

Although recent accumulating data have suggested that gut microbiota associates with Alzheimer disease (AD) pathogenesis, it is not yet clear how alterations in the gut microbiota mechanistically contribute to different aspects of AD pathology. Here, Wang et al. identified that infiltrating peripheral immune cells facilitated by gut microbiota imbalance in mouse models of amyloidosis are associated with changes in behavior and AD-related pathology.
Alzheimer disease (AD) is a neurodegenerative disorder characterized by a progressive decline in memory and thinking severe enough to interfere with daily life. As the population ages, the worldwide incidence of AD will markedly increase unless disease-modifying treatments are developed. Several clinical trials testing agents designed to reduce brain amyloid-β (Aβ) burden or neutralize Aβ toxicity during the symptomatic phase of AD have failed. The reasons for this are not yet clear though by the time individuals have cognitive decline due to AD, Aβ has already been accumulating for 15–20 years and several other pathological changes in the brain such as tau accumulation and inflammation have begun to ramp up. The public health challenge of AD has motivated scientists to develop candidate drugs acting on different aspects of AD pathogenesis as well as at different stages of disease.1
The immune system has a prominent role in AD pathogenesis with both innate and adaptive immunity in the central nervous system receiving attention. There is increasing interest in the role of the peripheral immune system in AD pathology. Some studies have shown that blood-derived leukocytes (e.g., lymphocytes, monocytes and neutrophils) are present in the brains of AD cases and animal models.2–4 For example, in a mouse model with amyloid deposition, the suppression of regulatory T (Treg) cells led to a decrease in Aβ.4 In a separate study, genetic ablation of peripheral immune cells, including T and B cells, led to a >2-fold increase in amyloid burden.3 Importantly, the peripheral immune system closely interacts with microbes in the gut-intestinal tract.5
With a growing appreciation for the importance of the microbiome to human health, there have been increasing attempts to find links among gut microbial dysbiosis, colonization with maladaptive or pathogenic microbiota, and AD pathogenesis.6–8 However, the underlying mechanisms explaining the connection between gut microbiota and AD pathogenesis have not yet been clarified. In a paper recently published in Cell Research, Wang et al.9 elegantly demonstrate that a gut microbiota imbalance facilitates peripheral immune cells to infiltrate the brain, resulting in enhanced microglial activation that contributes to cognitive impairment and Aβ burden in mouse models of Aβ amyloidosis. In addition, the authors examine the effects of GV-971, a marine-derived oligosaccharide that has recently shown promise in improving cognition in individuals with dementia of the Alzheimer type in China. In their new work, they show that GV-971 decreases Aβ-related pathologies by reconditioning the gut microbiota, providing further evidence that gut-targeted interventions may serve as novel strategies to tackle AD.
The authors began by evaluating the gut microbiota profile of a mouse model of Aβ-amyloidosis (5xFAD transgenic mice). In this model, Aβ aggregation and accumulation begin by 2 months of age followed by hippocampal synaptic loss and cognitive impairment in spatial discrimination tasks at 7–9 months. Over several months, the gut microbiota composition of the 5xFAD mice underwent a remarkable shift, while that of wild-type control mice remained relatively stable during this time. When comparing the major phyla Bacteroidetes and Firmicutes in the gut, the ratio of Firmicutes to Bacteroidetes greatly increased at 7 months of age in the 5xFAD model. This has previously been observed in other studies and supports the concept that Aβ accumulation is accompanied by shifts in the gut microbial population.10 On the basis of these observations, the authors then investigated how this shift may influence Aβ-linked pathogenesis. The authors noticed that as Aβ and its downstream consequences such as synaptic degeneration and gut dysbiosis increased, pro-inflammatory types of microglia also strongly increased by 7 months. Interestingly, as activated microglia increased, so did the number and pattern of infiltrating peripheral pro-inflammatory T helper type 1 (Th1) cells. This observation suggested the hypothesis that AD-related gut dysbiosis may somehow stimulate the infiltration of peripheral immune cells into the brain and thereby contribute to Aβ-related pathogenesis through enhancing neuroinflammation. To advance beyond correlational links, Wang et al. tested this hypothesis. Indeed, 5xFAD animals treated with antibiotic drugs that perturbed their gut microbiota had fewer activated microglia and less infiltrating Th1 cells. Similarly, increased infiltrating Th1 cells were present in wild-type mice that were co-housed with 5xFAD mice (i.e., sharing their gut microbiota via feces) or that received fecal microbiota transplantation (FMT) from 5xFAD animal donors, compared to controls.
Next, the authors extended their understanding of GV-971 in attenuating Aβ-related pathology and cognitive decline. Using another amyloidosis animal model (APP/PS1 transgenic mice), the authors found that GV-971 could ameliorate cognitive impairment partly by modulating the gut microbiota. Furthermore, 5xFAD mice treated orally with GV-971 showed gut microbiota alterations alongside a reduction in Aβ plaques, the number of brain Th1 cells, and the number of microglia. Intriguingly, in metabolomic analysis, the authors found that levels of phenylalanine and isoleucine (Phe/Ile) were higher in both the feces and blood of 5xFAD mice compared to controls. Notably, GV-971 treatment normalized Phe/Ile to control levels in 5xFAD mice. The authors further showed that Phe/Ile act on T cell proliferation and differentiation. Demonstrating potential translational relevance, elevated Phe/Ile levels were also observed in patients with mild cognitive impairment due to AD.
Taken together, these analyses suggest the idea that gut dysbiosis contributes to Phe/Ile elevation, which drives the proliferation/differentiation and brain infiltration of Th1 cells (Fig. 1). These infiltrating Th1 cells may then further activate microglia and contribute to Aβ-related pathogenesis. Whether this potential mechanism represents an AD-specific process is not clear, since there is great overlap in immunological changes and gut dysbiosis with other diseases (e.g., peripheral immune cell involvement in multiple sclerosis and increased ratio of Firmicutes to Bacteroidetes in obesity). In addition, although this study reveals that gut reconditioning may be one mechanism of action of the drug GV-971, it does not rule out other possible mechanisms. For example, GV-971 may attenuate AD pathogenesis by directly inhibiting neuroinflammation or amyloid-β fibril formation. However, there is no question that this data further supports the emerging idea that modulation of the gut microbiome via treatments such as GV-971 or other strategies should be further explored as novel strategies to slow the progression of AD.
Fig. 1.
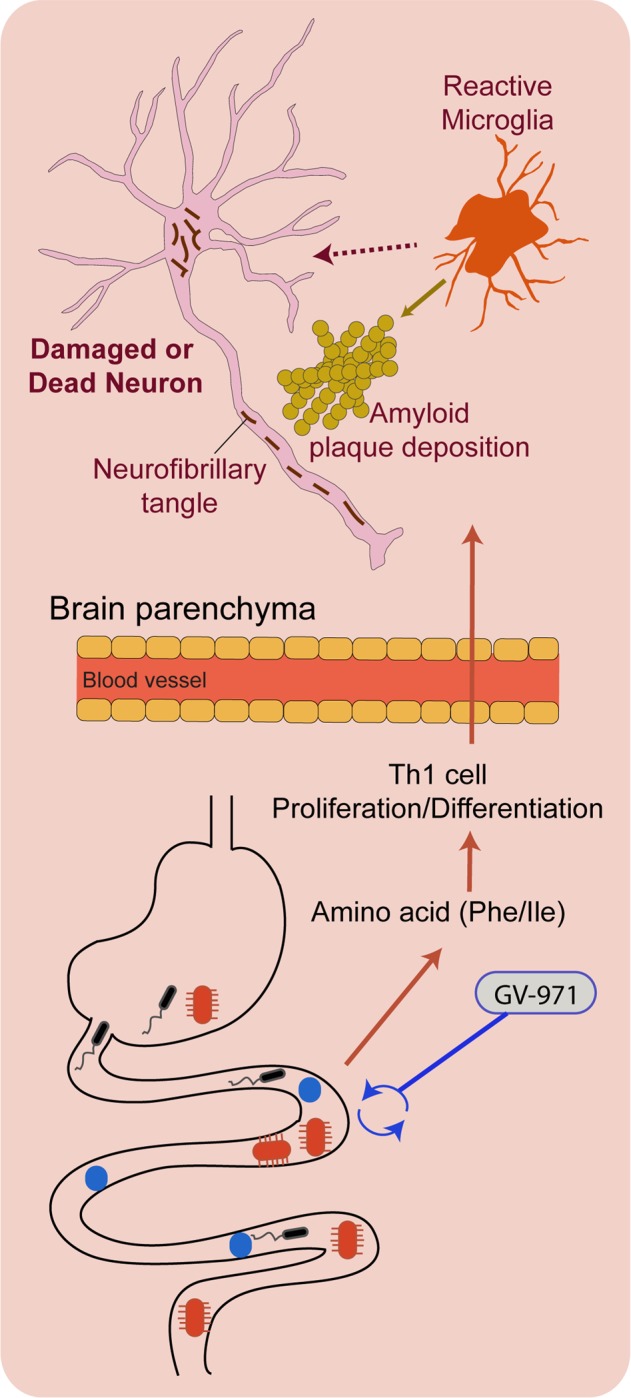
Schematic diagram of dysregulated gut-brain axis and its association with AD pathology. Dysbiosis of the gut microbiota contributes to AD progression by altering amino acid metabolism and peripheral adaptive immunity. Elevated phenylalanine and isoleucine (Phe/Ile) amplify the abundance of plasma T helper type 1 (Th1) cells, which invade the brain parenchyma. These promote neuroinflammation (i.e., increase pro-inflammatory microglia abundance) and contribute to AD pathogenesis: amyloid-β (Aβ) deposition and neurofibrillary tangles (not investigated in the current study). Treatment with GV-971, a novel marine-derived oligosaccharide, reconditions the gut microbiota and limits the contribution of the altered peripheral immunity to AD pathogenesis
Competing interests
David M. Holtzman is a co-founder of C2N Diagnostics LLC. He is on the scientific advisory board of Genentech, Denali, and C2N Diagnostics. He consults for Idorsia. His lab receives research grants from the National Institutes of Health, Cure Alzheimer’s Fund, Tau Consortium, the JPB Foundation, and C2N Diagnostics.
References
- 1.Holtzman DM, Morris JC, Goate AM. Sci. Transl. Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zenaro E, et al. Nat. Med. 2015;21:880–886. doi: 10.1038/nm.3913. [DOI] [PubMed] [Google Scholar]
- 3.Marsh SE, et al. Proc. Natl Acad. Sci. USA. 2016;113:E1316–E1325. doi: 10.1073/pnas.1525466113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baruch K, et al. Nat. Commun. 2015;6:916. doi: 10.1038/ncomms8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fung TC, Olson CA, Hsiao EY. Nat. Neurosci. 2017;20:145–155. doi: 10.1038/nn.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogt NM, et al. Sci. Rep. 2017;7:13537. doi: 10.1038/s41598-017-13601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harach T, et al. Sci. Rep. 2017;7:41802. doi: 10.1038/srep41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minter MR, et al. Sci. Rep. 2017;7:10411. doi: 10.1038/s41598-017-11047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang, X. et al. Cell Res. 10.1038/s41422-019-0216-x (2019).
- 10.Brandscheid C, et al. J. Alzheimers Dis. 2017;56:775–788. doi: 10.3233/JAD-160926. [DOI] [PubMed] [Google Scholar]