi Biotec) as per the manufacturer's instructions. The antitumor activity against GBM-NS was tested in congruence with a locally approved protocol (Istituto Neurologico Carlo Besta, Milan, Italy) and in accordance to the Italian Principle of Laboratory Animal Care (D. Lgs. 26/2014) and European Communities Council Directives (86/609/EEC and 2010/63/UE). Nude (CD1 (HO): CD1-Foxn1nu, from Charles River Laboratories, Calco, Italy female mice ages six weeks were injected intracranially at the coordinates described above with 1 × 105 GBM-NS cells in 2 μL PBS and, 15 days later, with 2 × 106 CAR-T cells in 5 μL PBS intratumorally (using the same stereotaxic coordinates used for the tumor cell implantation). Mice were then monitored every 2–3 days and sacrificed when showing neurological symptoms and/or reduced body condition.
2.7. Statistical analysis
Statistical significance of the difference of means between two groups was assessed using a 2-tailed, nonparametric Mann-Whitney U test. The difference of means between more than two groups was assessed using one-way or two-way ANOVA with Bonferroni's post-test correction. The difference in overall survival in Kaplan-Meier curves was determined using the log-rank Mantel-Cox test. All statistical analyses were performed in GraphPrism (Version 5.03).
2.8. Data availability statement
The data that support the findings in this study are available upon request.
3. Results
3.1. B7-H3 is expressed in GBM specimens
To assess the expression profile of B7-H3 in GBM, we first evaluated the B7-H3 mRNA expression (Gene name: CD276) in The Cancer Genome Atlas (TCGA) dataset. GBM showed relatively high B7-H3 expression, with about 77% of samples lying above the mean B7-H3 expression of all TCGA tumors (Supplementary Fig. S2). Importantly, B7-H3 was found expressed at similarly elevated levels in primary (“P”) and recurrent (“R”) GBM relative to normal (“N”) brain tissue (Fig. 1a). In addition, B7-H3 mRNA levels also had the smallest variance as compared to the other molecules such as CSPG4, EPHA2, ERBB2, and IL13RA2 that are targeted with CAR-T cells (Fig. 1a). Since mRNA levels do not necessarily directly predict protein expression, we evaluated the B7-H3 protein expression by immunohistochemistry in a cohort of 46 GBM specimens previously studied for the expression of CSPG4 [14]. We observed a diffuse positivity of B7-H3 expression, with 76% (35/46) of the specimens displaying a strong immunoreactivity for B7-H3, and 22% (10/46) showing detectable but low expression (Fig. 1b-e and Table 1). B7-H3 was not detected in one specimen. B7-H3 expression was mainly located into the plasma membrane of tumor cells. Among GBM with high B7-H3 expression, 41% were assigned to the classical subtype, 34% to the mesenchymal subtype and 25% to the proneural subtype (Table 1). Of note, tumor cells recruited in the vicinity of blood vessels, as well as adjacent normal-infiltrating tumor cells, were intensely positive for B7-H3 (Supplementary Fig. S3). To further validate these results, we also stained commercially available human tissue microarrays containing GBM specimens (n = 32) and normal brain (n = 69) spanning various central nervous system structures as well as the optic nerve. All GBM samples overexpressed B7-H3 relatively to normal brain samples (Fig. 1f,g).
Fig. 1.
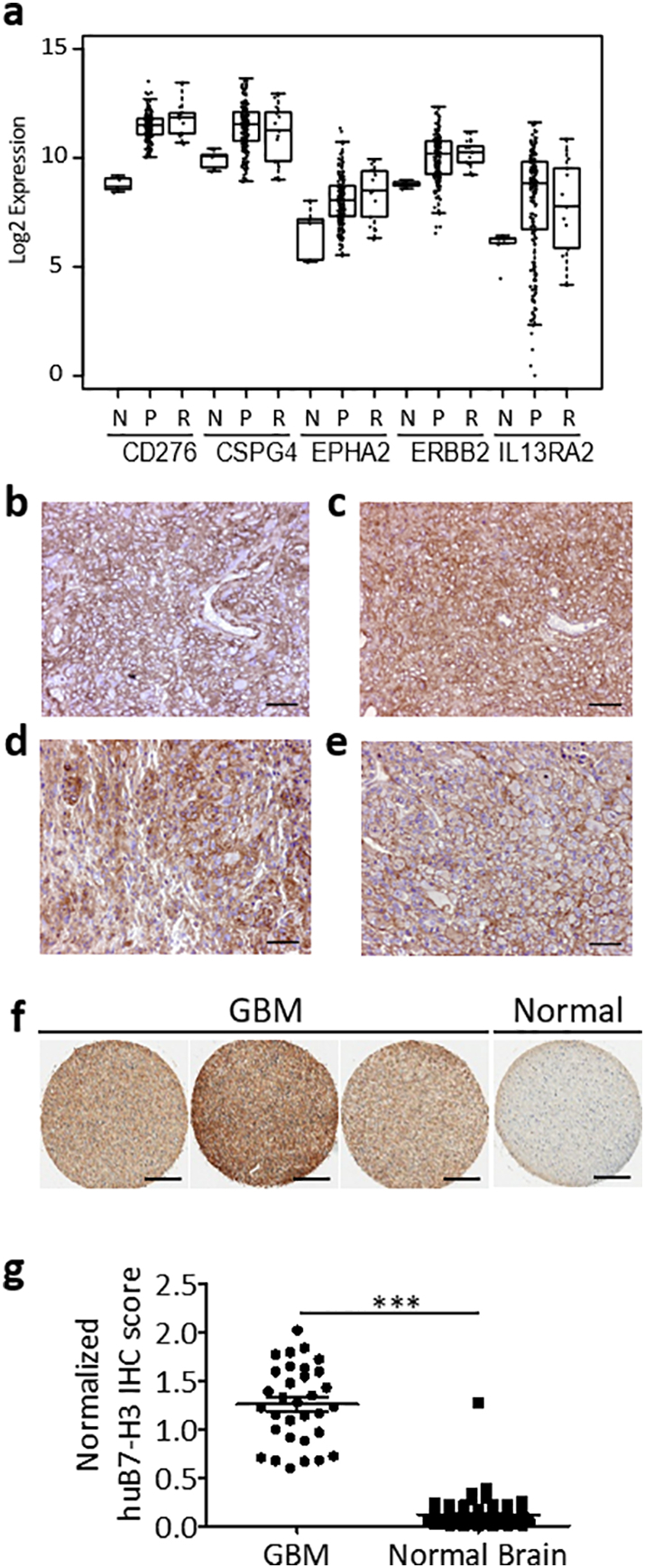
B7-H3 expression in GBM.
(a) Normalized mRNA levels of CD276 (B7-H3), CSPG4, EPHA2, ERBB2, and IL13RA2 in adjacent normal (“N”), primary GBM (“P”), and recurrent GBM (“R”) samples in TCGA. Each point represents a different TCGA sample. (b-e). Representative examples of B7-H3 immunostaining in classical (b), proneural (c), mesenchymal GBM (d), and a rare case of giant cell GBM (e). (f) Representative B7-H3 immunostaining of commercially available GBM tissue arrays and normal brain. (g) B7-H3 expression scores of the tissue microarrays of GBM (n = 32) and normal brain cores (n = 69) quantified via color deconvolution algorithm. Statistical analysis of difference in means was performed using two-tailed nonparametric Mann-Whitney U test (***p < 0·0001). Scale bar (b)-(e), 50 μm. Scale bar (f), 400 μm.
Table 1.
GBM specimen and NS classification and B7-H3 expression.
| Patient ID | B7-H3 expression | GBM-NS subtype | B7-H3 expression in GBM-NS (%) |
|---|---|---|---|
| BT137 | + | PN | NE |
| BT140 | + | MES | NE |
| BT150 | + | CLAS | NE |
| BT202 | + | MES | NE |
| BT205 | + | PN | 98.0 |
| BT211 | + | MES | NE |
| BT219 | + | MES | NE |
| BT235 | + | CLAS | NE |
| BT241 | + | NA | NE |
| BT245 | + | PN | NE |
| BT248 | + | NA | NE |
| BT302 | + | MES | 99.0 |
| BT308 | + | PN | 98.0 |
| BT328 | + | MES | 92.3 |
| BT337 | + | PN | NE |
| BT347 | + | NA | NE |
| BT379 | + | CLAS | 94.8 |
| BT417 | + | CLAS | 98.0 |
| BT422 | + | CLAS | 98.4 |
| BT423 | + | CLAS | 94.6 |
| BT462 | + | CLAS | 99.8 |
| BT482 | + | MES | 99.9 |
| BT483 | + | PN | 98.0 |
| BT513 | + | MES | NE |
| BT517 | + | CLAS | 97.7 |
| BT138 | ++ | CLAS | NE |
| BT168 | ++ | MES | 99.9 |
| BT175 | ++ | NA | NE |
| BT275 | ++ | CLAS | 97.1 |
| BT283 | ++ | CLAS | NE |
| BT261 | ++ | CLAS | NE |
| BT358 | ++ | MES | 92.1 |
| BT373 | ++ | PN | 89.0 |
| BT480 | ++ | MES | NE |
| BT500 | ++ | CLAS | NE |
| BT487 | +/− | MES | 96.4 |
| BT326 | +/− | PN | 90.1 |
| BT273 | +/− | CLAS | 97.8 |
| BT299 | +/− | PN | 95.5 |
| BT155 | +/− | CLAS | NE |
| BT209 | +/− | PN | NE |
| BT206 | +/− | CLAS | NE |
| BT334 | +/− | CLAS | NE |
| BT274 | +/− | PN | NE |
| BT279 | +/− | CLAS | NE |
| BT157 | − | MES | NE |
Abbreviations: GBM-NS: glioblastoma-neurospheres; PN: proneural; CLAS: classical; MES: mesenchymal; NA: not available; NE: not evaluated. A total of 20 GBM-NS were assessed by flow cytometry for B7-H3 expression, which is indicated as percentage of positive cells.
3.2. B7-H3-redirected CAR-T cells target human GBM cell lines in vitro and in vivo
We generated B7-H3-redirected CAR (B7-H3.CAR) T cells encoding either CD28 or 4-1BB endodomains (B7-H3.CD28 and B7-H3.41BB, respectively) [22], and CD19-redirected CAR (CD19.CD28) T cells [23] as control through retroviral transduction of activated T cells from healthy donors (Fig. 2a, b). CAR expression was assessed to be 93% ± 3%, 62% ± 14%, and 51% ± 8% in CD19.CD28, B7-H3.CD28, and B7-H3.41BB CAR-T cells, respectively. CAR-T cells were then tested for antitumor activity against the U-87 MG and U-138 MG human GBM tumor cell lines, which express B7-H3, while the B7-H3-negative HL-60 leukemia cell line served as a negative control (Fig. 2c). B7-H3.CAR-T cells showed complete or near complete elimination of U-87 MG and U-138 MG cells five days after seeding at 1:5 and 1:10 E:T ratios, as measured by the number of tumor cells remaining, with no statistically significant difference between B7-H3.CD28 and B7-H3.41BB CAR-T cells (vs. U-87 MG: 2311 ± 3639 and 1483 ± 927 at 1:5 E:T, 14908 ± 18,548 and 32,345 ± 27,580 at 1:10 E:T; vs. U-138 MG: 384 ± 338 and 1260 ± 1175 at 1:5 E:T, 7376 ± 5402 and 8835 ± 5716 at 1:10 E:T, tumor cells remaining against B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively). There was no evidence of killing in wells seeded with CD19.CD28 CAR-T cells or control non-transduced T cells (cT) and in wells seeded with HL-60 cells (Fig. 2d, e). We measured cytokine release by CAR-T cells in the coculture supernatant after 24 h and CAR-T cell proliferation in response to antigen stimulation. The effector cytokines IFNγ and IL-2 were detected in the supernatant only when B7-H3.CAR-T cells were cocultured with U-87 MG or U-138 MG cells, with B7-H3.CD28 CAR-T cells showing higher cytokine release as compared to B7-H3.41BB CAR-T cells (vs. U-87 MG: 791 ± 607 and 96 ± 110 IFNγ, 541 ± 220 and 167 ± 139 IL-2; vs. U-138 MG: 131 ± 140 and 0 ± 0 IFNγ, 193 ± 74 and 26 ± 30 IL-2, pg/mL/2 × 104 B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively) (Fig. 3a, b). To examine the proliferative capacity of B7-H3.CAR-T cells in response to antigen stimulation, we labeled the T cells with CFSE and cocultured them with U-87 MG, U-138 MG, or HL-60 cells at a 1:1 E:T ratio. After five days in coculture, B7-H3.CAR-T cells proliferated only in response to U-87 MG and U-138 MG cells, with no statistical difference seen between B7-H3.CD28 and B7-H3.41BB CAR-T cells (vs. U-87 MG: 97% ± 3% and 97% ± 1%; vs. U-138 MG: 97% ± 2% and 93% ± 5%, CFSE-diluted B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively). No proliferation was observed in CD19.CD28 CAR-T and cT cells (Fig. 3c, d).
Fig. 2.

B7-H3.CAR-T cells target human GBM cell lines in vitro.
(a) Representative plots showing the CAR expression in CD19.CD28, B7-H3.CD28 and B7-H3.41BB CAR-T cells as assessed by flow cytometry. Expression is represented relative to non-transduced control T cells (cT). (b) Summary of CAR expression. Data represent mean ± SD (n = 6 donors). (c) Expression of B7-H3 in U-87 MG, U-138 MG, and HL-60 cells stained with the 376.96 mAb and APC goat anti-mouse and assessed by flow cytometry. Expression is represented relative to staining with APC goat anti-mouse alone. (d) Representative flow cytometry plots of CAR-T cells (CD3+) cocultured with U-87 MG cells (CSPG4+), U-138 MG cells (CSPG4+), or HL-60 cells (CD33+) at 1:5 or 1:10 effector-to-target (E:T) ratios for five days. (e) The number of remaining U-87 MG, U-138 MG, or HL-60 cells in 1:5 and 1:10 E:T coculture experiments in (d). Tumor cell numbers were calculated using counting beads. Data represent mean ± SD (n = 3–4 donors). Each donor was tested once for each experiment. Difference in means was assessed through one-way ANOVA with Bonferroni's post-test correction (*p < 0·05, **p < 0·01).
Fig. 3.

B7-H3.CAR-T cells release effector cytokines and proliferate in response to B7-H3+ GBM cell lines.
(a-b) Quantification of IFNγ (a) and IL-2 (b) by ELISA in the supernatant 24 h after coculture of CAR-T cells or cTs with U-87 MG, U-138 MG, or HL-60 cells at 1:5 E:T ratio. Data represent mean ± SD (n = 5–6 donors). Difference in means was assessed through one-way ANOVA with Bonferroni's post-test correction (*p < 0·05, ***p < 0·0001). (c) Representative CFSE dilution of CFSE-labeled CAR-T cells cocultured at a 1:1 E:T ratio with U-87 MG, U-138 MG, or HL-60 cells for five days. The percent of CFSE dilution was measured relative to CFSE-labeled CAR-T cells cultured alone. (d) Summary of CFSE-dilution assays. Data represent mean ± SD (n = 5–6 donors). Each donor was tested once for each experiment. Difference in means was assessed through one-way ANOVA with Bonferroni's post-test correction (***p < 0·0001).
The antitumor activity of B7-H3.CAR-T cells was then tested in a xenograft murine model. We first generated U-87 MG cells stably expressing GFP-FFLuc (U87-GFP-FFLuc) (Supplementary Fig. S1). U87-GFP-FFLuc were then injected intracranially into the caudate nucleus of nu/nu mice. One week later, mice were treated intratumorally with CD19.CD28, B7-H3.CD28 or B7-H3.41BB CAR-T cells. Tumor growth was monitored by BLI (Fig. 4a). In each experiment, mice were assigned a treatment such that the pre-treatment tumor BLI distribution was comparable in each treatment group. Among mice with initial tumor burden <1 × 108 p/s/cm2/sr total flux, sustained tumor regression was seen in 86% (6/7) and 100% (5/5) of those treated with B7-H3.CD28 CAR-T cells or B7-H3.41BB CAR-T cells, respectively (Fig. 4b, c). B7-H3.CAR-T cell treatment conferred a significant and prolonged survival benefit, with no significant difference between B7-H3.CD28 (6 of 11 mice survive >120 days) and B7-H3.41BB (6 of 10 mice survive >120 days) CAR-T cells. (Fig. 4d). In mice with tumor recurrence observed after day 40, harvested tumors from mice treated with B7-H3.CAR-T cells revealed retention of the B7-H3 expression in tumor cells (Fig. 4e) indicating that tumor recurrence is not caused by antigen loss. No treatment-related adverse effects were observed. We could not assess the effects of B7-H3.CAR-T cells in mice engrafted with U-138 MG cells because this tumor cell line had inconsistent engraftment, and when engrafted was characterized by very slow tumor growth with no morbidity even months after engraftment. To further prove that B7-H3.CAR-T cells can target tumor cells in situ, we developed a xenograft rat brain slice coculture model in which U87-GFP-FFLuc or GBM-NS-GFP cells are seeded onto rat brain slices and treated one day later with either CD19.CAR-T cells, B7-H3.CAR-T cells, or no cells (Fig. 4f, g). Cytokine quantification of supernatant 24 h after CAR-T cell treatment reveals IFNγ and IL-2 release only in the presence of B7-H3.CAR-T cells, with B7-H3.CD28 CAR-T cells releasing more IFNγ and IL-2 in 2 out of 3 U87-GFP-FFLuc-seeded brain slices and more IL-2 in 2 out of 2 GBM-NS-GFP-seeded brain slices. Similar levels of IFNγ were released by B7-H3.CD28 and B7-H3.41BB CAR-T cells in all GBM-NS-GFP-seeded slices (vs. U87-GFP-FFLuc: 882 ± 497 and 469 ± 81 IFNγ, 266 ± 122 and 111 ± 14 IL-2 pg/mL/2.5 × 104; vs. GBM-NS: 254 ± 70 and 213 ± 49 IFNγ, 95 ± 10 and 16 ± 9 IL-2, pg/mL/6.3 × 103 B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively) (Fig. 4h, i).
Fig. 4.

B7-H3.CAR-T cells control the growth of human GBM cell lines in vivo.
(a) Schema of the xenograft murine model used to test the antitumor effects of B7-H3.CAR-T cells in vivo. U87-GFP-FFLuc GBM cells (1 × 105 cells) were inoculated into the caudate nucleus and tumor growth was monitored weekly by bioluminescence (BLI) imaging. CAR-T cells (2 × 106 cells) were injected intratumorally one week after tumor inoculation. (b) Representative images of tumor BLI. (c) Log-transformed BLI values showing the kinetics of tumor growth of each mouse (n = 9–11 mice/group across 3 donors of T cells). (d) Kaplan-Meier analysis of overall survival of the treated mice. Statistical analysis was performed using the Mantel-Cox log rank test (***p < 0·0001). (e) Representative flow cytometry plots showing the expression of GFP and B7-H3 in U87-GFP-FFLuc tumors explanted from mice treated with CAR-T cells. (f) Experimental setup of xenograft rat brain slice coculture model. (g) Representative images demonstrating U87-GFP-FFLuc and GBM-NS-GFP cells seeded on rat brain slice. (h,i) Quantification of IFNγ (h) and IL-2 (i) by ELISA in the supernatant 24 h after coculture of CAR-T cells with U87-GFP-FFLuc cells or GBM-NS-GFP at 1:2 E:T ratio on rat brain slides. Data represent one donor tested against U87-GFP-FFLuc cells or one GBM-NS-GFP line on 1–3 different rat brain slices. Each donor was tested once for each experiment. Difference in means was assessed through one-way ANOVA with Bonferroni's post-test correction (**p < 0·01).
3.3. B7-H3.CAR-T cells target GBM-NS in vitro and in vivo
Since GBM established tumor cell lines do not fully recapitulate the molecular subtypes of human GBM and do not mimic human CSCs, we sought to test B7-H3.CAR-T cells against patient-derived GBM-NS [25]. We measured the expression of B7-H3 in 20 GBM-NS representative of the most common GBM molecular subtypes. We found high expression levels of B7-H3 (ranging from 89% to 100% of cells) in all GBM-NS, independently of their molecular subtype (Fig. 5a, b and Table 1). We tested the antitumor activity of B7-H3.CAR-T cells against five representative GBM-NS. The killing ability of B7-H3.CAR-T cells was evaluated by flow cytometry in a coculture assay at different time points using 1:5 E:T ratio. B7-H3.CAR-T cells encoding CD28 showed prominent antitumor efficacy already at 24 h as compared to those encoding 4-1BB. However, at 48 h B7-H3.CD28 and B7-H3.41BB CAR-T cells equally eliminated GBM-NS from the culture (residual GBM-NS < 2%). In contrast, GBM-NS continued to grow in the presence of CD19.CD28 CAR-T cells (Fig. 5c). The Th1 cytokine profile in the supernatant in response to GBM-NS revealed no difference between the levels of IFNγ released by B7-H3.CD28 and B7-H3.41BB (965 ± 161 and 921 ± 828 pg/mL/1 × 105 B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively). However, the elevated IFNγ release by B7-H3.41BB CAR-T cells in 2 out of 5 GBM-NS tested correlates with the upregulation of PD-L1 in GBM-NS upon exposure to IFNγ, as we have previously reported (Fig. 5d) [14]. Nevertheless, B7-H3.CAR-T cells encoding CD28 released higher amount of IL-2, consistent with the in vitro studies with U-87 MG and U-138 MG cell lines (641 ± 175 and 407 ± 116 pg/mL/1 × 105 B7-H3.CD28 and B7-H3.41BB CAR-T cells, respectively) (Fig. 5e).
Fig. 5.

B7-H3.CAR-T cells control the growth of human GBM-NS in vitro and in vivo.
(a) Representative flow plots and (b) summary plots showing B7-H3 expression in human GBM-NS as assessed by flow cytometry. GBM-NS were subdivided by molecular subtypes (MES: mesenchymal, n = 6; PN: proneural, n = 6; CLAS: classical, n = 8). (c) CAR-T cells were cocultured with GBM-NS at 1:5 E:T ratio and residual tumor cells (B7-H3+) were quantified by flow cytometry at different time points. Data represent the average of two donors of CAR-T cells tested against five GBM-NS. Difference in means was assessed through two-way ANOVA with Bonferroni's post-test correction (***p < 0·0001). (d,e) Quantification of IFNγ (d) and IL-2 (e) release by B7-H3.CAR-T cells and CD19.CAR-T cells in the supernatant collected 24 h after coculture with GBM-NS. BT166-NS and BT462-NS released higher levels of IFNγ compared to BT308-NS, BT302-NS and BT273-NS [14]. Data represent the average of two donors of CAR-T cells tested against five GBM-NS. Difference in means was assessed through one-way ANOVA with Bonferroni's post-test correction (*p < 0·05). (f) Schema of the xenograft murine experimental model used to test the antitumor effects of CAR-T cells against GBM-NS. GBM-NS (1 × 105 cells) were inoculated into the caudate nucleus and CAR-T cells from one donor (2 × 106 cells) were injected intratumorally two week later. (g) Kaplan-Meier curves showing the overall survival of the treated mice (n = 6 mice/group). Overall survival comparison was measured using the Mantel-Cox log rank test (***p < 0·0001). (h,i) Tissue morphology assessed by hematoxylin and eosin staining and B7-H3 expression assessed by immunohistochemistry of xenograft gliomas collected from tumor-bearing mice. (h) Gliomas collected from mice treated with CD19.CAR-T cells show high cellularity and high B7-H3 expression. (i) Gliomas collected from mice treated with B7-H3.CAR-T cells show necrotic zones with fibrotic areas (marked by asterisks) and disrupted tumor architecture. Delimited area shows dense cellularity at the borders of necrosis. Scale bar, 100 μm. Each CAR-T cell donor was tested once for each experiment.
To test in vivo the antitumor activity of B7-H3.CAR-T cells against GBM-NS, nu/nu mice were implanted with GBM-NS into the brain, and CAR-T cells or control T cells were injected intratumorally 15 days later (Fig. 5f). B7-H3.CAR-T cells encoding either CD28 or 4-1BB equally controlled the tumor growth and prolonged the survival in 50% of the treated mice as compared to mice treated with control T cells (3 of 6 mice for both B7-H3.CD28 and B7-H3.41BB-treated groups) (Fig. 5g). Morphological analyses performed on explanted brains showed high cellularity in control xenograft gliomas. On the contrary, in B7-H3.CAR-T cell-treated xenograft gliomas the architecture was disrupted by large damaged and necrotic areas (Fig. 5h, i). As observed in the tumor model with GBM tumor cell line, recurrent tumors harvested from mice treated with B7-H3.CAR-T cells retained B7-H3 expression in tumor cells (Fig. 5i).
4. Discussion
One of the greatest barriers to developing effective immune therapies for GBM is the heterogeneity of antigen expression within and across tumors, which highlights the need of the identification of novel targets. B7-H3 is a promising therapeutic target in GBM and other solid tumors due to its high expression in tumor cells and low expression in normal tissues [16,22]. In this report, we confirmed that B7-H3 is highly expressed in GBM and provided comprehensive evidence that GBM can be effectively targeted using B7-H3-specific CAR-T cells. Furthermore, we found that B7-H3 expression is highly conserved in patient-derived GBM-NS supporting the potential of the proposed strategy to eradicate tumor cells with high tumor regenerative capacity.
Clinical trials of CAR-T cells in GBM patients have demonstrated tumor regression after either systemic or intracranial delivery routes [[10], [11], [12]]. Nonetheless, these trials also highlighted how tumor antigen escape due to the heterogeneous expression of the antigens hinders the durability of clinical responses. A bioactivity and safety study of IL13Rα2-targeted CD8+ CAR-T cells by Brown et al. documented recurrence of IL13Rα2-negative tumors [10,26]. Similarly, O'Rourke and colleagues described recurrence of antigen-negative tumors in patients with GBM treated with EGFRvIII-directed CAR-T cells [11]. The identification of antigens that are invariably expressed by tumor cells, but not by the counterpart normal cells remains a high priority to target solid tumors, including GBM, with CAR-T cells. We have previously reported that targeting CSPG4 in GBM partially addresses the issue, because CSPG4 is highly expressed in 67% of the GBM specimens we tested, and is also expressed in tumor-associated vessels, but not in normal cerebral vessels and normal brain [14]. Furthermore, we observed that CSPG4 expression can be induced by TNFα released by resident microglia, and potentially by TNFα secreted by CAR-T cells upon activation [14]. In this manuscript, we report that B7-H3 is also highly expressed in 76% of GBM, but not in normal brain tissue, and that targeting B7-H3 via CAR-T cells promotes antitumor activity both in vitro and in xenograft models highlighting the clinical relevance of B7-H3 as a molecular target in GBM. However, as previously observed for CSPG4 [14], a third of the GBM specimens analyzed show low expression of B7-H3, and this level of expression may be insufficient to promote effective killing by CAR-T cells as we have recently reported [22]. Nevertheless, the data presented in this report demonstrate that B7-H3 is almost invariably expressed in patient-derived GBM-NS spanning all molecular GBM subtypes. Furthermore, GBM-NS are effectively targeted by B7-H3-redirected CAR-T cells both in vitro and in vivo. GBM-NS are generated from human GBM samples using the same growth factors employed for neural stem cells and are enriched in putative CSCs [19]. Since several lines of evidence indicate that CSCs have a critical role in causing tumor recurrence [18,27,28], effectively targeting these cells with B7-H3.CAR-T cells offers greater potential to control tumor recurrence even if some differentiated tumor cells may not be eliminated because they express B7-H3 at low levels. Due to its high expression in GBM specimens and GBM-NS and lack of expression on normal brain, B7-H3 may thus represent an ideal target in a high proportion of patients as single antigen or in combination with other validated targets to offer CAR-T cells to the majority of patients with GBM [[32], [33], [34]].
Occurrence of cerebral edema and cytokine release syndrome has been attributed to CAR-T cells in hematologic malignancies [29]. Clinical studies of CAR-T cells in GBM patients have shown a relatively favorable safety profile, with no dose-limiting toxicities seen for HER2- and EGFRvIII-redirected CAR-T cells [11,12], and manageable grade 3 events (headache, neurologic deficits) in patients treated with high dose anti-IL13Rα2 CAR-T cells intracranially [10,26]. We have previously demonstrated that B7-H3 is not expressed in normal brain [22]. Furthermore, in a clinical study in which the 131I-conjugated B7-H3 Ab was administered intracranially to patients with metastatic neuroblastoma, no toxicity was encountered [30]. The B7-H3.CAR we have developed shows cross-reactivity to murine B7-H3, but no toxicity was observed in mice either immunodeficient or immunocompetent when B7-H3.CAR-T cells were infused systemically [22].. In this report, B7-H3.CAR-T cells injected directly intracranially in murine models of GBM also did not cause toxicity attributable to on target off tumor recognition of B7-H3. These data suggest that on-target but off-tumor toxicity may not occur in human subjects.
We observed only limited differences in the antitumor activity of B7-H3.CAR-T cells encoding either CD28 or 4-1BB co-stimulatory domains both in vitro and in vivo. As previously reported [31], we observed that CD28 co-stimulation provides faster antitumor effects as compared to 4-1BB co-stimulation, but this did not translate into enhanced antitumor activity in the experimental conditions we have used in vitro and in vivo. The intratumor delivery route of the CAR-T cells may confer immediate exposure of the CAR-T cells to tumor cells and overcome the difference in cytokine release we observed in vitro between CD28 and 4-1BB co-stimulation, leading to equivalent antitumor efficacy in vivo. Of note, nearly all the mice with large initial tumor burden failed treatment irrespective of the provided co-stimulation, highlighting the importance of additional studies aimed at enhancing the persistence of CAR-T cells within the tumor environment to eradicate large tumors or the need of early detection of tumor recurrence and early treatment.
In conclusion, our study shows that B7-H3 is an attractive target in GBM since it is highly expressed in 76% of the GBM specimens tested across all subtypes and is invariably expressed in GBM-NS that contain CSCs, while it is not detectable in normal brain. B7-H3-redirected CAR-T cells control tumor growth in multiple human GBM models, and B7-H3 expression is retained at tumor recurrence indicating that strategies aimed at enhancing the CAR-T cell delivery and persistence within the tumor may further increase the efficacy profile of B7-H3-redirected CAR-T cells in GBM.
Funding sources
This study was funded by the Alex's Lemonade Stand Foundation, Il Fondo do Gio Onlus, the National Cancer Institute, the Borroughs Wellcome Fund. None of these funding institutions had a role in the study design; collection, analysis, and interpretation of the data; the writing or editing of the manuscript; or in the decision to submit this work for publication. The corresponding authors have full access to all the data used to in the study and hold the final decision to submit this manuscript for publication.
Author contributions
Conceptualization: DN, BS, SP, GD; Methodology: DN, MDI, SM, HD, MP, BP, JJHP, DED, DSE, JSD, HH; Writing-Original Draft: DN, SP, GD; Writing-Review & Editing: all authors; Resources: SF, GF; Supervision: SRF, SP, BS, GD.
Declaration of Competing Interest
GD HD and SF filed a patent for B7-H3-redirected CAR T cells. No other others have conflicts of interest to declare.
Acknowledgements
The authors would like to thank the funders of this study: Alex's Lemonade Stand Foundation (A18-0829-001 to GD); Il Fondo di Gio Onlus (SP); National Cancer Institute (1R21CA226483-01A1 to SRF, 5P50CA190991-05 to SRF); and Burroughs Wellcome Fund Career Award for Medical Scientists (SRF). The authors would also like to thank the Translational Pathology Lab at UNC for staining and scoring the TMAs, Cristina Corbetta for helping with the in vivo experiments at Istituto Neurologica Carlo Besta, colleagues from the Department of Neurosurgery of the Istituto Neuroogico Carlo Besta, the SOL Group Spa, Italy for the cryo-management service and the technical assistance and Ana de Carvalho from the Henry Ford Health System for providing the GBM-NS for the xenograft rat brain slice coculture experiments.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.08.030.
Contributor Information
Serena Pellegatta, Email: serena.pellegatta@istituto-besta.it.
Gianpietro Dotti, Email: gdotti@med.unc.edu.
Appendix A. Supplementary data
Supplementary material
References
- 1.Weller M., Butowski N., Tran D.D. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017 Oct;18(10):1373–1385. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W.P., van den Bent M.J. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005 Mar 10;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Friedman H.S., Prados M.D., Wen P.Y. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009 Oct 1;27(28):4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 4.Brennan C.W., Verhaak R.G., McKenna A. The somatic genomic landscape of glioblastoma. Cell. 2013 Oct 10;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel A.P., Tirosh I., Trombetta J.J. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Touat M., Idbaih A., Sanson M., Ligon K.L. Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann Oncol. 2017 Jul 1;28(7):1457–1472. doi: 10.1093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dotti G., Gottschalk S., Savoldo B., Brenner M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014 Jan;257(1):107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochenderfer J.N., Dudley M.E., Kassim S.H. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015 Feb 20;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maude S.L., Frey N., Shaw P.A. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014 Oct 16;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown C.E., Alizadeh D., Starr R. Regression of Glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016 Dec 29;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Rourke D.M., Nasrallah M.P., Desai A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017 Jul 19;9:399. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed N., Brawley V., Hegde M. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017 Aug 1;3(8):1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chow K.K., Naik S., Kakarla S. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013 Mar;21(3):629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pellegatta S., Savoldo B., Di I.N. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR-T cell therapy. Sci Transl Med. 2018 Feb 28;10(430) doi: 10.1126/scitranslmed.aao2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picarda E., Ohaegbulam K.C., Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 2016 Jul 15;22(14):3425–3431. doi: 10.1158/1078-0432.CCR-15-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C., Zhang Z., Li F. Large-scale analysis reveals the specific clinical and immune features of B7-H3 in glioma. Oncoimmunology. 2018;7(11) doi: 10.1080/2162402X.2018.1461304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De B.F., Casanova E., Medico E. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012 Sep 1;72(17):4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- 18.Orzan F., De B.F., Crisafulli G. Genetic evolution of glioblastoma stem-like cells from primary to recurrent tumor. Stem Cells. 2017 Nov;35(11):2218–2228. doi: 10.1002/stem.2703. [DOI] [PubMed] [Google Scholar]
- 19.Lee J., Kotliarova S., Kotliarov Y. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006 May;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 20.Louis D.N., Perry A., Reifenberger G. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016 Jun;131(6):803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 21.Finocchiaro G., Pellegatta S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies. Cancer Immunol Immunother. 2016 Jan;65(1):101–109. doi: 10.1007/s00262-015-1754-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du H., Hirabayashi K., Ahn S. Antitumor responses in the absence of toxicity in solid Tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 2019 Feb 11;35(2):221–237. doi: 10.1016/j.ccell.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaconu I., Ballard B., Zhang M. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. 2017 Mar 1;25(3):580–592. doi: 10.1016/j.ymthe.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramos C.A., Savoldo B., Torrano V. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest. 2016 Jul 1;126(7):2588–2596. doi: 10.1172/JCI86000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pellegatta S., Poliani P.L., Corno D. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006 Nov 1;66(21):10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 26.Brown C.E., Badie B., Barish M.E. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015 Sep 15;21(18):4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007 Jan;19(1):61–64. doi: 10.1097/CCO.0b013e328011a8d6. [DOI] [PubMed] [Google Scholar]
- 28.Esparza R., Azad T.D., Feroze A.H., Mitra S.S., Cheshier S.H. Glioblastoma stem cells and stem cell-targeting immunotherapies. J Neurooncol. 2015 Jul;123(3):449–457. doi: 10.1007/s11060-015-1729-x. [DOI] [PubMed] [Google Scholar]
- 29.Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016 Jun 30;127(26):3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramer K., Kushner B.H., Modak S. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol. 2010 May;97(3):409–418. doi: 10.1007/s11060-009-0038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Z., Condomines M., van der Stegen S.J. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015 Oct 12;28(4):415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrov J.C., Wada M., Pinz K.G. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018 Jun;32(6):1317–1326. doi: 10.1038/s41375-018-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedge M., Corder A., Chow K.K. Combinatorial targeting offsets antigen escape and enhances effector function of adoptively transferred T cells in glioblastoma. Mol Ther. 2013 Nov;21(11):2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao J., Song Y., Liu D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J Hematol Oncol. 2019 Feb 14;12(1):17. doi: 10.1186/s13045-019-0705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
The data that support the findings in this study are available upon request.