

Abstract
The basolateral complex of amygdala (BLA) processes emotionally arousing aversive and rewarding experiences. The BLA is critical for acquisition and storage of threat-based memories and the modulation of the consolidation of arousing explicit memories, that is, the memories that are encoded and stored by the medial temporal lobe. In addition, in conjunction with the medial prefrontal cortex (mPFC), the BLA plays an important role in fear memory extinction. The BLA develops relatively early in life, but little is known about the molecular changes that accompany its development. Here, we quantified relative basal expression levels of sets of plasticity, synaptic, glia, and connectivity proteins in the rat BLA at various developmental ages: postnatal day 17 (PN17, infants), PN24 (juveniles), and PN80 (young adults). We found that the levels of activation markers of brain plasticity, including phosphorylation of CREB at Ser133, CamKIIα at Thr286, pERK1/pERK2 at Thr202/Tyr204, and GluA1 at Ser831 and Ser845, were significantly higher in infant and juvenile compared with adult brain. In contrast, age increase was accompanied by a significant augmentation in the levels of proteins that mark synaptogenesis and synapse maturation, such as synaptophysin, PSD95, SynCAM, GAD65, GAD67, and GluN2A/GluN2B ratio. Finally, we observed significant age-associated changes in structural markers, including MAP2, MBP, and MAG, suggesting that the structural connectivity of the BLA increases over time. The biological differences in the BLA between developmental ages compared with adulthood suggest the need for caution in extrapolating conclusions based on BLA-related brain plasticity and behavioral studies conducted at different developmental stages.
The biological changes that accompany brain development and the functional maturation of the brain are topics of intense interest in neuroscience. The mammalian brain undergoes prolonged postnatal development, and both pre- and postnatal developmental processes are remarkably well conserved across species (Semple et al. 2013).
Critical brain functions that influence most, if not all, behaviors are learning and memory. Although our understanding of how behavioral responses change over the course of development has advanced over the past 50 years, the biological details underlying learning, memory formation, and storage at early developmental ages, as well as how these biological changes progress over time, remain poorly understood. The brain is thought to be programmed to drive behavior, but we still do not know how the biology underlying these programs originates, develops, and functions.
Moreover, plasticity mechanisms have been largely studied in early developmental brain preparations (e.g., slices from young postnatal brains or cell cultures) and the results obtained have been often considered to be in principle applicable to the adult brain. Although these investigations provided important early insights into plasticity and memory mechanisms, it has become increasingly clear that a deeper understanding of the brain, as well as learning and memory processes recruited at different ages, will require a far more comprehensive and age-specific knowledge.
One obvious question is whether and how the brain systems engaged in learning and memory change their biology over the course of development, particularly during behavioral maturation. In previous studies, we determined that the dorsal hippocampus (dHC) (Travaglia et al. 2016a) and the medial prefrontal cortex (mPFC) (Jia et al. 2018), two brain regions that play critical roles in the formation of hippocampus-dependent memories, exhibit significant developmental changes in the expression levels of proteins involved in synaptic plasticity, glia, and connectivity. Our assessment of numerous markers of these functions revealed dramatic changes in rat dHC and mPFC between postnatal day 17 (PN17), PN24, and PN80. These ages were chosen for investigation because at PN17, rats can learn episodic contextual experiences but exhibit very rapid forgetting, a phenomenon typical of infancy that has been associated with infantile amnesia (Campbell and Spear 1972; Alberini and Travaglia 2017; Ramsaran et al. 2019). Our findings showed that rapid forgetting by PN17 rats reflects a critical period for hippocampal learning, during which the hippocampus-dependent memory system matures in response to experience (Travaglia et al. 2016b, 2018; Alberini and Travaglia 2017). On the other hand, by PN24, rats have acquired the ability to express long-lasting memories, a typical behavioral response of adulthood (PN80) (Travaglia et al. 2016a,b). According to anatomical and connectivity comparative studies, PN17 in the rat corresponds to ∼2–3 yr of age in humans, PN24 to 7–10 yr of age, and PN80 to postadolescence (Madsen and Kim 2016; Alberini and Travaglia 2017). Hence, our assessments of PN17 and PN24 were aimed at comparing the brain regions of two relatively close developmental ages that are significantly different with respect to functional development, and then comparing both of these early stages to young adulthood, when memory functions are fully matured.
In this study, we expanded our previous work on the dHC and mPFC by adding analyses of the same classes of proteins in the basolateral amygdala (BLA). The amygdala, and particularly the BLA, modulates the encoding, consolidation, and expression of contextual, spatial, and episodic memories (Killcross et al. 1997; LeDoux 2000; Maren 2003; McIntyre et al. 2003; McGaugh 2004; Janak and Tye 2015). Although the hippocampal complex and amygdala can function as two independent memory systems, in the context of complex arousing experiences the amygdala- and hippocampus-dependent systems work together and interact (McGaugh et al. 2002; LeDoux 2003; Phelps 2004). Moreover, the hippocampal system, via the mPFC, processes emotionally charged episodic representations and influences the response of the amygdala to emotional stimuli (McGaugh 2004; Richardson et al. 2004).
To increase the understanding of which molecular mechanisms change during development in the rat BLA, we assessed posttranslational modifications (i.e., phosphorylation) and the relative concentrations of numerous biological mechanisms involved in synapse maturation, plasticity, glia, and connectivity. To this end, we performed relative densitometric western blot analyses to compare PN17, PN24, and PN80. We then related the results to those previously obtained in the dHC and mPFC at the same ages for discussion purposes.
Results
Levels of Arc/Arg3.1, but not c-Fos and Zif268, are significantly higher in early postnatal development than in adulthood
In our analysis of the BLA (Fig. 1), we first assessed the developmental changes in levels of the activity-dependent immediate-early genes (IEGs) c-Fos (Tischmeyer and Grimm 1999), activity-regulated-cytoskeletal-associated-protein (Arc/Arg3.1) (Bramham et al. 2008), and Zif268 (also known as EGR1 or NGFI-A) (Jones et al. 2001). These IEGs are induced by neural activity, and, are commonly considered as markers of cellular activation and neuronal plasticity. In the adult brain, the mRNA and protein levels of these IEGs are generally low under basal conditions, but they undergo rapid transient increases in response to high-frequency stimulations that evoke long-term potentiation (LTP) and long-term depression (LTD), as well as behavioral experiences leading to long-term memory (Morgan et al. 1987; Cole et al. 1989; Worley et al. 1993; Tischmeyer and Grimm 1999; Jones et al. 2001; Plath et al. 2006; Bramham et al. 2008). c-Fos and Zif268 regulate the transcription of downstream target genes that drive long-term synaptic plasticity and long-term memory formation (Lanahan and Worley 1998; Jones et al. 2001), whereas Arc/Arg3.1 is involved in the remodeling of dendritic spine morphology and endocytosis of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (Steward and Worley 2001; Bramham et al. 2008; Korb and Finkbeiner 2011; Okuno et al. 2018). In addition, Arc may function as a neuronal transposon (Ashley et al. 2018; Shepherd 2018).
Figure 1.
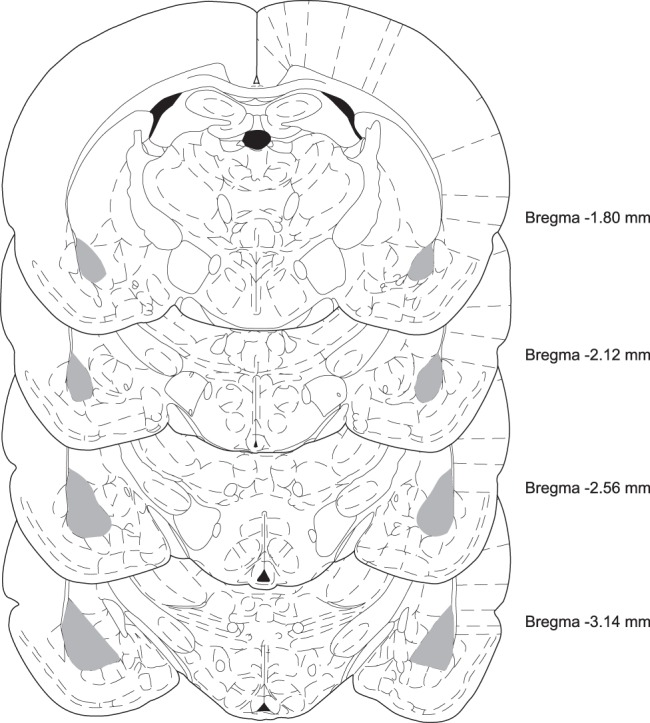
Coordinates of rat basolateral amygdala (BLA) (modified from Paxinos and Watson 2005), indicating the area isolated by micropunches (in gray) in adult (PN80) brains. The BLAs at all ages were identified and collected using the same anatomical references in each coronal section: the shape of dHC, medial eminence, third ventricle, rhinal fissure, external capsule, and optic tract.
As shown in Figure 2, one-way ANOVA followed by Newman–Keuls post-hoc tests revealed no significant difference in the level of c-Fos (F(2,13) = 2.409; P = 0.1288) among the three age groups, and a nonsignificant trend toward a decrease in Zif268 with age (F(2,14) = 2.055; P = 0.1650). In contrast, Arc expression was significantly higher at PN17 than at PN24 and PN80 (F(2,14) = 12.02; P = 0.0009) and decreased significantly both between PN17 and PN24 (P < 0.05) and between PN24 and adulthood (P < 0.05). Thus, the BLA at PN17 expresses the highest level of Arc; along with the decreasing trend in Zif268 with age, this suggests that overall cellular activation is higher in the infantile vs. adult BLA.
Figure 2.

The level of Arc, but not Zif268 or c-Fos, decreased from early development to adulthood. Densitometric western blot analyses were performed on total protein extracts of BLA obtained from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 7), or PN80 (adult; black, n = 4). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (*) P < 0.05; (***) P < 0.001.
The ratios of phosphorylated to total CREB, CaMKIIα, and ERK, but not TrkB, are significantly higher in early development than in adulthood
Next, we determined the levels of tropomyosin-related kinase B receptor (TrkB), measuring both total protein and the phosphorylated form at Tyr816 (phospho-TrkB). Phosphorylation of TrkB at this residue leads to phosphorylation of the N-methyl-d-aspartate receptor (NMDA) and AMPA receptor, both of which are critical for synaptic plasticity (Martínez et al. 1998; Minichiello 2009; Andero et al. 2014). In addition, we assessed the phosphorylation and total levels of synaptic plasticity-–coupled kinases calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα) (Lisman et al. 2002) and extracellular signal-regulated kinases ERK1/2 (Sweatt 2004), as well as their target transcription factor cAMP response element-binding protein (CREB) (Yin and Tully 1996; Silva et al. 1998; Mayr and Montminy 2001; Alberini 2009; Kandel 2012). The phosphorylation of all these proteins induces their functional activation. CaMKIIα and ERK activate the transcription factor CREB, which in many different systems and species exerts a fundamental role in long-term plasticity and memory formation (Frank and Greenberg 1994; Yin and Tully 1996; Silva et al. 1998; Barco et al. 2003; Alberini and Kandel 2015). Specifically, we monitored: (i) autophosphorylation of CaMKIIα at Thr286 (pCamKIIα), which is crucial for the persistent enzymatic activation of CAMKIIα and induction of LTP (Lisman et al. 2002, 2012); (ii) phosphorylations of ERK1 and ERK2 at Thr 202/Tyr204 (pERK1/2), which are necessary to activate ERK, and thus the phosphorylation and activation of downstream targets such as CREB (Sweatt 2004; Thomas and Huganir 2004); and, finally, (iii) the phosphorylation at Ser133 of CREB (pCREB), which is critical for CREB's function as a transcriptional regulator (Mayr and Montminy 2001).
As depicted in Figure 3, one-way ANOVA followed by Newman–Keuls post-hoc tests revealed a nonsignificant trend toward a decrease in the phosphorylation of TrkB at Tyr816 from PN17 to adult (F(2,14) = 3.035; P = 0.0804). Total levels of TrkB were similar among all three ages (F(2,14) = 2.251; P = 0.1420); consequently, there was no significant difference in the pTrkB/TrkB ratio (F(2,14) = 0.2636; P = 0.7720).
Figure 3.

pCREB/CREB, pCamKII/CamKII, pERK1/ERK1, and pERK2/ERK2 ratios decrease massively from early development to adulthood. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 6–7), or PN80 (adult; black, n = 4–5). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
We observed a significant and dramatic (∼1000%) decrease in the level of pCREB between PN17 and adulthood (F(2,14) = 11.54; P = 0.0011), which was statistically significant between PN17 and PN24 (P < 0.05, decrease of 400%–500%) and between PN24 and PN80 (P < 0.05, decrease of ∼500%). Conversely, the level of total CREB was slightly but significantly higher in adults compared with both PN17 and PN24 rats (F(2,14) = 5.142; P = 0.0212) (Fig. 3). Hence, the pCREB/CREB ratio significantly and dramatically decreased from PN17 to PN24 and from PN24 to adulthood (F(2,14) = 10.96; P = 0.0014) (Fig. 3).
The level of pCaMKIIα was reduced ∼1500-fold (F(2,14) = 13.03; P = 0.0006) from PN17 and PN24 to adults (P < 0.01 and P < 0.001, respectively), with no significant difference between the two youngest ages (P > 0.05). Total CaMKIIα levels did not change across age groups (F(2,14) = 0.5090; P = 0.6118), resulting in a significant and robust decrease in the pCaMKIIα/CaMKIIα ratio from PN17 and PN24 to adulthood (F(2,14) = 15.17; P = 0.0003) (Fig. 3).
Finally, there was no significant difference among the three age groups in pERK1 (F(2,15) = 2.386; P = 0.1260) and pERK2 (F(2,15) = 0.2429; P = 0.7874). In contrast, the total level of ERK1 (F(2,15) = 16.95; P = 0.0001) and ERK2 (F(2,15) = 29.51; P < 0.0001) significantly increased from both PN17 and PN24 to adult age, and ERK2 also significantly increased from PN17 to PN24 (P < 0.05) (Fig. 3). Consequently, the pERK1/ERK1 ratio significantly diminished (F(2,15) = 20.25; P < 0.0001) from PN17 to PN24 (P < 0.05), PN17 to adult age (P < 0.001) and PN24 to adulthood (P < 0.01). Similarly, the pERK2/ERK2 ratio significantly decreased (F(2,15) = 5.697; P = 0.0144) from both PN17 and PN24 to adult age (P < 0.05), with no significant difference between PN17 and PN24 (P > 0.05) (Fig. 3).
Collectively, these results show that, in the BLA, activation of kinases and transcription factors with fundamental roles in synaptic plasticity mechanisms is highest during early postnatal development and decreases toward adulthood.
The GluN2A/GluN2B ratio, as well as GluA1 and GluA2 levels, increase over development, whereas GluA1 phosphorylation dramatically decreases
Next, we examined the protein levels of the NMDA receptor (NMDAR) regulatory subunits GluN2A and GluN2B, whose ratio affects the kinetics of the NMDA excitatory postsynaptic current (EPSCs), the interactions of NMDARs with postsynaptic density proteins such as CaMKIIα, and the efficacy of synaptic plasticity (Sheng et al. 1994; Flint et al. 1997; Sanhueza et al. 2011; Paoletti et al. 2013). One-way ANOVA followed by Newman–Keuls post-hoc tests revealed no significant changes in GluN2A level across development (F(2,17) = 3.247; P = 0.0718) but a significant decrease in GluN2B (F(2,17) = 10.51; P = 0.0011); specifically, GluN2B levels were higher at PN17 and PN24 than in adulthood (P < 0.01), but did not differ significantly between PN17 and PN24 (P > 0.05) (Fig. 4). Consequently, there was a significant increase in the GluN2A/GluN2B ratio (F(2,17) = 23.00; P < 0.0001) between PN17 and both PN24 and adulthood (P < 0.001), as well as between PN24 and adults (P < 0.05) (Fig. 4).
Figure 4.
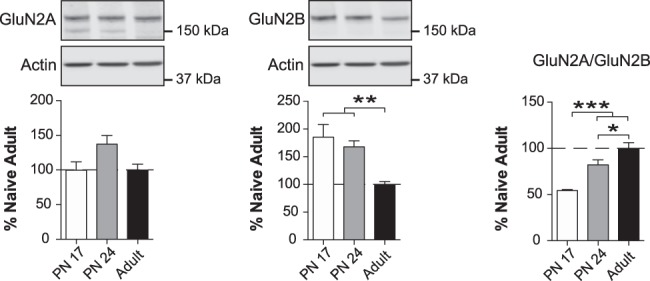
The GluN2A/GluN2B NMDAR subunit ratio increases over the course of development. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 7), or PN80 (adult; black, n = 7). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
The increase in GluN2A/GluN2B ratio over development has been well established in other areas of the postnatal rat brain, including the cortices (Sheng et al. 1994; Yashiro and Philpot 2008; Paoletti et al. 2013), hippocampus (Travaglia et al. 2016a), and mPFC (Jia et al. 2018). This switch in NMDAR composition occurs throughout the CNS; is evolutionarily conserved from amphibians to mammals, significantly influences the functional properties of the synapses; and occurs during a time window that coincides with synapse maturation, circuit refinement, and acquisition of learning abilities (Sheng et al. 1994; Flint et al. 1997; Dumas 2005; Paoletti et al. 2013).
Next, we determined the phosphorylation and total levels of AMPA receptor subunits GluA1 and GluA2. Both are fundamental and predominant AMPA receptor subunits, which control the majority of rapid, excitatory synaptic transmission and most of the depolarizing effects of glutamate. We determined the phosphorylation of GluA1 in Ser831 and Ser 845, as these phosphorylations are critical for delivery of AMPARs to synapses, single-channel conductivity of the GluR1 subunit (Derkach et al. 1999; Hayashi et al. 2000), and synaptic maturation and strength via the opening probability of AMPA receptors (Barria et al. 1997), respectively.
One-way ANOVA followed by Newman–Keuls post-hoc tests revealed that the PN17 BLA had a significantly lower level of GluA1 (F(2,17) = 5.941; P = 0.0111) and GluA2 (F(2,17) = 9.241; P = 0.0019) than that of adult rats (P < 0.01, respectively), and that the PN24 BLA also had a lower level of GluA2 than adult BLA (P < 0.01) (Fig. 5). No significant difference was observed between PN17 and PN24 BLAs for either subunit (P > 0.05). The level of Ser831 GluA1 phosphorylation was significantly and massively higher (more than 500% from PN17 to adult age) at PN17 vs. PN24 and at PN24 vs. adulthood (F(2,17) = 72.56; P < 0.0001, Fig. 5). The phosphorylation level of GluA1 at Ser 845 was similar between PN17 and PN24, but significantly decreased in adults (F(2,17) = 16.06; P = 0.0001, Fig. 5). The ratios of pGluA1(Ser831)/GluA1 (F(2,17) = 121.8; P < 0.0001) and pGluA1(Ser845)/GluA1 (F(2,17) = 43.74; P < 0.0001) decreased even more dramatically, dropping by 250%–650% between PN17 and adulthood (P < 0.0001) (Fig. 5).
Figure 5.
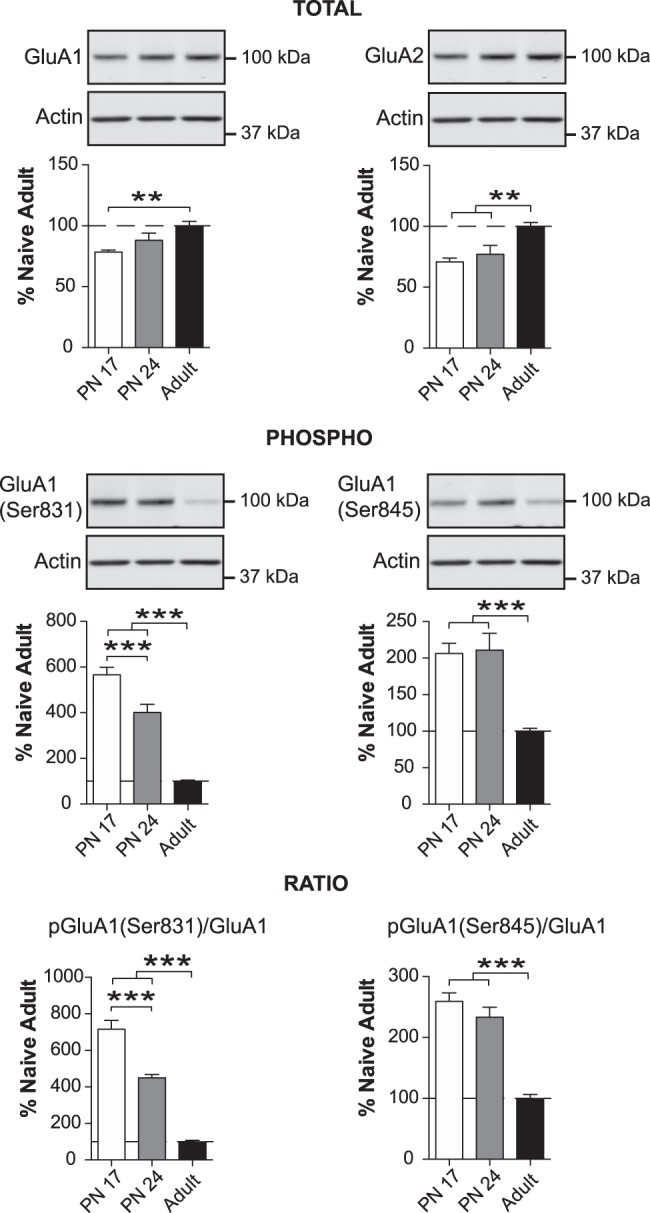
GluA1 and GluA2 AMPAR subunit levels increase, whereas Ser831 and Ser845 phosphorylation of GluA1 decrease, over the course of development. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 7) or PN80 (adult; black, n = 7). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (**) P < 0.01; (***) P < 0.001.
Thus, although GluA1 was less abundant in the BLA of PN17 rats, phosphorylation at Ser831 and Ser845 was significantly higher than in PN24 and/or of adult rats, suggesting that early development is accompanied by a very strong AMPAR activation.
Markers of excitatory and inhibitory synapse maturation increase over development
Excitatory and inhibitory synapses are major components of the brain and its functions. Here we investigated the levels of pre- and postsynaptic building proteins of excitatory or inhibitory synapse formation.
First, we assessed the levels of synaptophysin (Wiedenmann and Franke 1985; Frick and Fernandez 2003; Kwon and Chapman 2011) and postsynaptic density protein 95 (PSD-95) (Cho et al. 1992; Hunt et al. 1996; Cline 2005), which are markers of excitatory synaptic formation. Synaptophysin regulates exo- and endocytosis of neurotransmitter vesicles (Bähler et al. 1990; Kwon and Chapman 2011), whereas PSD95 guides assembly, clustering, and recycling of AMPARs and their interactive partners (Cline 2005; Good et al. 2011). As shown in Figure 6, one-way ANOVA followed by Newman–Keuls post-hoc tests revealed that the BLA has significantly less synaptophysin at PN17 (F(2,14) = 154.3; P < 0.0001) than at PN24 or in adulthood (P < 0.001). There was also a significant increase in the level of PSD95 (F(2,14) = 10.30; P = 0.0018) from PN17 to both PN24 and adulthood (P < 0.01), with no significant difference between PN24 and adult rats (P > 0.05) (Fig. 6). Similarly, BLA had significantly less SynCAM at PN17 (F(2,16) = 34.94; P < 0.0001) than in PN24 or adulthood (P < 0.01 and P < 0.001, respectively).
Figure 6.

Excitatory and inhibitory synapse maturation markers increase over the course of development. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 7), or PN80 (adult; black, n = 4–6). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (**) P < 0.01; (***) P < 0.001.
Inhibitory transmission shapes excitatory/inhibitory circuitry and is necessary for neuronal development (Ben-Ari 2002; Chamberland and Topolnik 2012). Accordingly, we measured two markers of inhibitory synapses, GAD65 and GAD67; these proteins are isomorphs of the enzyme glutamic acid decarboxylase (GAD), which synthesizes the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Erlander et al. 1991; Bu et al. 1992; Soghomonian and Martin 1998). GAD65 is mainly concentrated in axon terminals bound to synaptic vesicles and catalyzes GABA synthesis upon the evoked neuronal activity, whereas GAD67 is widely distributed throughout the cell body and catalyzes the synthesis of GABA under resting conditions (Erlander et al. 1991; Bu et al. 1992; Soghomonian and Martin 1998). Both mRNA and protein levels of GAD65 and GAD67 increase gradually during postnatal development (Soghomonian and Martin 1998; Frahm and Draguhn 2001; Popp et al. 2009). We also determined the levels of gephyrin, a scaffold protein that anchors the postsynaptic GABA receptors and is thought to be involved in the plasticity of inhibitory synapses (Yu et al. 2007; Fritschy et al. 2008; Tretter et al. 2008). By interacting with numerous cytoskeletal regulatory proteins, gephyrin regulates GABA receptor trafficking to the synapse, thereby influencing inhibitory synaptic plasticity (Tyagarajan and Fritschy 2014). GABA is the main inhibitory neurotransmitter in the adult brain, but early in development (i.e., before PN10 in rats) GABAergic synaptic transmission is actually excitatory (Ben-Ari 2002). One-way ANOVA followed by Newman–Keuls post-hoc tests revealed that BLA levels of both GAD65 and GAD67 were similar (P > 0.05) and significantly lower in PN17 and PN24 than in adulthood (GAD65: F(2,13) = 16.67; P = 0.0003 and GAD67: F(2,13) = 7.821; P = 0.0059) (respectively, P < 0.001; P < 0.01) (Fig. 6). Gephyrin levels significantly increased from PN17 and PN24 to adulthood (F(2,14) = 73.42; P < 0.0001), with no difference between PN17 and PN24 (P > 0.05).
Thus, during postnatal development in the BLA, there is a significant, gradual increase in the levels of excitatory synapses and a delayed increase in markers of inhibitory synapse maturation.
Neuronal morphogenesis markers decrease significantly over the course of development
Next, we assessed markers of neuronal morphogenesis, a fundamental process that guides the formation of neuronal networks. We surveyed two major proteins involved in this process: cofilin, a ubiquitous actin-binding factor that regulates actin filament polymerization via phosphorylation at Ser 3 (Yang et al. 1998; Bramham 2007) and microtubule-associated protein 2 (MAP2), a neuronal protein that regulates the structure and stability of microtubules and neuronal morphogenesis (Sánchez et al. 2000).
As shown in Figure 7, one-way ANOVA followed by Newman–Keuls post-hoc tests revealed no significant difference in the levels of cofilin among the three age groups (F(2,14) = 0.7610; P = 0.4856). However, the level of cofilin phosphorylated at Ser3 (p-cofilin) in the BLA was markedly and significantly higher at PN17 (F(2,14) = 32.62; P < 0.0001) than at PN24 and adulthood (P < 0.0001). PN24 and adult rats had similar levels of p-cofilin (P > 0.05). Consequently, the ratio of p-cofilin to cofilin significantly and massively (>1500%) decreased (F(2,14) = 24.90; P < 0.0001) from PN17 to both PN24 and adulthood (P < 0.001) (Fig. 7). The level of MAP2 did not differ significantly among the three age groups (F(2,16) = 2.024; P = 0.1646), although there was a nonsignificant trend toward an increase from PN17 and PN24 to adulthood (P > 0.05).
Figure 7.

Neurite connectivity markers change over the course of development. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 5–6), PN24 (gray, n = 7) or PN80 (adult; black, n = 4–6). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (**) P < 0.01; (***) P < 0.001.
We next focused on the pleiotropic kinase mechanistic target of rapamycin (mTOR) (Brown et al. 1994; Sabatini et al. 1994; Laplante and Sabatini 2012), a master regulator of growth and neuronal differentiation that senses cellular oxygen, nutrition, and energy levels. mTOR regulates diverse metabolic functions, including activity-dependent protein translation (Hoeffer and Klann 2010; Costa-Mattioli and Monteggia 2013) autophagy, lipid synthesis, cell survival and proliferation, and oligodendrocyte differentiation and myelination (Mizushima et al. 2008; Narayanan et al. 2009; Laplante and Sabatini 2012; Wahl et al. 2014). mTOR activation correlates with its phosphorylation at Ser2448 (Scott et al. 1998; Navé et al. 1999; Inoki et al. 2002; Reynolds et al. 2002), which we measured alongside total mTOR levels.
One-way ANOVA followed by Newman–Keuls post-hoc tests revealed significant increases in pmTOR levels (F(2,13) = 11.33; P = 0.0014) from PN17 and PN24 to adulthood (P < 0.01; P < 0.001, respectively). Total mTOR levels followed a similar pattern (F(2,13) = 27.09; P < 0.0001), increasing from both PN17 and PN24 to adulthood (P < 0.001) (Fig. 7). Hence, the pmTOR/mTOR ratio did not significantly change among the three age groups (F(2,13) = 1.631; P = 0.2333) (Fig. 7).
Collectively, these data suggest that neuronal remodeling is significantly more active during early development and then slows in adulthood when differentiated cell response regulation is higher.
Levels of myelination markers change significantly over the course of development
Finally, we determined the levels of the myelination markers myelin basic protein (MBP) (Eylar et al. 1971; Readhead et al. 1990; Boggs 2006; Harauz and Boggs 2013), myelin-associated glycoprotein (MAG) (McKerracher et al. 1994; Mukhopadhyay et al. 1994; Quarles 2007), neurite outgrowth inhibitor (Nogo-A), and Nogo Receptor (Nogo-R) (Fournier et al. 2001). Myelin is a spiral extension of the oligodendroglial cell membrane that increases axonal conduction velocity and efficiency, provides metabolic support to axons, and stabilizes axonal projections in the developing and adult mammalian brain (Almeida and Lyons 2017; Stassart et al. 2018). MBP is one of several essential proteins present in the myelin sheath (Harauz and Boggs 2013). The formation and survival of myelin sheaths are regulated by other myelin components such as MAG (Quarles 2007) and Nogo-A, as well as their receptor Nogo-R (Fournier et al. 2001). Nogo-R-mediated myelin inhibition of axonal outgrowth contributes to the closure of the critical period for monocular deprivation-induced ocular dominance plasticity in the visual cortex (McGee et al. 2005; Akbik et al. 2012) and could contribute to restriction of experience-driven plasticity in brain regions involved in learning and memory.
One-way ANOVA followed by Newman–Keuls post-hoc tests revealed that MBP levels in BLA dramatically increased over the course of development: PN17 rats had a significantly lower level of MBP (F(2,14) = 64.63; P < 0.0001) than both PN24 (∼25-fold P < 0.01) and adult rats (∼90-fold, P < 0.001) (Fig. 8). Moreover, PN24 rats also had a significantly lower level of MBP than adults (>70-fold, P < 0.001). Similarly, the level of Nogo-R (F(2,14) = 99.06; P < 0.0001) significantly increased from PN17 to PN24 (P < 0.05), and from both PN17 and PN24 to adulthood (P < 0.001) (Fig. 8), whereas Nogo-A remained stable across the three age groups (F(2,14) = 0.004905; P = 0.9951) (Fig. 8). The level of MAG (F(2,15) = 30.87; P < 0.0001) was significantly higher in PN24 rats than in both PN17 (P < 0.001) and adult rats (P < 0.05), and were also dramatically lower (>60%) in PN17 rats relative to adults (P < 0.001) (Fig. 8).
Figure 8.

Myelination markers change over the course of development. Densitometric western blot analyses were performed on total protein extracts of BLA from rats euthanized at PN17 (white, n = 6), PN24 (gray, n = 7) or PN80 (adult; black, n = 4–5). Data are expressed as mean percentage ± SEM of adult rat levels. One-way ANOVA followed by Newman–Keuls post-hoc tests: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Together, these results suggest that myelination substantially increases in the BLA during early postnatal development and reaches maturation in adulthood.
Discussion
Elucidating the molecular changes of brain regions involved in learning and memory during development is essential to improving our understanding of the biological mechanisms underlying maturation of cognitive functions. In this study, we focused on the BLA, a region critically involved in the encoding and expression of both recent and remote fear memories, as well as modulation of episodic salient memories, through dynamic and temporally regulated interactions with other brain regions, including the hippocampus and the mPFC (McGaugh et al. 2002; Bocchio et al. 2017; Kitamura et al. 2017; Ressler and Maren 2019).
We found that the levels of many synaptic and neuronal proteins in the BLA undergo significant developmental changes between PN17 and PN24 compared with PN80 (young adult). Activity and plasticity markers, including Arc, Ser133-phospho-CREB, Thr286-phospho-CaMKIIα, Ser3-phospho-cofilin, Thr202- and Tyr204-phosphoERK1/2, Ser831- and Ser845-phosphoGluA1 AMPAR subunits, and the NMDAR subunit GluN2B were all highest at PN17, and then decreased significantly with age. In contrast, the levels of other proteins increased significantly over the course of development: total ERK1/2, total GluA1 and GluA2, both excitatory and inhibitory synapse maturation markers, synaptophysin, PSD95, SynCAM, GADs, gephyrin, and markers of myelination such as MBP, MAG, and Nogo-R.
Some of these proteins, and particularly pCREB, pCaMKIIα, pcofilin, showed a 1000-fold decrease between PN17 and adult age, suggesting the possibility of activation of diverse signaling pathways at different ages. However, the significance and roles for these large differences in marker levels remain to be understood.
We suggest that the significantly higher levels of activity/plasticity markers at PN17 and/or PN24 relative to adulthood imply a considerably higher degree of cellular activation and plasticity during early development as a result of novel experience. This hypothesis is consistent with our previous studies showing that the early hippocampus is highly sensitive to learning stimuli (Travaglia et al. 2016b). Based on those observations, we postulated that experiences during early development mature the biological substrates in all brain regions involved in processing those experiences, leading to the acquisition of functional competences (Alberini and Travaglia 2017).
The levels of the excitatory synapse maturation proteins synaptophysin, PSD95, and SynCAM in the BLA increased gradually from PN17 to PN24, and then to PN80. In contrast, the levels of markers of inhibitory synapses GAD65 and GAD67, as well as gephyrin, remained unchanged between PN17 and PN24, but then significantly increased between PN24 and adulthood. These data suggest that inhibitory synapses in the BLA develop after excitatory synapses. As previously mentioned, the two early developmental ages compared in this study, PN17 and PN24, correspond to a temporal window of brain maturation during which rats gain functional competence to express long-term hippocampus-dependent memories (Travaglia et al. 2016b). At PN17, the amygdala memory system has acquired the ability to form and express cued fear conditioning (Camp and Rudy 1988; Dumas 2005; Moriceau and Sullivan 2006; Moriceau et al. 2006); however, our data reveal that significant changes in the BLA occur between PN17 and PN24: (i) a decrease in markers of cellular activation and plasticity, including the level of Arc/Arg3.1 and the ratios pCREB/CREB, pERK/ERK1, pGluA1(Ser831)/GluA1, and pcofilin/cofilin; (ii) an increase in the GluN2A/GluN2B ratio and in markers of excitatory synapse maturation such as synaptophysin, PSD95, and SynCAM; and finally (iii) an increase in the axonal maturation and myelination markers MBP, MAG, and Nogo-R. These changes may reflect the maturation of amygdala functions and its involvement in increasingly complex behavioral responses, such as modulation of hippocampal-cortical-dependent memories.
In agreement with this hypothesis, our findings show that numerous maturation-associated changes in the BLA over development occur in concert with those of the dHC and mPFC (Travaglia et al. 2016a; Jia et al. 2018). In adulthood, the BLA, mPFC, and dHC are highly interconnected and all critically recruited in the encoding, consolidation and expression of medial-temporal lobe-dependent memories (McGaugh et al. 2002; Preston and Eichenbaum 2013; Eichenbaum 2017; Tonegawa et al. 2018); thus, it is plausible that the maturation of BLA, mPFC, and dHC follow similar developmental trajectories. We observed that in the BLA, as for the previously noted dHC and mPFC, the level of MBP substantially increase between PN17 and adult age, and MAG substantially up-regulates at PN24 relative to both PN17 and adulthood in all three regions, suggesting a coordinated regulation of myelination. Our results revealed also that the GluN2A subunit remains unchanged with age in the BLA and dHC (Travaglia et al. 2016b) but increases between PN17 and PN24 in the mPFC. However, in the BLA, as in the mPFC (Jia et al. 2018) and dHC (Travaglia et al. 2016b), the GluN2A/GluN2B ratio increases over the course of development, and that this increase is most prominent between PN17 and PN24, during an interval that coincides with synapse maturation, circuit refinement, and acquisition of learning abilities (Dumas 2005; Paoletti et al. 2013). This developmental increase in GluN2A/GluN2B suggests that a change in the biophysical properties and signaling of the NMDAR complex takes place over ages. As GluN2B-containing receptors display relatively slower EPSC decay time course (Vicini et al. 1998), higher levels of GluN2B in early stages of development implies a longer time for detection of synaptic coincidence and/or an increase in synaptic efficacy, therefore, potentially an enhanced memory function during early-life. At the same time, the synaptic enrichment of GluN2B may prevent efficient long-term memory storage by restricting synapse stabilization (Dumas 2005; Gambrill and Barria 2011, Finnie and Nader 2012).
On the other hand, some proteins exhibited differential kinetics during development in the BLA vs. the dHC and mPFC (Fig. 9). First, MAP2 did not significantly change with age in the BLA. This pattern is different to that observed in the mPFC and dHC: in mPFC, MAP2 level decreased significantly between PN17 and PN24, and then increased between PN24 and adulthood, whereas in the dHC, MAP2 level tended to remain stable between PN17 and PN24 and then dropped significantly in adulthood. Two other markers that exhibit distinctive regulation with age in the BLA relative to mPFC and dHC were mTOR and pmTOR: they both remained unchanged between PN17 and PN24 and then significantly dropped and increased in adulthood in the dHC and the BLA, respectively. In contrast, in the mPFC, they did not change significantly across development. In addition, although pmTOR/mTOR ratio remained unchanged throughout development in the BLA, as in the dHC, it decreased significantly between PN17 and PN24 in the mPFC. Because mTOR and MAP2 levels may reflect neuron-specific cytoskeletal changes in dendrites, these data lead us to speculate that developmental regulation of dendritic structures follows distinct developmental kinetics in different brain regions. Also, the increase in the PSD95 level in the BLA followed the same kinetic as that observed in the mPFC (Jia et al. 2018): a significant increase between PN17 and PN24 and a plateau between PN24 and adulthood. However, this kinetic differs from those observed previously in the dHC (Travaglia et al. 2016a), which exhibited an elevation of PSD95 between PN24 and adulthood (Fig. 9), suggesting that excitatory synapse maturation occurs slightly later in the dHC than in the BLA and mPFC.
Figure 9.

Graphic representation depicting increases or decreases of protein levels at PN17 and PN24 relative to PN80 in the rat BLA, dHC (Travaglia et al. 2016a,b), and mPFC (Jia et al. 2018). The black lines denote the level of each marker at PN80, which was used as a relative reference. Blue and green triangles depict PN17 and PN24 markers, respectively. A triangle placed on the PN80 black line indicates no significant change compared with PN80 and the directions of the arrows indicate significant up-regulation or down-regulation of the protein levels. Only markers that exhibit differential progression over ages in the different regions are shown.
The second group of proteins that varied with age in the dHC and mPFC, but did not significantly vary across ages in the BLA, were c-Fos, Zif268, Nogo-A, and the pTrkB/TrkB ratio. These data suggest that IEG markers, which are considered as general readouts of cellular activation and plasticity, are differentially regulated in different brain regions. In fact, in the BLA, while the Arc/Arg3.1 level was significantly higher at PN17 than at PN24 and PN80, c-Fos and Zif268 levels did not significantly change, whereas in the dHC and mPFC they followed kinetics similar to those of Arc/Arg3.1. Moreover, the pTrkB/TrkB ratio remained unchanged with age in the BLA but decreased between PN17 and PN24 in the dHC and mPFC (Travaglia et al. 2016b; Jia et al. 2018). Hence, it appears that mechanisms of cellular activation and plasticity are regulated in distinctive manners in different brain regions.
Other plasticity markers, including Arc/Arg3.1, the GluN2B subunit, and the ratios pCREB/CREB, pGluA1(Ser831)/GluA1 and pGluA1(Ser845)/GluA1, followed the same decreasing trajectory with age in all three regions, but with different temporal kinetics. In the BLA, the Arc/Arg3.1 level decreased more gradually between PN17 and adulthood but remained similarly elevated at PN17 and PN24 in both the dHC and the mPFC and decreased only after PN24 (Fig. 9). These observations suggest that the plasticity promoted by Arc/Arg3.1, which is likely related to AMPAR receptor endocytosis and changes in dendritic spine morphology (Lanahan and Worley 1998; Plath et al. 2006; Bramham et al. 2008; Korb and Finkbeiner 2011), remains elevated for a longer time in the dHC and mPFC, whereas in the BLA it decreases more rapidly to adult levels. In contrast, the pCREB/CREB and pGluA1(Ser831)/GluA1 ratios followed similar temporal trajectories, with a gradual significant decrease between PN17 and adulthood in the BLA, whereas in both the dHC and mPFC the same markers decreased between PN17 and PN24 but then remained unchanged between PN24 and adulthood. In the BLA, the GluN2B subunit and the pGluA1(Ser845)/GluA1 ratio were stable between PN17 and PN24 and then decreased between PN24 and adulthood. In contrast, GluN2B underwent a gradual significant decrease between PN17 and adulthood in the mPFC, whereas in the dHC it decreased between PN17 and PN24 but then remained unchanged between PN24 and adulthood. Finally, in the dHC and mPFC, the pGluA1(Ser845)/GluA1 ratio decreased between PN17 and PN24 but remained stable between PN24 and adult age (Fig. 9). These markers of plasticity and AMPAR activation suggest that in the BLA, as in the dHC and mPFC, the degree of plasticity is highest at PN17 and decreases thereafter, although with different slopes in the three regions.
Another group of markers followed the same kinetics in the BLA and mPFC, but an opposite pattern in the dHC: (i) the ratios pCamKIIα/CamKIIα and pERK1,2/ERK1,2, which decreased between PN17 and adulthood in the BLA and mPFC but increased in the dHC and (ii) the level of Nogo-R, which increased between PN17 and adulthood in the BLA and mPFC, but decreased in the dHC. Finally, gephyrin levels significantly increased between PN24 and adulthood in BLA, but remained unchanged in the mPFC across ages.
These differences in both the trajectory and the kinetics of molecular changes among dHC, mPFC, and BLA likely reflect the differential cell-type composition and specific circuitry development of the three regions, as well as differences in their state of maturation, which changes in response to experience.
In summary, our biochemical analyses revealed that over the course of development, the BLA undergoes a remarkable increase in the levels of proteins related to synaptogenesis and synapse maturation, along with a dramatic decrease in markers of cell activation and significant changes in the levels of structural markers. Together, these observations suggest that structural stabilization is taking place with age.
As for the dHC and mPFC, the substantial differences in the levels of plasticity, activity, and neural structure markers in the BLA at different developmental stages suggest using caution when extrapolating from the conclusions of brain plasticity studies conducted in animals of different ages. Synaptic plasticity studies have often used tissues from early development to reach general conclusions about the mechanisms of learning and memory in adulthood. However, it is clear from our observations and many other studies that the infant brain is not a small version of the adult brain, but rather a distinct biological system.
Materials and Methods
Animals
Seventeen (PN17) and 24-d-old (PN24) male and female pups were obtained from pregnant Long–Evans female rats (Charles River Laboratories). Rats were housed in 30.8 × 40.6 × 22.2 cm plastic cages containing ALPHA-dri bedding under a 12 h light/dark cycle (light on at 07.00 a.m.), with food and water ad libitum. All experiments were carried out during the light cycle. The birth date was considered PN0, and litters were culled to 10–12. Only one male and female per litter was used under any experimental condition. Rats were weaned at PN21. Adult (PN80) male Long-Evans rats (Envigo), weighing 200–250 g were housed two per cage. All procedures complied with the U.S. National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the New York University Animal Care Committees.
Western blot analysis
Western blot analysis was carried out as previously reported (Chen et al. 2012). Rats were euthanized, and their brains were rapidly removed and frozen in isopentane. Basolateral amygdala punches were obtained from frozen brains mounted on a cryostat using a 19-gauge neuro punch (Fine Science Tools). Samples were homogenized in ice-cold RIPA buffer (50 mM Tris base, 150 mM NaCl, 0.1% SDS, 0.5% Na-deoxycholate, 1% NP-40) with protease and phosphatase inhibitors (0.5 mM PMSF, 2 mM DTT, 1 mM EGTA, 2 mM NaF, 1 µM microcystine, 1 mM benzamidine, 1 mM sodium orthovanadate, and commercial protease and phosphatase inhibitor cocktails [Sigma-Aldrich]). Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Laboratories). Equal amounts of total protein (20 µg per well) were resolved on 4%–20% Criterion TGX gradient gels (Bio-Rad Laboratories) and transferred to Immobilon-FL Transfer membrane (Millipore) by electroblotting. Membranes were dried, reactivated in methanol, and washed with water before they were blocked in Odyssey blocking buffer (TBS) (LI-COR Bioscience) for 1 h at room temperature. Membranes were then incubated with primary antibody overnight at 4°C in solution according to manufacturer's suggestion. Primary antibodies against the following proteins were obtained from the indicated suppliers: Arc (1:10000, Synaptic System, cat# 156 003), c-Fos (1:200, Millipore, cat# PC05), Zif268 (1:1000, Cell Signaling Technology, cat# 4153S), pTrkB (Tyr816) (1:1000, Millipore, cat# ABN1381), TrkB (80E3) (1:1000, Cell Signaling Technology, cat# 4603S), pCREB (Ser133) (1:1000, Cell Signaling Technology, cat# 9198), CREB (1:1000, Cell Signaling Technology, cat# 9104), pCaMKIIα (Thr286) (1:1000, Cell Signaling Technology, cat# 3361S), CaMKIIα (1:1000, Millipore, cat# 05-532), pERK1/2 (pp44/42 MAPK) (Thr202/Tyr204) (1:2000, Cell Signaling Technology, cat# 9101S), ERK1/2 (p44/42 MAPK) (1:2000, Cell Signaling Technology, cat# 4695S), GluN2A (1:1000, Millipore, cat# 7-632), GluN2B (1:1000, Cell Signaling, cat# 4212S), GluA1 (1:2000, Millipore, cat# AB1504), GluA2 (1:1000, UC Davis/NIH NeuroMab Facility, cat# 75-002), pAMPA receptor GluA1 (Ser845) (1:1000, Cell Signaling Technology, cat# 8084S), pAMPA receptor GluA1 (Ser831) (1:1000, Abcam, cat# ab109464), PSD95 (1:1000, Cell Signaling Technology, cat# 2507S), synaptophysin (1:1000, Cell Signaling Technology, cat# 5467), GAD65 (1:1000, Millipore, cat# ABN101), GAD67 (1:1000, Millipore, cat# AB5406), Gephyrin (1:1000, Synaptic Systems, cat# 147-111), p-cofilin (Ser3) (1:1000, Abcam, cat# ab12866), cofilin (1:1000, Millipore, cat# AB3842), MAP2 (1:1000, Millipore, cat# MAB3418), mTOR (1:1000, Cell Signaling Technology, cat# 4517S), pmTOR (1:1000, Cell Signaling Technology, cat# 2971S), myelin basic protein (MBP) (1:1000, Millipore, cat# 05-675), Nogo-A (1:1000, Abcam, cat# 62024), Nogo-R (1:1000, Abcam, cat# 26291), myelin-associated glycoprotein (MAG) (1:1000, Cell Signaling Technology, cat# 9043S). The membranes were washed in TBS with 0.1% Tween-20 (TBST), and then incubated for 1 h at room temperature with a species-appropriate fluorescently conjugated secondary antibody (goat anti-mouse IRDye 680LT [1:10,000] or goat anti-rabbit IRDye 800CW [1:10,000] from LI-COR Bioscience [Lincoln, NE]). Membranes were again washed in TBST and scanned using the Odyssey Infrared Imaging system (Li-Cor Bioscience). Data were quantified from pixel intensities using the Odyssey software (Image Studio 4.0). Antibody against actin (1:20,000, Santa Cruz Biotechnology, Dallas, TX, USA, cat# sc-47778) was used to costain all membranes; this signal was used as the relative internal loading control for all western blots. When appropriate, membranes were stripped and reprobed with additional antibodies to target multiple proteins on the same blot. In those cases, the same actin control was used for the densitometric analysis of all proteins on the membrane.
Statistical analyses
All data analysis was performed in Prism 6 (GraphPad Software). Data were analyzed by one-way analysis of variance (ANOVA) followed by Newman–Keuls post-hoc tests. Results were considered statistically significant when P < 0.05. The intent of this study was not to investigate sex differences; therefore, we included both female and male rats in our prepuberal PN17 and PN24 groups after preliminary statistical analyses of separate sex groups (n = 3–4 each sex) yielded no significant difference (unpaired two-tailed Student's t-test, P > 0.05) and the range of individual values was similarly distributed. Only PN80 male rats were used, hence, we cannot exclude the possibility that there are differential expressions of the markers studied in adult females and males or changes related to the female reproductive cycle.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant MH065635 to C.M.A. We thank Gabriella Pollonini for technical assistance.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.049866.119.
References
- Akbik F, Cafferty WB, Strittmatter SM. 2012. Myelin associated inhibitors: a link between injury-induced and experience-dependent plasticity. Exp Neurol 235: 43–52. 10.1016/j.expneurol.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM. 2009. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev 89: 121–145. 10.1152/physrev.00017.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Kandel ER. 2015. The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol 7: a021741 10.1101/cshperspect.a021741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Travaglia A. 2017. Infantile amnesia: a critical period of learning to learn and remember. J Neurosci 37: 5783–5795. 10.1523/JNEUROSCI.0324-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RG, Lyons DA. 2017. On myelinated axon plasticity and neuronal circuit formation and function. J Neurosci 37: 10023–10034. 10.1523/JNEUROSCI.3185-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero R, Choi DC, Ressler KJ. 2014. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog Mol Biol Transl Sci 122: 169–192. 10.1016/B978-0-12-420170-5.00006-4 [DOI] [PubMed] [Google Scholar]
- Ashley J, Cordy B, Lucia D, Fradkin LG, Budnik V, Thomson T. 2018. Retrovirus-like Gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172: 262–274.e11. 10.1016/j.cell.2017.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler M, Benfenati F, Valtorta F, Greengard P. 1990. The synapsins and the regulation of synaptic function. Bioessays 12: 259–263. 10.1002/bies.950120603 [DOI] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. 2003. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets 7: 101–114. 10.1517/14728222.7.1.101 [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. 1997. Regulatory phosphorylation of AMPA-type glutamate receptors by CaMKII during long-term potentiation. Science 276: 2042–2045. 10.1126/science.276.5321.2042 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. 2002. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 3: 728–739. 10.1038/nrn920 [DOI] [PubMed] [Google Scholar]
- Bocchio M, Nabavi S, Capogna M. 2017. Synaptic plasticity, engrams, and network oscillations in amygdala circuits for storage and retrieval of emotional memories. Neuron 94: 731–743. 10.1016/j.neuron.2017.03.022 [DOI] [PubMed] [Google Scholar]
- Boggs JM. 2006. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci 63: 1945–1961. 10.1007/s00018-006-6094-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR. 2007. Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Prog Brain Res 163: 453–471. 10.1016/S0079-6123(07)63025-8 [DOI] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. 2008. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci 28: 11760–11767. 10.1523/JNEUROSCI.3864-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. 1994. A mammalian protein targeted by G1-arresting rapamycin receptor complex. Nature 369: 756–758. 10.1038/369756a0 [DOI] [PubMed] [Google Scholar]
- Bu DF, Erlander MG, Hitz BC, Tillakaratne NJ, Kaufman DL, Wagner-McPherson CB, Evans GA, Tobin AJ. 1992. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci 89: 2115–2119. 10.1073/pnas.89.6.2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp LL, Rudy JW. 1988. Changes in the categorization of appetitive and aversive events during postnatal development of the rat. Dev Psychobiol 21: 25–42. 10.1002/dev.420210103 [DOI] [PubMed] [Google Scholar]
- Campbell BA, Spear NE. 1972. Ontogeny of memory. Psychol Rev 79: 215–236. 10.1037/h0032690 [DOI] [PubMed] [Google Scholar]
- Chamberland S, Topolnik L. 2012. Inhibitory control of hippocampal inhibitory neurons. Front Neurosci 6: 165 10.3389/fnins.2012.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. 2012. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci 15: 1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. 1992. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9: 929–942. 10.1016/0896-6273(92)90245-9 [DOI] [PubMed] [Google Scholar]
- Cline H. 2005. Synaptogenesis: a balancing act between excitation and inhibition. Curr Biol 15: R203–R205. 10.1016/j.cub.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Cole AJ, Saffen DW, Baraban JM, Worley PF. 1989. Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature 340: 474–476. 10.1038/340474a0 [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Monteggia LM. 2013. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16: 1537–1543. 10.1038/nn.3546 [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. 1999. Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci 96: 3269–3274. 10.1073/pnas.96.6.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC. 2005. Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol 76: 189–211. 10.1016/j.pneurobio.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. 2017. Prefrontal–hippocampal interactions in episodic memory. Nat Rev Neurosci 18: 547–558. 10.1038/nrn.2017.74 [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. 1991. Two genes encode distinct glutamate decarboxylases. Neuron 7: 91–100. 10.1016/0896-6273(91)90077-D [DOI] [PubMed] [Google Scholar]
- Eylar EH, Brostoff S, Hashim G, Caccam J, Burnett P. 1971. Basic A1 protein of the myelin membrane. The complete amino acid sequence. J Biol Chem 246: 5770–5784. [PubMed] [Google Scholar]
- Finnie PS, Nader K. 2012. The role of metaplasticity mechanisms in regulating memory destabilization and reconsolidation. Neurosci Biobehav Rev 36: 1667–1707. 10.1016/j.neubiorev.2012.03.008 [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. 1997. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17: 2469–2476. 10.1523/JNEUROSCI.17-07-02469.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. 2001. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409: 341–346. 10.1038/35053072 [DOI] [PubMed] [Google Scholar]
- Frahm C, Draguhn A. 2001. GAD and GABA transporter (GAT-1) mRNA expression in the developing rat hippocampus. Brain Res Dev Brain Res 132: 1–13. 10.1016/S0165-3806(01)00288-7 [DOI] [PubMed] [Google Scholar]
- Frank DA, Greenberg ME. 1994. CREB: a mediator of long-term memory from mollusks to mammals. Cell 79: 5–8. 10.1016/0092-8674(94)90394-8 [DOI] [PubMed] [Google Scholar]
- Frick KM, Fernandez SM. 2003. Enrichment enhances spatial memory and increases synaptophysin levels in aged female mice. Neurobiol Aging 24: 615–626. 10.1016/S0197-4580(02)00138-0 [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Harvey RJ, Schwarz G. 2008. Gephyrin: where do we stand, where do we go? Trends Neurosci 31: 257–264. 10.1016/j.tins.2008.02.006 [DOI] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. 2011. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci 108: 5855–5860. 10.1073/pnas.1012676108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332: 680–686. 10.1126/science.1198701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, Boggs JM. 2013. Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J Neurochem 125: 334–361. 10.1111/jnc.12195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. 2000. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262–2267. 10.1126/science.287.5461.2262 [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. 2010. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33: 67–75. 10.1016/j.tins.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CA, Schenker LJ, Kennedy MB. 1996. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci 16: 1380–1388. 10.1523/JNEUROSCI.16-04-01380.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657. 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- Janak PH, Tye KM. 2015. From circuits to behaviour in the amygdala. Nature 517: 284–292. 10.1038/nature14188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Travaglia A, Pollonini G, Fedele G, Alberini CM. 2018. Developmental changes in plasticity, synaptic, glia, and connectivity protein levels in rat medial prefrontal cortex. Learn Mem 25: 533–543. 10.1101/lm.047753.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. 2001. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci 4: 289–296. 10.1038/85138 [DOI] [PubMed] [Google Scholar]
- Kandel ER. 2012. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5: 14–22. 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killcross S, Robbins TW, Everitt BJ. 1997. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature 388: 377–380. 10.1038/41097 [DOI] [PubMed] [Google Scholar]
- Kitamura T, Ogawa SK, Roy DS, Okuyama T, Morrissey MD, Smith LM, Redondo RL, Tonegawa S. 2017. Engrams and circuits crucial for systems consolidation of a memory. Science 356: 73–78. 10.1126/science.aam6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Finkbeiner S. 2011. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci 34: 591–598. 10.1016/j.tins.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SE, Chapman ER. 2011. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70: 847–854. 10.1016/j.neuron.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanahan A, Worley P. 1998. Immediate-early genes and synaptic function. Neurobiol Learn Mem 70: 37–43. 10.1006/nlme.1998.3836 [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. 2000. Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. 10.1146/annurev.neuro.23.1.155 [DOI] [PubMed] [Google Scholar]
- LeDoux J. 2003. The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23: 727–738. 10.1023/A:1025048802629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. 2002. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175–190. 10.1038/nrn753 [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. 2012. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13: 169–182. 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen HB, Kim JH. 2016. Ontogeny of memory: an update on 40 years of work on infantile amnesia. Behav Brain Res 298: 4–14. 10.1016/j.bbr.2015.07.030 [DOI] [PubMed] [Google Scholar]
- Maren S. 2003. The amygdala, synaptic plasticity, and fear memory. Ann N Y Acad Sci 985: 106–113. 10.1111/j.1749-6632.2003.tb07075.x [DOI] [PubMed] [Google Scholar]
- Martínez A, Alcántara S, Borrell V, Del Río JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, et al. 1998. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci 18: 7336–7350. 10.1523/JNEUROSCI.18-18-07336.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. 2001. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609. 10.1038/35085068 [DOI] [PubMed] [Google Scholar]
- McGaugh JL. 2004. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci 27: 1–28. 10.1146/annurev.neuro.27.070203.144157 [DOI] [PubMed] [Google Scholar]
- McGaugh JL, McIntyre CK, Power AE. 2002. Amygdala modulation of memory consolidation: interaction with other brain systems. Neurobiol Learn Mem 78: 539–552. 10.1006/nlme.2002.4082 [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. 2005. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 309: 2222–2226. 10.1126/science.1114362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CK, Power AE, Roozendaal B, McGaugh JL. 2003. Role of the basolateral amygdala in memory consolidation. Ann N Y Acad Sci 985: 273–293. 10.1111/j.1749-6632.2003.tb07088.x [DOI] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. 1994. Identification of myelin-associated glycoprotein as a major myelinderived inhibitor of neurite growth. Neuron 13: 805–811. 10.1016/0896-6273(94)90247-X [DOI] [PubMed] [Google Scholar]
- Minichiello L. 2009. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10: 850–860. 10.1038/nrn2738 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075. 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, Curran T. 1987. Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237: 192–197. 10.1126/science.3037702 [DOI] [PubMed] [Google Scholar]
- Moriceau S, Sullivan RM. 2006. Maternal presence serves as a switch between learning fear and attraction in infancy. Nat Neurosci 9: 1004–1006. 10.1038/nn1733 [DOI] [Google Scholar]
- Moriceau S, Wilson DA, Levine S, Sullivan RM. 2006. Dual circuitry for odor-shock conditioning during infancy: corticosterone switches between fear and attraction via amygdala. J Neurosci 26: 6737–6748. 10.1523/JNEUROSCI.0499-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. 1994. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13: 757–767. 10.1016/0896-6273(94)90042-6 [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. 2009. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci 29: 6860–6870. 10.1523/JNEUROSCI.0232-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navé BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. 1999. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344: 427–431. 10.1042/bj3440427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Minatohara K, Bito H. 2018. Inverse synaptic tagging: an inactive synapse-specific mechanism to capture activity-induced Arc/arg3.1 and to locally regulate spatial distribution of synaptic weights. Semin Cell Dev Biol 77: 43–50. 10.1016/j.semcdb.2017.09.025 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14: 383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. 2005. The rat brain in stereotaxic coordinates. Compact 6th ed Academic, New York. [Google Scholar]
- Phelps EA. 2004. Human emotion and memory: interactions of the amygdala and hippocampal complex. Curr Opin Neurobiol 14: 198–202. 10.1016/j.conb.2004.03.015 [DOI] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, et al. 2006. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52: 437–444. 10.1016/j.neuron.2006.08.024 [DOI] [PubMed] [Google Scholar]
- Popp A, Urbach A, Witte OW, Frahm C. 2009. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS One 4: e4371 10.1371/journal.pone.0004371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston AR, Eichenbaum H. 2013. Interplay of hippocampus and prefrontal cortex in memory. Curr Biol 23: R764–R73. 10.1016/j.cub.2013.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles RH. 2007. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem 100: 1431–1448. 10.1111/j.1471-4159.2006.04319.x [DOI] [PubMed] [Google Scholar]
- Ramsaran AI, Schlichting ML, Frankland PW. 2019. The ontogeny of memory persistence and specificity. Dev Cogn Neurosci 36: 100591–. 10.1016/j.dcn.2018.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead C, Takasashi N, Shine HD, Saavedra R, Sidman R, Hood L. 1990. Role of myelin basic protein in the formation of central nervous system myelin. Ann N Y Acad Sci 605: 280–285. 10.1111/j.1749-6632.1990.tb42401.x [DOI] [PubMed] [Google Scholar]
- Ressler RL, Maren S. 2019. Synaptic encoding of fear memories in the amygdala. Curr Opin Neurobiol 54: 54–59. 10.1016/j.conb.2018.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds TH IV, Bodine SC, Lawrence JC Jr. 2002. Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem 277: 17657–17662. 10.1074/jbc.M201142200 [DOI] [PubMed] [Google Scholar]
- Richardson MP, Strange BA, Dolan RJ. 2004. Encoding of emotional memories depends on amygdala and hippocampus and their interactions. Nat Neurosci 7: 278–285. 10.1038/nn1190 [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. 1994. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78: 35–43. 10.1016/0092-8674(94)90570-3 [DOI] [PubMed] [Google Scholar]
- Sánchez C, Díaz-Nido J, Avila J. 2000. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61: 133–168. 10.1016/S0301-0082(99)00046-5 [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. 2011. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci 31: 9170–9178. 10.1523/JNEUROSCI.1250-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC Jr. 1998. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci 95: 7772–7777. 10.1073/pnas.95.13.7772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. 2013. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106-107: 1–16. 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. 1994. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147. 10.1038/368144a0 [DOI] [PubMed] [Google Scholar]
- Shepherd JD. 2018. Arc—an endogenous neuronal retrovirus? Semin Cell Dev Biol 77: 73–78. 10.1016/j.semcdb.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. 1998. CREB and memory. Annu Rev Neurosci 21: 127–148. 10.1146/annurev.neuro.21.1.127 [DOI] [PubMed] [Google Scholar]
- Soghomonian JJ, Martin DL. 1998. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci 19: 500–505. 10.1016/S0165-6147(98)01270-X [DOI] [PubMed] [Google Scholar]
- Stassart RM, Möbius W, Nave KA, Edgar JM. 2018. The Axon-Myelin unit in development and degenerative disease. Front Neurosci 12: 467–476. 10.3389/fnins.2018.00467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Worley PF. 2001. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 30: 227–240. 10.1016/S0896-6273(01)00275-6 [DOI] [PubMed] [Google Scholar]
- Sweatt JD. 2004. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311–317. 10.1016/j.conb.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. 2004. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173–183. 10.1038/nrn1346 [DOI] [PubMed] [Google Scholar]
- Tischmeyer W, Grimm R. 1999. Activation of immediate early genes and memory formation. Cellular Mol Life Sci 55: 564–574. 10.1007/s000180050315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonegawa S, Morrissey MD, Kitamura T. 2018. The role of engram cells in the systems consolidation of memory. Nat Rev Neurosci 19: 485–498. 10.1038/s41583-018-0031-2 [DOI] [PubMed] [Google Scholar]
- Travaglia A, Bisaz R, Cruz E, Alberini CM. 2016a. Developmental changes in plasticity, synaptic, glia and connectivity protein levels in rat dorsal hippocampus. Neurobiol Learn Mem 135: 125–138. 10.1016/j.nlm.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglia A, Bisaz R, Sweet ES, Blitzer RD, Alberini CM. 2016b. Infantile amnesia reflects a developmental critical period for hippocampal learning. Nat Neurosci 19: 1225–1233. 10.1038/nn.4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglia A, Steinmetz AB, Miranda JM, Alberini CM. 2018. Mechanisms of critical period in the hippocampus underlie object location learning and memory in infant rats. Learn Mem 25: 176–182. 10.1101/lm.046946.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Jacob TC, Mukherjee J, Fritschy JM, Pangalos MN, Moss SJ. 2008. The clustering of GABAA receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor α2 subunits to gephyrin. J Neurosci 28: 1356–1365. 10.1523/JNEUROSCI.5050-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Fritschy JM. 2014. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci 15: 141–156. 10.1038/nrn3670 [DOI] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. 1998. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol 79: 555–566. 10.1152/jn.1998.79.2.555 [DOI] [PubMed] [Google Scholar]
- Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. 2014. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci 34: 4453–4465. 10.1523/JNEUROSCI.4311-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenmann B, Franke WW. 1985. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41: 1017–1028. 10.1016/S0092-8674(85)80082-9 [DOI] [PubMed] [Google Scholar]
- Worley PF, Bhat RV, Baraban JM, Erickson CA, McNaughton BL, Barnes CA. 1993. Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long-term enhancement. J Neurosci 13: 4776–4786. 10.1523/JNEUROSCI.13-11-04776.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393: 809–812. 10.1038/31735 [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. 2008. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55: 1081–1094. 10.1016/j.neuropharm.2008.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Tully T. 1996. CREB and the formation of long-term memory. Curr Opin Neurobiol 6: 264–268. 10.1016/S0959-4388(96)80082-1 [DOI] [PubMed] [Google Scholar]
- Yu W, Jiang M, Miralles CP, Li RW, Chen G, de Blas AL. 2007. Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci 36: 484–500. 10.1016/j.mcn.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]