

Abstract
Background
The complement and kallikrein-kinin systems (KKS) are activated during vascular inflammation. The aim of this study was to investigate if blockade of the KKS can affect complement activation on the endothelium during inflammation.
Methods
Complement deposition on endothelial microvesicles was assayed in vasculitis patient plasma samples and controls. Plasma was perfused over glomerular endothelial cells and complement deposition assayed by flow cytometry. The effect of the kinin system was assessed using kinin receptor antagonists and C1-inhibitor. The in vivo effect was assessed in kidney sections from mice with nephrotoxic serum-induced glomerulonephritis treated with a kinin receptor antagonist.
Findings
Vasculitis patient plasma had significantly more C3- and C9-positive endothelial microvesicles than controls. Perfusion of patient acute-phase plasma samples over glomerular endothelial cells induced the release of significantly more complement-positive microvesicles, in comparison to remission or control plasma. Complement activation on endothelial microvesicles was reduced by kinin B1- and B2-receptor antagonists or by C1-inhibitor (the main inhibitor of the classical pathway and the KKS). Likewise, perfusion of glomerular endothelial cells with C1-inhibitor-depleted plasma induced the release of complement-positive microvesicles, which was significantly reduced by kinin-receptor antagonists or C1-inhibitor. Mice with nephrotoxic serum-induced glomerulonephritis exhibited significantly reduced glomerular C3 deposition when treated with a B1-receptor antagonist.
Interpretation
Excessive complement deposition on the endothelium will promote endothelial injury and the release of endothelial microvesicles. This study demonstrates that blockade of the KKS can reduce complement activation and thereby the inflammatory response on the endothelium.
Funding
Full details are provided in the Acknowledgements/Funding section.
Keywords: Vasculitis, Endothelial microvesicles, Complement, Kinin, Kidney, Mouse
Research in context.
Evidence before this study
Excessive complement activation on the vascular wall leads to endothelial cell damage and during this injurious process endothelial cell-derived microvesicles will be released. Both the complement and kallikrein-kinin systems are activated during vascular inflammation. The interaction between these two proinflammatory systems has not been investigated in vascular inflammatory disease.
Added value of this study
We demonstrate that blockade of the kallikrein-kinin system, by blocking kinin B1 and/or B2 receptors, or by incubation with C1 inhibitor, decreases complement deposition on glomerular endothelial microvesicles induced by exposure to vasculitis patient plasma. Furthermore, a B1 receptor antagonist decreased complement deposition in murine glomerular capillaries in rapidly progressive glomerulonephritis caused by nephrotoxic serum-induced glomerulonephritis.
Implications of all the available evidence
Blocking the kallikrein-kinin system can reduce complement-mediated endothelial cell injury and may thereby have therapeutic potential in vascular inflammation. C1 inhibitor and B2 receptor antagonists are commercially available.
Alt-text: Unlabelled Box
1. Introduction
The complement system is activated during vascular inflammation [1]. Complement is involved in the host's natural defence against invading microbes, clearance of debris and immune complexes, as well as enhancement of the adaptive immune response [2]. Overwhelming complement activation may potentiate an inflammatory response due to the release of degradation products that induce anaphylaxis, opsonisation and chemotaxis [3]. Extensive activation on the endothelium promotes thrombosis, leukocyte recruitment, vascular permeability and vascular wall injury [1,4].
In addition to the complement system, the contact/kallikrein-kinin system (KKS) is also activated during vascular inflammation [5]. KKS activation results in the liberation of kinin peptides from high-molecular weight kininogen, such as bradykinin [6], or PR3-kinin, the latter cleaved from high-molecular weight kininogen by proteinase 3 (PR3) [7]. These kinins activate a proinflammatory signal by binding to their receptors. Des-arg [9]-bradykinin (a stable derivate of bradykinin) and PR3-kinin bind to the B1-receptor (B1R) while bradykinin binds to the B2-receptor (B2R) [7,8]. B1R and B2R activation induces vascular permeability [9] as well as neutrophil recruitment [10] and thus contributes to the inflammatory state.
Both the complement and kallikrein-kinin systems are activated during vasculitides [4,7,9,[11], [12], [13]]. Vasculitis is characterized by massive inflammation in and around vessel walls, affecting multiple organs. The most common vasculitis in childhood is IgA-vasculitis (Henoch-Schönlein purpura), which is often transient, while adults are more frequently affected by chronic anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) [14,15]. ANCA are primarily directed against neutrophil-derived proteases such as PR3 and myeloperoxidase (MPO) [16]. Activation of the complement and kallikrein-kinin systems may contribute to the severe vascular inflammation occurring during vasculitis [11,17].
Our previous studies have shown that patients with vasculitides, both children and adults, exhibit increased degradation of high molecular kininogen and high levels of kinins in plasma [7,11], as well as enhanced kinin deposition in inflamed tissues such as the kidneys and skin [11]. Furthermore, in patient kidneys neutrophil-derived microvesicles (MVs) bearing the kinin B1R were demonstrated to dock onto the glomerular endothelium [18]. Neutrophil-derived MVs could transfer the kinin B1R to recipient cells and thereby promote inflammation [18].
MVs are shed from endothelial cells during cellular activation and apoptosis [19]. Patients with vasculitis were shown to have circulating MVs derived from endothelial cells [10,[20], [21], [22], [23]]. These MVs exhibited KKS activation as they were B1R-positive, thereby inducing neutrophil chemotaxis [10], an effect blocked by C1-inhibitor, the main inhibitor of the KKS and of the classical pathway of complement [10].
The aim of this study was to investigate if activation of the KKS can affect complement activation on the endothelium during vascular inflammation, as reflected by complement deposits on endothelial-derived microvesicles (EMVs) and glomerular capillaries, and if complement deposition was modulated by kinin receptor antagonists. As both these pro-inflammatory systems are activated during vasculitis, plasma from vasculitis patients was used as a model of vascular inflammation associated with complement and KKS activation. The mechanism of complement activation on glomerular endothelial cells was investigated using vasculitis patient plasma, C1-inhibitor-depleted plasma, or kininogen-depleted plasma, perfused over the cells, in the absence of blood cells. The impact of kinin B1R and B2R antagonists and C1-inhibitor, was studied, in order to determine if blocking KKS activation affects complement deposition on EMVs. The effect of a B1R antagonist on complement deposition in vivo was assessed in the kidneys of mice with nephrotoxic serum-induced glomerulonephritis in which B1R antagonism was previously shown to reduce glomerulonephritis injury [24].
2. Material and methods
2.1. Patients and controls
Blood samples were available from 22 children and adults (11 females, 11 males, median age 61 years) with vasculitis treated at the Department of Nephrology and the Department of Pediatrics, section of Pediatric Nephrology, Skåne University Hospital, Lund and Malmö, and the Department of Nephrology, Linköping University Hospital Sweden. The patients and their Birmingham Vasculitis Activity Score (BVAS) are presented in Table 1. Vasculitis was defined according to the Chapel Hill consensus paper [25]. Some patients were previously described (n = 17) [10]. Samples from the acute phase of disease were available from all patients and none of the patients were on hemodialysis at sampling. Samples from 5 of the patients were also available during remission (Table 1). Blood samples were obtained from 21 healthy controls (14 females, 7 males, median age 41 years) not using any medications. The blood samples were used for flow cytometry analysis and perfusion experiments. The study was conducted with the approval of the Regional Ethics review board of Lund and Linköping Universities and the written informed consent of the patients or their parents and the controls.
Table 1.
Description of patients included in this study.
| Patient number | Sex | Age at sampling | Diagnosis | ANCA | Creatinine | BVAS score |
|---|---|---|---|---|---|---|
| 1a | M | 75 | AAV | PR3 | 92 | 16 |
| 2a | M | 61 | AAV | PR3 | 237 | 17 |
| 3a | F | 49 | AAV | PR3 | 49 | 6 |
| 4a | M | 70 | AAV | PR3 | 90 | 6 |
| 5a | M | 69 | AAV | PR3 | 188 | 12 |
| 6b | M | 43 | IgAV | – | 89 | 15 |
| 7b | M | 16 | IgAV | – | 69 | 3 |
| 8b | M | 80 | AAV | MPO | 259 | 23 |
| 9b | M | 62 | AAV | PR3 | 61 | 6 |
| 10b | F | 19 | SLE | – | 62 | 17 |
| 11b | F | 76 | AAV | PR3 | 74 | 14 |
| 12b | F | 12 | AAV | PR3/MPO | 312 | 15 |
| 13b | F | 61 | AAV | MPO | 54 | 13 |
| 14b | M | 10 | IgAV | – | 47 | 7 |
| 15b | F | 51 | AAV | PR3 | 132 | 8 |
| 16b | M | 73 | AAV | MPO | 173 | 15 |
| 17b | F | 68 | AAV | PR3 | 86 | 13 |
| 18b | F | 67 | AAV | MPO | 148 | 22 |
| 19b | F | 9 | IgAV | – | 51 | 18 |
| 20b | M | 13 | AAV | PR3 | 84 | 24 |
| 21b | F | 17 | AAV | MPO | 86 | 26 |
| 22b | F | 13 | AAV | PR3 | 48 | 17 |
ANCA: anti-neutrophil cytoplasmic antibody; AAV: ANCA-associated vasculitis; PR3: proteinase 3; MPO: myeloperoxidase; IgAV: IgA-vasculitis (Henoch-Schönlein purpura); SLE: Systemic Lupus Erythematosus.
Samples available from the acute phase and remission.
These patients were previously shown to have increased B1R on endothelial microvesicles [10].
2.2. Blood samples from vasculitis patients
Whole blood from patients and controls was taken by venipuncture into 2·7 mL vacutainer tubes containing 0·5 mL of 0·129 M sodium citrate (Becton Dickinson, Franklin Lakes, NJ). The blood samples underwent serial centrifugation steps to obtain platelet-free plasma as previously described [18]. Plasma samples containing MVs were obtained by washing platelet-free plasma in Hank's balanced salt solution (HBSS without Ca2+, Invitrogen, Carlsbad, CA) and centrifuged for an additional 10 min at 20800g. The pellet was washed twice in HBSS and centrifugation was repeated as above resulting in a MV-enriched suspension.
Certain plasma samples underwent further centrifugation to reduce the EMV content to 7·5% of the original, as previously described [10]. Certain samples were IgG-depleted (to deplete the samples of ANCA) by adsorption onto a protein G Sepharose column (Amersham Biosciences, Uppsala, Sweden). Comparison to the same patients' plasma was carried out after adjusting for the dilution factor, accounted for by measurement of the protein concentrations by spectrophotometry (NanoDrop 1000, NanoDrop Technologies, Wilmington, DE). All buffers used for MV-preparations were pre-filtered to remove pre-existent particles (0·2 μm filters, Schleicher-Schuell Dassel, Germany). The plasma samples were stored in aliquots at −80 °C until used.
2.3. Primary glomerular endothelial cells
Primary glomerular endothelial cells (PGECs) were cultured as previously described [10] and used in perfusion experiments. PGECs were previously shown to express endogenous B1R [18] and glomerular endothelial cells also express B2R [26].
2.4. Perfusion of plasma over PGECs
Complement expression on EMVs was studied using a semi-automated microfluidic perfusion system (VenaFlux, Cellix, Dublin, Ireland). Briefly, microcapillary channels (Vena8 Endothelial+ biochips, Cellix) were pre-coated with fibronectin (100 μg/mL, Sigma-Aldrich, St. Louis, MO) and the PGEC suspension allowed to attach. A Mirus Evo nanopump (Cellix) was used to flow 200 μM histamine (Sigma-Aldrich) to pre-stimulate the PGEC at a shear stress of 5 dynes/cm2. These histamine pre-stimulated PGECs released more EMVs, as previously described [10,27].
Plasma samples from patients and controls, as well as C1-inhibitor-depleted or kininogen-depleted plasma (both from Milan Analytica, Rheinfelden, Switzerland) were centrifuged at 10000g for 5 min before perfusion (to remove cell debris and protein aggregates) and diluted 1:1 in filtered Dulbecco's phosphate buffered saline (DPBS, PAA Laboratories). Samples were perfused over PGEC at a shear stress of 2–5 dynes/cm2 for 5 min. To prevent fibrin polymerization, Gly-Pro-Arg-Pro (10 μM, Sigma-Aldrich) was added to the plasma before perfusion. The levels of kinins and EMVs in the perfused C1-inhibitor-depleted plasma were previously shown to be elevated compared to control plasma [10]. In some experiments C1-inhibitor (final concentration 1 IU, Berinert, CSL Behring, Marburg Germany), the B1R antagonist R715 (1 μM, Tocris Bioscience, Bristol, UK) or the B2R antagonist HOE-140 (1 μM, Sigma-Aldrich) were added to the plasma sample just before perfusion. Both the pre-sample (plasma before perfusion over PGEC) and the samples after perfusion were centrifuged for 5 min at 10000g. The dilution factor was accounted for by measurement of the protein concentrations by spectrophotometry, before storage of the supernatant at −80 °C. EMVs in the samples were assayed by flow cytometry.
2.5. Detection of microvesicles derived from PGECs positive for complement C3 and C9
Detection of EMVs in the plasma samples and the samples obtained from the PGEC perfusion experiments was carried out as previously described [10] using mouse anti-human CD144 (conjugated with phycoerythrin (PE), 1:200, peridinin chlorophyll protein-cyanin5.5 (PerCP-Cy™5.5) 1:600 or fluorescein isothiocyanate (FITC) 1:200) and mouse anti-human CD105:PerCP-Cy™5.5, 1:800. All antibodies, including irrelevant antibodies, were from BD Biosciences, San Jose, CA, except the PE-conjugated antibodies which were from eBioscience, San Diego CA.
To detect surface-bound C3 or C9 on the EMVs mouse anti-human C3 (directed to the neoepitope formed on the cleavage fragments of C3b, iC3b, and C3c, (Hycult Biotech Catalogue # HM2168, RRID: AB_533007) or mouse anti-human C9 neoepitope ((Hycult Biotech Cat# HM2264, RRID: AB_1953581, both at 1:100, Hycult Biotech, Plymouth Meeting, PA) were incubated with the plasma or perfusion samples for 20 min at rt. in the dark. Mouse IgG1 (1:100; Hycult, Biotechnology) was used as the control antibody. The secondary antibody was goat anti-mouse:FITC (1:500, Dako, Glostrup, Denmark, Agilent Cat# F047902, RRID: AB_578665). Before analysis suspensions were washed in DPBS to remove unbound antibodies.
2.6. Detection of complement regulators on EMVs
Patient plasma samples were analysed for the presence of cell-bound complement regulators CD46 and CD55 on EMVs before and after perfusion over PGEC, using mouse anti-human CD46 BB700 (1:800, BD Biosciences Cat# 746019, RRID: AB_2743413) and anti-CD55 BB515 (1:2000, BD Biosciences Cat# 564585, RRID: AB_2732068). Microvesicles were identified as endothelial by using mouse anti-human CD144:PE (1:800, eBioscience).
2.7. Acquisition and interpretation of flow cytometry data
Flow cytometry was performed using two instruments: BD FACSCanto Cytometer using FACSDiva Software version 6.0 (Becton Dickinson Immunocytometry Systems, San Jose, CA) or CyFlow® Cube 8 flow cytometer in which samples were run at a flow rate of 0·2 μL/s (Sysmex, Norderstedt, Partec, Germany) with FCS Express 4 Flow Research Edition software version 4.07.0003 (De Novo Software, Glendale, CA). The latter flow cytometer detects smaller submicron particles and thus identifies more microvesicles.
2.8. Renal tissue from mice
Kidney sections were obtained from mice with nephrotoxic serum (NTS)-induced glomerulonephritis, which were previously described [24]. In this model 6-week-old CD-1 mice were preimmunized by subcutaneous injection of normal sheep IgG (200 μg) in Freunds complete adjuvant (both from Sigma). Five days later the mice were injected intravenously with NTS on three consecutive days, inducing rapidly progressive glomerulonephritis. Certain mice were treated with an oral B1R antagonist (SSR240612, 10 mg/kg every other day) starting two weeks after NTS-injection and continued until mice were sacrificed six weeks later. Mice treated with the B1R antagonist exhibited reduced crescent formation and tubular atrophy, less renal inflammation and improved renal function, compared to untreated mice [24]. Control mice were orally treated with the vehicle consisting of 0·01% DMSO.
All mouse experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local animal care and use committee (Toulouse, France).
2.9. Immunofluorescence staining of C3 in murine renal tissue
Paraffin-embedded tissues were dried for 30 min at 60 °C and deparaffinized by subsequent washing in xylene and ethanol. For antigen retrieval, sections were boiled in citrate buffer (10 mM, pH 6, Merck, Darmstadt, Germany) for 20 min. Unspecific staining was blocked by 5% bovine serum albumin (Sigma-Aldrich) for 1 h at rt., followed by incubation with rabbit anti-C3 (4 μg/mL, Hycult Biotech Cat# HP8012, RRID: AB_533006) at 4 °C overnight, or control rabbit IgG (Dako) at the same concentration. Incubation with secondary goat anti-rabbit:Alexa488 (Invitrogen, Cat# R37116, RRID:AB_2556544, Thermo Fisher Scientific, Carlsbad, California), diluted 1:400 for 1 h at rt. was followed by application of ProLong™ Diamond Antifade Mountant with DAPI (Invitrogen). Kidney sections were visualized using a super resolution microscope system (Nikon Ti-E microscope with N-SIM E), equipped with Hamamatsu Flash 4 camera, using a 20× objective.
Tissues were analysed in blinded fashion for the presence of C3 deposition in glomeruli, and the level of intensity was graded using a scoring system of no staining (0), low (1+), medium (2+) or high (3+) intensity (Supplementary Fig. S1). All glomeruli in each section were counted and a degree of C3 intensity assigned each glomerulus. The degree of C3 intensity (0−1−2−3) was multiplied by the number of glomeruli with a specific C3 intensity and the total level of intensity in each kidney section was thereby calculated. Immunofluorescence was performed on untreated, B1R antagonist-treated as well as healthy control mice.
2.10. Statistics
The Mann-Whitney U test was used to compare EMV levels between patient and control samples, in plasma as well as in the perfusion experiments, and for comparison of C3 intensity in murine kidney sections. Multivariate analysis (perfusion experiments to which inhibitors were added) was carried out using the Kruskal-Wallis multi-comparison test followed by specific comparisons carried out with the Dunn procedure. A P value of ≤0·05 was considered significant. Statistical analysis was performed using GraphPad prism software (GraphPad Software, Version 8, La Jolla, Ca).
3. Results
3.1. Endothelial microvesicles in vasculitis plasma are positive for complement C3 and C9
Flow cytometry was used to analyse plasma from patients with vasculitis (n = 13) and healthy controls (n = 17) for the presence of EMVs, defined as microvesicles positive for CD105 and/or CD144. Significantly more EMVs were positive for C3 and C9 in patient plasma compared to controls (Fig. 1).
Fig. 1.
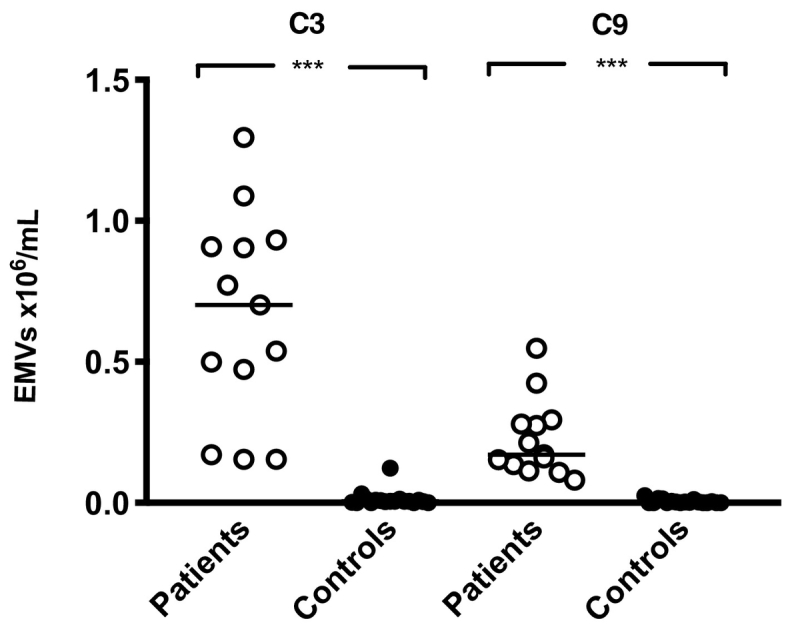
Endothelial microvesicles in vasculitis plasma were positive for complement C3 and C9.
Plasma samples from patients with vasculitis (n = 13, Patients 6–8, 10–18, and 22 in Table 1) exhibited significantly higher levels of circulating endothelial microvesicles (EMVs, positive for CD105 and/or CD144) expressing complement C3 and C9 compared to healthy controls (n = 17) (median 5 × 103/mL and 3 × 103/mL, respectively). ***: P < 0·001. The bar depicts the median. Samples were run using the BD FACSCanto Cytometer.
3.2. Release of C3- and C9-positive EMVs from primary glomerular endothelial cells
Plasma from vasculitis patients (n = 6) and controls (n = 6) was perfused over PGECs (Cell Systems, Kirkland WA) using a microfluidic perfusion system. Patient plasma induced a significant increase in the release of C3- and C9-positive EMVs compared to controls (Fig. 2A and B). Fig. 2A–B depict results of EMV release after perfusion from which microvesicles in the pre-perfusion sample were subtracted (ΔEMVs). EMVs in pre-perfusion and perfusion samples are presented in Supplementary Fig. S2. The percentage of complement-positive EMVs was also higher from PGECs perfused with patient plasma compared to controls, as shown in Supplementary Fig. S3. The findings were confirmed in 5 additional vasculitis patients also perfused over PGECs taken from the acute phase and remission using another flow cytometer to enable detection of smaller microvesicles (Fig. 2C–E). The results showed higher total EMV release from perfused samples taken during the acute phase, compared to remission, and more acute phase EMVs were C9-positive. Results showing absolute values in the pre-perfusion and perfused samples are presented in Supplementary Fig. S4. The effect on the release of complement-positive EMVs was abrogated by reduction of the microvesicle content of vasculitis plasma in the sample before perfusion (Fig. 2F).
Fig. 2.

Release of C3- and C9-positive endothelial microvesicles from primary glomerular endothelial cells during perfusion.
Patient samples were perfused over primary glomerular endothelial cells and shed endothelial microvesicles (EMVs) positive for C3 and C9 were detected by flow cytometry. In panels A-E EMVs in the perfused sample are presented after subtraction of EMVs in the pre-perfused sample. A) Patient samples from vasculitis patients (Patients 7–9, 12, 19, and 22 in Table 1) released significantly higher levels of C3-positive EMVs compared to controls (the lowest value of C3-positive EMVs in control plasma was 97 × 103/mL, see Supplementary Fig. S2 for absolute values). B) Perfused patient samples released more C9-positive EMVs than controls (the lowest value of C9-positive EMVs in control plasma was 9 × 104/mL see Supplementary Fig. S2 for absolute values). C) Perfused plasma from patients with vasculitis (Patients 1–5 in Table 1) released significantly more EMVs during the acute phase of disease compared to the same patient samples taken during remission and perfused over PGECs (lowest value in remission was 3·2 × 108/mL). D) The acute samples had C3 deposits on EMVs but not significantly more than at remission (lowest value in remission was 8·5 × 107/mL). E) The acute samples exhibited significantly more C9-positive EMVs compared to samples from remission (lowest value in remission was 1·3 × 107/mL). F) Patient plasma samples in which microvesicles were reduced did not induce the release of C3- and C9-positive EMVs after perfusion. **: P < 0·01. *: P < 0·05. ns: not significant. The bar represents the median. Samples were analysed using a FACSCanto Cytometer in panels A and B and a CyFlow Cube 8 flow cytometer in panels C, D, E, and F.
As patient samples perfused over PGECs induced the release of C3- and C9-positive EMVs we examined if the cell-bound complement regulators CD46 and CD55 were reduced under these conditions. Perfusion did not affect the expression of these complement regulators (Supplementary Fig. S5).
3.3. Kinin-receptor antagonists and C1-inhibitor decrease release of complement-positive EMVs from PGECs
Vasculitis patient plasma samples (n = 5) were pretreated with the kinin B1R antagonist R715 and the B2R antagonist HOE-140 (alone or in combination), or C1-inhibitor, before perfusion over PGECs. Results, presented after deduction of EMVs in the pre-perfusion sample, show that combined B1R and B2R inhibition as well as C1-inhibitor significantly decreased the total number of shed EMVs as well as those with C3 and C9 deposits (Fig. 3). Results showing absolute values of EMVs in the pre-perfusion and perfused samples are presented in Supplementary Fig. S6.
Fig. 3.

Kinin-receptor antagonists and C1-inhibitor decreased release of complement-positive endothelial microvesicles from primary glomerular endothelial cells.
Plasma from vasculitis patients (Patients 1–5 in Table 1) was perfused over primary glomerular endothelial cells in the presence of the kinin B1R antagonist R715 and the B2R antagonist HOE-140 (alone or in combination), or C1-inhibitor. Released C3- and C9-positive endothelial microvesicles (EMVs) were measured by flow cytometry and individual patient samples are marked in color. EMVs in pre-perfusion samples have been deducted from perfused samples. A) Adding kinin-receptor antagonists in combination or C1-inhibitor significantly reduced the total amount of EMVs released. B) The release of C3-positive EMVs was significantly reduced by pre-treating the plasma with kinin-receptor antagonists in combination or C1-inhibitor. C) The release of C9-positive EMVs was significantly reduced by adding kinin-receptor antagonists in combination or C1-inhibitor. **: P < 0·01. *: P < 0·05. ns: not significant. C1INH: C1-inhibitor. The median is represented by the bar. Samples were run using CyFlow Cube 8 flow cytometer.
3.4. IgG-depletion of vasculitis plasma did not affect EMV shedding from PGECs
Samples from 2 patients were IgG-depleted in order to remove ANCA. IgG-depletion did not reduce the total number of EMVs or the C3- and C9-positive EMVs shed from perfused PGECs (data not shown).
3.5. Kinin-receptor blockade reduced complement-positive EMVs after perfusion with C1-inhibitor-depleted plasma
EMVs were released from PGECs perfused with C1-inhibitor-depleted plasma (Fig. 4A, presented after deduction of the pre-perfusion sample). Addition of the B2R antagonist HOE-140, alone or in combination with the B1R antagonist R715, significantly reduced EMV shedding, compared to cells perfused with C1-inhibitor-depleted plasma alone. C1-inhibitor-depleted plasma perfused over PGECs induced the release of EMVs coated with complement C3 and C9. PGECs exposed to C1-inhibitor-depleted plasma in the presence of either or both B1R- and B2R antagonists exhibited significantly lower C3-positive EMVs (Fig. 4B). Similar results were obtained when detecting C9 on the released EMVs although the reduction was not significant using the B1R antagonist alone (Fig. 4C). Results showing absolute values of released EMVs in samples before and after perfusion are presented in Supplementary Fig. S7. The results suggest that kinin-receptor antagonists reduce the total number of EMVs as well as the level of complement C3 and C9 deposits on the EMVs in C1-inhibitor-depleted plasma.
Fig. 4.

Kinin-receptor antagonists reduced release of C3- and C9-positive endothelial microvesicles after perfusion of C1-inhibitor-depleted plasma over primary glomerular endothelial cells.
C1-inhibitor-depleted plasma was perfused over primary glomerular endothelial cells (PGECs) alone or after addition of the B1R antagonist R715, the B2R antagonist HOE-140, or both antagonists combined and released C3- and C9-positive endothelial microvesicles (EMVs) were detected by flow cytometry. EMVs in pre-perfusion samples have been deducted from perfused samples. A) The total amount of EMVs released from PGECs perfused with C1-inhibitor-depleted plasma after addition of the B2R antagonist HOE-140, alone or in combination with the B1R antagonist R715, was significantly reduced, compared to cells perfused with C1-inhibitor-depleted plasma alone. The B1R antagonist R715 alone did not significantly reduce released EMVs from the cells. B) PGECs exposed to C1-inhibitor-depleted plasma in the presence of either or both the B1R- and B2R antagonists exhibited significantly lower C3-positive EMVs. C) PGECs exposed to C1-inhibitor-depleted plasma exhibited less C9 on the released EMVs when the plasma was incubated with the B2R antagonist, alone or in combination with the B1R antagonist, but the reduction was not significant using the B1R antagonist alone. **: P < 0·01, *: P < 0·05. ns: not significant. The bar represents the median. Samples were run using a CyFlow Cube 8 flow cytometer.
3.6. C1-inhibitor reduced complement-positive EMVs after perfusion with C1-inhibitor-depleted plasma
Addition of C1-inhibitor (Berinert) to the C1-inhibitor-depleted plasma significantly reduced the shedding of EMVs positive for complement C3 and C9 from PGECs (Fig. 5A and B). Absolute values of released EMVs in samples before and after perfusion are presented in Supplementary Fig. S8.
Fig. 5.

C1-inhibitor reduced the release of C3- and C9-positive endothelial microvesicles after perfusion of C1-inhibitor-depleted plasma over primary glomerular endothelial cells.
C1-inhibitor-depleted plasma was perfused over primary glomerular endothelial cells (PGECs) and shed C3- and C9-positive endothelial microvesicles (EMVs) were detected by flow cytometry. EMVs in pre-perfusion samples have been deducted from perfused samples. A) C1-inhibitor-depleted plasma perfused over PGECs released EMVs positive for C3 that were higher than control plasma (median EMVs in control plasma was 5·9 × 107/mL). Addition of C1-inhibitor to the C1-inhibitor-depleted plasma significantly reduced the C3-positive EMVs. B) C1-inhibitor-depleted plasma perfused over PGECs released EMVs positive for C9 that were higher than control plasma (median in control plasma 5·1 × 106/mL). Addition of C1-inhibitor to the C1-inhibitor-depleted plasma significantly reduced the C9-positive EMVs. **: P < 0·01, *: P < 0·05. C1INH: C1-inhibitor. Dpl: depleted. The bar represents the median. Samples were analysed using a CyFlow Cube 8 flow cytometer.
3.7. Complement deposits on EMVs did not increase in kininogen-depleted plasma
PGEC were perfused with kininogen-depleted plasma (to which prekallilrein was supplemented and its activity tested by the manufacturer). The total number of EMVs did not increase significantly. Likewise, C3- and C9-positive EMVs did not increase compared to control plasma (Fig. 6). The contribution of KKS to release of C3- and C9-positive EMVs during perfusion was assessed by comparing the release of EMVs from C1-inhibitor-depleted plasma (median C3 1·76 × 109/mL, median C9 1·02 × 109/mL) to kininogen-depleted plasma (median C3 5·64 × 105/mL, median C9 1·36 × 106/mL, P < 0·01 for both).
Fig. 6.

C3- and C9 expression on endothelial microvesicles did not increase when kininogen-depleted plasma was perfused over primary glomerular endothelial cells.
Kininogen-depleted (kininogen-dpl) plasma was perfused over primary glomerular endothelial cells (PGECs) and the total (A), C3-positive (B) or C9-positive (C) endothelial microvesicles (EMVs) were detected in the plasma by flow cytometry, and compared to control plasma. C3- and C9-positive EMVs did not increase compared to control samples. ns: not significant. The median is depicted by the bar. Samples were analysed using a CyFlow Cube 8 flow cytometer.
3.8. B1R antagonist reduced complement in murine kidneys with nephrotoxic serum-induced glomerulonephritis
Kidney sections from mice with nephrotoxic serum (NTS)-induced glomerulonephritis exhibited C3 deposition in glomerular capillary walls as presented in Supplementary Fig. S1. The intensity of C3 deposition in glomeruli was quantified and found to be significantly lower in mice treated with the oral B1R antagonist SSR240612 compared to untreated mice sacrificed 6 weeks after treatment (P < 0·05, Fig. 7 and Table 2). Healthy control mice exhibited negligible C3 deposition (Table 2).
Fig. 7.
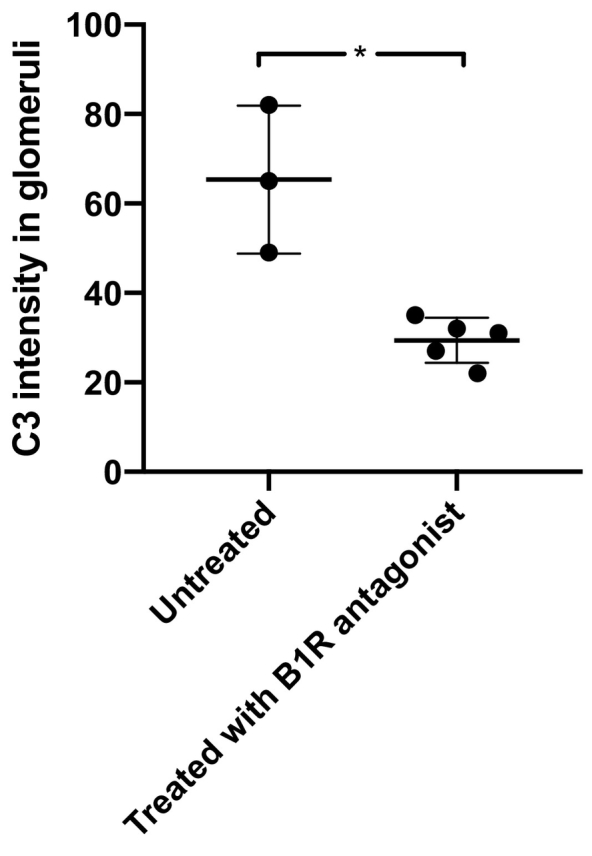
C3 deposition in glomeruli of mice with nephrotoxic serum-induced glomerulonephritis decreased when treated with a B1 receptor antagonist.
Mice with nephrotoxic serum-induced glomerulonephritis were left untreated (n = 3) or treated with the oral B1R antagonist SSR240612 (n = 5). Renal tissues were stained for C3 and the degree of intensity was calculated as per Table 2. Significantly more C3 deposition was seen in mice given vehicle, compared to mice treated with the B1R-antagonist, *: P < 0·05. The bar represents the median.
Table 2.
Intensity of C3 deposition in kidneys of mice with glomerulonephritis and controls.
| Mouse | Intensity of C3 staining |
|||||
|---|---|---|---|---|---|---|
| Total number of glomeruli | 0 (%) | 1 (%) | 2 (%) | 3 (%) | Totala | |
| Untreated | ||||||
| 1 | 48 | 8 (16·7) | 23 (47·9) | 9 (18·8) | 8 (16·7) | 65 |
| 2 | 46 | 21 (45·7) | 11 (23·9) | 4 (8·7) | 10 (21·7) | 49 |
| 3 | 51 | 5 (9·8) | 21 (41·2) | 14 (27·5) | 11 (21·6) | 82 |
| Treated with the B1R antagonist | ||||||
| 4 | 56 | 38 (67·9) | 7 (30·4) | 1 (1·8) | 1 (1·8) | 22 |
| 5 | 55 | 31 (56·4) | 15 (27·3) | 7 (12·7) | 2 (3·6) | 35 |
| 6 | 42 | 17 (40·5) | 22 (52·4) | 0 (0) | 3 (7·1) | 31 |
| 7 | 47 | 25 (53·2) | 15 (31·9) | 4 (8·5) | 3 (6·4) | 32 |
| 8 | 50 | 30 (60) | 13 (26) | 7 (14) | 0 (0) | 27 |
| Controls | ||||||
| 9 | 39 | 34 (87·2) | 4 (10·3) | 1 (2·6) | 0 (0) | 6 |
| 10 | 42 | 28 (66·7) | 15 (35·7) | 4 (9·5) | 0 (0) | 23 |
| 11 | 46 | 37 (80·4) | 7 (15·2) | 2 (4·3) | 0 (0) | 11 |
| 12 | 51 | 38 (74·5) | 10 (19·6) | 3 (5·9) | 0 (0) | 16 |
All glomeruli in each section were counted and graded in blinded fashion after intensity as follows: no staining (0), low (1+), medium (2+) and high (3+) as depicted in Supplementary Fig. S1. The total level of intensity was calculated for each section as the number of glomeruli multiplied by the level of intensity.
4. Discussion
Kinin peptides are released from high-molecular weight kininogen during activation of the KKS and bind to their receptors, B1R and B2R, present on the endothelium [28,29] inducing a prothrombotic [30] and proinflammatory state with neutrophil recruitment and vascular leakage [10,31]. The kallikrein-kinin and complement systems can be triggered in parallel on the endothelium during inflammation [29] and both systems are activated, locally and systemically, during vasculitis [7,11,13,32]. In this study we found that blocking kinin signaling on the endothelium had a modulating effect on complement activation, as reflected in decreased circulating complement-coated endothelial-derived microvesicles and decreased C3 deposition in glomerular capillary walls. Blockade of vasculitis plasma or C1-inhibitor-depleted plasma with a combination of B1R and B2R antagonists, or addition of C1-inhibitor, diminished complement deposition on EMVs in vitro and a B1R antagonist reduced complement deposition in glomeruli in rapidly progressive glomerulonephritis in vivo, suggesting that these interventions may decrease complement-mediated vascular inflammation and thereby have therapeutic potential.
There are several links between the complement system and KKS. Both systems can be activated on the gC1q-receptor present on the endothelium, which binds both C1q and high-molecular weight kininogen [33], and both the classical pathway of complement and the KKS are inhibited by C1-inhibitor [9]. Furthermore, kallikrein, that cleaves high-molecular weight kininogen and releases bradykinin, has also been shown to cleave and activate C3 in vitro [34]. C3a is an important anaphylatoxin byproduct of C3 cleavage, that is further degraded and inactivated by carboxypeptidase N [35], which also degrades bradykinin [36]. Although these two systems share multiple points of association no previous study has shown that reducing KKS signaling on the endothelium decreases complement activation, as shown here, using B1R and B2R antagonists as well as C1-inhibitor.
We have demonstrated that complement components C3 and C9 are present on circulating endothelial cell-derived microvesicles in vasculitis. This finding could be reproduced when vasculitis plasma was perfused over glomerular endothelial cells, inducing the release of C3- and C9-positive EMVs. A similar finding was noted when C1-inhibitor depleted plasma was perfused over the cells. Lack of C1-inhibitor will allow uninhibited activation of the KKS, however, patients with vasculitis were found to have normal levels of C1-inhibitor [11]. Complementing C1-inhibitor-depleted plasma with C1-inhibitor blocked excess complement activation on EMVs. C1-inhibitor blocks the activity of plasma kallikrein but not tissue kallikrein [37] and thus the effect demonstrated was most likely related to inhibition of plasma kallikrein.
C1-inhibitor also blocks activation of the classical pathway of complement [38]. Previous studies have shown that complement activation in vasculitis primarily occurs via activation of the alternative pathway [13,39,40]. Deposition of the factor B cleavage product Bb in patient glomeruli correlated with the severity of renal disease [41]. In an animal model of AAV factor B- and C5-deficiency were protective, whereas C4-deficiency was not [40]. In patients with IgA-vasculitis (Henoch Schönlein purpura) activation of the alternative pathway was reported [42] whereas in systemic lupus erythematosus activation of the classical pathway appears to be of predominant importance [43]. ANCA-stimulated neutrophils activate complement and generate C3a in serum [40] followed by generation of C5a. C5a in turn activates more neutrophils, by receptor binding, and thus this mechanism of activation is amplified [44]. This concept requires the presence of neutrophils and/or ANCA for complement activation.
The current study demonstrates that complement activation can occur in the absence of neutrophils and ANCA. Vasculitis plasma activated complement on the endothelium, or on the EMVs, in the absence of blood cells. The presence of MVs in patient plasma may contain neutrophil-derived MVs, as our group, and others, have shown [18,45]. Potentially these MVs could have the same complement-activating effect as the neutrophils themselves. As reduction of MVs in patient plasma decreased complement activation on EMVs it is plausible that leukocyte-derived MVs in the circulation contribute to complement activation on the endothelium. IgG removal, albeit only in a limited number of patients, did not reduce complement deposits. Thus, we assume that ANCA did not promote complement deposition on the EMVs, a finding that requires corroboration in a larger patient sample.
The effects of kinin-receptor antagonism or C1-inhibitor suggest that KKS activation triggers complement activation on the endothelium. In line with these findings complement deposition did not increase in kininogen-depleted plasma. Interestingly, C1-inhibitor has been suggested as a treatment for renal injury in a model of renal ischemia-reperfusion [46] and in patients that develop antibody-mediated rejection after renal transplantation [47]. C1-inhibitor is commercially available but its use has not been reported in patients with vasculitis. Although complement activation in vasculitis usually occurs via the alternative pathway, our findings suggest that C1-inhibitor will block kinin signaling, and thereby regulate complement activation.
Our results do not allow us to differentiate between complement deposition on the endothelium itself, followed by shedding onto EMVs or direct complement activation on the EMVs. Possibly both phenomena occur. The in vivo data from the NGS-induced glomerulonephritis model suggest complement activation on the endothelium. This may occur when activation exceeds the protective effect of regulators, or if the effect of complement regulators is diminished in the presence of vasculitis/glomerulonephritis plasma. Membrane-bound complement regulators (CD59, CD46 and CD55) have been detected on the endothelium suggesting that they could be detached with the shed EMVs [[48], [49], [50]]. We detected CD46 and CD55 on the EMVs in patient plasma but their expression was not affected by perfusion in a manner that could explain complement deposition on patient EMVs after perfusion.
Complement activation in vasculitis is being addressed in the clinical trial of CCX168 (avacopan), a small molecule inhibitor of C5aR [51,52]. In the current study we suggest that blocking kinin signaling will have a beneficial effect on complement activation. We speculate that signaling via kinin B2 or B1 receptors, expressed constitutively, or upregulated during inflammation, will trigger an endothelial cell response allowing complement to deposit on the endothelium. Blocking these receptors, or the degradation of high-molecular weight kininogen, will diminish the inflammatory response, as demonstrated here in vitro and in vivo. These data are strengthened by perfusion experiments in the presence of kininogen-depleted plasma that did not show enhanced complement deposition.
Kinin-receptor antagonists and C1-inhibitor both blocked complement deposition on EMVs. As complement activation via the alternative pathway seems more important in the forms of vasculitis studied here we assume that C1-inhibitor mediated its affect via blocking the kinin system. Our previous results showed that C1-inhibitor blocked KKS activation on EMVs and their chemotactic effect [10] and thus this potent inhibitor could have a beneficial effect on the vasculature during inflammation, as it blocks both kinin and complement activation. Importantly, the B2R antagonist, Icatibant, and C1-inhibitor are both available therapeutics in the clinic that should be investigated for treatment of vascular inflammation in future studies.
Funding sources
The study was supported by The Swedish Research Council (K2015-99X-22877-01-6 and 2017-01920), The Knut and Alice Wallenberg Foundation (Wallenberg Clinical Scholar 2015.0320), The Torsten Söderberg Foundation, Skåne Centre of Excellence in Health, IngaBritt och Arne Lundberg's Research Foundation, Crown Princess Lovisa's Society for Child Care, Region Skåne and The Konung Gustaf V:s 80-årsfond (all to DK). Alfred Österlund Foundation (to LMFLL and RK). The Wallenberg Center for Molecular Medicine, The Swedish Rheumatism Association, The Anna-Greta Crafoord Foundation, Greta and Johan Kock's Foundation, the Samariten Foundation, Fanny Ekdahl foundation, the Jerring foundation and the Thelma Zoegas Foundation (to RK). JPS and JK were partially funded by a grant from the “Fondation pour la Recherche Médicale” (grant number DEQ20170336759). MS was funded by The Swedish Rheumatism Association and the Ingrid Asp Foundation. The funding sources had no role in the study design, the collection, analysis, and interpretation of data, the writing of the paper and in the decision to submit the paper for publication.
Author contributions
I.L.F, A-L.S, M.M, R.T. A-C.K., J-L.B., J.K., J.P.S., M.S., D.K. designed the study, I.L.F., A-L.S., M.M, R.T., A-C.K. J-L.B., J.K. carried out experiments, I.L.F., A.L.S., M.M, R.T. A-C.K., R.K, J-L.B., J.K., J.P.S., D.K analysed the data, I.L.F, A-L.S., M.M. made the figures, I.L.F., M.M., D.K. drafted the manuscript, all authors approved of the final version of the manuscript.
Declaration of Competing Interest
Mårten Segelmark has received consultancy fees from ChemoCentryx. The other authors report no conflicts of interest.
Acknowledgements
The authors thank Dr. Caroline Heijl, Dept of Cardiology, Skåne University Hospital Lund, for samples from four patients treated at the Dept of Nephrology, Skåne University Hospital. A preliminary version of this paper appeared in the PhD thesis of Dr. Maria Mossberg.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2019.08.020.
Appendix A. Supplementary data
Supplementary figures.
References
- 1.Fischetti F., Tedesco F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity. 2006;39:417–428. doi: 10.1080/08916930600739712. [DOI] [PubMed] [Google Scholar]
- 2.Walport M.J. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Ricklin D., Hajishengallis G., Yang K., Lambris J.D. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karpman D., Ståhl A.L., Arvidsson I. Complement interactions with blood cells, endothelial cells and microvesicles in thrombotic and inflammatory conditions. Adv Exp Med Biol. 2015;865:19–42. doi: 10.1007/978-3-319-18603-0_2. [DOI] [PubMed] [Google Scholar]
- 5.Ekdahl K.N., Teramura Y., Hamad O.A. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev. 2016;274:245–269. doi: 10.1111/imr.12471. [DOI] [PubMed] [Google Scholar]
- 6.Colman R.W., Schmaier A.H. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. [PubMed] [Google Scholar]
- 7.Kahn R., Hellmark T., Leeb-Lundberg L.M. Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol. 2009;182:7906–7915. doi: 10.4049/jimmunol.0803624. [DOI] [PubMed] [Google Scholar]
- 8.Leeb-Lundberg L.M., Marceau F., Müller-Esterl W., Pettibone D.J., Zuraw B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 9.Bossi F., Peerschke E.I., Ghebrehiwet B., Tedesco F. Cross-talk between the complement and the kinin system in vascular permeability. Immunol Lett. 2011;140:7–13. doi: 10.1016/j.imlet.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mossberg M., Ståhl A.L., Kahn R. C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J Am Soc Nephrol. 2017;28:2472–2481. doi: 10.1681/ASN.2016060637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahn R., Herwald H., Müller-Esterl W. Contact-system activation in children with vasculitis. Lancet. 2002;360:535–541. doi: 10.1016/S0140-6736(02)09743-X. [DOI] [PubMed] [Google Scholar]
- 12.Kallenberg C.G., Heeringa P. Complement system activation in ANCA vasculitis: a translational success story? Mol Immunol. 2015;68:53–56. doi: 10.1016/j.molimm.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Chen M., Jayne D.R.W., Zhao M.H. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. 2017;13:359–367. doi: 10.1038/nrneph.2017.37. [DOI] [PubMed] [Google Scholar]
- 14.Eleftheriou D., Dillon M.J., Brogan P.A. Advances in childhood vasculitis. Curr Opin Rheumatol. 2009;21:411–418. doi: 10.1097/BOR.0b013e32832c49f2. [DOI] [PubMed] [Google Scholar]
- 15.Watts R.A., Mahr A., Mohammad A.J., Gatenby P., Basu N., Flores-Suarez L.F. Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant. 2015;30(Suppl. 1):i14–i22. doi: 10.1093/ndt/gfv022. [DOI] [PubMed] [Google Scholar]
- 16.Segelmark M., Wieslander J. IgG subclasses of antineutrophil cytoplasm autoantibodies (ANCA) Nephrol Dial Transplant. 1993;8:696–702. doi: 10.1093/ndt/8.8.696. [DOI] [PubMed] [Google Scholar]
- 17.Wester Trejo M.A.C., Trouw L.A., Bajema I.M. The role of complement in antineutrophil cytoplasmic antibody-associated vasculitis. Curr Opin Rheumatol. 2019;31:3–8. doi: 10.1097/BOR.0000000000000557. [DOI] [PubMed] [Google Scholar]
- 18.Kahn R., Mossberg M., Ståhl A.L. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. 2017;91:96–105. doi: 10.1016/j.kint.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Dignat-George F., Boulanger C.M. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. 2011;31:27–33. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 20.Brogan P.A., Shah V., Brachet C. Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. 2004;50:927–936. doi: 10.1002/art.20199. [DOI] [PubMed] [Google Scholar]
- 21.Brogan P.A., Dillon M.J. Endothelial microparticles and the diagnosis of the vasculitides. Intern Med. 2004;43:1115–1119. doi: 10.2169/internalmedicine.43.1115. [DOI] [PubMed] [Google Scholar]
- 22.Kumpers P., Erdbrugger U., Grossheim M. Endothelial microparticles as a diagnostic aid in Churg-Strauss vasculitis-induced cardiomyopathy. Clin Exp Rheumatol. 2008;26:S86–S89. [PubMed] [Google Scholar]
- 23.Clarke L.A., Hong Y., Eleftheriou D. Endothelial injury and repair in systemic vasculitis of the young. Arthritis Rheum. 2010;62:1770–1780. doi: 10.1002/art.27418. [DOI] [PubMed] [Google Scholar]
- 24.Klein J., Gonzalez J., Decramer S. Blockade of the kinin B1 receptor ameloriates glomerulonephritis. J Am Soc Nephrol. 2010;21:1157–1164. doi: 10.1681/ASN.2009090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jennette J.C., Falk R.J. Bacon PA, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 26.Cerrato B.D., Carretero O.A., Janic B., Grecco H.E., Gironacci M.M. Heteromerization between the Bradykinin B2 receptor and the angiotensin-(1-7) mas receptor: functional consequences. Hypertension. 2016;68:1039–1048. doi: 10.1161/HYPERTENSIONAHA.116.07874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tati R., Kristoffersson A.C., Ståhl A.L. Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J Immunol. 2013;191:2184–2193. doi: 10.4049/jimmunol.1301221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regoli D. Neurohumoral regulation of precapillary vessels: the kallikrein-kinin system. J Cardiovasc Pharmacol. 1984;6(Suppl. 2):S401–S412. doi: 10.1097/00005344-198406002-00015. [DOI] [PubMed] [Google Scholar]
- 29.Ghebrehiwet B., Kaplan A.P., Joseph K., Peerschke E.I. The complement and contact activation systems: partnership in pathogenesis beyond angioedema. Immunol Rev. 2016;274:281–289. doi: 10.1111/imr.12469. [DOI] [PubMed] [Google Scholar]
- 30.Maas C., Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131:1903–1909. doi: 10.1182/blood-2017-04-569111. [DOI] [PubMed] [Google Scholar]
- 31.de Maat S., Tersteeg C., Herczenik E., Maas C. Tracking down contact activation - from coagulation in vitro to inflammation in vivo. Int J Lab Hematol. 2014;36:374–381. doi: 10.1111/ijlh.12222. [DOI] [PubMed] [Google Scholar]
- 32.Xing G.Q., Chen M., Liu G. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–291. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 33.Joseph K., Ghebrehiwet B., Peerschke E.I., Reid K.B., Kaplan A.P. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor that binds to the globular "heads" of C1q (gC1q-R) Proc Natl Acad Sci U S A. 1996;93:8552–8557. doi: 10.1073/pnas.93.16.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irmscher S., Doring N., Halder L.D. Kallikrein cleaves C3 and activates complement. J Innate Immun. 2018;10:94–105. doi: 10.1159/000484257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell W.D., Lazoura E., Okada N., Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2002;46:131–134. doi: 10.1111/j.1348-0421.2002.tb02669.x. [DOI] [PubMed] [Google Scholar]
- 36.Kuoppala A., Lindstedt K.A., Saarinen J., Kovanen P.T., Kokkonen J.O. Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma. Am J Physiol Heart Circ Physiol. 2000;278:H1069–H1074. doi: 10.1152/ajpheart.2000.278.4.H1069. [DOI] [PubMed] [Google Scholar]
- 37.Bhoola K.D., Misso N.L., Naran A., Thompson P.J. Current status of tissue kallikrein inhibitors: importance in cancer. Curr Opin Investig Drugs. 2007;8:462–468. [PubMed] [Google Scholar]
- 38.Petersen S.V., Thiel S., Jensen L., Vorup-Jensen T., Koch C., Jensenius J.C. Control of the classical and the MBL pathway of complement activation. Mol Immunol. 2000;37:803–811. doi: 10.1016/s0161-5890(01)00004-9. [DOI] [PubMed] [Google Scholar]
- 39.Noone D., Hebert D., Licht C. Pathogenesis and treatment of ANCA-associated vasculitis-a role for complement. Pediatr Nephrol. 2018;33:1–11. doi: 10.1007/s00467-016-3475-5. [DOI] [PubMed] [Google Scholar]
- 40.Xiao H., Schreiber A., Heeringa P., Falk R.J., Jennette J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gou S.J., Yuan J., Wang C., Zhao M.H., Chen M. Alternative complement pathway activation products in urine and kidneys of patients with ANCA-associated GN. Clin J Am Soc Nephrol. 2013;8:1884–1891. doi: 10.2215/CJN.02790313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y.H., Tsai I.J., Chang C.J., Chuang Y.H., Hsu H.Y., Chiang B.L. The interaction between circulating complement proteins and cutaneous microvascular endothelial cells in the development of childhood Henoch-Schonlein Purpura. PLoS One. 2015;10 doi: 10.1371/journal.pone.0120411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen M., Daha M.R., Kallenberg C.G. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276–J286. doi: 10.1016/j.jaut.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber A., Xiao H., Jennette J.C., Schneider W., Luft F.C., Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daniel L., Fakhouri F., Joly D. Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int. 2006;69:1416–1423. doi: 10.1038/sj.ki.5000306. [DOI] [PubMed] [Google Scholar]
- 46.Castellano G., Melchiorre R., Loverre A. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176:1648–1659. doi: 10.2353/ajpath.2010.090276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montgomery R.A., Orandi B.J., Racusen L. Plasma-derived C1 esterase inhibitor for acute antibody-mediated rejection following kidney transplantation: results of a randomized double-blind placebo-controlled pilot study. Am J Transplant. 2016;16:3468–3478. doi: 10.1111/ajt.13871. [DOI] [PubMed] [Google Scholar]
- 48.McNearney T., Ballard L., Seya T., Atkinson J.P. Membrane cofactor protein of complement is present on human fibroblast, epithelial, and endothelial cells. J Clin Invest. 1989;84:538–545. doi: 10.1172/JCI114196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asch A.S., Kinoshita T., Jaffe E.A., Nussenzweig V. Decay-accelerating factor is present on cultured human umbilical vein endothelial cells. J Exp Med. 1986;163:221–226. doi: 10.1084/jem.163.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamilton K.K., Ji Z., Rollins S., Stewart B.H., Sims P.J. Regulatory control of the terminal complement proteins at the surface of human endothelial cells: neutralization of a C5b-9 inhibitor by antibody to CD59. Blood. 1990;76:2572–2577. [PubMed] [Google Scholar]
- 51.Jayne D.R.W., Bruchfeld A.N., Harper L. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated Vasculitis. J Am Soc Nephrol. 2017;28:2756–2767. doi: 10.1681/ASN.2016111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kettritz R. Vasculitis: A CLEAR argument for targeting complement in ANCA vasculitis. Nat Rev Nephrol. 2017;13:448–450. doi: 10.1038/nrneph.2017.69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures.