

Abstract
Background
One of the biggest challenge in Alzheimer's disease (AD) is to identify pathways and markers of disease prediction easily accessible, for prevention and treatment. Here we analysed blood samples from the INveStIGation of AlzHeimer's predicTors (INSIGHT-preAD) cohort of elderly asymptomatic individuals with and without brain amyloid load.
Methods
We performed blood RNAseq, and plasma metabolomics and lipidomics using liquid chromatography-mass spectrometry on 48 individuals amyloid positive and 48 amyloid negative (SUVr cut-off of 0·7918). The three data sets were analysed separately using differential gene expression based on negative binomial distribution, non-parametric (Wilcoxon) and parametric (correlation-adjusted Student't) tests. Data integration was conducted using sparse partial least squares-discriminant and principal component analyses. Bootstrap-selected top-ten features from the three data sets were tested for their discriminant power using Receiver Operating Characteristic curve. Longitudinal metabolomic analysis was carried out on a subset of 22 subjects.
Findings
Univariate analyses identified three medium chain fatty acids, 4-nitrophenol and a set of 64 transcripts enriched for inflammation and fatty acid metabolism differentially quantified in amyloid positive and negative subjects. Importantly, the amounts of the three medium chain fatty acids were correlated over time in a subset of 22 subjects (p < 0·05). Multi-omics integrative analyses showed that metabolites efficiently discriminated between subjects according to their amyloid status while lipids did not and transcripts showed trends. Finally, the ten top metabolites and transcripts represented the most discriminant omics features with 99·4% chance prediction for amyloid positivity.
Interpretation
This study suggests a potential blood omics signature for prediction of amyloid positivity in asymptomatic at-risk subjects, allowing for a less invasive, more accessible, and less expensive risk assessment of AD as compared to PET studies or lumbar puncture.
Fund
Institut Hospitalo-Universitaire and Institut du Cerveau et de la Moelle Epiniere (IHU-A-ICM), French Ministry of Research, Fondation Alzheimer, Pfizer, and Avid.
Keywords: Alzheimer, Asymptomatic, Amyloid PET, Biomarkers, Prediction, Multi-omics
Research in context.
Evidence before this study
The INSIGHT-preAD cohort designed to investigate Alzheimer's predictors in subjective memory complainers has been published in 2018 in Lancet Neurology (PMID 29500152). No study combining multi-omics had been performed on this cohort, or on other similar cohorts worldwide. We searched PubMed with the terms “preclinical Alzheimer(’s) disease”, “presymptomatic Alzheimer(’s) disease”, and “asymptomatic Alzheimer(’s) disease” together with “multiomics”, “multi-omics” for articles published up to June 14, 2019, without any language restrictions.
Added value of the study
Our study is to our knowledge the first to combine transcriptomic, metabolomics and lipidomic analyses to uncover a new blood biomarker signature of early amyloid deposition in asymptomatic individuals at risk for Alzheimer's disease with 99·4% chance prediction.
Implications of all the available evidence
Finding blood biomarker signatures predicting early amyloid deposition in the brain, avoiding PET scan with injection of radio ligand or lumbar puncture is highly relevant to human health. It is also paramount for future presymptomatic treatments in individuals at risk for Alzheimer's disease.
Alt-text: Unlabelled Box
1. Introduction
Alzheimer's disease (AD) is the most common cause of dementia and a major health problem due to number of cases (>130 million worldwide anticipated in 2050) and unmet medical needs. Clinical trials conducted in patients with either dementia, subjective cognitive decline, or mild cognitive impairments (MCI) have been unsuccessful so far, while studies in cohorts of individuals at risk either carrying familial mutations (DIAN) or the APOEε4 allele are still ongoing [1]. Besides pathological amyloid peptides, phosphorylated tau, and genetic risk factors, many biological pathways are modified in AD (HSA 05010 KEGG pathway) [[2], [3], [4], [5]]. Metabolic changes such as impaired brain glucose uptake, altered lipid metabolism, synaptic dysfunction, mitochondrial, endolysosomal and autophagy alterations, and inflammation can occur before the onset of clinical symptoms and are not always correlated with pathological hallmarks or cognitive deficits [6,7]. Most of these changes are found in the brain of patients with AD after autopsy and few can be assessed before death. Even fewer are detected before clinical symptoms although it is noticeable that brain glucose hypometabolism occurs many years before symptom onset in presymptomatic carriers of PSEN1, PSEN2 and APP mutations, underlying early metabolic changes in the disease process [8]. Still, changes in the peripheral fluids remain to be discovered [9]. Thus, one of the biggest challenges in AD is to identify pathways and markers of disease progression, which can be easily accessible, in asymptomatic at-risk individuals. Here we analysed blood samples from The Investigation of Alzheimer's Predictors in Subjective Memory Complainers (INSIGHT-preAD) study designed to identify risk markers of progression to clinical AD in asymptomatic at-risk individuals [10]. We selected 48 amyloid (+) and 48 amyloid (−) subjects and performed extensive omics analyses: transcriptomics using RNAseq, metabolomics, and lipidomics. Besides univariate and multivariate methods, we applied multi-block approaches for integrating amyloid imaging to metabolomics, lipidomics, and transcriptomics datasets to unravel new panels of biomarkers predicting amyloid deposition in elderly asymptomatic individuals.
2. Materials and methods
2.1. Cohort
The INSIGHT-preAD cohort is formed by 318 volunteers who were included after consulting at the Pitié-Salpêtrière University Hospital for memory complaints, but who presented no objective clinical impairment after extensive neuropsychological evaluation [10]. The participants were followed in a longitudinal manner, undergoing neuroimaging and APOE allele examination. All the individuals from the cohort underwent 18F-florbetapir PET scan to assess their brain amyloid status in order to classify them as amyloid (+) or amyloid (−). We considered global SUVr (Standard Uptake Value ratio) and used the more liberal threshold at 0·7918 described earlier [10,11]. We selected 48 amyloid (+) and 48 amyloid (−) subjects for which we had fasting blood samples at 12 or 18 months after inclusion with the most extreme SUVr values. SUVr values ranged from 0·538 to 0·681 for the amyloid (−) subjects and from 0·797 to 1·577 for the amyloid (+) subjects. The time interval between the 18F-florbetapir PET scan and blood sampling was 11·1+/− 2·8 months. Additionally, data on age, sex, weight, body mass index, APOE genotype, treatments, education and place of residence were collected for all selected individuals. For the longitudinal study we used available fasting blood samples obtained 36 months after inclusion from a subset of 22 subjects included in the initial analysis (9 amyloid (+) and 13 amyloid (−)).
The ethics committee of the Pitie-Salpetriere University Hospital approved the study protocol. All participants signed an informed consent form, given and explained to them 2 weeks before enrolment. Neither the participants nor the investigators were aware of participants' amyloid β status.
2.2. Chemical reagents
All Liquid Chromatography Mass Spectrometry (LC-MS) grade reference solvents, acetonitrile (ACN), water (H2O) and methanol (MeOH) were from VWR International (Plainview, NY). LC grade ammonium formate, chloroform (CHCl3), 2-propanol (IPA) and formic acid were from Sigma-Aldrich (Saint Quentin Fallavier, France). A pool of plasma samples was purchased from Biopredic and used as interbatch quality control (Rennes, France). Stock solutions of stable isotope-labeled mix (Algal amino acid mixture-13C,15N) for metabolomic approach were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). All internal standards (IS), previously described [12], used in lipidomic approach were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL).
2.3. Sample preparation
2.3.1. Metabolomics
Eight volumes of frozen acetonitrile (−20 °C) containing internal standards (labeled mixture of amino acids at 12·5 μg/mL) were added to 100 μL plasma samples and vortexed. The resulting samples were then sonicated during 10 min and centrifuged during 2 min at 10000 xg at 4 °C. Supernatants were incubated at 4 °C during 1 h for slow protein precipitation process. Samples were centrifuged for 20 min at 20000 ×g at 4 °C. Supernatants were dried and stored at −80 °C prior to LC-MS analyses. Pellets were diluted 3-fold and reconstituted in H2O/ACN (95/05) for pentafluorophenylpropy (PFPP) column and in H2O/ACN (40/60) for hydrophilic interaction liquid chromatography (HILIC) column.
2.3.2. Lipidomics
Plasma samples were extracted using adapted method to that previously described [12]. A volume of 100 μL of plasma was added to 490 μL of CHCl3/MeOH 1:1 (v/v) and 10 μL of internal standard mixture. Samples were vortexed for 60 s, sonicated for 30 s using an ultrasonic probe (Bioblock Scientific Vibra Cell VC 75185, Thermo Fisher Scientific Inc., Waltham, MA, USA) and incubated for 2 h at 4 °C with mixing. Seventy-five μL of H2O was then added and samples were vortexed for 60 s before centrifugation at 15000 g for 15 min at 4 °C. The upper phase (aqueous phase), containing gangliosides, lysoglycerophospholipids, and short chain glycerophospholipids, was transferred into a glass tube and dried under a stream of nitrogen. The protein disk interphase was discarded and the lower rich-lipid phase (organic phase) was pooled with the dried upper phase and the mixture dried under nitrogen. Samples were resuspended with 100 μL of a solution CHCl3/MeOH 1:1 (v/v). 10 μL was 100-fold diluted in a solution of MeOH/IPA/H2O 65:35:5 (v/v/v) before injections.
2.3.3. Metabolomics and lipidomics analyses and data processing
Three LC-MS experiments were performed using HILIC, Sequant ZIC-pHILIC column 5 μm, 2·1 × 150 mm at 15 °C (Merck, Darmstadt, Germany), and pFPP, Discovery HS F5-PFPP column, 5 μm, 2·1 × 150 mm (Sigma, Saint Quentin Fallavier, France), chromatographic columns for metabolomics analyses, and using a reverse phase chromatographic column, kinetex C8 2·6 μm, 2·1 × 150 mm (Phenomenex, Sydney, NSW, Australia) for lipidomics analyses. Q-Exactive and Fussion mass spectrometers (Thermo Scientific, San Jose, CA) were used for metabolomics and lipidomics respectively. Experimental settings for metabolomics and lipidomics global approaches by LC-HRMS were carried out as detailed in Garali et al. and Seyer et al. 12, respectively.
All processing steps were carried out using the R software [13] . LC-MS raw data were first converted into mzXML format using MSconvert tool [14]. Peak detection, correction, alignment and integration were processed using XCMS R package with CentWave algorithm [15,16] and workflow4metabolomics platforms [17]. The resulted datasets in both approaches were Log-10 normalised, filtered and cleaned based on quality control (QC) samples [18]. The remaining features were annotated based on their mass over charge ratio (m/z) and retention time using an “in house” database as described previously [19], and also putatively annotated based solely on their m/z using public databases such as the human metabolome database HMDB [20] and the Kyoto Encyclopedia of Genes and Genomes database, KEGG [21]. Lipidomic features were then annotated thanks to an in silico lipid database and based on accurate measured masses, retention time windows and relative isotopic abundance (RIA) of lipid species as described previously [12].
2.3.4. RNA sequencing
Total RNA was extracted from 2·6 mL total blood using Nucleospin RNA Blood Midi (Macherey Nagel, Hoerdt, France) according to the manufacturer's instructions. RNA quality was assessed on Agilent 4200 Tapestation, Santa Clara, USA. Libraries were prepared with 500 ng of total RNA from each individual using the TruSeq Stranded Total RNA with Ribo-Zero Globin (Illumina, Evry, France) according to the manufacturer's instructions. Libraries were analysed on the Agilent 4200 Tapestation and sequenced on a NextSeq500 (Illumina) as 75-bp paired-end reads with a sequencing depth of 30 million reads.
2.3.5. Univariate and multivariate omics data analyses
The large and heterogeneous datasets generated from these high-throughput technologies were analysed using appropriate computational solutions [22,23]. For the RNA-Seq dataset, all genes with fewer than 10 raw read counts across 75% of the samples (low expressed genes) were filtered out from the analysis. Expression data was then normalised using the rlog (regularised logarithm) function in the R package DESeq2 [24]. Analyses were carried out using the R software version 3·4·4. The Student's t-test and the chi-square test were respectively used to compare age and sex differences between the amyloid (+) and amyloid (−) groups. The distribution of both APOE ε2 and ε4 carriers was compared between amyloid groups using Fisher's exact tests. The Wilcoxon rank-sum test was performed to assess whether there were age differences between APOE ε4 carriers from the amyloid (+) and amyloid (−) groups. The metabolomics and lipidomics data used for analysis were log10-transformed intensity values. To control for potential effects of sex and APOE ε4 status, an additional normalization step was applied to the omics datasets using the ComBat method [25].
Omics datasets were first investigated separately to identify features (among genes, metabolites and lipids) that best discriminate between amyloid (+) and amyloid (−) groups. This was done by combining the results of different feature selection techniques in an attempt to overcome the limitation of individual methods and thus retain the most consistent features [26]. Therefore, only features selected by at least two methods were considered. Therefor we used more permissive p value threshold of 0·05 thus minimizing false negatives. Two methods were applied to all datasets: 1) non-parametric Wilcoxon rank-sum test (WT) using the Benjamini-Hochberg (BH) correction for multiple testing and 2) a parametric selection using parametric selection based on correlation-adjusted t-scores (CAT scores) [27], as implemented in the R package sda. The CAT score, which is a multivariate modification of the Student t-statistic, has the advantage of taking into account the correlation structure among features. Following a procedure described previously [28], selection from CAT scores was optimised using a 5-fold cross validation with 20 repeats to assess the feature selection stability over a total of 100 rounds. In each round, 1/5 of the training dataset was left out and the CAT scores were calculated from the remaining 4/5 of the subjects with a Local False Discovery Rate (LFDR) (indicating the posterior probability of a feature being non-informative given its CAT score) for each tested feature. Only features that had a LFDR smaller than 0·2 over >50 rounds were retained. Additionally, as this is the most commonly used approach for transcriptomics data, the selected genes were also overlapped with the most differentially expressed genes identified by DESeq2 [24] with the BH adjustment. This latter approach applied to RNA-Seq data can be considered as a second parametric selection strategy using the negative binomial distribution. Since WT and DESeq2 mostly yield non-significant p-values, a more permissive criteria based on a raw p-value <0·05 was adopted to allow comparison among the different methods and further gene-set analysis. Gene ontology (GO) enrichment analyses for selected genes were conducted using the Enrichr web tool [29].
2.4. Multi-omics data integration
2.4.1. Multi-block analysis
Beside univariate and multivariate common statistical methods, we used regularised generalised canonical correlation analysis (RGCCA) [30] that provides a common framework to access a variety of multi-block data analysis methods. The term ‘multi-block’ refers to the datasets organised in blocks of variables for the same group of individuals, preserving the block structure of each dataset (typically, one or more blocks per data type). To address variable selection, sparse GCCA (SGCCA) was proposed to extend the RGCCA model using the Lasso method [31] as successfully applied to a case study of spinocerebellar ataxia (SCA) [32]. Multi-omics data integration was conducted on the 78 participants (41 amyloid (+) and 37 amyloid (−)) for which data were available on the three complete omics datasets. In doing this, the objective was to simultaneously examine in a unified framework the complex relationship between the different sources of omics data (transcriptomics, metabolomics and lipidomics) and the amyloid status (amyloid (+) and amyloid (−)) both at the block and the variable levels. Analyses were performed using RGCCA and SGCCA for multi-block data [32] as implemented in the block.splsda function of the R package mixOmics [34]. Using a component-based analysis strategy, RGCCA and SGCCA offer an effective solution for dimension reduction by searching a small set of orthogonal latent variables (constructed as linear combinations of the manifest variables) to summarise each high-dimensional dataset. sPLS-DA (sparse partial least squares-discriminant analysis) is a special case of SGCCA that combines Partial Least Squares (PLS) regression for discriminant analysis, and Lasso penalization for selecting the most discriminative variables. Supervised integration of multi-omics data, involving one block per omics dataset and a dummy block matrix indicating the levels of amyloid (+) or (−), was determined by a weighted design matrix to define a trade-off between the double-objective of maximising the covariance between the blocks of omics, and maximising the covariance with the dummy block (i.e. discriminating between the amyloid groups). In order to prioritise this second objective, the weights were set to 0·1 between the blocks of omics and to 1 between the blocks of omics and the block amyloid. The optimal number of components was determined by the balanced error rate based on centroids distance, averaged over 50 repeats of a 5-fold cross-validation, for each tested number of components up to 10 units per function. An optimal number of variables to be kept for each component was chosen over a grid of candidate values, ranging from 1 to 12, using the lowest average balanced classification error rate based on centroids distance as calculated by the function tune.block.splsda with 3 components and 100 repeated 5-fold cross-validations. For these optimal parameters, 9 sparse components (3 components per block) were extracted with sPLS-DA, each component representing the latent structure of its omics block that is associated with the amyloid levels. The weight coefficients for each block-wise component were considered to prioritise the genes, metabolites, and lipids selected by the model. In order to assess the stability of the variable selection, the model was run for 1000 bootstrap replicates of the blocks to calculate stability frequency scores for the selected omics variables.
2.4.2. Principal component analysis
In an attempt to derive a global molecular signature of early amyloid deposition, a ‘superblock’ was built in grouping the 9 block-wise sparse components (3 components for each dataset: transcriptomic, lipidomic, and metabolomics). A principal component analysis (PCA) was then performed on the superblock to extract two so-called ‘superscores’, defined by the two first principal components with all the selected omics variables. For data visualization, the superscores were used to generate a common space to observe the distribution of subjects in amyloid groups; and which were the omics variables that most influenced this distribution.
2.4.3. Discriminant power of selected omics features
Finally, the discriminant potential of the selected omics features was examined through a Receiver Operating Characteristic (ROC) curve analysis using the R package cvAUC [35]. With this aim, sPLS-DA was repeated for the blocks of omics restricted to the top ten most stable features as determined by the bootstrap procedure and the addition of 3 blocks for the pairwise combination of the omics data (no feature selection; 1 component per block). The prediction of the amyloid status was then calculated in a leave-one-out analysis using the function perf for the first component of each block. The derived ROC curves and area under the curve (AUC) estimates were used to rank the tested classifiers and to assess the potential of a multi-omics signature for better characterising the earlier stages of amyloid deposition.
3. Results
3.1. Sample description
A total of 94 individuals were included in the study after exclusion of two subjects with APOE ε2/ε4 genotypes based on the observation that those two alleles show differential effects on amyloid deposition [36,37]. To consider additional effect of APOE, subjects were also grouped according to their APOEε4 status.
Among those, 81 passed the RNAseq quality test and were included in the transcriptomics study (44 amyloid (+) and 37 amyloid (−)). In the case of lipidomics, one individual was excluded for being an outlier and there was no data available for three. We thus had lipidomics data on 45 amyloid (+) and 45 amyloid (−). For metabolomics, all non APOE ε2/ε4 subjects were included (47 amyloid (+) and 47 amyloid (−)). For each of the datasets, all data available were used for the univariate analysis. Demographics of the samples are described in Table 1. Neither sex nor age significantly differed between the amyloid groups. The distribution of the APOE genotype in our study population did not differ from the one described in general the French population [38]. There was a clear and expected difference in genotype frequency, with significantly fewer ε4 (p = 0·012, one-sided Fisher's exact test) and a trend to more ε2 carriers (p = 0·064, one-sided Fisher's exact test) in the amyloid (−) group as compared to the amyloid (+) subjects. Because there were fewer ε4 carriers in the amyloid (−) group and amyloid deposition is age-related [39], we analysed whether there was an age difference between ε4 carriers in both amyloid groups. We did not find any significant difference of age between ε4 carriers in both groups.
Table 1.
Demographic description of the datasets.
| Metabolomics (n = 94) |
Lipidomics (n = 90) |
Transcriptomics (n = 81) |
|||||
|---|---|---|---|---|---|---|---|
| Amyloid – (n = 47) | Amyloid + (n = 47) | Amyloid – (n = 45) | Amyloid + (n = 45) | Amyloid – (n = 44) | Amyloid + (n = 37) | ||
| Sex | Male | 20 (42.6%) | 20 (42.6%) | 19 (42.2%) | 19 (42.2%) | 19 (43.3%) | 13 (35.1%) |
| Female | 27 (57.4%) | 27 (57.4%) | 26 (57.7%) | 26 (57.7%) | 25 (56.8%) | 24 (64.9%) | |
| Age | Mean (±SD) | 76.4 ± 3.61 | 76.4 ± 3.47 | 76.5 ± 3.66 | 76.4 ± 3.37 | 76.6 ± 3.48 | 76.1 ± 3.82 |
| Range | 70–84 | 70–84 | 70–84 | 70–84 | 70–84 | 70–84 | |
| ApoE genotype | ε2/ε2 | 1 (2.1%) | 0 (0%) | 1 (2.2%) | 0 (0%) | 1 (2.3%) | 0 (0%) |
| ε2/ε3 | 8 (17%) | 3 (6.4%) | 8 (17.7%) | 3 (6.7%) | 7 (15.9%) | 3 (8.1%) | |
| ε3/ε3 | 33 (70.2%) | 26 (55.3%) | 31 (68.8%) | 25 (55.5%) | 25 (56.8%) | 24 (64.9%) | |
| ε3/ε4 | 5 (10.6%) | 17 (36.2%) | 5 (11.1%) | 17 (37.7%) | 5 (11.4%) | 15 (40.5%) | |
| ε4/ε4 | 0 (0%) | 1 (2.1%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (2.7%) | |
3.2. Univariate analyses of omics data
3.2.1. Metabolomics
PCA of the 830 features from metabolomics analyses showed a strong effect of sex while APOE and brain amyloid deposition did not interfere with blood metabolite profiles (Fig. 1). Because of this sex-dependency and of the significant increase of APOEε4 carriers in the amyloid (+) group, metabolomics data were corrected for sex and APOE genotype. Differential analysis was carried out by combining the results obtained from the CAT scores and the Wilcoxon rank sum test. For CAT scores, only five annotated features passed the threshold ≥50% of LFDR ≤0·2, while the WT selected a set of 112 annotated features at uncorrected p-values ≤0·05, 18 of which remained significant after BH correction. Coherently, the five metabolites from the CAT score selection also came out in the Wilcoxon list, and were thus retained as the most significant differential features between amyloid (+) and amyloid (−) subjects (Fig. 2 and Supplementary Table 1). These features were attributed to four medium-chain fatty acids (MCFA) and 4-nitrophenol, all increased in the amyloid (+) group as compared to the amyloid (−) group. Spearman's rank correlations between the levels of the five most significant metabolites and amyloid load in the brain (SUVr) were all significant. However when considering amyloid (+) and amyloid (−) subjects as separate groups, no correlations were observed (Supplementary Fig. 1).
Fig. 1.

PCAs of the metabolomics dataset in amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD. a: Distribution of males and females according to the two principal components (females in hollow triangles, males in filled in triangles); b: Distribution of APOEε4 carriers and APOEε4 non carriers according to the two principal components (APOEε4 negative in hollow squares, APOEε4 positive in filled in squares); c: Distribution of amyloid (+) and amyloid (−) subjects according to the two principal components (hollow circles for amyloid (−) filled in circles for amyloid (+) after correction for APOE genotype and sex.
Fig. 2.

Differential analysis of metabolites between amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD. a: Venn diagram depicting the overlap of significant metabolites between CAT score and Wilcoxon rank sum test applied on 830 metabolites detected in blood samples. b: Table listing the overlapping features and their variation in amyloid (+) and amyloid (−) subjects.
3.2.2. Lipidomics
PCA of the 373 features from lipidomics data showed no effect of sex, APOE, or amyloid status (Fig. 3). The same statistical approach described for metabolomics was performed on lipidomics. Differential analysis of the lipidomics dataset highlighted 129 annotated lipids selected by the WT. However, none passed the threshold for CAT scores and consequently no feature was retained as different in the plasma of amyloid (+) versus amyloid (−) subjects (Supplementary Table 2).
Fig. 3.

PCAs of the lipidomics dataset in amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD. a: Distribution of males and females according to the two principal components (females in hollow triangles, males in filled in triangles); b: Distribution of APOEε4 carriers and APOEε4 non carriers according to the two principal components (APOEε4 negative in hollow squares, APOEε4 positive in filled in squares); c: Distribution of amyloid (+) and amyloid (−) subjects according to the two principal components (hollow circles for amyloid (−) filled in circles for amyloid (+) after correction for APOE genotype and sex.
3.2.3. Transcriptomics
Similarly to the lipids dataset, PCA of the 15,616 transcripts expressed in peripheral blood in the 83 subjects analysed showed no effect of sex, APOE, or amyloid status (Fig. 4). Differential gene expression analysis between amyloid (+) and amyloid (−) subjects was performed using three methods: WT, CAT score and DESeq2. This led to the selection of 330 differentially expressed genes by WT, 133 by CAT scores, and 389 by DESeq2 (raw p < 0·05). Finally, a core set of 64 overlapping genes was extracted from these three lists (Fig. 5 and Supplementary Table 3). Of these 64 genes, 31 were upregulated and 33 were downregulated in the amyloid (+) group. The main biological processes related to these 64 genes were tested by a GO enrichment analysis, which highlighted genes involved in inflammation and fatty acid metabolism (Fig. 5 and Supplementary Table 4).
Fig. 4.

PCAs of the transcriptomics dataset in amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD. a: Distribution of males and females according to the two principal components (females in hollow triangles, males in filled in triangles); b: Distribution of APOEε4 carriers and APOEε4 non carriers according to the two principal components (APOEε4 negative in hollow squares, APOEε4 positive in filled in squares); c: Distribution of amyloid (+) and amyloid (−) subjects according to the two principal components (hollow circles for amyloid (−) filled in circles for amyloid (+) after correction for APOE genotype and sex.
Fig. 5.
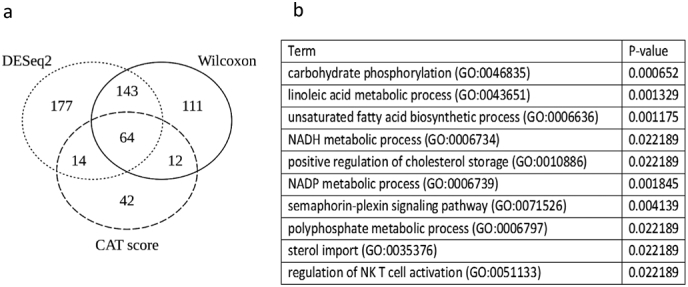
Differentially expressed transcripts between amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD. a: Venn diagram depicting the overlap of significant metabolites between CAT score, Wilcoxon rank sum test and DESeq2 analysis applied to 15,616 transcripts expressed in blood cells. b: Table listing the ten first GO terms obtained through GO Enrichment analysis.
3.2.4. Multi-omics data integration
In order to investigate how the omics can complement each other to explain the differences between amyloid (+) and amyloid (−) subjects, we performed an integrative analysis using sPLS-DA for multi-block data whereby ‘multi-block’ refers to datasets organised in blocks of variables for the same group of individuals, hence preserving the block structure of each dataset. This required the organisation of the omics datasets into three blocks of explanatory variables (metabolomics, lipidomics and transcriptomics) based on the amyloid status added as a dummy block matrix (positive or negative). Parameter tuning performed prior to sPLS-DA suggested the use of three components for each omics dataset. The number of selected variables to be retained on the three components after adding sparsity constraint were respectively set to 4, 3, and 3 for the metabolomics block, to 12, 12, and 3 for the lipidomics block and to 3, 3, and 12 for the transcriptomics block. Fig. 6 shows the list of features for the two first components for all three omics datasets with their corresponding loading plots. Metabolites could efficiently separate amyloid (+) and amyloid (−) subjects, while transcripts showed trends towards separation, and lipidomics data did not discriminate amyloid (+) and amyloid (−) subjects. The weak performance of the lipidomics block confirmed the results of the previous univariate selection showing no difference in the levels of lipids according to the level of amyloid in the brain of the subjects. Bootstrap procedure was also performed to assess the robustness of the feature selections. This analysis highlighted a short set of three metabolites (X280, X75 and X5), one lipid (V300), and three genes (ACSBG2, RASL11A, RNU12) that were much more frequent over the repeats and could correspond to the most discriminant features. Notably, these variables mostly contributed to the first component of their respective block (Supplementary Table 5).
Fig. 6.

Integrative analysis for multi-block data in amyloid (+) and amyloid (−) asymptomatic individuals at-risk for AD using sparse partial least squares-discriminant analysis (sPLS-DA). a: PCA for the metabolomics block (green circles amyloid (−) individuals, red triangles amyloid (+) individuals). Percentages of the variance for the two axis are indicated in parenthesis; b: List of metabolites for the three first components with their corresponding loading plots indicating the most relevant metabolites. Each graph represents one of the components and the weight of metabolites; c: PCA for the lipidomics block (green circles amyloid (−) individuals, red triangles amyloid (+) individuals. Percentages of the variance for the two axis are indicated in parenthesis); d: List of lipids for the three first components with their corresponding loading plots indicating the most relevant lipids. Each graph represents one of the components and the weight of lipids; e: PCA for the transcriptomics block (green circles amyloid (−) individuals, red triangles amyloid (+) individuals). Percentages of the variance for the two axis are indicated in parenthesis; f: List of transcripts for the three first components with their corresponding loading plots indicating the most relevant transcripts. Each graph represents one of the components and the weight of transcripts. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In order to derive a multi-omics signature combining these top features, all the block-wise sparse components were grouped in a so-called super-block. PCA was then applied to this superblock for the extraction of two superscores defined by the two first principal components that combined the top selected omics features. These superscores were used to generate a common space to visualize all amyloid (+) and amyloid (−) subjects together with the relative contribution of the omics variables. The individual factor map obtained with the combined omics data slightly improved the separation of the two groups compared to the one achieved with the metabolomics block alone (Fig. 7). The corresponding variable factor map also confirmed the relative importance of the three types of omics. Similarly to what was observed with the single blocks, the best omics contributor was the metabolomics block followed by a rather similar influence of both transcriptomics and lipidomics blocks.
Fig. 7.

Multi-omics signature of amyloid positivity in asymptomatic individuals at-risk for AD by grouping block-wise sparse components in a super-block. a: Graphic representation of the common space associated with the two superscores corresponding to the two first principal components combining the top omics features (green circles amyloid (−) individuals, red triangles amyloid (+) individuals); b: Variable factor map generated by the two superscores (metabolic variables in blue, genes in green and lipids in dark red). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To assess the potential of multi-omics signature to detect amyloid status we performed a ROC analysis. We retained the top frequent features from each omics dataset as assessed by the bootstrap procedure (Supplementary Table 5), which notably included several of the best features identified by the multivariate analysis. With these stable features, we repeated the sPLS-DA with the three omics blocks and three additional blocks that were built as a pairwise combination of the individual omics blocks. For this analysis, we also considered the first component of each block without feature selection. Based on these components, we performed ROC analysis using a leave-one-out procedure for the six blocks, to obtain the potential predictive power of each block and the possible gain of combining them. These analyses confirmed the higher discriminant power of the metabolomics block (AUC = 0·978), followed by the transcriptomics (AUC = 0·881), and the lipidomic ones (AUC = 0·732). The most discriminant combinations were metabolomics and transcriptomics (AUC = 0·994), followed by metabolomics and lipidomics (AUC = 0·978), and lipidomics and transcriptomics (AUC = 0·866) (Fig. 8).
Fig. 8.
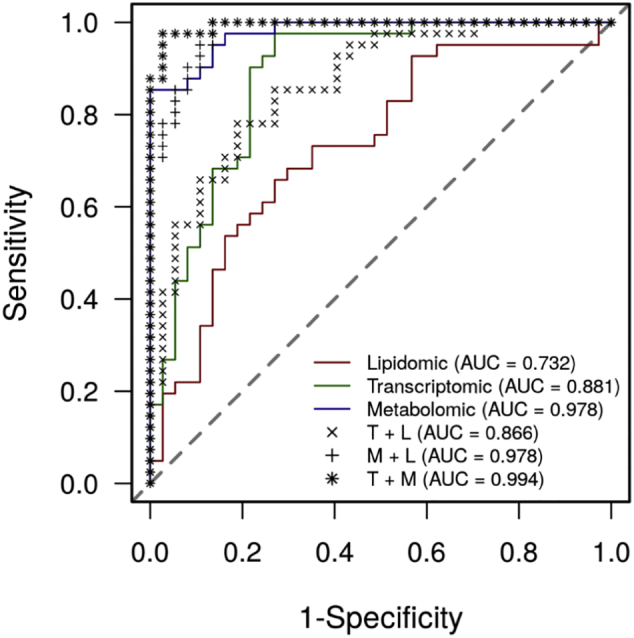
Discriminant power of the block-wise components and the superscores for detecting brain amyloid deposition in asymptomatic individuals at-risk for AD. ROC curves plots for (a) the first component of each block, (b) the combination of the first two components. The black bold dashed line represents the AUC for the superscore, blue is for the metabolomics block, green for the transcriptomics, and red for lipidomics. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2.5. Correlations between the levels of the five most significant metabolites, brain amyloid burden and cerebrospinal fluid biomarkers
We collected the available data on 20 subjects from our omics study who underwent lumbar puncture: 9 amyloid (−) and 11 amyloid (+). Supplementary Table 6 indicates that in the amyloid (+) subjects, Florbetapir SUVr was correlated with cerebrospinal fluid (CSF) phospho-tau (p = 0·00062 and p = .051) but not with total tau. Such correlations were not found in the amyloid (−) subjects.
Finally, we analysed the correlations between the four medium-chain fatty acids and 4-nitrophenol, brain amyloid load and tau, phospho-tau and Aβ42 cerebrospinal fluid (CSF) levels in amyloid (+) and amyloid (−) subjects, separately or altogether. When analyzing the amyloid (+) and (−) subjects together, strong correlations were found between tau and phospho-tau and between metabolites and Florbetapir SUVr, as well as between metabolites themselves. Aβ42 was inversely correlated with Florbetapir SUVr and the levels of all five metabolites, suggesting a strong link between the five metabolites and brain amyloid burden. Correlations were less consistent when analyzing amyloid (+) and amyloid (−) subjects separately, which suggests that the correlation is driven by the differences in the levels of metabolites between the groups rather than by a real correlation (Supplementary Table 6). Finally, six years after the first inclusion, six subjects from our selection converted to AD-MCI as defined previously [10]. All of these converters were in the amyloid (+) group, they had high Florbetapir SUVr, high levels of the three medium chain fatty acids and 4-nitrophenol (Supplementary Fig. 2 green dots).
3.2.6. Longitudinal analysis of the most relevant metabolites in a subset of individuals
In order to study the evolution of the five most discriminant metabolites from the initial study (Fig. 2, (X3, X5, X6, X75, and X280)), we performed a double-blind study in a subset of 22 subjects (9 amyloid (+) and 13 amyloid (−)) for whom we had plasma samples 36 months following their inclusion. We compared the two values obtained in the same individuals at two different time points and found significant correlation for the three MCFA X3 (p = 0·028), X5 (p = 0·02) and X6 (p = 0·041) while significance was not reached for X75 (p = 0·729) and X280 (p = 0·1053) (Supplementary Fig. 3). However, 4-nitrophenol and nonanoic acid were not associated to the amyloid status in this sub cohort. Although the number of subjects was low, comparison between amyloid (+) and amyloid (−) using WT showed a significant difference for X3 (p = 0·03) (Fig. 9) but not for the other four metabolites. These results suggest that differences at baseline are also present at follow-up, indicating that the identified metabolites are stable in time. Additionally, amyloid burden was also stable during this time range. All amyloid (−) subjects stayed amyloid (−) (maximum percentage of change 3·16%) while the highest percentage of increase of SUVr in the amyloid (+) group was only 4·37% (Supplementary Fig. 4).
Fig. 9.
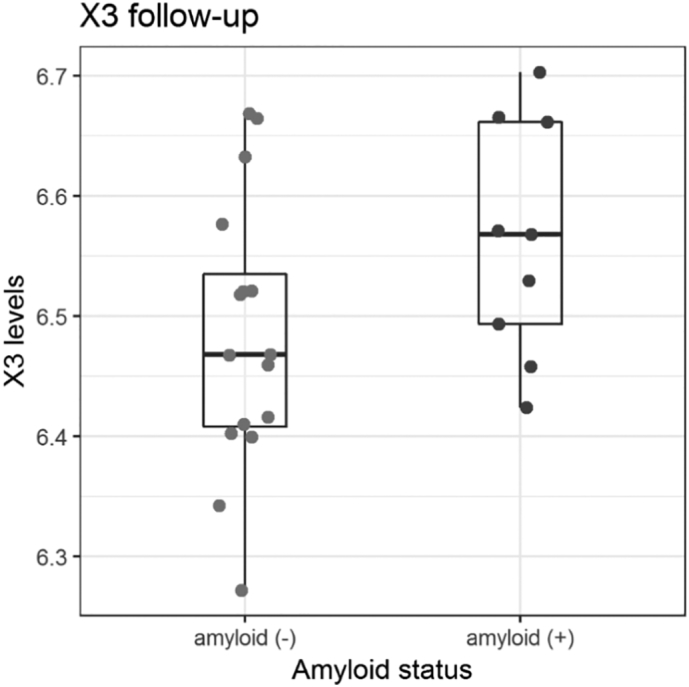
Validation of X3 (undecanoic acid) as a marker of amyloid positivity in asymptomatic individuals at-risk for AD. Box plot analysis of the values obtained for X3 metabolite identified as undecanoic acid in a follow-up longitudinal study of 22 subjects. Wilcoxon p = 0·03.
4. Discussion
We report the successful application of a multi-omics approach to find a peripheral blood biomarker signature of brain amyloid deposition in a cohort of asymptomatic individuals at risk for AD. To our knowledge, this is the first multi-omics study focusing on such a population instead of patients with sporadic late-onset AD or familial early-onset AD [40].
Our omics approach was based on the analysis of three different datasets – metabolomics, lipidomics, and transcriptomics – obtained from two groups of individuals amyloid (+) and amyloid (−), either separately or in combination. We used two differential statistical approaches, a univariate method and a multivariate one. The univariate method allows to detect features in each dataset that are differential between groups. The multivariate strategy integrates several datasets to find a multi-omics signature of amyloid deposition. Similar approaches have previously proven successful in other neurodegenerative diseases such as Parkinson's disease [41] or spinocerebellar ataxia [32]. ROC analysis of the combined omics data applied to the INSIGHT-preAD cohort showed that combining metabolomics and transcriptomics features was slightly more predictive than each omics taken separately. However, it was clear that the contribution of each dataset was not equivalent, the metabolomics dataset providing the best performance. Some specific features such as nonanoic acid and 4-nitrophenol were both highly differential in the univariate analysis and had an important impact on the group separation in the integrative superblock analysis. The transcriptomics dataset showed a moderate discriminatory power, but slightly more than the lipidomics one. Still, these two datasets should not be discarded as they were part of the final multi-block signature, highlighting the particular power of this multivariate approach in the search of biomarkers.
A strength of multi-omics approaches is their ability to analyse the interplay between different datasets. Here, we were particularly interested in the relationship between the levels of metabolites in plasma and gene expression in blood cells. We focused on fatty acids as they proved to differentiate between groups in the metabolomics univariate analysis and because the GO enrichment analysis of the univariate transcriptomics analysis pointed out to an enrichment of genes involved in fatty acid metabolism (Fig. 5B). However, we were not able to establish any correlation between gene expression and metabolite levels (data not shown). One possible solution would be to use exosome preparation from blood samples, as they can be classified depending on specific markers related to their organ of origin [42], such as the brain, and are known to contain both RNA and proteins [43].
4-Nitrophenol is an exogenous metabolite of environmental origin obtained from the metabolism of organophosphates by the enzyme praoxonase-1 [44], and a degradation product of parathion pesticides. It is currently detected in drinking water and urine [45], therefore we were intrigued by its presence as a differential metabolite in our cohort. Due to its relation to pesticides, we evaluated whether it could be linked to the place of residence of the individuals. However, our sample was rather homogenous, as most volunteers originated from the region of Île-de-France (around Paris), and it was not possible to draw a relationship between the presence of 4-nitrophenol and participant's place of residence and, thus, a possible exposition to pesticides (data not shown). Likewise, additional data on occupation or medication did not indicate any environmental origin of 4-nitrophenol (data not shown). Therefore, we looked into the correlation between blood expression of PON-1, the gene encoding paroxonase-1, and the levels of 4-nitrophenol. We found no association (data not shown), although we measured gene expression and not enzymatic activity and it is still possible that protein expression and activity are subject to translational regulation and possible post-translational modifications. Nevertheless because of the evidence suggesting an association between pesticides and Parkinson's disease [46] and since many insecticides act on the nervous system, attention should be given to clarify the putative role of pesticides in AD [47].
As already stated, no previous studies have analysed peripheral blood metabolomics and lipidomics differences between asymptomatic subjects with and without brain amyloid deposition. Most studies have focused on the differences between controls and either MCI or AD and the evolution of the disease. In the blood of AD patients, these studies showed various metabolic alterations, especially lipid metabolism and transport [[48], [49], [50]], aminoacid metabolism [50], and mitochondrial function [48]. In fact, fatty acids were one of the most prominent differential features when comparing controls to both AD and MCI [50,51], together with desmoterol [52], sphingomyelins [48,53,54], phosphatidylcholines [48,54], ceramides [53], biliary acids [49], and lysophospholipid 18:1 [52]. In addition, some metabolites have been linked to disease progression such as 2,4-dihydroxybutanoic acid [48] and phosphatidylcholines [55]. Still, our results point out to a different dysregulation of fatty acid metabolism, with increased blood levels of four MCFA distinct from the ones previously correlated with brain amyloid deposition in AD and MCI [54]. Thus, metabolic changes during amyloid deposition in asymptomatic individuals appear to be different to the changes observed after symptom onset.
In an attempt to confirm the five most significant metabolites, we ran a double blind longitudinal study on a sub sample of 22 individuals with 9 amyloid (+) and 13 amyloid (−). The three MCFA (octanoic, undecanoic and hydroxyl-nonanoic acids) showed correlation over time in a follow-up plasma sample obtained 18 to 24 months after the initial blood sampling, while the two others did not, including 4-nitrophenol.
Medium-chain triglycerides (MCT) are hydrolysed in the intestine to produce MCFA, mainly decanoic and octanoic acids that are metabolised to ketones in the liver. AD is associated with brain glucose hypometabolism [56], and replacing glucose with ketones by administration of MCT to patients with AD or MCI has been shown to improve short-term memory in patients APOEε4 (−) [57] and reviewed in Augustin et al. [58]. Increase of MCFA in asymptomatic individuals at-risk for AD could be a compensatory mechanism in response to brain amyloid deposition leading to higher ketones thus preventing glucose hypometabolism. However, we did not find any difference in ketone bodies levels between amyloid (+) and amyloid (−) groups. Finally, difference in MCFA levels are unlikely to be due to variations in blood sampling or specific diets since subjects were fasted at the time of blood collection and the levels of MCFA were correlated longitudinally at 18 to 24 months intervals.
Regarding transcriptomics, previous studies explored blood gene expression in patients with AD with the objective of finding a common signature that could help identifying those individuals at risk of AD. A differential expression profile has been observed not only when comparing AD to controls [[59], [60], [61], [62], [63], [64], [65], [66]], or MCI to controls [64], but also when comparing rapidly progressing AD patients to slowly progressing patients [67], and patients with AD to patients with vascular dementia [61]. Those differentially expressed genes were grouped according to biological functions such as inflammation [60,68,69], immune response [61,65], cell cycle and apoptosis [61,65,68,69], gene expression [65,68], cytoskeleton [59,65], and interaction with the extracellular matrix [59,70]. In our cohort, we found 64 differentially expressed genes between amyloid (+) and amyloid (−) groups but the biological functions enriched in our analysis do not mirror any of those found in previous studies in AD, as ours are mostly related to fatty acid metabolism and inflammation and may reflect initial dysregulation in the amyloid disease process (Fig. 5B).
Our result does not preclude from evaluating the potential biomarkers identified here in an independent cohort of asymptomatic individuals at-risk for AD. For example the AD Neuroimaging Initiative has nearly 500 control subjects with Florbetapir PET scans accessible. However no blood metabolomics data are available yet (http://adni.loni.usc.edu/). Of note, even though amyloid deposition is a risk factor for AD, it does not always lead to the development of dementia, thus our amyloid (+) population is not comparable to an AD population. Regarding the applicability of our findings in the general population, studies showed that 17 to 76·6% subjects have memory complaints depending on tests and age range [[71], [72], [73]]. The INSIGHT cohort would then be representative of these highly variable percentages of the general population. Grading subjective cognitive decline with sensitive tests remains a challenge.
In addition, other pathologies of the brain are possibly associated with amyloid deposition such as neuroinflammation and vascular changes. PET imaging in AD patients with specific ligands of activated microglia have shown that distinct dynamic profiles of microglial activation are associated with clinical decline in AD but no correlation with amyloid PET [74]. Vascular changes associated to altered amyloid clearance could indeed be a confounding factor in our study. Vascular risk and amyloid burden synergistically accelerate cognitive decline in clinically normal older individuals [75].
Nevertheless, our main objective in this study was to find a blood signature indicative of amyloid deposition, a major risk factor for AD. We indeed found correlations between blood levels of the most significant metabolites and brain amyloid load when considering all subjects (Supplementary Fig. 1). However no correlation was observed when selecting only amyloid (+) or amyloid (−) subjects. This lack of correlation in the subgroups could be due to the initial selection of 48 amyloid (+) and 48 amyloid (−) subjects with the most extreme SUVr values and possibly to floor effect in the amyloid (−) group.
Finally it will be interesting to test reliability of our multi-omics approach in the entire spectrum from asymptomatic subjects to patients during follow-up of the INSIGHT cohort and conversion to AD-MCI and AD with dementia. The relatively small sample size analysed here does not allow for considering conversion to AD. Furthermore, we need to acknowledge that amyloid deposition is not the best marker for cognitive decline [76]. However, at the present time and 6 years after the first inclusion, only 12 subjects from the whole INSIGHT cohort converted to AD-MCI (3·8%) and five to other types of dementia. In our selection of subject with extreme SUVrs, only six subjects converted to AD-MCI (6·2%) and four to other types of dementia. This 1·6-fold increase of the percentage of conversion in the population selected for the omics study could be due to the increase of florbetapir positivity (50% in the omics selection while 27·7% in the INSIGHT cohort) [10]. Although this current low frequency of conversion to AD-MCI does not allow any correlation studies, we can still notice that subjects who converted to AD-MCI so far are all amyloid (+), six of them with SUVr values above the mean. As for the selection of the top-five metabolites, levels tend to be above the 25 percentiles (Supplementary Fig. 2).
In conclusion, our results show a potential omics signature with 99·4% chance prediction between individuals according to their brain amyloid positivity, allowing – if confirmed in an independent cohort – for a less invasive, more accessible, and less expensive risk assessment of AD as compared to PET studies or lumbar puncture. Although the results of our study look very promising, sample size and geographic distribution did not allow for an exposome analysis in order to establish whether the levels of 4-nitrophenol could be related to place of residence. Amyloid positivity was assessed from the SUVr values that were very narrow in the amyloid (−) group, thus limiting correlative studies in this group. Following our results, we will explore changes in fatty acids and 4-nitrophenol in mouse models of Aβ deposition. Similarly, it would be interesting to do a detailed exposome to determine possible environmental risk factors of amyloid deposition.
The following are the supplementary data related to this article.
Spearman's rank correlations between the levels of metabolites and florbetapir SUVr. Correlation plots of the values obtained for X3, X5, X6, X75 and X280 in the plasma and the florbetapir SUVr for all selected subjects (dark blue lines) and for amyloid (+) and amyloid (−) subjects separately (green and red lines respectively). Correlation coefficients and p-values are indicated on the graphs.
Florbetapir global SUVr in the amyloid (+) subjects and levels of the five most significant metabolites in the amyloid (+) and amyloid (−) subjects from the INSIGHT cohort. Subjects who converted to AD-MCI or to other types of dementia are green and blue respectively. Non-converters are in red.
Comparison of the longitudinal measures of three discriminant metabolites for amyloid positivity in asymptomatic individuals at-risk for AD. Correlation between values obtained in the same individuals at two different time points for three medium chain fatty acids: X3 (p = 0·028), X5 (p = 0·02) and X6 (p = 0·041).
Evolution of florbetapir SUVr in the amyloid (−) (A) and amyloid (+) (B) subjects from the INSIGHT cohort. Time lapse between the two PET scans are normalised to one year.
List of differentially expressed metabolomics features according to the univariate analysis. The sheet “Overlap” includes the list of the features that were significant in both Wilcoxon (p < .05) and CatScore (>50), detailed respectively on the sheets “Wilcoxon p < .05” and “CatScore 50”.
List of differentially expressed lipidomics features according to the univariate analysis. The sheet “Wilcoxon p < .05” contains the list of variables that are significant in the Wilcoxon Rank Sum Test at p < 0·05. In this case no CatScore >50 or Overlap sheets are available because no features were significant for CatScore.
List of differentially expressed metabolomics features according to the univariate analysis. The sheet “Overlap” includes the list of the features that were significant in DeSeq2 (p < 0·05), Wilcoxon (p < 0·05), and CatScore (>50), detailed respectively on the sheets “DeSeq2 p < 0·05”, “Wilcoxon p < 0·05” and “CatScore 50”.
List of all the GO Biological terms obtained from the enrichment analysis. Results presented include GO Term, p-value, adjusted p-value, z-score, combined score, and the genes contributing to the GO term. For this study uncorrected p-values have been taken into account.
Top 10 bootstrap stability selection (1000 replicates) for the transcriptomic, metabolomics and lipidomic, and multiomic model selection for each dataset, with colour-coded increases and decreases in the amyloid (+) group.
Correlation matrix for the five most significant metabolites, florbetapir SUVr and the CSF levels of tau, phospho-tau and Aβ42 in amyloid (+) and amyloid (−) subjects from the INSIGHT cohort without and with Bonferroni correction.
Funding sources
Institut Hospitalo-Universitaire and Institut du Cerveau et de la Moelle Epiniere (IHU-A-ICM), Ministry of Research, Fondation Alzheimer, Pfizer, and Avid. The funders did not play a role in manuscript design, data collection, data analysis, interpretation nor writing of the manuscript.
Author contributions
M-CP, FM and BD designed the study. FI, BC, ML, GF, HB, M-OH, SE collected the data. LX, F-XL, AT, IL, FM and M-CP analysed the data. All authors contributed to the writing and revisions of the paper and approved the final version.
Declaration of Competing Interest
BD has received consultancy fees from Biogen, Boehringer Ingelheim and Eli Lilly, and grants for his institution from Merck, Pfizer, and Roche. SE has received grants from Eli Lilly and consul tant fees from Astellas Pharma. M-CP has received grants from Fondation Vaincre Alzheimer, Laboratoires Servier, Pfizer, and Roche. M-OH has received consultant fees from Eli Lilly and speaker fees from Piramal. The other authors declare no competing interests.
Acknowledgments
This study was supported by INSERM in collaboration with Institut du Cerveau et de la Moelle Epiniere (ICM), Institut Hospitalo-Universitaire-A ICM, and Pfizer, and was supported with funding from Pfizer and the Investissement d'Avenir (ANR-10-AIHU-06) that was used for the recruitment of clinical research assistants, neuropsychologists, and a study physician. The study was done in collaboration with the Centre Hospitalier Universitaire de Bordeaux (study number CIC EC7). Avid provided the 18F-florbetapir ligand for amyloid β PET.
Contributor Information
Fanny Mochel, Email: fanny.mochel@upmc.fr.
Marie-Claude Potier, Email: marie-claude.potier@upmc.fr.
References
- 1.Golde T.E., DeKosky S.T., Galasko D. Alzheimer's disease: the right drug, the right time. Science. 2018;362(6420):1250–1251. doi: 10.1126/science.aau0437. [DOI] [PubMed] [Google Scholar]
- 2.Jack C.R., Jr., Wiste H.J., Therneau T.M. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321(23):2316–2325. doi: 10.1001/jama.2019.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knopman D.S., Petersen R.C., Jack C.R., Jr. A brief history of "Alzheimer disease": multiple meanings separated by a common name. Neurology. 2019;92(22):1053–1059. doi: 10.1212/WNL.0000000000007583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanehisa M., Sato Y., Furumichi M., Morishima K., Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590–d5. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arenaza-Urquijo E.M., Przybelski S.A., Lesnick T.L. The metabolic brain signature of cognitive resilience in the 80+: beyond Alzheimer pathologies. Brain. 2019;142(4):1134–1147. doi: 10.1093/brain/awz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen R.C., Lundt E.S., Therneau T.M. Predicting progression to mild cognitive impairment. Ann Neurol. 2019;85(1):155–160. doi: 10.1002/ana.25388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon B.A., Blazey T.M., Su Y. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241–250. doi: 10.1016/S1474-4422(18)30028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molinuevo J.L., Ayton S., Batrla R. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol. 2018;136(6):821–853. doi: 10.1007/s00401-018-1932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubois B., Epelbaum S., Nyasse F. Cognitive and neuroimaging features and brain beta-amyloidosis in individuals at risk of Alzheimer's disease (INSIGHT-preAD): a longitudinal observational study. Lancet Neurol. 2018;17(4):335–346. doi: 10.1016/S1474-4422(18)30029-2. [DOI] [PubMed] [Google Scholar]
- 11.Habert M.O., Bertin H., Labit M. Evaluation of amyloid status in a cohort of elderly individuals with memory complaints: validation of the method of quantification and determination of positivity thresholds. Ann Nucl Med. 2018;32(2):75–86. doi: 10.1007/s12149-017-1221-0. [DOI] [PubMed] [Google Scholar]
- 12.Seyer A., Boudah S., Broudin S., Junot C., Colsch B. Annotation of the human cerebrospinal fluid lipidome using high resolution mass spectrometry and a dedicated data processing workflow. Metabolomics. 2016;12:91. doi: 10.1007/s11306-016-1023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.R Core Team . 2018. R: A language and environment for statistical computing.https://www.R-project.org/ [Google Scholar]
- 14.Kessner D., Chambers M., Burke R., Agus D., Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith C.A., Want E.J., O'Maille G., Abagyan R., Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 16.Tautenhahn R., Patti G.J., Rinehart D., Siuzdak G. XCMS online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84(11):5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giacomoni F., Le Corguille G., Monsoor M. Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics. 2015;31(9):1493–1495. doi: 10.1093/bioinformatics/btu813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn W.B., Broadhurst D., Begley P. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–1083. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- 19.Boudah S., Olivier M.F., Aros-Calt S. Annotation of the human serum metabolome by coupling three liquid chromatography methods to high-resolution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;966:34–47. doi: 10.1016/j.jchromb.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Wishart D.S., Feunang Y.D., Marcu A. HMDB 4.0: The human metabolome database for. Nucleic Acids Res 2018. 2018;46(D1) doi: 10.1093/nar/gkx1089. (D608-D17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1) doi: 10.1093/nar/gkw1092. (D353-D61) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bersanelli M., Mosca E., Remondini D. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. 2016;17(Suppl. 2):15. doi: 10.1186/s12859-015-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang S., Chaudhary K., Garmire L.X. More is better: recent Progress in multi-omics data integration methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (Oxford, England) 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 26.Stanley E., Delatola E.I., Nkuipou-Kenfack E. Comparison of different statistical approaches for urinary peptide biomarker detection in the context of coronary artery disease. BMC Bioinforma. 2016;17(1):496. doi: 10.1186/s12859-016-1390-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuber V., Strimmer K. Gene ranking and biomarker discovery under correlation. Bioinformatics. 2009;25(20):2700–2707. doi: 10.1093/bioinformatics/btp460. [DOI] [PubMed] [Google Scholar]
- 28.Iniesta R., Hodgson K., Stahl D. Antidepressant drug-specific prediction of depression treatment outcomes from genetic and clinical variables. Sci Rep. 2018;8(1):5530. doi: 10.1038/s41598-018-23584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen E.Y., Tan C.M., Kou Y. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenenhaus A., Tenenhaus M. Regularized generalized canonical correlation analysis. Psychometrika. 2011;76(2):257. doi: 10.1007/s11336-017-9573-x. [DOI] [PubMed] [Google Scholar]
- 31.Tenenhaus A., Philippe C., Guillemot V., Le Cao K.A., Grill J., Frouin V. Variable selection for generalized canonical correlation analysis. Biostatistics (Oxford, England) 2014;15(3):569–583. doi: 10.1093/biostatistics/kxu001. [DOI] [PubMed] [Google Scholar]
- 32.Garali I., Adanyeguh I.M., Ichou F. A strategy for multimodal data integration: application to biomarkers identification in spinocerebellar ataxia. Brief Bioinform. 2018;19(6):1356–1369. doi: 10.1093/bib/bbx060. [DOI] [PubMed] [Google Scholar]
- 34.Rohart F., Gautier B., Singh A., Le Cao K.A. mixOmics: An R package for 'omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13(11):e1005752. doi: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeDell E., Petersen M., van der Laan M. Computationally efficient confidence intervals for cross-validated area under the ROC curve estimates. Electronic J Statistics. 2015;9(1):1583–1607. doi: 10.1214/15-EJS1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim Y.Y., Mormino E.C. APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology. 2017;89(10):1028–1034. doi: 10.1212/WNL.0000000000004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grothe M.J., Villeneuve S., Dyrba M., Bartres-Faz D., Wirth M. Multimodal characterization of older APOE2 carriers reveals selective reduction of amyloid load. Neurology. 2017;88(6):569–576. doi: 10.1212/WNL.0000000000003585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genin E., Hannequin D., Wallon D. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–907. doi: 10.1038/mp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigue K.M., Kennedy K.M., Devous M.D., Sr. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78(6):387–395. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marttinen M., Paananen J., Neme A. A multiomic approach to characterize the temporal sequence in Alzheimer's disease-related pathology. Neurobiol Dis. 2019;124:454–468. doi: 10.1016/j.nbd.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 41.Glaab E., Trezzi J.P., Greuel A. Integrative analysis of blood metabolomics and PET brain neuroimaging data for Parkinson's disease. Neurobiol Dis. 2019;124:555–562. doi: 10.1016/j.nbd.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Athauda D., Gulyani S., Karnati H. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-PD trial. JAMA Neurol. 2019;76(4):420–429. doi: 10.1001/jamaneurol.2018.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M., Zeringer E., Barta T., Schageman J., Cheng A., Vlassov A.V. Analysis of the RNA content of the exosomes derived from blood serum and urine and its potential as biomarkers. Philos Trans R Soc Lond B Biol Sci. 2014;369(1652) doi: 10.1098/rstb.2013.0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chambers J.E., Chambers H.W., Meek E.C. Novel nucleophiles enhance the human serum paraoxonase 1 (PON1)-mediated detoxication of organophosphates. Toxicol Sci. 2015;143(1):46–53. doi: 10.1093/toxsci/kfu205. [DOI] [PubMed] [Google Scholar]
- 45.Du F., Fung Y.S. Dual-opposite multi-walled carbon nanotube modified carbon fiber microelectrode for microfluidic chip-capillary electrophoresis determination of methyl parathion metabolites in human urine. Electrophoresis. 2018;39(11):1375–1381. doi: 10.1002/elps.201700470. [DOI] [PubMed] [Google Scholar]
- 46.Ball N., Teo W.P., Chandra S., Chapman J. Parkinson's disease and the environment. Front Neurol. 2019;10:218. doi: 10.3389/fneur.2019.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eid A., Mhatre I., Richardson J.R. Gene-environment interactions in Alzheimer's disease: a potential path to precision medicine. Pharmacol Ther. 2019;199:173–187. doi: 10.1016/j.pharmthera.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oresic M., Hyotylainen T., Herukka S.K. Metabolome in progression to Alzheimer's disease. Transl Psychiatry. 2011;1 doi: 10.1038/tp.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trushina E., Dutta T., Persson X.M., Mielke M.M., Petersen R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer's disease using metabolomics. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G., Zhou Y., Huang F.J. Plasma metabolite profiles of Alzheimer's disease and mild cognitive impairment. J Proteome Res. 2014;13(5):2649–2658. doi: 10.1021/pr5000895. [DOI] [PubMed] [Google Scholar]
- 51.Conquer J.A., Tierney M.C., Zecevic J., Bettger W.J., Fisher R.H. Fatty acid analysis of blood plasma of patients with Alzheimer's disease, other types of dementia, and cognitive impairment. Lipids. 2000;35(12):1305–1312. doi: 10.1007/s11745-000-0646-3. [DOI] [PubMed] [Google Scholar]
- 52.Sato Y., Suzuki I., Nakamura T., Bernier F., Aoshima K., Oda Y. Identification of a new plasma biomarker of Alzheimer's disease using metabolomics technology. J Lipid Res. 2012;53(3):567–576. doi: 10.1194/jlr.M022376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X., Rozen S., Boyle S.H. Metabolomics in early Alzheimer's disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One. 2011;6(7) doi: 10.1371/journal.pone.0021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toledo J.B., Arnold M., Kastenmuller G. Metabolic network failures in Alzheimer's disease: a biochemical road map. Alzheimers Dement. 2017;13(9):965–984. doi: 10.1016/j.jalz.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mapstone M., Cheema A.K., Fiandaca M.S. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 2014;20(4):415–418. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rice L., Bisdas S. The diagnostic value of FDG and amyloid PET in Alzheimer's disease-a systematic review. Eur J Radiol. 2017;94:16–24. doi: 10.1016/j.ejrad.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 57.Rebello C.J., Keller J.N., Liu A.G., Johnson W.D., Greenway F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: a randomized controlled trial. BBA Clin. 2015;3:123–125. doi: 10.1016/j.bbacli.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Augustin K., Khabbush A., Williams S. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018;17(1):84–93. doi: 10.1016/S1474-4422(17)30408-8. [DOI] [PubMed] [Google Scholar]
- 59.Naughton B.J., Duncan F.J., Murrey D.A. Blood genome-wide transcriptional profiles reflect broad molecular impairments and strong blood-brain links in Alzheimer's disease. J Alzheimers Dis. 2015;43(1):93–108. doi: 10.3233/JAD-140606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukhamedyarov M.A., Rizvanov A.A., Yakupov E.Z. Transcriptional analysis of blood lymphocytes and skin fibroblasts, keratinocytes, and endothelial cells as a potential biomarker for Alzheimer's disease. J Alzheimers Dis. 2016;54(4):1373–1383. doi: 10.3233/JAD-160457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo H., Han G., Wang J. Common aging signature in the peripheral blood of vascular dementia and Alzheimer's disease. Mol Neurobiol. 2016;53(6):3596–3605. doi: 10.1007/s12035-015-9288-x. [DOI] [PubMed] [Google Scholar]
- 62.Lunnon K., Sattlecker M., Furney S.J. A blood gene expression marker of early Alzheimer's disease. J Alzheimers Dis. 2013;33(3):737–753. doi: 10.3233/JAD-2012-121363. [DOI] [PubMed] [Google Scholar]
- 63.Kalman J., Kitajka K., Pakaski M. Gene expression profile analysis of lymphocytes from Alzheimer's patients. Psychiatr Genet. 2005;15(1):1–6. doi: 10.1097/00041444-200503000-00001. [DOI] [PubMed] [Google Scholar]
- 64.Booij B.B., Lindahl T., Wetterberg P. A gene expression pattern in blood for the early detection of Alzheimer's disease. J Alzheimers Dis. 2011;23(1):109–119. doi: 10.3233/JAD-2010-101518. [DOI] [PubMed] [Google Scholar]
- 65.Bai Z., Stamova B., Xu H. Distinctive RNA expression profiles in blood associated with Alzheimer disease after accounting for white matter hyperintensities. Alzheimer Dis Assoc Disord. 2014;28(3):226–233. doi: 10.1097/WAD.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Antonell A., Llado A., Sanchez-Valle R. Altered blood gene expression of tumor-related genes (PRKCB, BECN1, and CDKN2A) in Alzheimer's disease. Mol Neurobiol. 2016;53(9):5902–5911. doi: 10.1007/s12035-015-9483-9. [DOI] [PubMed] [Google Scholar]
- 67.Chong M.S., Goh L.K., Lim W.S. Gene expression profiling of peripheral blood leukocytes shows consistent longitudinal downregulation of TOMM40 and upregulation of KIR2DL5A, PLOD1, and SLC2A8 among fast progressors in early Alzheimer's disease. J Alzheimers Dis. 2013;34(2):399–405. doi: 10.3233/JAD-121621. [DOI] [PubMed] [Google Scholar]
- 68.Fehlbaum-Beurdeley P., Sol O., Desire L. Validation of AclarusDx, a blood-based transcriptomic signature for the diagnosis of Alzheimer's disease. J Alzheimers Dis. 2012;32(1):169–181. doi: 10.3233/JAD-2012-120637. [DOI] [PubMed] [Google Scholar]
- 69.Delvaux E., Mastroeni D., Nolz J. Multivariate analyses of peripheral blood leukocyte transcripts distinguish Alzheimer's, Parkinson's, control, and those at risk for developing Alzheimer's. Neurobiol Aging. 2017;58:225–237. doi: 10.1016/j.neurobiolaging.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 70.Chen K.D., Chang P.T., Ping Y.H., Lee H.C., Yeh C.W., Wang P.N. Gene expression profiling of peripheral blood leukocytes identifies and validates ABCB1 as a novel biomarker for Alzheimer's disease. Neurobiol Dis. 2011;43(3):698–705. doi: 10.1016/j.nbd.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 71.Mitchell A.J. The clinical significance of subjective memory complaints in the diagnosis of mild cognitive impairment and dementia: a meta-analysis. Int J Geriatr Psychiatry. 2008;23(11):1191–1202. doi: 10.1002/gps.2053. [DOI] [PubMed] [Google Scholar]
- 72.Vlachos G.S., Cosentino S., Kosmidis M.H. Prevalence and determinants of subjective cognitive decline in a representative Greek elderly population. Int J Geriatr Psychiatry. 2019;34(6):846–854. doi: 10.1002/gps.5073. [DOI] [PubMed] [Google Scholar]
- 73.Snitz B.E., Wang T., Cloonan Y.K. Risk of progression from subjective cognitive decline to mild cognitive impairment: the role of study setting. Alzheimers Dement. 2018;14(6):734–742. doi: 10.1016/j.jalz.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamelin L., Lagarde J., Dorothee G. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain. 2018;141(6):1855–1870. doi: 10.1093/brain/awy079. [DOI] [PubMed] [Google Scholar]
- 75.Rabin J.S., Schultz A.P., Hedden T. Interactive associations of vascular risk and beta-amyloid burden with cognitive decline in clinically normal elderly individuals: findings from the Harvard Aging Brain Study. JAMA Neurol. 2018;75(9):1124–1131. doi: 10.1001/jamaneurol.2018.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nelson P.T., Alafuzoff I., Bigio E.H. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spearman's rank correlations between the levels of metabolites and florbetapir SUVr. Correlation plots of the values obtained for X3, X5, X6, X75 and X280 in the plasma and the florbetapir SUVr for all selected subjects (dark blue lines) and for amyloid (+) and amyloid (−) subjects separately (green and red lines respectively). Correlation coefficients and p-values are indicated on the graphs.
Florbetapir global SUVr in the amyloid (+) subjects and levels of the five most significant metabolites in the amyloid (+) and amyloid (−) subjects from the INSIGHT cohort. Subjects who converted to AD-MCI or to other types of dementia are green and blue respectively. Non-converters are in red.
Comparison of the longitudinal measures of three discriminant metabolites for amyloid positivity in asymptomatic individuals at-risk for AD. Correlation between values obtained in the same individuals at two different time points for three medium chain fatty acids: X3 (p = 0·028), X5 (p = 0·02) and X6 (p = 0·041).
Evolution of florbetapir SUVr in the amyloid (−) (A) and amyloid (+) (B) subjects from the INSIGHT cohort. Time lapse between the two PET scans are normalised to one year.
List of differentially expressed metabolomics features according to the univariate analysis. The sheet “Overlap” includes the list of the features that were significant in both Wilcoxon (p < .05) and CatScore (>50), detailed respectively on the sheets “Wilcoxon p < .05” and “CatScore 50”.
List of differentially expressed lipidomics features according to the univariate analysis. The sheet “Wilcoxon p < .05” contains the list of variables that are significant in the Wilcoxon Rank Sum Test at p < 0·05. In this case no CatScore >50 or Overlap sheets are available because no features were significant for CatScore.
List of differentially expressed metabolomics features according to the univariate analysis. The sheet “Overlap” includes the list of the features that were significant in DeSeq2 (p < 0·05), Wilcoxon (p < 0·05), and CatScore (>50), detailed respectively on the sheets “DeSeq2 p < 0·05”, “Wilcoxon p < 0·05” and “CatScore 50”.
List of all the GO Biological terms obtained from the enrichment analysis. Results presented include GO Term, p-value, adjusted p-value, z-score, combined score, and the genes contributing to the GO term. For this study uncorrected p-values have been taken into account.
Top 10 bootstrap stability selection (1000 replicates) for the transcriptomic, metabolomics and lipidomic, and multiomic model selection for each dataset, with colour-coded increases and decreases in the amyloid (+) group.
Correlation matrix for the five most significant metabolites, florbetapir SUVr and the CSF levels of tau, phospho-tau and Aβ42 in amyloid (+) and amyloid (−) subjects from the INSIGHT cohort without and with Bonferroni correction.