

Abstract
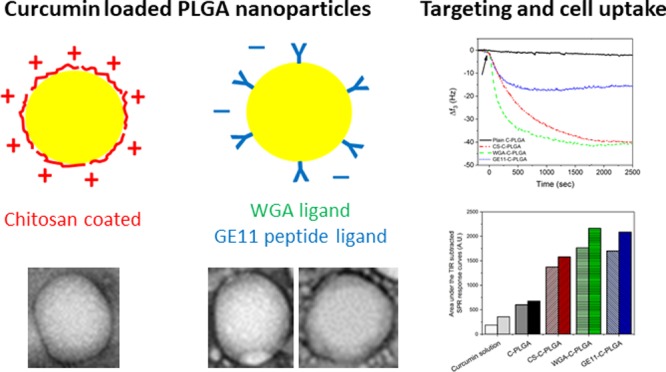
The exploitation of curcumin for oral disease treatment is limited by its low solubility, poor bioavailability, and low stability. Surface-functionalized poly-lactic-co-glycolic acid (PLGA) nanoparticles (NPs) have shown promising results to ameliorate selective delivery of drugs to the gastro-intestinal tract. In this study, curcumin-loaded PLGA NPs (C-PLGA NPs) of about 200 nm were surface-coated with chitosan (CS) for gastro-intestinal mucosa adhesion, wheat germ agglutinin (WGA) for colon targeting or GE11 peptide for tumor colon targeting. Spectrometric and zeta potential analyses confirmed the successful functionalization of the C-PLGA NPs. Real-time label-free assessment of the cell membrane-NP interactions and NP cell uptake were performed by quartz crystal microbalance coupled with supported lipid bilayers and by surface plasmon resonance coupled with living cells. The study showed that CS-coated C-PLGA NPs interact with cells by the electrostatic mechanism, while both WGA- and GE11-coated C-PLGA NPs interact and are taken up by cells by specific active mechanisms. In vitro cell uptake studies corroborated the real-time label-free assessment by yielding a curcumin cell uptake of 7.3 ± 0.3, 13.5 ± 1.0, 27.3 ± 4.9, and 26.0 ± 1.3 μg per 104 HT-29 cells for noncoated, CS-, WGA-, and GE11-coated C-PLGA NPs, respectively. Finally, preliminary in vivo studies showed that the WGA-coated C-PLGA NPs efficiently accumulate in the colon after oral administration to healthy Balb/c mice. In summary, the WGA- and GE11-coated C-PLGA NPs displayed high potential for application as active targeted carriers for anticancer drug delivery to the colon.
1. Introduction
Curcumin is a natural product with a wide range of pharmacological activities, including anti-inflammatory,1,2 antioxidant,3,4 and antimicrobial effects.5,6 The most attractive property of curcumin is, however, its ability to inhibit cell proliferation and induce apoptotic cell death of a wide variety of tumor cells.7,8 Unfortunately, the exploitation of curcumin as anticancer agent is hampered by its low solubility and rapid decomposition in aqueous medium, photo-instability, and poor bioavailability.
Nanoparticles (NPs) have widely been studied as drug carriers for anticancer drug delivery9,10 because they can protect drugs from harsh environments and enable sustained drug release or tumor targeting. The surface properties of NPs, namely charge, hydrophilic/lipophilic features, and targeting agents, dictate their interaction with cells and consequently their bioselectivity. For example, negatively charged NPs are poorly taken up by cells compared to positively charged NPs.11,12 On the other hand, surface-coated NPs with targeting moieties have been developed to enhance the cell selectivity and the intracellular delivery of drugs with a narrow therapeutic window and low cell membrane permeability.13
Poly-lactic-co-glycolic acid (PLGA) NPs decorated with mucoadhesive materials, including polymers, lectins,14 and peptides,15 have been developed for oral administration to selectively deliver drugs to the gastro-intestinal tract (GIT). Chitosan (CS) coating can endow NPs with mucoadhesive properties and enhanced transmucosal penetration.16−18 CS can also promote coulombic interactions with negatively charged cancer cells, which translocate negatively charged constituents of the inner layer of the cell membrane (e.g., phosphatidylserine, anionic phospholipids, glycoproteins, and proteoglycans) to the cell surfaces.19
Small molecules and macromolecules have also been exploited to generate targeted NPs with specific mucoadhesivity or cell surface selectivity. Lectins, the second generation mucoadhesives,20 enable site-specific targeting to mucosal cells through noncovalent carbohydrate residue binding.21,22 For example, wheat germ agglutinin (WGA) is a dietary lectin that recognizes N-acetyl-d-glucosamine and sialic acid exposed on the cell membranes of human colonocytes, colon cancer cells, and prostate cancer cells.23 EGF analogue peptides such as GE11,24 a peptide devoid of bioactivity, have been used to produce drug delivery systems that selectively target the epidermal growth factor receptor overexpressed on tumor cells,25,26 including gastric, colorectal, oesophageal, nonsmall cell lung cancer, and ovarian cancer cells.27,28
The biopharmaceutical and pharmacokinetic (PK) characterization of tailored NPs is a critical step in the development of efficient drug delivery systems. Real-time label-free in vitro analytical techniques can provide complementary information to conventional label-based biochemical and in vitro cell culture assays. This can improve the cost- and time effectiveness of the development of NPs by providing new complementary tools for selecting candidates with the biopharmaceutical requisites for further in vivo testing, as well as for building a better comprehension of their PK behavior.
Quartz crystal microbalance (QCM) combined with solid supported lipid bilayers (SLBs)29,30 and living cell sensing with surface plasmon resonance (SPR)31−35 are two real-time label-free in vitro surface-sensitive technologies providing rapid information on interfacial processes while obviating the need for additional labeling processes. Although, SPR and QCM have been used since the 1990s for biomolecular interaction studies, they are still unexplored for research and development of pharmaceutical nanosystems. The QCM is a mechanical measuring technique providing nanoscale sensitivity for mass and viscoelasticity measurements by utilizing the changes in resonant frequency and dissipation of quartz crystal resonators upon the deposition of mass on the sensor surface. The SPR technique, on the other hand, relies on the SPR phenomena and measures the changes in the refractive index in the close vicinity of SPR metal sensors upon mass deposition on the sensor surfaces.
The main objective of the present study was to investigate the targeting of curcumin-loaded PLGA (C-PLGA) NPs to human colorectal adenocarcinoma cells (HT-29). Accordingly, C-PLGA NPs were surface coated with CS, WGA, and GE11 in order to improve the cell uptake of the NPs and consequently ameliorate the delivery of curcumin to the cells. The behavior of these three platforms was investigated with new label-free approaches and traditional methods. Real-time label-free QCM and SPR measurements were used to assess the cell membrane surface interactions and targeting capability of noncoated and coated C-PLGA NPs to HT-29 cells. Complementary physicochemical characterization of noncoated and coated C-PLGA NPs were performed through particle size, zeta potential, drug entrapment efficiency, and in vitro drug release kinetics measurements. Traditional in vitro cell studies were also performed to determine in vitro cell uptake of curcumin and cell viability of HT-29 cells. Finally, a preliminary in vivo study with healthy Balb/c mice was performed to evaluate the accumulation of noncoated and targeted C-PLGA NPs in the colon after oral administration.
2. Experimental Section
2.1. Materials
Curcumin was purchased from Cayman chemical company (China, purity 91%). Acid-terminated poly(d,l-lactide-co-glycolide) (PLGA, 50:50, 7–17 kDa MW, Resomer RG 502H), polyvinyl alcohol (PVA, 31–50 kDa MW; 87–89% hydrolyzed), chitosan (CS, medium MW), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetra-acetic acid (EGTA, purity ≥ 97%), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS, purity ≥ 98%), Triton X-100 (purity for molecular biology), N-hydroxysuccinimide (NHS, purity 98%), mannitol (purity ≥ 98%), sodium taurocholate (purity ≥ 95%), trypan blue solution (purity suitable for cell culture), and the MTT-based in vitro toxicology assay kit were purchased from Sigma-Aldrich (Finland). WGA was purchased from Vector laboratories, Inc. (USA). Mucin extracted from porcine stomach (Type III) and pepsin from porcine gastric mucosa were purchased from LEE Biosolutions (USA). Pectinase from Aspergillus niger was purchased from Sigma-Aldrich Life Science (Denmark). N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, purity ≥ 99%) was purchased from Fluka Analytical (Japan). Cyanine 5.5 NHS ester (Cy5.5-NHS, purity 95%) was purchased from Lumiprobe GmbH (Germany). The GE11-PEG-NH2 and the Cy5.5-PEG-NH2 were synthesized according to the protocols set up by Balasso et al. to conjugate the peptide PreS1 and the fluorescent probe rhodamine, respectively, to NH2-PEG-NH2.36l-α-Phosphatidylcholine (eggPC, purity 95%), 1,2-dioleoyl-sn-glycero-3-[phospho-l-serine] (sodium salt) (DOPS, purity > 99%), and 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC, purity > 99%) were purchased from Avanti Polar Lipids (USA). Dulbecco’s Modified Eagle Medium (DMEM), Dulbecco phosphate-buffered saline (DPBS), McCoy’s 5A medium, OPTI-MED-reduced serum medium, fetal bovine serum (FBS), TrypLE express, and the μ-b bicinchoninic acid protein assay kit (Pierce μ-BCA kit) were purchased from Thermo Fischer Scientific (Finland). Hellmanex II was obtained from Hellma GmbH (Germany). High purity water (18.2 MΩ cm) from a Millipore Milli-Q system (USA) was used for the preparation of all buffer and water-based solutions. All other salts and solvents were from Sigma-Aldrich (Finland, purity of at least Reagent grade).
2.2. Preparation and Characterization of the Formulations
2.2.1. Preparation of the Curcumin-Loaded PLGA NPs (C-PLGA NPs)
Noncoated curcumin-loaded PLGA NPs (C-PLGA NPs) were prepared as described previously.37 Briefly, 176 mg of PLGA was dissolved in 5 mL of dichloromethane (DCM) and 16.6 mg of curcumin was added to the solution. The organic curcumin/PLGA solution was rapidly dropped into a glass tube containing 10 mL of 2% PVA in high purity water under vortexing. The mixture was vortexed for further 10 s at a high setting and sonicated for 7 min at 40% amplitude in an ice-water bath by using a probe sonicator (Vibra-Cell VCX 750 sonicator, Sonics & Materials Inc., Newtown, CT, USA). The resulting fine O/W emulsion was immediately poured into 30 mL of 0.5% PVA in high purity water under rapid stirring with a magnetic stirrer. DCM was then evaporated under magnetic stirring at 800 rpm for 3 h and the NPs were collected by centrifugation at 48 000g for 15 min and washed 3 times with high purity water. The supernatant was collected, desiccated under vacuum, and the encapsulation efficiency of curcumin was evaluated as described in Section 2.2.6.
2.2.2. Chitosan Functionalization of C-PLGA NPs
C-PLGA NPs coated with chitosan (CS-C-PLGA NPs) were prepared as described above, provided that 5 mL of CS solutions (0.25, 0.50, or 1% w/v) in 1% glacial acetic acid were added to 5 mL PVA aqueous solution prior the emulsification with the curcumin/PLGA organic solution.17
2.2.3. Preparation of Lectin or Peptide Ligand-Coated C-PLGA NPs
C-PLGA NPs were surface-coated with WGA lectin or GE11 peptide according to a two-step EDC/NHS method.38,39 Briefly, 5 mL aliquots of noncoated C-PLGA NP suspensions in high purity water were washed with 20 mL 0.1 M MES buffer, pH 5.8, and then the noncoated C-PLGA NPs were separated by centrifugation at 15 000g. The noncoated C-PLGA NPs were incubated with approximately 0.1 M EDC and 0.15 M NHS in 0.1 M MES buffer, pH 5.8, for 1 h at room temperature (RT). The EDC excess was quenched by washing three times with 20 mL of 20 mM phosphate, 0.15 M (PBS), pH 7.4. The activated C-PLGA NPs were resuspended in 5 mL PBS, pH 7.4, and added of 1 mL of 1 mg/mL WGA lectin or GE11 peptide in the same buffer and the suspension was maintained overnight under gentle agitation. The resulting WGA-coated NPs (WGA-C-PLGA NPs) and GE11-coated NPs (GE11-C-PLGA NPs) were added of 1 mL of 50 mM glycine. After 1 h, the WGA-C-PLGA and GE11-C-PLGA NPs were separated by centrifugation, washed twice with PBS, pH 7.4, and finally collected by centrifugation.
2.2.4. Determination of the Amount of Bound WGA and GE11 to C-PLGA NPs
A water-based system comprised 2 mL DMSO and 10 mL of 0.05 N NaOH containing 0.5% SDS was used to dissolve the WGA-C-PLGA and GE11-C-PLGA NPs for determining the amount of ligands conjugated on the C-PLGA NPs. The amount of WGA lectin or GE11 peptide ligand in the dissolved NP solutions was quantified by colorimetric determination of the WGA lectin/GE11 peptide content by using a μ-BCA protein assay. The WGA lectin or GE11 peptide conjugation density and efficiency was calculated from the following equations
| 1 |
| 2 |
2.2.5. Physicochemical Characterization of Noncoated and Coated C-PLGA NPs
The particle size, polydispersity index (PDI), and zeta potential of noncoated and chitosan, WGA-, and GE11-coated C-PLGA NPs were measured at RT by using a ZetaSizer Nano ZS instrument (Malvern Instruments Ltd, Worcestershire, UK) after NP dispersion in high purity water. Morphological analyses were performed with a JEOL 1200 EX II transmission electron microscope (JEOL Ltd., Tokyo, Japan).
2.2.6. Encapsulation Efficiency of Curcumin in Noncoated and Coated C-PLGA NPs
The encapsulation efficiency of curcumin in noncoated and chitosan, WGA-and GE11-coated C-PLGA NPs was determined by dissolving the NPs in acetonitrile and determining the amount of curcumin by ultraperformance liquid chromatography (AcquityTM, Waters Corp., Milford, Massachusetts, USA) at λmax = 426 nm (according to the procedure reported in the literature,40 2011), with minor changes.37 The drug encapsulation efficiency (EE %) for each NP was calculated by using the following equation:
| 3 |
2.2.7. In Vitro Curcumin Release from Noncoated and Coated C-PLGA NPs
The in vitro release of curcumin from noncoated and chitosan, WGA- and GE11-coated C-PLGA NPs was determined by the dialysis bag method reported by Zanotto-Filho et al.,41 with slight modifications. A three-stage approach with three different pH release media was used in order to follow the recommendations on methods for dosage form testing by US Pharmacopeia 36,42 as described earlier.37 The media and time intervals used for the three different stages were the following: simulated gastric fluid (SGF; 0.2% NaCl, 0.2% pepsin, 0.7% hydrochloric acid, pH 1.2) between 1 and 2 h, simulated intestinal fluid (SIF; 0.68% KH2PO4, 3 mM sodium taurocholate, pH 6.8) between 3 and 5 h, and simulated colonic fluid (SCF; PBS, pH 7.4) between 6 and 24 h. The NP suspensions were placed into dialysis bags with a molecular cutoff of 12–14 kDa and dialyzed in the dark against 150 mL of the corresponding media for each time interval. Samples from the solution in the external buffer were taken at scheduled times and analyzed by ultraperformance liquid chromatography (AcquityTM, Waters Corp., Milford, Massachusetts, USA) at λmax = 426 nm, as described earlier.37
2.2.8. In Vitro Stability of Noncoated and Coated C-PLGA NPs
The stability of noncoated and chitosan, WGA-, and GE11-coated C-PLGA NPs in high purity water was assessed at two different temperatures (4 °C and RT) for up to four months. Stability of NPs in SGF, SIF, and SCF at 37 °C was assessed by incubation of 1 mg of NPs in 1 mL of the corresponding fluids for appropriate times for simulating GI-tract residence times. At scheduled time, samples were taken and analyzed by ZetaSizer Nano ZS (Malvern Instruments Ltd, Worcestershire, UK). The experimental data were elaborated according to the method used previously.37
2.2.9. In Vitro Mucin Adsorption to Noncoated and Coated C-PLGA NPs
Adsorption of pig mucin (PM) on the surface of noncoated and chitosan, WGA-, and GE11-coated C-PLGA NPs was determined by slightly modifying the method reported by Yin et al.43 Briefly, 1 mL of mucin suspension (1 mg/mL) in PBS with pH 7.4 was stirred with 1 mL of each C-PLGA NP formulation for 2 h at 37 °C. Then, the suspensions were centrifuged at 25 000g for 1 h. The amount of free PM in the supernatant was determined by measuring the absorbance value at 260 nm by using a UV spectrophotometer (UV Visible Spectrophotometer, Cary 100 Conc, Australia). The amount of adsorbed PM was calculated by using a standard curve measured for known amounts of PM in PBS. The calibration curve for mucin in PBS was determined with a series of mucin standard solutions with concentrations of 30, 60, 125, 250, 500, 750, and 850 μg/mL. The PM binding efficiency of the different C-PLGA NPs was calculated from the following equation
| 4 |
where C0 is the initial concentration of PM used for incubation and Cs is the concentration of free PM determined in the supernatant.
2.3. QCM Studies
2.3.1. QCM Sensor Preparation
The silica-coated QCM sensor crystals (Biolin Scientific, Q-Sense, Gothenburg, Sweden) was cleaned by boiling them in a cleaning solution (water/hydrogen peroxide/ammonium hydroxide; 5/1/1, v/v/v) for 5 min followed by rinsing with copious amounts of ion-exchanged water and finally blow-drying with nitrogen. Prior and after each QCM measurement, the SiO2-coated QCM sensor crystal was cleaned in situ by flushing the flow channel with sequential 5 min injections of 20 mM CHAPS, 2% Hellmanex II, 95% ethanol, and high purity water.
2.3.2. Preparation of EggPC and DOPC/DOPS Cell Model Membranes on QCM Sensors
Vesicle rupture of EggPC or DOPC/DOPS (75/25 mol %) vesicles was used to prepare SLBs on the silica-coated QCM sensor crystals as simple cell model membranes. Phospholipids were dissolved separately in a defined amount in chloroform and stored at −20 °C until used for preparation of vesicles for SLB formation. Vesicles were prepared by evaporating the solvent from EggPC or a mixture of DOPC/DOPS in chloroform to dryness under a stream of nitrogen. The formed lipid films were then hydrated with a 20 mM Hepes/150 mM NaCl buffer solution with pH 7.4 at RT by vortexing to obtain vesicle suspensions with a total lipid content of 10 mg/mL. The vesicle suspensions were sonicated by a tip sonicator (Vibra-Cell VCX 750 sonicator, Sonics & Materials Inc., Newtown, CT, USA) until clear solutions were obtained. The vesicle solutions were then centrifuged for 1 min at 15 000g to remove titanium particles. The particle size, PDI index, and ZP of the vesicle solutions were determined by using a ZetaSizer Nano ZS instrument (Malvern Instruments Ltd, Worcestershire, UK). The vesicle solutions were stored at 4 °C and used within two weeks. Just before preparing the SLBs for QCM measurements, the vesicle solutions were diluted with a 20 mM HEPES/150 mM NaCl/5 mM CaCl2 buffer solution with pH 7.4 to a total lipid content of 0.1 mg/mL.
2.3.3. HT-29 Cell Membrane Extraction and Use for SLB Formation on QCM Sensors
HT-29 cell pellets (cells cultured as described in Section 2.5.1.) collected from 8 cell culture flasks were suspended in 10 mL of a harvest buffer (50 mM Tris-HCl, 300 mM mannitol, pH 7). The obtained cell suspension was centrifuged at 800g for 5 min at 4 °C and the supernatant was thrown away. This step was repeated twice. The residue was further suspended in 10 mL of a membrane buffer (50 mM Tris-HCl, 50 mM mannitol, and 2 mM EGTA, pH 7). The obtained cell suspension was transferred into a cell homogenizer and homogenized by 40 strokes followed by incubation on ice for 1 h. The cells homogenized were transferred into a Falcon tube and centrifuged at 800g for 10 min at 4 °C. The supernatant from this step was taken and centrifuged at 15 000g for 1 h at 4 °C. The supernatant was again taken and further centrifuged at 100 000g for 1.15 h at 4 °C. Finally, the supernatant was removed and the pellet formed by the cell membrane extract was weighed and stored at −75 °C until used. Just before use in QCM measurements, the HT-29 cell membrane extract pellet was suspended in a 20 mM Hepes/150 mM NaCl buffer solution with pH 7.4 to reach a concentration of 10 mg/mL of the membrane extract. This suspension was further mixed by using a 27G needle by filling and emptying the syringe with the whole suspension. Hereafter, the suspension was sonicated for 20 min until the solution no longer appeared cloudy and used immediately. A qualitative test for the presence of proteins in the extracted cell membrane suspension was performed using a BCA Protein Assay Reagent kit, which verified that the cell membrane extract contained a substantial amount of protein. The HT-29 cell membrane extract was diluted as such or in a mixture with different mass ratios of EggPC vesicles (1/1, 1/1.5 1/2, and 1/2.5, respectively) in order to find the optimized cell membrane extract/EggPC ratio for SLB formation. The HT-29 cell membrane extract and cell membrane extract/EggPC mixtures were diluted with a 20 mM HEPES/150 mM NaCl/5 mM CaCl2 buffer solution with pH 7.4 to a cell membrane extract/lipid concentration of 0.14 mg/mL just before performing the QCM measurements for verifying SLB formation.
2.3.4. QCM Measurements
QCM measurements were performed at 20 °C by using an impedance-based QCM instrument (QCM-Z500, KSV Instruments, Helsinki, Finland). Measurement data for frequency (f) and dissipation (D) were acquired at the fundamental frequency (5 MHz) and several overtone frequencies (15, 25, 35, 45, and 55 MHz). The interaction of the noncoated and chitosan, WGA- and GE11-coated C-PLGA NPs with DOPC/DOPS and the HT-29 cell membrane extract/EggPC SLBs were performed by monitoring QCM signal responses for a fixed time of 30 min when 1 mg/mL of the NPs were injected into the QCM flow channel. The flow rate during the QCM interaction measurements was kept constant at 50 μL/min, and a 20 mM HEPES/150 mM NaCl buffer solution with pH 7.4 was used as the running buffer during the measurements. The frequency and dissipation changes reported for the QCM measurements are the normalized signal changes for the 3rd overtone frequency, if not otherwise stated.
2.4. SPR Studies
2.4.1. Immobilization of Cells on SPR Sensors
Gold-coated SPR sensor slides (Bionavis Ltd., Tampere, Finland) were cleaned by boiling in a solution of high purity water, 30% hydrogen peroxide, and 30% ammonium hydroxide solution (5/1/1; v/v/v) for 5 min. After boiling, the sensor slides were rinsed thoroughly with high purity water and dried by aspirating them. The SPR sensors were hereafter autoclaved before cell immobilization. The immobilization of HT-29 cells (cultured as described in Section 2.5.1.) on the SPR sensor slides was performed by first treating confluent cell layers in cell culture flasks with trypsin in DPBS after which the cells were resuspended in the cell culture medium. The SPR sensor slide was then placed in a cell culturing polystyrene Petri dish with a cell growth area of 8.8 cm2 and 2 mL of the cell suspension was pipetted on the SPR sensor slide. The optimum seeding density for culturing the HT-29 cells on the SPR sensor slides was ∼1 × 104 cells/Petri dish. The cells were allowed to grow on the SPR sensor slides until they reached 90–100% confluence.
2.4.2. SPR Measurements
The interaction between noncoated and chitosan, WGA-, and GE11-coated C-PLGA NPs and immobilized HT-29 cells at 37 °C were monitored by using a Multi-Parametric SPR instrument (MP-SPR Navi 200, BioNavis Ltd, Tampere, Finland) equipped with a single flow channel with two detection spots. The signal responses in the SPR measurements were recorded simultaneously with a laser wavelength of 785 nm in two different positions in the flow channel (upper and lower flow channel positions) on the same sensor covered with a cell monolayer. The NP sample solutions for SPR measurements were prepared by diluting the H2O-based stock formulations to a final concentration of 1 mg/mL of the corresponding C-PLGA NP formulation with the running medium (DMEM supplemented with 10 mM HEPES, pH: 7.4), just before performing each SPR experiment. The baseline for the SPR measurements was measured for ∼60 min by flowing the running medium through the SPR flow channel at a constant flow rate of 10 μL/min. After the baseline signal was stabilized, the sample solution was injected into the SPR flow channel by replacing the running medium with the sample solution. The interaction of the NP formulations with HT-29 cells were monitored at a constant flow speed of 10 μL/min by recording the full SPR spectra between 60 and 78° every 3 s for up to 90 min. Because the full SPR angular spectrum was recorded, the bulk effect caused by the difference in the refractive index of the DMEM running buffer and the samples (i.e., diluted H2O-based stock solutions of the NPs in DMEM) was compensated for by subtracting the contribution of the total internal reflection angle (TIR; sensitive for bulk refractive index changes) from the overall change in the main SPR peak angular position (SPR PAP). The corrected SPR responses were then used for further analysis. Light microscope images (Leica DM IL LED microscope with 5× and 10× magnifications) of the HT-29 cell monolayers were taken immediately before and after each SPR experiment in order to evaluate the integrity of the cell monolayer.
2.5. In vitro Cell Studies with Noncoated and Coated C-PLGA NPs
2.5.1. Cell Culture and Cell Preparation
Human Caucasian colon adenocarcinoma (HT-29) obtained from the European Collection of Authenticated Cell Cultures (ECACC) were cultured in a McCoy’s 5A medium supplemented with 10% FBS at a density of about 2 × 106 cells/mL in a 75 cm2 culture flask. The cells were maintained at 37 °C, 95% relative humidity, and 5% CO2. The cells were harvested weekly from plastic flasks (75 cm2) with TrypLE Express and the medium was changed twice per week.
2.5.2. Cellular Drug Uptake Assay
HT-29 cells were cultured in a 12-well plate with a cell density of 1 × 104 cells/per well and incubated in a 5% CO2 incubator at 37 °C for 24 h before cellular drug uptake studies. The cell association assay was carried out at 37 °C in OPTI-MED-reduced serum medium containing enough pure curcumin or the noncoated or coated C-PLGA NP formulations so that each well contained the same absolute amount of 200 μg curcumin. Washing the wells with ice-cold PBS three times terminated incubation of the cells with pure curcumin or the NPs. The washed cells were lysed in PBS containing 0.5% Triton X-100, then vortexed for 3 min followed by centrifugation of the cell lysate for 10 min at 1200g to remove insoluble materials. The supernatant was transferred to another tube and the amount of curcumin was determined by diluting the supernatant with a mobile phase mixture (composed of (A) 38% acetonitrile and (B) 62% of a 0.05% phosphoric acid solution) and measured with an AcquityTM ultraperformance liquid chromatography instrument at λmax = 426 nm (Waters Corp., Milford, Massachusetts, USA).
2.5.3. In Vitro Cytotoxicity of Noncoated and Coated C-PLGA NPs
Cytotoxicity assays were performed by incubating HT-29 cancer cell lines with curcumin in solution, noncoated and chitosan, WGA- and GE11-coated C- PLGA NPs. Cells were treated with the same absolute amount of curcumin (200 μg). HT-29 cells were seeded in 96-well plates and after the attachment of cells; the plates were washed twice with 100 μL of PBS. After washing, the sample solutions were added to the wells. The wells were then incubated for 2 h. After incubation, the samples were removed by pipetting and 10 μL of MTT solution was added to each well. The plates were further incubated for 3 h and the MTT solution was removed. The blue crystals formed in each well were dissolved with 200 μL of DMSO. Absorbance values from the wells were measured at 550 nm with a Multiscan GO Microplate Spectrophotometer (ThermoFischer Scientific, Finland). Cell viability was calculated based on the absorbance values.
2.6. In Vivo Experiments and Optical Imaging
2.6.1. Mice
Six to eight week old female BALB/c mice were obtained from and housed in the IOV-IRCCS Specific Pathogen Free (SPF) animal facility. Procedures involving animals and their care were in conformity with institutional guidelines (D.L. 26/2014, and subsequent implementing circulars), and the experimental protocol was approved by the local “Organismo Preposto al Benessere Animale” (OPBA) of Padua University, and authorized by the Italian Ministry of Health (Authorization n.1050/2015-PR).
2.6.2. In Vivo Biodistribution of Noncoated and Ligand Coated C-PLGA NPs
The noncoated and coated C-PLGA NPs were labeled with a near-IR dye, that is Cy5.5, by using Cy5.5-PEG-NH2. The labeling of the NPs was carried out by using a similar two-step EDC/NHS method as described for WGA lectin or GE11 peptide functionalization of the C-PLGA NPs, as described in Section 2.2.3. In vivo optical imaging studies were conducted to evaluate the biodistribution of the labeled NPs in the GIT of mice. BALB/c mice were placed on a low-manganese diet to reduce autofluorescence from normal mouse chow. Five days later, animals were randomly divided into three groups of four animals each, and treated with Cy5.5-coated noncoated C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs. The Cy5.5-labeled NP formulations were administered by oral gavage in a total volume of 200 μL. The in vivo total body scanning was performed at different time points using an Explore Optix instrument (ART, Advanced Research Technologies, Montreal, Canada) by setting the excitation pulsing laser at 650 nm and recording the emission at 710 nm, thus performing a time course scanning of animals (spatial resolution/scan step fixed at 1.5 mm, exposure time 0.1 s, and laser power automatically adjusted for each scan session). At 0, 1, 2, 4, 6, 24, and 48 h the animals were anesthetized with a continuous flow of 5% isoflurane/oxygen mixture and placed inside a 37 °C heated camera box and a longitudinal whole body fluorescence image was recorded. Before imaging, the anesthesia was maintained using a nose cone delivery system for the duration of image acquisition. One mouse from each group was imaged at 2 h post administration and then sacrificed, and the abdominal cavity was opened to visualize the GIT at place. Thereafter, the entire GIT was removed and imaged alone, followed by imaging of the carcass.
2.7. Statistical Analysis
Results are expressed as a mean ± standard deviation (SD). Statistical analyses were performed by using ANOVA followed by Tukey’s test. A difference at P < 0.05 was considered to be significant.
3. Results and Discussions
3.1. Preparation and Biopharmaceutical Characterization of Curcumin-Loaded PLGA NPs
3.1.1. NP Preparation
Curcumin-loaded PLGA NPs (C-PLGA NPs) were coated with chitosan (CS), WGA or GE11 peptide (GE11) for selective or targeted uptake of the NPs to HT-29 colon cancer cells in order to improve the delivery of curcumin to the cells.
CS, a polycationic mucoadhesive polysaccharide, was adsorbed on the negatively charged C-PLGA NP surface by coulombic interaction. The NP decoration was carried out by using 0.25, 0.50, and 1% CS solutions that yielded different CS densities on the NP surface, that is, 0.25% CS-C-PLGA, 0.50% CS-C-PLGA, and 1% CS-C-PLGA NPs, respectively. WGA, a mucoadhesive glycoprotein, was chemically anchored to the carboxyl groups on the C-PLGA NP surface through a condensation process to yield WGA-C-PLGA NPs coated with 33.0 ± 1.4 μg WGA/mg C-PLGA. NPs targeting colorectal cancer cells (GE11-C-PLGA NPs) were obtained through chemical conjugation of GE1144 peptide to C-PLGA NPs which resulted in 38.1 ± 1.0 μg GE11 peptide/mg C-PLGA. The degree of NP functionalization calculated based on the C-PLGA NP size (220 ± 2 nm) and the PLGA NP density (1.2 mg/cm3)45 was found to correspond to a mean of 3.7 WGA molecules/NP and 82 GE11 peptides/NP. The coupling efficiency of WGA and the GE11 peptide ligand was calculated by using eq 2 and was 74.5% and 82.7%, respectively.
3.1.2. Physicochemical Characterization of Noncoated and Coated C-PLGA NPs
The dynamic light scattering (DLS) data reported in Table 1 show that the CS coating significantly increased the size of the noncoated C-PLGA NPs, while the PDI remained constant. The high negative zeta potential of the noncoated C-PLGA NPs originates from the presence of a high density of terminal carboxyl groups of PLGA. Coating the C-PLGA NPs with CS solutions with different polysaccharide concentrations (i.e., 0.25, 0.5 and 1%) yielded NPs with similar particle sizes and PDIs, while the zeta potential values increased with increasing CS concentration. Thus, the use of increasing CS concentrations for coating C-PLGA NPs obviously resulted in an increased CS deposition on the NP surface. The 1% CS-C-PLGA NPs exhibited a significantly higher positive zeta potential compared to the other CS-coated NPs, which could result in high mucoadhesive properties. The WGA and GE11 decoration did not substantially affect the size of the coated C-PLGA NPs probably because of the limited number of protein and peptide molecules conjugated on the particle surfaces. However, WGA and GE11 conjugation reduced the absolute value of the negative zeta-potential of the NPs, but the particles still maintained a negative zeta potential. The slight quenching of the negative zeta potential obtained by WGA and GE11 conjugation can be attributed to the limited number of carboxyl groups engaged in the protein conjugation and to the overall positive charge of these polypeptidic molecules with isoelectric points of 9 and 9.76, respectively. In this regard, it is interesting to note that in the case of WGA-C-PLGA NPs bearing only ∼4 lectin molecules/NP, the zeta potential is highly negative −20.5 mV, while in the case of GE11-C-PLGA NPs bearing about 82 peptide molecules/NP the zeta potential was −10.8 mV. These results indicate that the noncoated C-PLGA NPs were successfully modified with both ligands.
Table 1. Particle Size, Size Distribution, Zeta Potential, and Encapsulation Efficiency of Noncoated and Coated Curcumin-Loaded PLGA NPs (Mean ± SD, n = 3).
| formula | particle size [nm] | PDI | ζ-potential [mV] | encapsulation efficiency (EE %) | |
|---|---|---|---|---|---|
| C-PLGA | 220 ± 2 | 0.186 ± 0.023 | –36.8 ± 0.0 | 74 ± 4 | |
| CS-C-PLGA | 0.25% | 245 ± 4 | 0.172 ± 0.024 | 2.5 ± 2.1 | 64 ± 2 |
| 0.50% | 251 ± 3 | 0.276 ± 0.002 | 4.8 ± 1.2 | 62 ± 1 | |
| 1% | 252 ± 3 | 0.126 ± 0.033 | 32.9 ± 1.7 | 54 ± 2 | |
| WGA-C-PLGA | 227 ± 3 | 0.195 ± 0.011 | –20.5 ± 0.8 | 66 ± 1 | |
| GE11-C-PLGA | 209 ± 2 | 0.140 ± 0.044 | –10.8 ± 0.9 | 62 ± 2 | |
The TEM images reported in Figure 1 show that all NPs had regular spherical shapes indicating that the surface coating did not influence the particle morphology. The slightly smaller size (i.e., <200 nm) observed by TEM compared to DLS is in agreement with the dry state of NPs, while DLS provides the hydrodynamic diameter of NPs. Table 1 shows that the noncoated C-PLGA NPs have a high curcumin encapsulation efficiency (EE 74%). The encapsulation efficiency of the coated NPs was slightly lower compared to the noncoated C-PLGA NPs which may be ascribed to partial curcumin release during the coating process.14,38,39
Figure 1.

TEM images of noncoated and coated C-PLGA NPs (scale bar 200 nm). (A) Noncoated C-PLGA NPs, (B) 1% CS-C-P-PLGA NPs, (C) WGA-C-PLGA NPs, and (D) GE11-C-PLGA NPs.
3.1.3. In Vitro Drug Release
Curcumin release was assessed by NP incubation in three different pH release media simulating: gastric fluid (SGF), small intestine fluid (SIF), and colon fluid (SCF) (Figure S1). The sequential incubation of noncoated C-PLGA NPs in SGF (1–2 h in Figure S1) and SIF (3–5 h in Figure S1) yielded a 10% curcumin release within 5 h (Figure S1). Hereafter, when the noncoated C-PLGA NPs were incubated in SIF media a biphasic profile with a burst release with 56% of curcumin released within the first 24 h was followed by a slow release over a period of 48 h yielding a final curcumin release of 78% (6–24 h in Figure S1). These results are in agreement with the higher curcumin solubility at pH 7.4 with respect to acidic conditions.46
Noncoated C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs showed similar curcumin release profiles, suggesting that the surface modification of C-PLGA NPs with a few protein or peptide molecules does not remarkably affect the drug release properties of the formulation. In SCF during the first burst phase, the WGA-C-PLGA and GE11-C-PLGA NPs released about 62 and 54% of the loaded drug, respectively. Then, the release slowly continued up to 76% for WGA-C-PLGA and 84% for GE11-C-PLGA NPs in 48 h. The CS coating had a stronger effect on the drug release from the NPs compared to WGA and GE11. The 1% CS-C-PLGA NPs showed in fact a slower curcumin release compared to the other formulations: about 42 and 58% of the loaded curcumin was released in 24 h and in 48 h, respectively. The slower release observed after CS coating may be because of the CS layer on the NP surface that represents a barrier to the diffusion of curcumin.47 Furthermore, the affinity of curcumin for CS may also reduce the drug availability.
The kinetic data obtained by the elaboration of the in vitro release profiles (Table S1) show that the curcumin release from all the C-PLGA NPs fits the Higuchi diffusion model,48,49 which is in agreement with the fact that degradation of PLGA can last from weeks to months.50
3.1.4. In Vitro Adsorption of Crude PM
The mucoadhesive properties of the NP formulations were evaluated by adsorption of PM on the surface of C-PLGA, 0.25, 0.5, and 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs. The noncoated C-PLGA NPs adsorbed 22.29% of PM after 2 h incubation at 37 °C (Figure S2). According to its well-known mucoadhesivity, chitosan-coated NPs showed high PM adsorption, which was 3.0, 3.2, and 3.4-fold compared to C-PLGA NPs in the case of 0.25, 0.50, and 1% CS-C-PLGA NPs, respectively. These results indicate that in this coating range the density of chitosan and the positive charge on the NP surface has a limited effect on their final mucoadhesivity. This may be due to a surface saturation by PM even at low chitosan density.51 PM adsorption on WGA-C-PLGA was 2.3-fold compared to the noncoated C-PLGA NPs, indicating that a limited amount of this protein has a remarkable effect on mucoadhesion properties of the NPs. The PM interaction with WGA-C-PLGA NPs is attributed to specific binding with the N-acetyl-d-glucosamine and sialic acid residues of PM glycoproteins.52 Unexpectedly, also GE11, a nonmucoadhesive peptide, was found to increase the PM adsorption on the NPs of about 2-fold. The GE11-C-PLGA NPs/PM association probably occurs through nonspecific coulombic GE11/mucin interactions.
3.1.5. In Vitro Stability of Different C-PLGA NPs
Stability studies of the C-PLGA NPs were carried out by storing the NPs in high purity water at RT and 4 °C for up to 4 months. Table S2 shows that the particle size and PDI of noncoated C-PLGA, 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs did not change over time indicating that the NPs have a good long-term stability. On the contrary, the 0.25 and 0.50% CS-C-PLGA NPs showed a significant size and PDI increase after 1 month storage. The lower stability of 0.25 and 0.5% CS-PLGA NPs compared to the other NPs can be attributed to the low zeta potential (+2.5 and +4.5 mV, respectively, Table 1). According to these results, the 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs were chosen for further studies.
In view of oral delivery, the colloidal stability of the NPs was also investigated by incubating noncoated C-PLGA, 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs at 37 °C for up to 2 h in SGF, up to 6 h in SIF, and up to 24 h in SCF to simulate GI-tract residence times. All NPs except 1% CS-C-PLGA NPs were stable in the three fluids without relevant particle size and PDI changes (Table S3). The size increase of 1% CS-C-PLGA NPs in SGF may be due to a change of the hydrophilic/hydrophobic surface balance that results in a more hydrophobic surface. This hypothesis seems to be confirmed by the high stability observed when the 1% CS-C-PLGA NPs were incubated in SIF, which contains the bile acid taurocholate that apparently stabilizes the 1% CS-C-PLGA NPs because of its surfactant properties.
3.2. Interaction of Different C-PLGA NPs with Biomembranes and Cells
3.2.1. QCM Analyses with Supported Lipid Bilayer
The interaction of noncoated C-PLGA, 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs with cell model membranes were investigated by using the QCM technique in combination with SLBs composed of a simple two-component phospholipid mixture (DOPC/DOPS, 75/25 mol %) or a mixture of HT-29 cell membrane extract and EggPC. DOPC/DOPS SLBs were used to mimic the negative charge of natural cell membranes. The HT-29 cell membrane extract incorporated into the EggPC SLB was used to evaluate the interaction of NPs with biological targets. The DOPC/DOPS and the HT-29 cell membrane extract/EggPC vesicle mixtures were spread on the silica-coated QCM sensor to form the SLBs according to standard protocols reported in the literature.29,30
Figure 2 shows the adsorption-bursting behavior typically observed in QCM responses during vesicle spreading when SLBs are formed. Table 2 reports the size and zeta potential of the vesicles used to form the SLBs, as well as the equilibrium frequency change, Δf, for the SLB. The size of the vesicles obtained with the HT-29 cell membrane extract/EggPC resembled the size of plain EggPC vesicles, while the zeta potential was close to the average of the components. This result suggests that the HT-29 cell membrane extract components and EggPC are incorporated in the hybrid vesicles. The equilibrium frequency values obtained with the SLBs in this study are well in line with the values of typical SLBs reported in the literature.30
Figure 2.
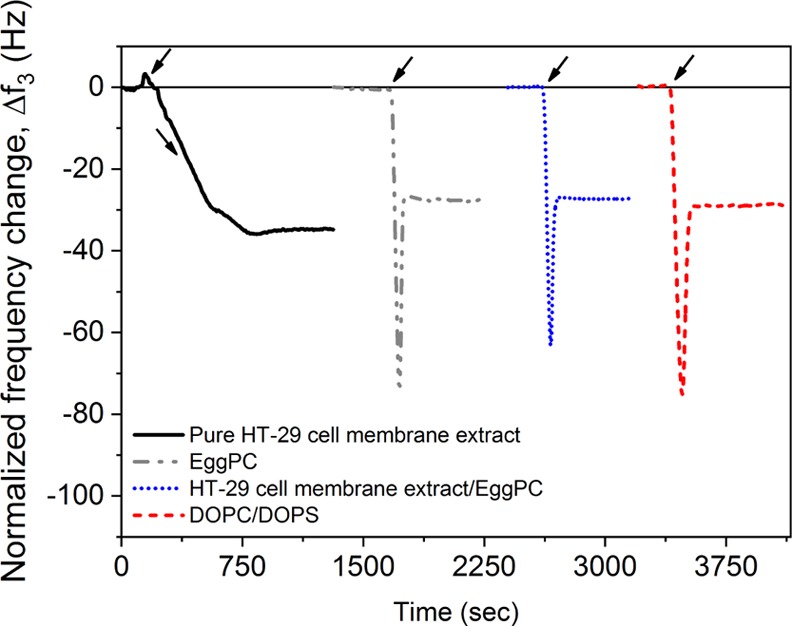
QCM signal responses for the pure HT-29 cell membrane extract, and SLB formation through vesicle spreading of EggPC vesicles, HT-29 cell membrane extract/EggPC (1/1 mass ratio) vesicles and DOPC/DOPS (75/25 mol %) vesicles. Arrows indicate time of injection of vesicles. Each QCM signal response correspond to a separate measurement and have been shifted in time for clarity.
Table 2. Vesicle Size and ζ-Potential of Vesicles Used for SLB Formationa.
| SLB composition | vesicle size (nm) | ζ-potential (mV) | Δf3 (Hz) |
|---|---|---|---|
| DOPC/DOPS (75/25 mol %) | 71.5 ± 1.3 | –27.6 ± 1.3 | –27.3 ± 1.4 |
| pure EggPC | 100.7 ± 1.0 | –3.7 ± 0.1 | –28.0 ± 3.2 |
| pure HT-29 membrane extract | 224.5 ± 4.6 | –15.5 ± 1.7 | |
| HT-29 membrane extract/EggPC (1/1 mass ratio) | 109.9 ± 2.6 | –8.5 ± 0.1 | –27.5 ± 4.9 |
Equilibrium frequency change, normalized Δf3, for the final SLB of the DOPC/DOPS (75/25 mol %) mixture, pure EggPC and the mixture of membrane extract/EggPC (1/1 mass ratio). Mean ± SD, n = 3.
Figure 3 shows the changes in QCM frequency and dissipation at the 3rd overtone during the interaction of various NPs with DOPC/DOPS SLBs. The NPs were injected over the SLBs at time t = 0. After 30 min, buffer was injected to rinse the system and clear out the NPs that did not interact with the SLBs (vertical dashed line). The frequency and dissipation changes indicate the extent and the type of the NP interaction with the SLBs. The QCM measurements show that the various NPs underwent similar reversible interaction with the DOPC/DOPS SLBs, except for the 1% CS-C-PLGA NPs that showed a slightly higher dissipation change during sample injection compared to the other NPs. Additionally, the frequency and dissipation signals in the case of 1% CS-C-PLGA NPs do not return to the starting level when flushing with running buffer, as observed with the other NPs. This is probably a consequence of electrostatic interactions between the highly positively charged 1% CS-C-PLGA NPs (zeta potential ≈ +33 mV) and the negatively charged DOPC/DOPS SLB (∼−27 mV).
Figure 3.
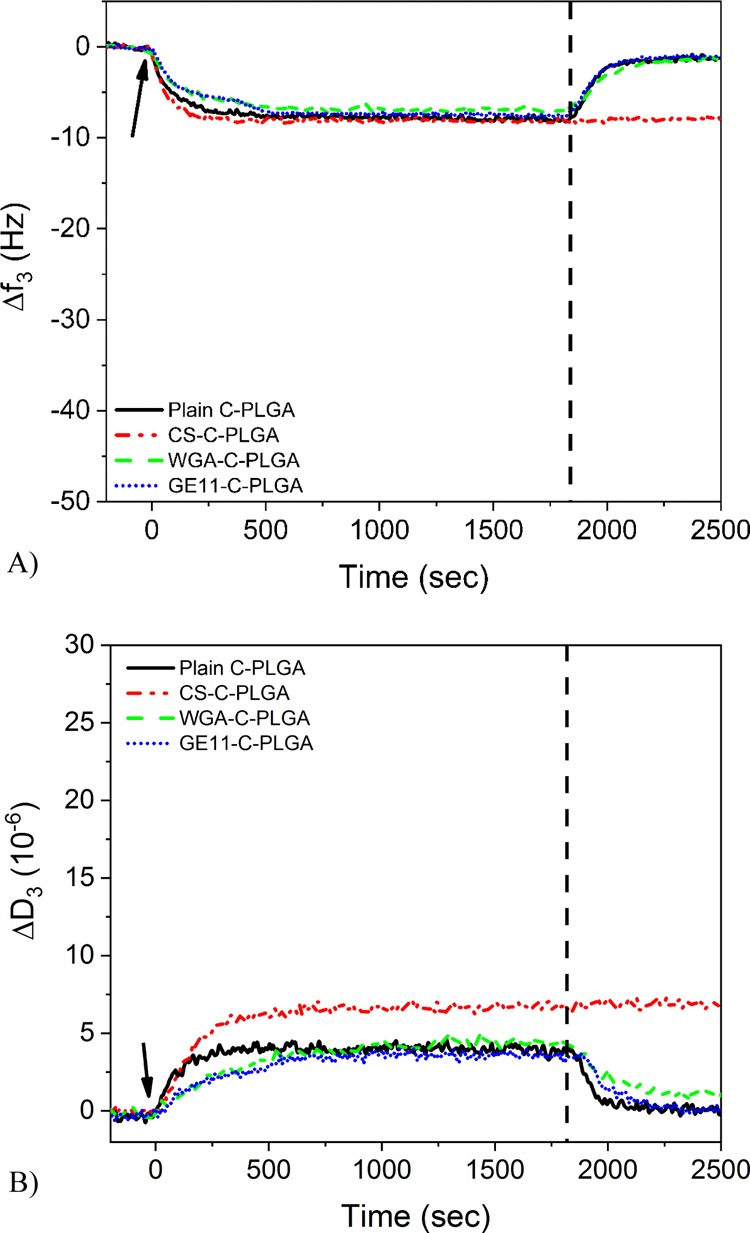
(A) Normalized QCM frequency changes at the 3rd overtone and (B) dissipation changes at the 3rd overtone during interaction of noncoated and coated C-PLGA NPs with a DOPC/DOPS (75:25 mol %) SLB. Arrows indicate time point for injection of different PLGA NPs and the vertical dashed line indicates the time point for flushing with the running buffer.
In the case of the HT-29 cell membrane extract/EggPC SLBs the small frequency (∼3 Hz) and dissipation (∼1 × 10–6) changes at the 3rd overtone during the interaction of the noncoated C-PLGA NPs reported in Figure 4 indicate that NP/SLB interactions occurs according to weak associations. The 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs showed higher frequency changes (40, 40, and 15 Hz, respectively) and dissipation changes (25 × 10–6, 20 × 10–6 and 10 × 10–6, respectively) compared to noncoated C-PLGA NPs (Figure 4). The 1% CS-C-PLGA displayed the highest QCM responses corresponding to over 10 times higher signal responses compared to the ones obtained with noncoated C-PLGA NPs. This strong adsorption of 1% CS-C-PLGA NPs on the HT-29 cell membrane extract/EggPC SLBs can be ascribed to nonspecific electrostatic interactions between the negatively charged SLBs and the positively charged NPs. Interestingly, the QCM responses obtained with WGA-C-PLGA NPs were similar to those obtained with 1% CS-C-PLGA NPs despite the fact that the former possessed a highly negative surface charge. The GE11-C-PLGA NPs induced lower QCM responses compared to 1% CS-C-PLGA and WGA-C-PLGA NPs, but still significantly higher (over 5 times higher) compared to the noncoated C-PLGA NPs. Because both WGA-C-PLGA and GE11-C-PLGA NPs are negatively charged, the NP adsorption on these SLBs occurs through specific binding. The WGA-C-PLGA NPs can specifically interact with glycoproteins and -lipids on the cell membrane surface, namely, N-acetyl-d-glucosamine and N-acetyl-d-neuraminic acid residues, and GE11-C-PLGA NPs interact with the HT-29 cell receptors. The weaker interaction of GE11-C-PLGA NPs compared to WGA-C-PLGA NPs could be ascribed to a difference in density of the receptors for the two targeting agents on the HT-29 cell membrane extract/EggPC SLB or different binding affinities.
Figure 4.
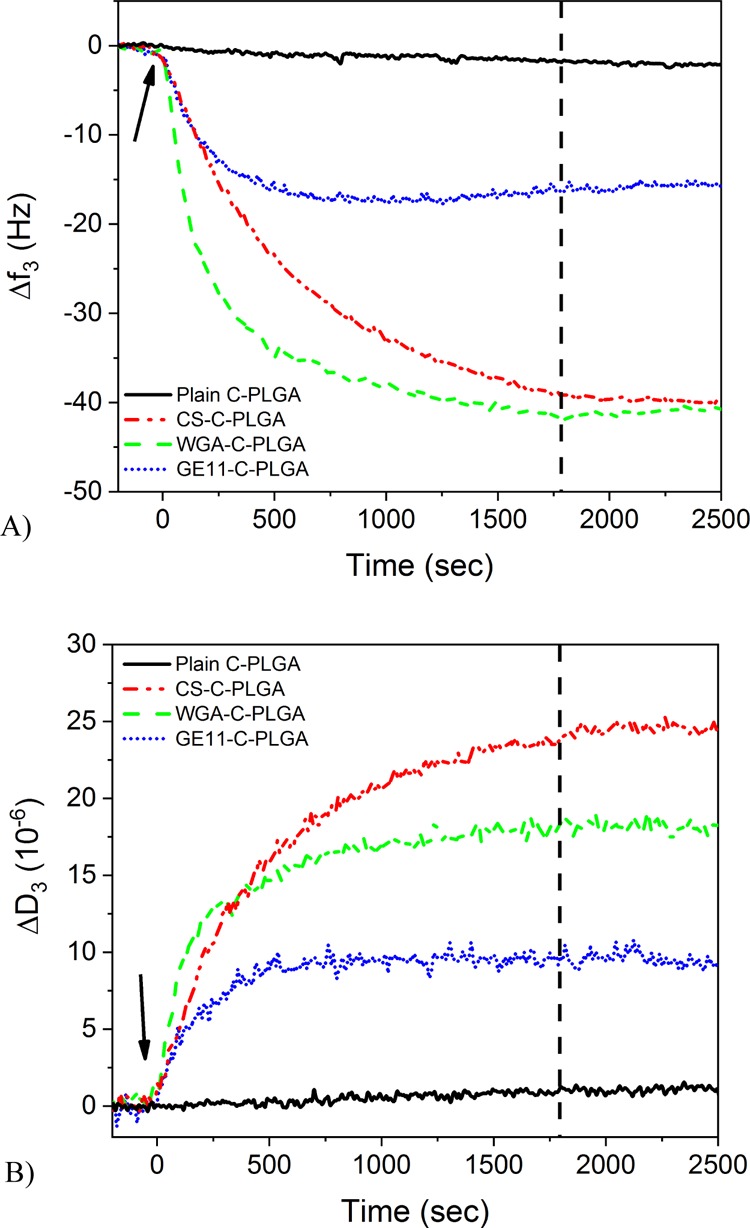
(A) Normalized QCM frequency changes and (B) dissipation changes of the 3rd overtone for different C-PLGA NPs during the interaction with an SLB composed of the HT-29 membrane extract/EggPC (1:1 mass ratio). Arrows indicate time point for injection of the different PLGA NPs and the vertical dashed line indicates the time point for flushing with the running buffer.
3.2.2. SPR Analyses with HT-29 Cell Monolayers
In order to elucidate the mechanism of the NP interaction with living cells, SPR measurements were carried out with confluent colon cancer HT-29 cell monolayers. The immobilization of HT-29 cell monolayers on the SPR sensors was validated before each interaction measurement by measuring the full SPR angular spectrum of the cell monolayer. Figure 5A shows that the main SPR PAP and the shape of the TIR region of the full SPR angular spectrum when the HT-29 cell monolayer is immobilized are substantially shifted to higher angles with respect to the spectrum obtained with a pure gold-coated SPR sensor (from ∼66° to ∼69°). A SPR PAP at about 69° and the smooth shape of the TIR area confirmed the confluence of the HT-29 cell layer.34,35,53 The HT-29 cell monolayer confluence on the SPR sensors was also confirmed by optical microscopy. The microscopy images reported in Figure 5B,C show the cell confluence and cell integrity without changes in the morphology after SPR measurements.
Figure 5.

(A) Full SPR angular spectra of a pure gold-coated SPR sensor (dashed black line) and a confluent HT-29 cell monolayer-covered SPR sensor (solid blue line). The large shift in the SPR PAP and the TIR region to higher angles, and the smoother shape in the TIR region confirms the presence of a confluent cell monolayer. (B) Optical microscope image of a confluent HT-29 cell monolayer on a gold-coated SPR sensor. (C) Optical microscope image of the HT-29 cell monolayer after an SPR interaction measurement demonstrating the preserved cell monolayer integrity.
The SPR signals obtained during the injection of pure curcumin solution reported in Figure S3A show an initial negative shift followed by a leveling out and slow increase of the SPR PAP. The noncoated and coated C-PLGA NPs showed also an initial negative shift with a minimum SPR PAP signal in the range of 3–10 min followed by an increase (>10 min) (Figure S3B–E). Initial negative shifts were observed in the TIR angular position as well (Figure S3). In contrast to the SPR PAP, the TIR angular position remained constant after the initial negative shift. This can be attributed to the fact that the TIR angular position is only sensitive to changes in bulk refractive indexes, while the SPR PAP is sensitive to refractive index changes in the close vicinity of the sensor surface. Thus, the initial negative decrease of the SPR responses can be attributed to optical differences (i.e., different refractive indexes) between the running buffer (DMEM supplemented with 10 mM HEPES, pH 7.4) and the injected sample solutions, which were actually formulations in aqueous medium diluted in the running buffer. The initial decrease in both SPR signals is due to an increase of water content in the samples because water has a lower refractive index than the running buffer. However, the increase in the SPR PAP responses after the initial decrease, which is not seen in the TIR angular position responses, can be attributed to NP-cell surface interactions and NP-cell uptake.
The TIR angular position responses were subtracted from the corresponding SPR PAP responses to evaluate the targeting and cell uptake events of the curcumin formulations (Figure S3). This removed any contribution of optical differences between the curcumin formulations and the running buffer, and provided an SPR response that only reflects the targeting and cell uptake events of the different curcumin formulations.
Figure 6A shows that the interaction kinetics of the NPs obtained by the TIR-subtracted SPR responses measured in the lower position of the flow channel are in good agreement with the results obtained by the QCM combined with the HT-29 cell membrane extract/EggPC SLB analysis. The targeting and cell uptake of the various NPs could then be estimated by integrating the area under the TIR-subtracted SPR responses reported in Figure 6B. The integrated areas show that the HT-29 cell interaction and internalization of 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs are clearly higher compared to those obtained with curcumin in solution and noncoated C-PLGA NPs.
Figure 6.
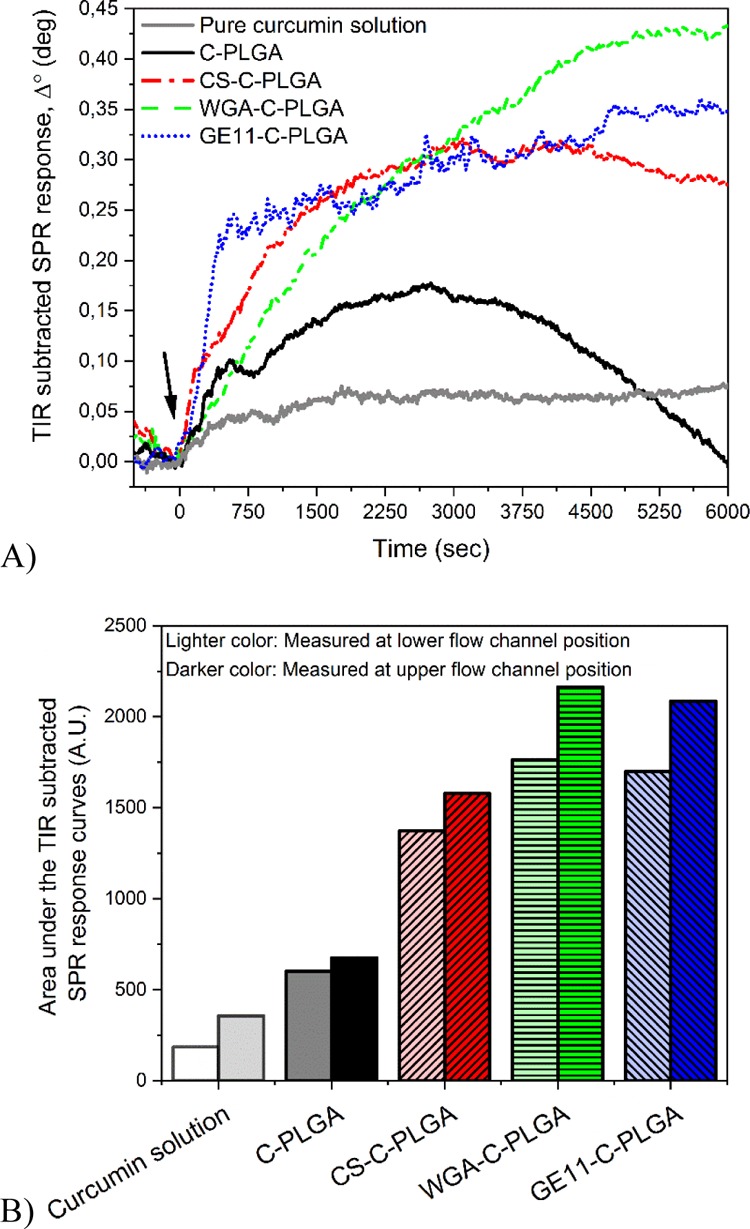
(A) TIR-subtracted SPR response during the interaction of pure curcumin solution and different C-PLGA NPs with HT-29 cells (measured in the lower flow channel position of the cell covered sensor). Arrow indicates time point for injection of the different PLGA NPs. (B) Integrated area under the TIR-subtracted SPR response curve for pure curcumin solution and different C-PLGA NPs measured on two separate positions (lower and upper flow channel positions) on the same sensor with confluent HT-29 cell monolayers. SPR measurements with each formulation were performed on different HT-29 cell-covered SPR sensors.
3.2.3. In Vitro Cell Uptake and Cytotoxicity Studies
In order to corroborate the real-time label-free QCM and SPR results, in vitro cell uptake and cytotoxicity assays were performed using HT-29 human colorectal adenocarcinoma cells. Figure 7A shows the HT-29 cell uptake of curcumin after 2 h incubation. The cell uptake of curcumin in solution was 3.7 ± 0.6 μg/104 cells, while the curcumin uptake from noncoated C-PLGA, 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs was 7.0 ± 0.3, 13.5 ± 1.0, 26.0 ± 1.3, and 27.3 ± 4.9 μg/104 cells, respectively. These results are in good agreement with the cell interaction data obtained by QCM and SPR analysis reported above, and demonstrate that the selective NP interaction achieved with WGA-C-PLGA and GE11-C-PLGA is much more efficient than nonselective cell interaction obtained with 1% CS-C-PLGA. It should be noted that CS is a highly positively charged mucoadhesive material. Consequently, the interactions of 1% CS-C-PLGA NPs with mucin (Figure S2) and the HT-29 membrane extract/EggPC SLB are primarily electrostatic in nature (Figure 4). However, the interaction of 1% CS-C-PLGA NPs with HT-29 cells measured by SPR was lower compared to WGA-C-PLGA and GE11-C-PLGA NPs (Figure 6). Furthermore, the nonselective interaction of 1% CS-C-PLGA NPs with cells may result in a different cell-uptake mechanism compared to the targeted NPs, which consequently could lead to differences in the intracellular delivery of curcumin. Accordingly, the 1% CS-C-PLGA NPs were less efficient in the intracellular delivery compared to WGA-C-PLGA and GE11-C-PLGA NPs.
Figure 7.
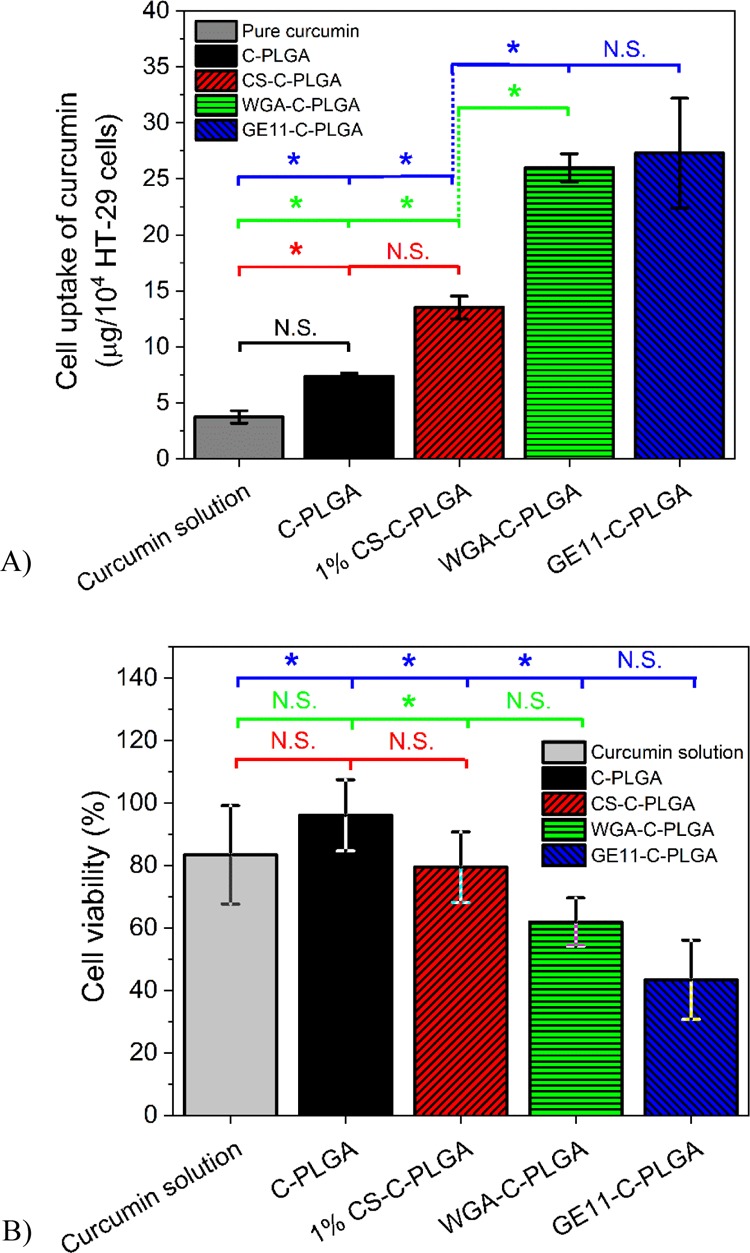
(A) In vitro cell uptake of curcumin and (B) cell viability after 2 h incubation with pure curcumin solution, noncoated C-PLGA, and coated C-PLGA NPs with HT-29 colorectal adenocarcinoma cells (mean ± SD, n = 3). Not significant (N.S.), * significant difference at the 0.05 level.
The cytotoxicity results reported in Figure 7B are in agreement with the cell uptake results shown in Figure 7A and confirm that curcumin is indeed released from the NPs. After 2 h, incubation with curcumin in solution or formulated with noncoated C-PLGA, 1% CS-C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs, the HT-29 cell viability was 83 ± 16, 96 ± 11, 79 ± 11, 62 ± 8, and 43 ± 13%, respectively. It is worth noting that the active targeting obtained with WGA-C-PLGA and GE11-C-PLGA NPs yields the highest cytotoxicity with a significant difference compared to the noncoated C-PLGA NPs.
Importantly, the QCM data (Figure 4) are in fair agreement with the cell uptake and cytotoxicity results obtained by NP incubation with HT-29 cells, demonstrating that this sophisticated technique can be properly exploited to obtain relevant information about the cell-targeting capability of colloidal drug delivery systems. Also, the SPR results (Figure 6) are in very good agreement with the higher curcumin uptake obtained by traditional in vitro cell uptake assay.
3.3. In Vivo Delivery of Noncoated and Targeted C-PLGA NPs
A preliminary in vivo comparative delivery study was carried out to verify that the targeted C-PLGA NPs with colon-selective agents, namely WGA and GE11, could withstand other GI tract conditions before reaching the colon, and to confirm their colon selectivity of these NPs. The study was performed by oral administration to mice of Cy5.5 labeled noncoated C-PLGA, WGA-C-PLGA, and GE11-C-PLGA NPs. The images reported in Figure 8 show that all NPs partially remained in the stomach up to 6 h from administration. However, noncoated C-PLGA and GE11-C-PLGA NPs were cleared out without deposition in the colon while high fluorescence signal corresponding to WGA-C-PLGA NPs was found in the colon, indicating that these NPs adsorb to this GI tract. These results are in fair agreement with the cell affinity of the NPs discussed above. WGA-C-PLGA NPs can in fact recognize the glycoproteins and glycolipids of the normal cells of colon. On the contrary, neither the noncoated C-PLGA nor the GE11-PLGA NPs have a specific target in the colon of healthy mice, and consequently they did not deposit in this GI tract as observed by the images reported in Figure 8.
Figure 8.

In vivo optical images of mice orally administered with (A) noncoated C-PLGA, (B) WGA-C-PLGA and (C) GE11-C-PLGA NPs labeled with Cy5.5. Left panels show one representative mouse per group imaged at different time points (30 min to 24 h) after gavage. An additional mouse from each group was imaged at 2 h post administration and then sacrificed, to expose and visualize the GIT at place for fluorescent NP detection. Thereafter, the entire GIT was removed and imaged alone, followed by imaging of the remaining carcass (right panels).
4. Conclusions
Curcumin-loaded PLGA NPs were successfully prepared and coated with chitosan, WGA and GE11 to yield drug carriers for ameliorating curcumin delivery to HT-29 colon cancer cells. The properties of the coated C-PLGA NPs were comparatively investigated by standard in vitro physicochemical techniques. Stability studies revealed a good colloidal stability of the coated C-PLGA NPs formulations in synthetic gastrointestinal fluids. The NP interactions and cell uptake were studied by means of innovative investigational techniques, namely, QCM combined with SLBs and SPR combined with living cells. The QCM and SPR techniques in combination with biomimetic sensing layers were found to be successful tools to investigate the targeting properties of NPs. These innovative techniques allowed in fact for elucidating a few aspects of NP/cell interactions and uptake providing for the correlation between the nature of the coating and the cell selectivity. The results showed that CS-coated C-PLGA NPs can interact with the cells by nonspecific electrostatic mechanisms, while WGA- and GE11-coated C-PLGA NPs mediate active recognition of specific cell targets.
Acknowledgments
We thank the Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki for providing laboratory facilities.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02086.
Table of kinetic parameters of in vitro release, tables of particle size related to stability, in vitro drug release curves, graph of PM binding, and original SPR response curves (PDF)
Author Present Address
# Drug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, P.O. Box 56, 00014 Helsinki, Finland.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was funded by Center for International Mobility CIMO, Egypt (grant #KM-14-9066) and Academy of Finland (grant #s 137053 and 263861) and partly supported by grants from the Finnish Cultural Foundation to A.K.-H., and the Italian Association for Cancer Research (AIRC, IG-17035 and Special Program Molecular Clinical Oncology 5 per mille ID 10016), and Progetto di Ricerca di Ateneo 2015, University of Padova, to AR.
The authors declare no competing financial interest.
Supplementary Material
References
- Jurenka J. S. Anti-Inflammatory Properties of Curcumin, a Major Constituent of Curcuma Longa: A Review of Preclinical and Clinical Research. Altern Med Rev 2009, 14, 141–153. [PubMed] [Google Scholar]
- Edwards R. L.; Luis P. B.; Varuzza P. V.; Joseph A. I.; Presley S. H.; Chaturvedi R.; Schneider C. The Anti-Inflammatory Activity of Curcumin Is Mediated by Its Oxidative Metabolites. J. Biol. Chem. 2017, 292, 21243–21252. 10.1074/jbc.RA117.000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan A. P.; Watanabe K.; Thandavarayan R. A.; Sari F. R.; Meilei H.; Soetikno V.; Arumugam S.; Giridharan V. V.; Suzuki K.; Kodama M. Curcumin Attenuates Hyperglycaemia-Mediated AMPK Activation and Oxidative Stress in Cerebrum of Streptozotocin-Induced Diabetic Rat. Free Radical Res. 2011, 45, 788–795. 10.3109/10715762.2011.579121. [DOI] [PubMed] [Google Scholar]
- Jha N. S.; Mishra S.; Jha S. K.; Surolia A. Antioxidant Activity and Electrochemical Elucidation of the Enigmatic Redox Behavior of Curcumin and Its Structurally Modified Analogues. Electrochim. Acta 2015, 151, 574–583. 10.1016/j.electacta.2014.11.026. [DOI] [Google Scholar]
- Sarkar A.; De R.; Mukhopadhyay A. K. Curcumin as a Potential Therapeutic Candidate for Helicobacter Pylori Associated Diseases. World J. Gastroenterol. 2016, 22, 2736–2748. 10.3748/wjg.v22.i9.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun D. G.; Lee D. G. Antibacterial Activity of Curcumin via Apoptosis-like Response in Escherichia Coli. Appl. Microbiol. Biotechnol. 2016, 100, 5505–5514. 10.1007/s00253-016-7415-x. [DOI] [PubMed] [Google Scholar]
- Imran M.; Ullah A.; Saeed F.; Nadeem M.; Arshad M. U.; Suleria H. A. R. Cucurmin, Anticancer, & Antitumor Perspectives: A Comprehensive Review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1271–1293. 10.1080/10408398.2016.1252711. [DOI] [PubMed] [Google Scholar]
- Rauf A.; Imran M.; Orhan I. E.; Bawazeer S. Health Perspectives of a Bioactive Compound Curcumin: A Review. Trends Food Sci. Technol. 2018, 74, 33–45. 10.1016/j.tifs.2018.01.016. [DOI] [Google Scholar]
- Shi J.; Kantoff P. W.; Wooster R.; Farokhzad O. C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson J.; Vaughan H. J.; Green J. J. Biodegradable Polymeric Nanoparticles for Therapeutic Cancer Treatments. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 105–127. 10.1146/annurev-chembioeng-060817-084055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foged C.; Brodin B.; Frokjaer S.; Sundblad A. Particle Size and Surface Charge Affect Particle Uptake by Human Dendritic Cells in an in Vitro Model. Int. J. Pharm. 2005, 298, 315–322. 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Vasir J.; Labhasetwar V. Biodegradable Nanoparticles for Cytosolic Delivery of Therapeutics. Adv. Drug Delivery Rev. 2007, 59, 718–728. 10.1016/j.addr.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen S. D.; Caldorera-Moore M.; Peppas N. A. A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. 10.1016/j.ejps.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N.; Tiwari S.; Vaidya B.; Agrawal G. P.; Vyas S. P. Lectin Anchored PLGA Nanoparticles for Oral Mucosal Immunization against Hepatitis B. J. Drug Targeting 2011, 19, 67–78. 10.3109/10611861003733946. [DOI] [PubMed] [Google Scholar]
- Garinot M.; Fiévez V.; Pourcelle V.; Stoffelbach F.; des Rieux A.; Plapied L.; Theate I.; Freichels H.; Jérôme C.; Marchand-Brynaert J.; et al. PEGylated PLGA-Based Nanoparticles Targeting M Cells for Oral Vaccination. J. Controlled Release 2007, 120, 195–204. 10.1016/j.jconrel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Hu F.-Q.; Meng P.; Dai Y.-Q.; Du Y.-Z.; You J.; Wei X.-H.; Yuan H. PEGylated Chitosan-Based Polymer Micelle as an Intracellular Delivery Carrier for Anti-Tumor Targeting Therapy. Eur. J. Pharm. Biopharm. 2008, 70, 749–757. 10.1016/j.ejpb.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Illum L. Chitosan and Its Use as a Pharmaceutical Excipient. Pharm. Res. 1998, 15, 1326–1331. 10.1023/a:1011929016601. [DOI] [PubMed] [Google Scholar]
- Parveen S.; Sahoo S. K. Long Circulating Chitosan/PEG Blended PLGA Nanoparticle for Tumor Drug Delivery. Eur. J. Pharmacol. 2011, 670, 372–383. 10.1016/j.ejphar.2011.09.023. [DOI] [PubMed] [Google Scholar]
- Danhier F.; Ansorena E.; Silva J. M.; Coco R.; Le Breton A.; Préat V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Controlled Release 2012, 161, 505–522. 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- Kompella U. B.; Lee V. H. L. Delivery Systems for Penetration Enhancement of Peptide and Protein Drugs: Design Considerations. Adv. Drug Delivery Rev. 2001, 46, 211–245. 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- Chowdary K. P. R.; Srinivasa Rao Y. Mucoadhesive Microspheres for Controlled Drug Delivery. Biol. Pharm. Bull. 2004, 27, 1717–1724. 10.1248/bpb.27.1717. [DOI] [PubMed] [Google Scholar]
- Naisbett B.; Woodley J. The Potential Use of Tomato Lectin for Oral Drug Delivery: 3. Bioadhesion in Vivo. Int. J. Pharm. 1995, 114, 227–236. 10.1016/0378-5173(94)00242-W. [DOI] [Google Scholar]
- Beckmann H. S. G.; Möller H. M.; Wittmann V. High-Affinity Multivalent Wheat Germ Agglutinin Ligands by One-Pot Click Reaction. Beilstein J. Org. Chem. 2012, 8, 819–826. 10.3762/bjoc.8.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genta I.; Chiesa E.; Colzani B.; Modena T.; Conti B.; Dorati R. GE11 Peptide as an Active Targeting Agent in Antitumor Therapy: A Minireview. Pharmaceutics 2018, 10, 2. 10.3390/pharmaceutics10010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong M.; Kwekkeboom D.; Valkema R.; Krenning E. P. Radiolabelled Peptides for Tumour Therapy: Current Status and Future Directions. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 463–469. 10.1007/s00259-002-1107-8. [DOI] [PubMed] [Google Scholar]
- Langer M.; Beck-Sickinger A. Peptides as Carrier for Tumor Diagnosis and Treatment. Curr. Med. Chem. Anti Cancer Agents 2001, 1, 71–93. 10.2174/1568011013354877. [DOI] [PubMed] [Google Scholar]
- Salomon D. S.; Brandt R.; Ciardiello F.; Normanno N. Epidermal Growth Factor-Related Peptides and Their Receptors in Human Malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Woodburn J. R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol. Ther. 1999, 82, 241–250. 10.1016/S0163-7258(98)00045-X. [DOI] [PubMed] [Google Scholar]
- Keller C. A.; Kasemo B. Surface Specific Kinetics of Lipid Vesicle Adsorption Measured with a Quartz Crystal Microbalance. Biophys. J. 1998, 75, 1397–1402. 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N.-J.; Frank C. W.; Kasemo B.; Höök F. Quartz Crystal Microbalance with Dissipation Monitoring of Supported Lipid Bilayers on Various Substrates. Nat. Protoc. 2010, 5, 1096–1106. 10.1038/nprot.2010.65. [DOI] [PubMed] [Google Scholar]
- Hide M.; Tsutsui T.; Sato H.; Nishimura T.; Morimoto K.; Yamamoto S.; Yoshizato K. Real-Time Analysis of Ligand-Induced Cell Surface and Intracellular Reactions of Living Mast Cells Using a Surface Plasmon Resonance-Based Biosensor. Anal. Biochem. 2002, 302, 28–37. 10.1006/abio.2001.5535. [DOI] [PubMed] [Google Scholar]
- Chabot V.; Cuerrier C. M.; Escher E.; Aimez V.; Grandbois M.; Charette P. G. Biosensing Based on Surface Plasmon Resonance and Living Cells. Biosens. Bioelectron. 2009, 24, 1667–1673. 10.1016/j.bios.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Yashunsky V.; Shimron S.; Lirtsman V.; Weiss A. M.; Melamed-Book N.; Golosovsky M.; Davidov D.; Aroeti B. Real-Time Monitoring of Transferrin-Induced Endocytic Vesicle Formation by Mid-Infrared Surface Plasmon Resonance. Biophys. J. 2009, 97, 1003–1012. 10.1016/j.bpj.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitala T.; Granqvist N.; Hallila S.; Raviña M.; Yliperttula M. Elucidating the Signal Responses of Multi-Parametric Surface Plasmon Resonance Living Cell Sensing: A Comparison between Optical Modeling and Drug-MDCKII Cell Interaction Measurements. PLoS One 2013, 8, e72192 10.1371/journal.pone.0072192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suutari T.; Silen T.; Karaman D. S.; Saari H.; Desai D.; Kerkelä E.; Laitinen S.; Hanzlikova M.; Rosenholm J. M.; Yliperttula M.; et al. Real-Time Label-Free Monitoring of Nanoparticle Cell Uptake. Small 2016, 12, 6289–6300. 10.1002/smll.201601815. [DOI] [PubMed] [Google Scholar]
- Balasso A.; Salmaso S.; Pontisso P.; Rosato A.; Quarta S.; Malfanti A.; Mastrotto F.; Caliceti P. Re-Programming Pullulan for Targeting and Controlled Release of Doxorubicin to the Hepatocellular Carcinoma Cells. Eur. J. Pharm. Sci. 2017, 103, 104–115. 10.1016/j.ejps.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Akl M. A.; Kartal-Hodzic A.; Oksanen T.; Ismael H. R.; Afouna M. M.; Yliperttula M.; Samy A. M.; Viitala T. Factorial Design Formulation Optimization and in Vitro Characterization of Curcumin-Loaded PLGA Nanoparticles for Colon Delivery. J. Drug Delivery Sci. Technol. 2016, 32, 10–20. 10.1016/j.jddst.2016.01.007. [DOI] [Google Scholar]
- Mo Y.; Lim L. Preparation and in Vitro Anticancer Activity of Wheat Germ Agglutinin (WGA)-Conjugated PLGA Nanoparticles Loaded with Paclitaxel and Isopropyl Myristate. J. Controlled Release 2005, 107, 30–42. 10.1016/j.jconrel.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Moulari B.; Béduneau A.; Pellequer Y.; Lamprecht A. Lectin-Decorated Nanoparticles Enhance Binding to the Inflamed Tissue in Experimental Colitis. J. Controlled Release 2014, 188, 9–17. 10.1016/j.jconrel.2014.05.046. [DOI] [PubMed] [Google Scholar]
- Tsai Y.-M.; Chien C.-F.; Lin L.-C.; Tsai T.-H. Curcumin and Its Nano-Formulation: The Kinetics of Tissue Distribution and Blood–Brain Barrier Penetration. Int. J. Pharm. 2011, 416, 331–338. 10.1016/j.ijpharm.2011.06.030. [DOI] [PubMed] [Google Scholar]
- Zanotto-Filho A.; Coradini K.; Braganhol E.; Schröder R.; de Oliveira C. M.; Simões-Pires A.; Battastini A. M. O.; Pohlmann A. R.; Guterres S. S.; Forcelini C. M.; et al. Curcumin-Loaded Lipid-Core Nanocapsules as a Strategy to Improve Pharmacological Efficacy of Curcumin in Glioma Treatment. Eur. J. Pharm. Biopharm. 2013, 83, 156–167. 10.1016/j.ejpb.2012.10.019. [DOI] [PubMed] [Google Scholar]
- US Pharmacopeia . USP 36—NF 31 The United States Pharmacopeia and National Formulary 2013 Main Edition plus Supplements 1 and 2; Deutscher Apotheker Verlag, 2013.
- Yin Y.; Chen D.; Qiao M.; Lu Z.; Hu H. Preparation and Evaluation of Lectin-Conjugated PLGA Nanoparticles for Oral Delivery of Thymopentin. J. Controlled Release 2006, 116, 337–345. 10.1016/j.jconrel.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Master A. M.; Sen Gupta A. EGF Receptor-Targeted Nanocarriers for Enhanced Cancer Treatment. Nanomedicine 2012, 7, 1895–1906. 10.2217/nnm.12.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cu Y.; Saltzman W. M. Controlled Surface Modification with Poly(Ethylene)Glycol Enhances Diffusion of PLGA Nanoparticles in Human Cervical Mucus. Mol. Pharm. 2009, 6, 173–181. 10.1021/mp8001254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.; Telny T.; Ravi T.; Kuppusamy S. Role of Surfactant and PH in Dissolution of Curcumin. Indian J. Pharm. Sci. 2009, 71, 139–142. 10.4103/0250-474X.54280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R.; Shim W.-S.; Cui F.-D.; Cheng G.; Han X.; Jin Q.-R.; Kim D.-D.; Chung S.-J.; Shim C.-K. Enhanced Electrostatic Interaction between Chitosan-Modified PLGA Nanoparticle and Tumor. Int. J. Pharm. 2009, 371, 142–147. 10.1016/j.ijpharm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Soppimath K. S.; Aminabhavi T. M.; Kulkarni A. R.; Rudzinski W. E. Biodegradable Polymeric Nanoparticles as Drug Delivery Devices. J. Controlled Release 2001, 70, 1–20. 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- Niwa T.; Takeuchi H.; Hino T.; Kunou N.; Kawashima Y. Preparations of Biodegradable Nanospheres of Water-Soluble and Insoluble Drugs with D,L-Lactide/Glycolide Copolymer by a Novel Spontaneous Emulsification Solvent Diffusion Method, and the Drug Release Behavior. J. Controlled Release 1993, 25, 89–98. 10.1016/0168-3659(93)90097-O. [DOI] [Google Scholar]
- Wischke C.; Schwendeman S. P. Principles of Encapsulating Hydrophobic Drugs in PLA/PLGA Microparticles. Int. J. Pharm. 2008, 364, 298–327. 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- Makhlof A.; Fujimoto S.; Tozuka Y.; Takeuchi H. Vitro and in Vivo Evaluation of WGA–Carbopol Modified Liposomes as Carriers for Oral Peptide Delivery. Eur. J. Pharm. Biopharm. 2011, 77, 216–224. 10.1016/j.ejpb.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Sheng Y.; He H.; Zou H. Poly(Lactic Acid) Nanoparticles Coated with Combined WGA and Water-Soluble Chitosan for Mucosal Delivery of β-Galactosidase. Drug Delivery 2014, 21, 370–378. 10.3109/10717544.2014.905653. [DOI] [PubMed] [Google Scholar]
- Baghirov H.; Karaman D.; Viitala T.; Duchanoy A.; Lou Y.-R.; Mamaeva V.; Pryazhnikov E.; Khiroug L.; de Lange Davies C.; Sahlgren C.; et al. Feasibility Study of the Permeability and Uptake of Mesoporous Silica Nanoparticles across the Blood-Brain Barrier. PLoS One 2016, 11, e0160705 10.1371/journal.pone.0160705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.