

Abstract
The interaction of cobalt phthalocyanine disodium disulfonate (CoPc) with calf thymus DNA in solutions was investigated by UV/vis spectrophotometry, circular dichroism (CD), and hydrodynamic methods (viscosity and flow birefringence). Two types of CoPc binding to DNA were observed. Fast CoPc interactions with DNA via external binding to phosphates were accompanied by the formation of stack-type phthalocyanine structures on the periphery of the DNA helix. The optical absorption spectra of such CoPc complexes with DNA were analyzed in order to obtain a binding constant K = (4.8 ± 0.4) × 104 M–1. CD spectra show the increasing optical activity of phthalocyanines bonded to DNA. DNA plays the role of a matrix, contributing to an increase in their stacking interactions. The CD spectrum of DNA varies slightly. The second type of cobalt-to-DNA binding manifests itself over a certain time. It can be associated with the reorganization of ligands in the cobalt coordination sphere by introducing DNA atoms. In our experiments, such binding was observed after storage of solutions for approximately 20 h at a temperature of 4 °C. It was shown that the minor groove of DNA remains free in CoPc–DNA complexes. CoPc does not bind with the most important group for metal coordinating to DNA in the major groove (N7 guanine). We completely excluded the intercalation binding model. The planes of phthalocyanines in CoPc–DNA complexes are oriented predominantly normal to the axis of the DNA helix. DNA rigidity (persistent length) does not change. This follows from the data on the measurement of the optical anisotropy and intrinsic viscosity of DNA in complexes.
Introduction
Phthalocyanines and their derivatives have attracted attention as promising materials for molecular electronics and photonics and in biomedical applications.1−4 Their interactions with proteins, DNA, and RNA have been studied intensively.5−8 Phthalocyanine metal complexes are used as fluorescent labels and effective photosensitizers in photodynamic therapy9−11 because they have an intense absorption in the long-wavelength region, where the transparency of biological tissues is high enough. The high sensitivity of the complexes to pH variations (tumor cells develop in a medium with a low pH) and the ability to generate singlet oxygen when they are being excited under irradiation can be effectively used in the treatment of cancer.12 Phthalocyanines are prone to aggregation in aqueous solutions, which reduces their photodynamic activity and affinity to biopolymers and makes it difficult for them to penetrate into the cell.13,14 The attachment of carboxy, phosphate, sulfo, or ammonium groups reduces their aggregation. Phthalocyanines can form coordination bonds with metals, and the nature of the metal largely determines the physicochemical properties of such compounds. Diamagnetic metals (zinc, silicon, and aluminum) promote the formation of luminescent complexes, and transition metals (cobalt, iron, and manganese) provide catalytic properties.
The flat heterocyclic geometry of phthalocyanines allows them to intercalate between the DNA nitrogenous bases. This ability is usually regarded as a sign of antitumor activity of different compounds.5 They also have low toxicity compared with other coordination metal compounds. In contrast to cationic phthalocyanine complexes,7,15 the interaction of anionic complexes with biological objects has been studied significantly less. In this research, DNA interaction with cobalt disulfophthalocyanine was evaluated. A model of complexes is proposed, the binding constant was evaluated, and the conformational changes of DNA in complexes with cobalt compounds were analyzed.
Results and Discussion
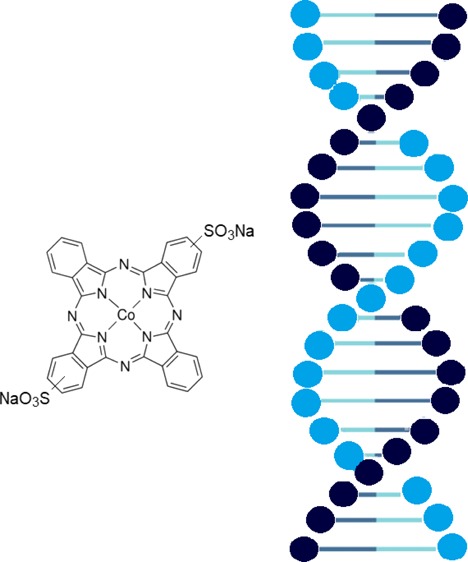
The absorption of CoPc (the structure of CoPc is shown in Figure 1) in a water solution partly intersects with the DNA absorption band (Figure 2a).
Figure 1.

Structure of [CoPc(NaSO3)2].
Figure 2.

Absorption spectra (a) of CoPc (1) and DNA (2) in 0.005 M NaCl with Tris-HCl and the dependence of CoPc absorption on C(CoPc) (b) at 663 nm (1) and 345 nm (2).
From this, it follows that we cannot correctly verify changes in the spectral properties of DNA upon binding. We can only analyze the change in CoPc absorption upon binding. Out of DNA absorption (at a wavelength above 300 nm), the CoPc spectrum includes a Soret or B band with a maximum at approximately 340 nm and an intense Q band with a maximum at 663 nm and a weak satellite near 595 nm. Phthalocyanines are prone to the formation of associates (aggregates) in aqueous solutions. For such systems, it is expected that dilution will change the shape of the Q band.16 We did not observe such changes in the spectrum of CoPc. The linear type of the concentration dependence of the optical density of CoPc solutions at 663 and 345 nm was observed (Figure 2b). However, we cannot completely exclude the presence of dimers or other very stable associates even at very low CoPc concentrations.
Preliminary experiments have shown that the absorption spectrum of CoPc varies noticeably in the presence of DNA.17 The result of spectrophotometric titration with different DNA concentrations [DNA] and constant [CoPc] = 2.5 × 10–5 M in 0.005 M NaCl is shown in Figure 3. The absorption spectra of CoPc in complexes with DNA were registered at 1 h after the preparation of the systems and the next day (about 20 h after preparation). Solutions were stored in the dark at a temperature of 4 °C. It was shown that at 1 h after preparation of the systems with different [DNA], the B and Q bands have isosbestic points, indicating one type of CoPc binding to DNA (Figure 3a,b).
Figure 3.

Results of spectrophotometric titration in 0.005 M NaCl: the Soret band (a) and the Q band (b), [CoPc] = 2.5 × 10–5 M, [DNA] is shown in the figure; the binding curves for two CoPc absorption bands at 330 and 663 nm (c) and Wolf–Shimmer plot (d) with the inset (the inset shows the Scatchard isotherm).
The next day experiment shows the same result for the Q band, but the isosbestic point of the B band is not detectable (spectra not shown). This finding indicates a restructuring of the complex over a longer time. Indeed, the second type of binding manifests itself over time, and the equilibrium in the DNA solution for that type of binding occurs substantially more slowly. It is reasonable to assume the formation of a new CoPc–DNA linkage (e.g., via the coordination of cobalt to DNA). In this case, the ligand in the cobalt coordination sphere should be replaced by an incoming DNA group. Other types of binding can also be implemented.
The hypochromism and small blue shift of maximum observed for the Q band with increasing DNA concentration in CoPc solution is accompanied by a hyperchromic effect and a more significant blue shift in the Soret (B) band. The unchanged view of the Q band with the isosbestic point in time indicates the stability of the first type of complex, for which the equilibrium is established quickly.
Let us analyze this type of binding (the isosbestic point is observed in both bands for the spectra registered at 1 h after preparation of the systems, and it remains to be observed for the Q band the next day). This binding affects the electronic structure of phthalocyanines and indicates that they are in direct contact with DNA. The isosbestic points and the limit spectrum (spectrum of the completely bound compound) make it possible to apply the Scatchard procedure to determine the binding constant. The binding curves were constructed using spectral changes recorded at 330 and 663 nm for two CoPc absorption bands (Figure 3c). The deviation of the linear dependence indicates the emergence of cooperative binding of CoPc to DNA at [CoPc] > 10–5 M from the analysis of the Soret band and at [CoPc] > 6 × 10–6 M for the Q band. As we believe, for phthalocyanines associated with DNA phosphates, cooperativity occurs at lower concentrations and to a greater degree (dependence for 663 nm) compared to the binding that affects the B band (the second type of binding appears over time in the Soret band). At low C [CoPc], both binding curves coincide. For this area, the apparent binding constant (Kapp) and the number of binding sites of CoPc per DNA base pair (n) were determined. Figure 3d shows two different ways for the binding constant definition: the method for determining Kapp according to the approach described in the literature18 and the construction of a Scatchard isotherm (see the inset in Figure 3d). The binding isotherm constructed in Scatchard coordinates19 using formula 1 gives the same values of Kapp and n for two bands: Kapp = (4.2 ± 0.4) × 104 M–1 and n = 1.
| 1 |
Here, r is the ratio of the concentration of the bound ligand to the total available binding sites on the DNA and n is the number of binding sites on the DNA (per base pair). [L] is the concentration of free ligands in the solution. The value n = 1 indicates that CoPc has only one binding site per base pair, and there are no excluded binding sites. This result indicates the absence of intercalation. Note that the Kapp value refers specifically to monomeric binding (before the occurrence of a cooperative effect).
Another widely used method for determining Kapp is eq 2(18)
| 2 |
Here, [DNA] is the DNA concentration in moles of base pairs, εa corresponds to the extinction coefficient of the compound in complexes at the selected wavelength within the analyzed band, and εb and εf are the extinction coefficients for free compounds and for compounds fully bonded to DNA, respectively. The plot of the dependence of [DNA]/|εa – εf| value on [DNA] gives the binding constant. Such an approach is valid only for a very small degree of binding. Indeed, eq 2 is a modification of the procedure reported earlier,20 which is valid for r → 0. The equilibrium binding constant Kapp can be determined in this way.21 The analysis of our spectral data with eq 2 for two spectral bands (for the first type of binding) gives the graphs shown in Figure 3d. In this case, Kapp = (4.8 ± 0.4) × 104 M–1 for D at 663 nm and Kapp = (4.0 ± 0.4) × 104 M–1 for D at 327 nm. One can see a good agreement in the estimation of this parameter, carried out in different ways. It should be emphasized that we considered the absorption bands of the compound outside the region of DNA absorption. Thus, UV spectrophotometric titration gives a binding constant value of approximately 104 and the number of binding sites on DNA n = 1, which absolutely excludes the intercalation model.
The second type of binding manifests itself in time and leads to the disappearance of the isosbestic point in the B band (such spectra are not shown). For this type of binding, the correct estimation of binding constant is not possible. There are several ways for CoPc to interact with DNA: coordination of cobalt to DNA with the replacement of the ligand in its coordination sphere, another binding, such as interaction via the hydrogen bonds with atomic groups of DNA, intercalation or major/minor groove binding, and electrostatic interactions (this binding requires a certain orientation of CoPc near DNA). We must remember that DNA has an extremely high negative charge density, which cannot be completely shielded in 0.005 M NaCl. However, we clearly see the binding of CoPc to the DNA molecule. It is possible that the binding provokes a partial replacement of the ligands in the coordination sphere of cobalt, leading to a change in the total charge of the complex ion.
The melting of free DNA and DNA in complex with CoPc in 5 mM NaCl with Tris-HCl was observed. The analysis of hyperchromism in DNA absorption at 260 nm (maximum of the DNA absorption band) with temperature was carried out after subtracting the CoPc absorption. All measurements were carried out 1 day after the preparation of complexes. Figure 4 shows melting curves normalized to the absorption at 25 °C and their first derivatives. It was found that no significant changes were observed in the absorption of free CoPc at 260 nm within the temperature range used (see the inset in Figure 4a). According to the experimental data, the DNA melting temperature increased insignificantly (∼3 °C), which reflects the high stability of the DNA double helix structure in the complexes. In addition, the slightly increased optical density at T > 80 °C for complexes may indicate the existence of CoPc associates on DNA (absorbing in the same area) that break down when heated. Such small shifts in DNA melting temperature indicate the absence of intercalation and provide the evidence of other modes of binding.
Figure 4.

Melting curves of DNA (dependences of DNA absorption at 260 nm on temperature) (a) and the first derivatives of melting curves (b) for DNA in complexes with CoPc (1) and for free DNA (2) in 5 mM NaCl with Tris-HCl. [DNA] = 7.6 × 10–5 M, [CoPc] = 5 × 10–5 M.
A widely used method for determining the localization of ligands on DNA in complexes is to study the competition for the binding site on DNA between the compound under investigation and well-known compounds. It is known that the 4′,6-diamidino-2-phenylindole (DAPI) dye interacts with DNA in a minor groove with preferential binding to A–T pairs. This binding with the binding constant K = (3.0 ± 0.5) × 106 M–1 and with the number of binding sites z = 0.02 (about one DAPI molecule per 50 DNA base pairs, z = [DAPI]/[DNA]) causes DAPI luminescence with a maximum at 460 nm (λex = 340 nm). An increase in dye concentration causes its additional binding to DNA phosphates with a maximum of DAPI luminescence at 540 nm (λex = 420 nm) with a low quantum yield (DAPI luminescence in the DNA groove has a much higher quantum yield).
In our research, three systems were prepared. First, three pairs of solutions in 0.005 M NaCl with Tris-HCl were mixed together (DNA and DAPI, DNA and CoPc, DAPI and CoPc). Second, after 2 h of incubation at room temperature (21 °C), the third solution (CoPc, DAPI, or DNA in 0.005 M NaCl with —Tris-HCl) was added. In this way, three systems with the same DNA, DAPI, and CoPc concentrations were prepared: (DNA + DAPI) + CoPc, (DNA + CoPc) + DAPI, and (DAPI + CoPc) + DNA. We also used (DNA + DAPI) and DAPI solutions with the same concentrations for comparison. All spectra were registered the next day after the addition of the last component (solutions were placed in the dark at a temperature of 4 °C). Because DAPI has two modes of binding to DNA with luminescence at different wavelengths, as noted above, we can test both the interaction of CoPc with DNA in the minor groove and the binding of CoPc to phosphates. It is necessary to emphasize that DAPI binding with phosphates is observed only at a high DAPI concentration in DNA solution (the emission has a maximal intensity at z = 0.3 and λex = 420 nm).22 Because of its much higher quantum yield, DAPI luminescence in the minor groove can also be observed at z = 0.3 at the excitation wavelength of 340 nm. In our experiment, we used λex = 380 nm because of the strong absorption of CoPc at 340 nm (see Figure 2). The luminescence of DAPI can be quenched when there is another compound that can displace the bound dye. We checked the competition between CoPc and DAPI for binding sites on DNA at z = 0.02 (DAPI luminescence is typical for groove binding) and z = 0.3 (we can observe DAPI luminescence for groove and phosphate modes).
In general, the presence of CoPc in a solution does not prevent DAPI location in a minor groove of DNA regardless of the order of CoPc addition to the solution (see shortwave peaks 1–4 in Figure 5a,b). The only peak is noticeably different from the other spectra (Figure 5a, spectrum 3), but over time, this spectrum becomes indistinguishable from the others. This spectrum is related to a DAPI solution with CoPc just after the addition of DNA. We can assume that CoPc can interact with DAPI and prevents further binding of DAPI to DNA with the location in a minor groove. Nevertheless, later, such complexes are fully formed (Figure 5b, spectrum 3).
Figure 5.

Influence of CoPc on DAPI luminescence in complexes with DNA immediately after the preparation of solutions (a) and after 2 days (b). (1–4) z = 0.02, λex = 380 nm; (5–8)—z = 0.3, λex = 420 nm; (1, 5)—(DAPI + DNA) without CoPc; (2, 6)—(DNA + CoPc) + DAPI for the addition of DAPI to CoPc–DNA complexes; (4, 8)—(DNA + DAPI) + CoPc for the addition of CoPc to DNA–DAPI complexes; (3, 7)—(DAPI + CoPc) + DNA for the addition of DNA to DAPI solution with CoPc. [DNA] = 1.5 × 10–5 M, [CoPc] = 5 × 10–6 M, [DAPI] = 3 × 10−7 M (for z = 0.02), [DAPI] = 5 × 10−6 M (for z = 0.3), pH 7. Note that the real intensity for the spectra (5–8) is 6 times greater than that shown in the figure.
The external binding of DAPI to phosphate groups can be characterized with long-wave peaks as shown in Figure 5. It is clear that the presence of CoPc in solutions partially prevents the binding of DAPI to DNA phosphates. Naturally, when CoPc is added to an already formed DAPI–DNA complex, such an effect is manifested only with time (compare spectrum 8 in Figure 5a,b).
This result supports the existence of such type of CoPc binding to DNA that requires a significantly long time. Overall, this experiment confirmed an interaction between CoPc and negatively charged DNA, which causes a change in the absorption spectrum of CoPc and weakly increases the DNA melting temperature. We emphasize that the minor groove of DNA remains accessible for DAPI after the formation of CoPc–DNA complexes.
To clarify the possibility of CoPc location in the major groove during binding, we used compounds that form complexes with N7 guanine (N7G): cis-DDP (cisplatin) and Mn2+ ions (Figure 6).
Figure 6.

Adsorption spectra: Soret (a,c), Q (b,d) bands and DNA absorption region (e) for CoPc–DNA complexes formed after and before the binding of compounds located in the DNA major groove: Mn2+ (a,b) and cis-DDP (c–e). Spectrum 8 in e demonstrates the sum of spectrum 1 and spectrum 6. The order of the addition of components is shown near the lines.
The binding of positively charged ligands to N7G causes specific changes in the DNA absorption: slight hypochromism that at high concentrations turns into hyperchromism at 260 nm and an emergence of a shoulder at λ > 270 nm.23−25 The experimental data indicate competition for the binding sites on DNA between CoPc and Mn2+ (Figure 6a,b).
Note that manganese ions are associated not only with DNA bases but also with phosphates. Therefore, this competition can manifest itself precisely because of the CoPc binding to DNA phosphates. Indeed, coordination of platinum to N7G does not prevent the binding of CoPc to DNA (Figure 6c,d). The coincidence of spectra 2, 3, and 4 in Figure 6e for different orders of addition of components in the region of DNA absorption suggests that CoPc does not bind to N7G in the major groove of DNA. N7 guanine is the most important group for metal coordinating to DNA. The spectra also indicate the absence of a CoPc interaction with Mn2+ and cis-DDP in our experiment.
It was also shown that CoPc in complexes with DNA has only a minor influence on the DNA CD spectrum (Figure 7a). A small decrease in the amplitude of the bands is observed, for example, for DNA complexes with positive ions that bind to phosphate groups.26,27 Thus, it is unlikely that cobalt binds with nitrogen bases.
Figure 7.

CD spectra of DNA in solutions with different CoPc concentrations (a) and CD spectra of CoPc without and with DNA in 0.005 M NaCl (b). Concentrations of components are shown near the curves.
Phthalocyanine compounds have optical activity.28 Indeed, CoPc has its own CD spectrum in the Q band region with zero near 663 nm at the maximum of CoPc absorption (Figure 7b). The addition of DNA causes more intensive CD spectra and a blue shift (zero shifts of the wavelength corresponding to the maximum of CoPc absorption in complexes with DNA).
The results of hydrodynamic experiments provide information on the state of the tertiary structure of DNA in a solution. Figure 8 shows the intrinsic viscosity determination result with and without CoPc in a solution (the measurements were carried out 1 day after the preparation of the systems). Nonetheless, only the intrinsic viscosity of DNA can clear up the question of the possibility of intercalation of the phthalocyanine ligands. Indeed, the relative, specific, and reduced viscosities of DNA solutions depend on the DNA concentration, and because of intermolecular interactions, they cannot precisely indicate the change in the size of the macromolecule. This is especially true for solutions containing compounds that are prone to association. Certainly, the intrinsic viscosity of DNA gives correct information about the main macromolecular parameters (coil volume, hydrodynamic length, and chain rigidity according to eq 4). The determination of the intrinsic viscosity of DNA (eq 3) requires the correct dilution with preservation of the equilibrium between bound and free CoPc in DNA solution (in other words, the volume of the macromolecule should not change during dilution). For that reason, we used a binding constant, as described in the literature.29 It was shown that the presence of CoPc in the concentrations used (measurements were carried out for r = 0.36 when filling the binding sites on DNA) does not affect the intrinsic viscosity (Figure 8), that is, the DNA molecular parameters, which excludes the intercalation mode of binding for CoPc–DNA complexes.
Figure 8.

Results of viscometric experiments: dependence of the reduced viscosity of DNA solutions in 0.005 M NaCl on DNA concentration at r = 0 (1) and r = 0.36 (2) for determining the DNA intrinsic viscosity by extrapolation of dependences to C([DNA)] = 0.
The combination of spectral and viscometric experimental data shows that intercalation is not observed when CoPc interacts with DNA. Fluorescence of DAPI in solutions with CoPc–DNA complexes shows that the minor groove of DNA is free and available for the dye binding. The major groove is also not occupied by CoPc. External binding with phosphates can be accompanied by the stacking of phthalocyanine ligands. This assumption was confirmed when measuring the optical anisotropy of DNA in the complexes. Figure 9 shows the dependence of the relative change in the optical anisotropy of the DNA statistical segment (Kuhn segment) on the concentration of CoPc in solutions (measured 1 day after preparation of the systems). This parameter was determined in an experiment according to eqs 5 and 6.
Figure 9.

Dependence of relative changes in optical anisotropy of the DNA statistical segment on [CoPc] in 0.005 M NaCl. C (DNA) = 0.007%.
It is known that the optical anisotropy of a statistical segment (α1 – α2) is determined as a product of the number of monomeric units in a Kuhn segment S (DNA base pairs) and the average optical anisotropy of a monomeric unit (base pair for DNA) Δβ. The first value (S) characterizes DNA rigidity (like DNA persistent length in a wormlike model), while the second parameter Δβ depends on the orientation of the base pair regarding the DNA helix. For DNA in B-form, Δβ has a maximum value. Thus, any change in the secondary structure of DNA can only lead to a decrease in the Δβ value. Because the viscometric data show that DNA rigidity does not change upon binding, it remains to be assumed that the experimentally detectable increase in optical anisotropy may be caused by the contribution of bound phthalocyanine ligands to the measured optical anisotropy of the DNA statistical segment. For that, a certain mutual orientation of phthalocyanines (in a stacked state) and their general orientation mainly parallel to the plane of DNA bases are necessary. These data confirm the assumption of the external binding of CoPc to DNA with the formation of stacked phthalocyanines outside the helix.
Conclusions
Our research shows that in 0.005 M NaCl, CoPc interacts with DNA. CoPc binding causes a blue shift with a hypochromic effect in the Q band and hyperchromism with a similar shortwave shift in the B band. These data indicate that phthalocyanines have direct contact with DNA. Two types of CoPc binding to DNA achieving fast and slow equilibrium were observed. First, CoPc interacts with DNA via external binding with DNA phosphates with the binding constant K= (4.8 ± 0.4) × 104 M–1. In such complexes, phthalocyanine associates form stack-type structures around the periphery of the DNA helix. Second, cobalt-to-DNA binding manifests itself over time (the isosbestic point in the Soret band disappears and the DAPI luminescence also indicates the slow binding of CoPc to DNA). The binding can be accompanied by the reorganization of ligands in the cobalt coordination sphere over a certain time via the entry of DNA atoms. CD and UV–vis spectra as well as hydrodynamic methods show that the second type of binding does not affect the first complexes (CoPc binding with DNA phosphates). The minor DNA groove remains available for the penetration of other molecules after the formation of CoPc–DNA complexes. All data clearly indicate the absence of an intercalation. The viscometry and optical anisotropy of DNA in the complexes point to the invariance of DNA rigidity (DNA persistent length) when binding. The flow birefringence method also indicates that the orientation of phthalocyanine ligands in complexes is predominantly parallel to the plane of the base pairs.
Materials and Methods
Sodium salt of high-molecular-weight calf thymus DNA (Sigma-Aldrich) with molecular mass M = 6 × 106 (determined from DNA intrinsic viscosity in 0.15 M NaCl) was used.30 DNA was dissolved in distilled water (10 mg in 100 mL). After storage for 5 days at a temperature of 4 °C and adding an NaCl solution of a given concentration, the resulting stock solution was filtered. DNA concentration in the stock solution was determined from the difference in absorption ΔD at 270 and 290 nm for hydrolyzed DNA in 0.005 M NaCl after 15 min boiling with 6% HClO4 at 100 °C.31 C(DNA) = [DNA] = 50 × ΔD (in g/dL or %). This method of determining the DNA concentration makes it possible to control the DNA double-stranded structure during the experiment by using the value of the molar extinction coefficient: E260(P) = 31.1 × D260/[DNA]. In this experiment, we have used DNA with an E260(P) value less than 6800 M–1·cm–1. Cobalt (II) phthalocyanine disodium disulfonate [CoPc(NaSO3)2] (we will denote it further as CoPc) was purchased from the Dmitrievsky chemical plant (Russia). A stock solution of CoPc in purified water type I also contained Tris-HCl buffer (pH 7) and 5 mM NaCl. To prevent CoPc aggregation in solution, we used a concentration not exceeding 10–4 M. CoPc complexes with DNA were prepared by mixing equal volumes of their solutions in 0.005 M NaCl with Tris-HCl buffer.
Absorption spectra were registered with Specord 200 Plus (Analytic Jena). The luminescence of DAPI (4′,6-diamidino-2-phenylindole) as a competing dye was registered with a Hitachi 850 spectrofluorimeter (Japan). Luminescence excitation and emission spectra were corrected for the spectral instrument sensitivity. Circular dichroism (CD) spectra were obtained with an autodichrograph Mark V (Jobin Yvon, France). Measurements were performed in a quartz cell with an optic path length of 1.0 cm. Spectra were recorded three times with subsequent averaging. Initial processing of the obtained spectra was performed using the software supplied with the instrument. CD spectra are shown as the difference in absorbance between left- and right-polarized beams ΔA = AL – AR or in the corresponding extinction coefficients Δε = εL – εR. Smoothing of the spectra was performed by the Savitsky–Golay method32 with a 15-point smoothing frame. For DNA melting experiments, two systems were prepared: a DNA solution in 0.005 M NaCl with and without CoPc. Tris-HCl buffer was used. Melting curves were obtained with Specord 200 Plus.
To control the tertiary structure of the DNA molecule during the formation of complexes with CoPc, we used hydrodynamic methods. To describe the behavior of high molecular DNA in a solution, one can use either the model of a freely joined chain or the model of the wormlike (persistence) chain. The universal conformational parameters of the macromolecules for these models are the mean-square distance between the ends of the chain (h̅2)1/2, the contour (hydrodynamic) length L, and the molecular mass M. The length of the statistical segment (Kuhn segment) A and the persistent length p for the wormlike chain describe the bending (equilibrium) rigidity of the macromolecule.
In our experiment, a relative viscosity ηr of DNA solutions in 0.005 M NaCl with and without CoPc was determined at 21 °C with a low gradient Zimm–Crothers-type rotation viscometer.33 The velocity gradients did not exceed 1 s–1. The intrinsic viscosity of DNA was obtained because of the extrapolation of the dependence of reduced viscosity (ηr – 1)/C on DNA concentration to C = 0
| 3 |
Here, C is the concentration of the polymer (in our research, we denote DNA concentration as [DNA]). For DNA in complexes with CoPc, the dilution was carried out taking into account the binding constant value.29 The main DNA parameters can be determined from the equation
| 4 |
where Φ is the Flory parameter, M is the DNA molecular mass, (h̅2)1/2 and (h̅02)1/2 are the mean-square distances between the ends of the DNA chain in the real and in the ideal solutions, respectively, α is the coefficient of linear swelling, α = (h̅2)1/2/(h̅02)1/2, L is the hydrodynamic length of the polymer chain, and A is the length of the Kuhn segment (chain rigidity parameter). For high molecular DNA samples, A = 2p.
The optical anisotropy of DNA was determined with the flow birefringence method. The difference in the polarizabilities of DNA statistical segments (α1 – α2) along (α1) and normal (α2) to the DNA helix axis was determined from the equation (for a negligible form effect)
| 5 |
Here, Δn is the measured birefringence value of DNA solution in the field of the flow gradient g, η and η0 are the viscosities of the solution and solvent, ns is the refractive index of the solvent, k is the Boltzmann constant, and T is the absolute temperature. The (α1 – α2) value is proportional to the DNA base pair optical anisotropy Δβ (difference in its polarizabilities along (β1) and normal (β2) to the DNA helix axis) and to the number of base pairs S in the DNA statistical segment
| 6 |
The S value also determines the DNA rigidity: S = A/l (l is the length of the base pair along the DNA helix). All measurements were carried out at 21 °C.
Acknowledgments
This research was partially carried out with the equipment at the Resource Centers of Saint Petersburg State University (SPbU). This work was funded by SPbU (project 11.19.198.2018) and RFBR (no. 18-08-01500).
The authors declare no competing financial interest.
References
- Lim C.-K.; Shin J.; Lee Y.-D.; Kim J.; Oh K. S.; Yuk S. H.; Jeong S. Y.; Kwon I. C.; Kim S. Phthalocyanine-aggregated polymeric nanoparticles as tumor-homing near-Infrared absorbers for photothermal therapy of cancer. Theranostics 2012, 2, 871–879. 10.7150/thno.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaku H.; Fujimoto T.; Murashima T.; Miyoshi D.; Sugimoto N. Phthalocyanines: a new class of G-quadruplex-ligands with many potential applications. Chem. Commun. 2012, 48, 6203–6216. 10.1039/c2cc31037f. [DOI] [PubMed] [Google Scholar]
- Sorokin A. B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. 10.1021/cr4000072. [DOI] [PubMed] [Google Scholar]
- Lukyanets E. A. Phthalocyanines as Photosensitizers in the Photodynamic Therapy of Cancer. J. Porphyr. Phthalocyanines 1999, 3, 424–432. . [DOI] [Google Scholar]
- Barut B.; Demirbaş Ü.; Şenocak A.; Özel A.; Kantekin H. Water soluble axially morpholine disubstituted silicon phthalocyanines: Synthesis, characterisation, DNA/BSA binding, DNA photocleavage properties. Synth. Met. 2017, 229, 22–32. 10.1016/j.synthmet.2017.05.006. [DOI] [Google Scholar]
- Georgiades S. N.; Abd Karim N. H.; Suntharalingam K.; Vilar R. Interaction of Metal Complexes with G-quadruplex DNA. Angew. Chem., Int. Ed. 2010, 49, 4020–4034. 10.1002/anie.200906363. [DOI] [PubMed] [Google Scholar]
- Kuznetsova A. A.; Chernonosov A. A.; Kuznetsov N.; Koval V. V.; Knorre D. G.; Fedorova O. S. Kinetic study of DNA modification by phthalocyanine derivative of the oligonucleotide. Bioinorg. Chem. Appl. 2006, 1, 23560. 10.1155/bca/2006/23560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bağda E.; Yabaş E.; Bağda E. Analytical approaches for clarification of DNA-double decker phthalocyanine binding mechanism: As an alternative anticancer chemotherapeutic. Spectrochim. Acta, Part A 2017, 172, 199–204. 10.1016/j.saa.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Li X.; Zheng B.-D.; Peng X.-H.; Li S.-Z.; Ying J.-W.; Zhao Y.; Huang J.-D.; Yoon J. Phthalocyanines as medicinal photosensitizers: Developments in the last five years. Coord. Chem. Rev. 2019, 379, 147–160. 10.1016/j.ccr.2017.08.003. [DOI] [Google Scholar]
- Hodgkinson N.; Kruger C. A.; Mokwena M.; Abrahamse H. Cervical cancer cells (HeLa) response to photodynamic therapy using a zinc phthalocyanine photosensitizer. J. Photochem. Photobiol., B 2017, 177, 32–38. 10.1016/j.jphotobiol.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Wong R. C. H.; Lo P.-C.; Ng D. K. P. Stimuli responsive phthalocyanine - based probes and photosensitizers. Coord. Chem. Rev. 2019, 379, 30–46. 10.1016/j.ccr.2017.10.006. [DOI] [Google Scholar]
- Wang A.; Chen X.; Zhang L.; Zhang G.; Zhou L.; Lu S.; Zhou J.; Wei S. Effects of pH on aggregation and photodynamic activities of cationic zinc phthalocyanines substituted with amides. J. Photochem. Photobiol., A 2014, 288, 1–12. 10.1016/j.jphotochem.2014.05.003. [DOI] [Google Scholar]
- Jeong J.; Kumar R. S.; Mergu N.; Son Y.-A. Photophysical, electrochemical, thermal and aggregation properties of new metal phthalocyanines. J. Mol. Struct. 2017, 1147, 469–479. 10.1016/j.molstruc.2017.06.125. [DOI] [Google Scholar]
- Lin D.; Wang Y.; Zhang Q.; Zhou J.; Zhou L.; Wei S. The substituted amino group type dependent sensitivity enhancing of cationic phthalocyanine derivatives for photodynamic activity. J. Photochem. Photobiol., A 2016, 315, 107–120. 10.1016/j.jphotochem.2015.09.017. [DOI] [Google Scholar]
- Çakir V.; Çakir D.; Piskin M.; Durmus M.; Biyiklioglu Z. New peripherally and non-peripherally tetra-substituted water soluble zinc phthalocyanines: Synthesis, photophysics and photochemistry. J Organomet Chem 2015, 783, 120–129. 10.1016/j.jorganchem.2015.02.021. [DOI] [Google Scholar]
- Nyokong T.; Isago H. The renaissance in optical spectroscopy of phthalocyanines and other tetraazaporphyrins. J. Porphyrins Phthalocyanines 2004, 8, 1083–1090. 10.1142/s1088424604000453. [DOI] [Google Scholar]
- Tikhomirov R.; Demidov V.; Kasyanenko N. Study of DNA interaction with cobalt disulfophthalocyanine. J. Phys.: Conf. Ser. 2018, 1038, 012026. 10.1088/1742-6596/1038/1/012026. [DOI] [Google Scholar]
- Wolfe A.; Shimer G. H. Jr.; Meehan T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. 10.1021/bi00394a013. [DOI] [PubMed] [Google Scholar]
- Scatchard G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. 10.1111/j.1749-6632.1949.tb27297.x. [DOI] [Google Scholar]
- Meehan T.; Gamper H.; Becker J. F. Characterization of reversible, physical binding of benzo[a]pyrene derivatives to DNA. J. Biol. Chem. 1982, 257, 10479–10485. [PubMed] [Google Scholar]
- Schmechel D. E. V.; Crothers D. M. Kinetic and hydrodynamic studies of the complex of proflavine with poly A-poly U. Biopolymers 1971, 10, 465–480. 10.1002/bip.360100304. [DOI] [PubMed] [Google Scholar]
- Kasyanenko N.; Bakulev V.; Perevyazko I.; Nekrasova T.; Nazarova O.; Slita A.; Zolotova Y.; Panarin E. Model system for multifunctional delivery nanoplatforms based on DNA-polymer complexes containing silver nanoparticles and fluorescent dye. J. Biotechnol. 2016, 236, 78–87. 10.1016/j.jbiotec.2016.08.010. [DOI] [PubMed] [Google Scholar]
- Kas’ianenko N. A.; Valueva S. V.; Smorygo N. A.; D’iachenko S. A.; Frisman E. V. Study of the interaction of a DNA molecule with coordination compounds of divalent platinum. I. Effect of cis-diaminodichloroplatinum on molecular parameters of DNA in solution. Mol. Biol. (Moscow) 1995, 29, 345–353. [PubMed] [Google Scholar]
- Kasyanenko N. A.; Prokhorova S. A.; Haya Enriquez E. F.; Sudakova S. S.; Frisman E. V.; Dyachenko S. A.; Smorygo N. A.; Ivin B. A. Interaction of protonated DNA with trans-dichlorodiammineplatinum(II). Colloids Surf., A 1999, 148, 121–128. 10.1016/s0927-7757(98)00599-8. [DOI] [Google Scholar]
- Kas’yanenko N. A.; Bartoshevich S. F.; Frisman E. V. Investigation on the influence of ph on DNA conformation. Mol. Biol. (Moscow) 1985, 19, 1386–1393. [PubMed] [Google Scholar]
- Kasyanenko N. A.; Zanina A. V.; Nazarova O. V.; Panarin E. F. DNA Interaction with Complex Ions in Solution. Langmuir 1999, 15, 7912–7917. 10.1021/la980980p. [DOI] [Google Scholar]
- Kasyanenko N.; Arikainen N.; Frisman E. Investigation of DNA complexes with iron ions in solution. Biophys. Chem. 1998, 70, 93–100. 10.1016/s0301-4622(97)00111-7. [DOI] [PubMed] [Google Scholar]
- Lu H.; Kobayashi N. Optically Active Porphyrin and Phthalocyanine Systems. Chem. Rev. 2016, 116, 6184–6261. 10.1021/acs.chemrev.5b00588. [DOI] [PubMed] [Google Scholar]
- Krivtsova M. A.; Moroshkina E. B.; Khamman Kh.; Glibin E. N.; Frisman E. V. Interaction of DNA with low molecular weight ligands of various structures 1 Complexes of DNA with actinomycin and its simple analogs. Mol. Biol. 1981, 15, 613–621. [PubMed] [Google Scholar]
- Eigner J.; Doty P. The native, denatured and renatured states of deoxyribonucleic acid. J. Mol. Biol. 1965, 12, 549–580. 10.1016/s0022-2836(65)80312-6. [DOI] [PubMed] [Google Scholar]
- Spirin A. S. Spectrophotometric Determination of Total Nucleic Acids. Biokhimiya (Moscow) 1958, 23, 656–662. [PubMed] [Google Scholar]
- Savitzky A.; Golay M. J. E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. 10.1021/ac60214a047. [DOI] [Google Scholar]
- Frisman E. V.; Schagina L. V.; Vorobiev V. I. A glass rotation viscometer. Biorheoogy 1965, 2, 189–194. [PubMed] [Google Scholar]