

Abstract

Proline- and arginine-rich peptide PR11 is an allosteric inhibitor of 20S proteasome. We modified its sequence inter alia by introducing HbYX, RYX, or RHbX C-terminal extensions (Hb, hydrophobic moiety; R, arginine; Y, tyrosine; X, any residue). Consequently, we were able to improve inhibitory potency or to convert inhibitors into strong activators: the former with an aromatic penultimate Hb residue and the latter with the HbYX motif. The PR peptide activator stimulated 20S proteasome in vitro to efficiently degrade protein substrates, such as α-synuclein and enolase, but also activated proteasome in cultured fibroblasts. The positive and negative PR modulators differently influenced the proteasome conformational dynamics and affected opening of the substrate entry pore. The resolved crystal structure showed PR inhibitor bound far from the active sites, at the proteasome outer face, in the pocket used by natural activators. Our studies indicate the opportunity to tune proteasome activity by allosteric regulators based on PR peptide scaffold.
Introduction
The ubiquitin–proteasome system (UPS) is one of the two main proteolytic pathways existing in human cells.1 It is involved in regulation of all aspects of cellular physiology, and aberrations in its function are intricately related to the well-being of cells, organs, and organisms.2 Proteasome inhibition became an efficient way to kill rapidly proliferating, and thus addicted to proteasome activity, cancer cells. Three FDA-approved drugs specifically inhibiting the proteasome, bortezomib, carfilzomib, and ixazomib, revolutionized the therapy of hematological cancers.3 Multiple other inhibitors are in clinical trials.4 However, killing oversensitive cells by abolition of the proteasome activity is not the only way to use proteasome-targeting compounds. For example, mild inhibition of the proteasome may protect muscle cells from excessive protein degradation that occurs in disease-related cachexia or aging-related sarcopenia.5 On the other hand, activation of the proteasome should help to attenuate these age-related diseases which result from the diminished proteasome activity.6
The 26S proteasome, which is a central element of the UPS, comprises one or two 19S regulatory particles and a barrel-shaped catalytic core called 20S proteasome or core particle (CP). The 19S module enables recognition of proteins marked for degradation by polyubiquitin chains, their deubiquitination, unfolding, and translocation into the catalytic core.7 The core consists of 28 subunits, which are arranged in a stack of four heptameric rings in an αββα fashion.8 The two outer α rings provide binding sites for activating or regulatory particles, whereas the catalytic activity resides within the inner β subunits. In eukaryotic proteasomes the catalytically active β subunits exhibit three different substrate cleavage preferences: caspase-like post acidic (C-L; β1/1′), trypsin-like post basic (T-L; β2/2′), and chymotrypsin-like post hydrophobic (ChT-L; β5/5′).8−10 The tightly packed N-termini of the α-subunits form the gate, which restricts access of substrates to the catalytic chamber. Opening of the gate is promoted by docking of proteasome activators: 19S (PAN in Archaea), 11S (PA28/REG), or PA200.11−14 All these additional modules attach to the surface of the α ring (the “α-face”), anchoring in the pockets between the α subunits, but through transduction of allosteric signals they can influence the performance of the active sites.7,13,14
Although most intracellular proteins are selectively targeted for degradation through ubiquitin tagging, an increasing number of proteins have been identified as undergoing ubiquitin-independent cleavage by the 20S core itself. The pool of its substrates includes proteins that have been partially or completely unfolded due to aging, mutations, or oxidation and also native proteins, which are intrinsically disordered or encompass large (>30 residues) disordered regions.15,16 A growing body of evidence indicates that the 20S core plays a major role in the clearance of proteins that can be precursors of toxic oligomeric species implicated in the pathogenesis of severe neurological disorders, such as Parkinson’s, Alzheimer’s, and Huntington’s diseases and amyotrophic lateral sclerosis.17−19
The extensive involvement of proteasome in human health and disease requires precise regulation of its activity and causes that small molecules with such capacity are of great interest. However, while the field of competitive inhibitors of the enzyme is mature, the concepts for noncompetitive allosteric regulation are only starting to emerge, mainly because of structural complexity of the proteasomes, which offers not only unique opportunities but also challenges to the rational design of allosteric regulators. Relatively few noncompetitive/allosteric small molecule regulators of the CP activities have been described so far. Among them are several compounds with a quinoline or imidazoline scaffold (inhibitors), derivatives of rapamycin (inhibitors), chlorpromazine (activators), or betulinic acid (inhibitors/activators).20−26 A promising direction in the design of proteasome modulators could be peptidic structures,27 since peptides and peptidomimetics can offer higher specificity and lower toxicity than low molecular weight compounds.28 Peptide modulators can be derived from the binding regions of proteins which are natural proteasome regulators. One example of such an approach is a short, 10-residue C-terminal fragment of the RP subunit, Rpt5, which has been reported to activate the core in trans.29 We designed a 14-mer peptide based on the C-terminal fragment of Blm10 (a yeast ortholog of PA200), which stimulated human 20S proteasome’s activity 3-fold at 1 μM concentration.30 The key feature of both these peptides is the three-residue C-terminal “HbYX” motif (hydrophobic-Tyr-any residue) through which the modulator docks in the pocket between α subunits and probably allosterically affects catalytic activity.13,29 Another example of the peptidic approach to the regulation of proteasome activity is PAP1 peptide, described by Dal Vechio et al.31 This peptide reportedly activated the proteasome and was able to prevent protein aggregation in a cellular model of amyotropic lateral sclerosis. The distinct group of peptidic modulators of the 20S proteasome are peptides and mimetics derived by us from the proteasome-binding viral protein HIV-1 Tat, which in vitro potently inhibited the core.32
Here, we focus our attention on the proline- and arginine-rich (PR) porcine cathelicidine PR39 peptide, which is a proteasome inhibitor with unique molecular and intracellular effects.33,34 Since it was reported that the 11-residue N-terminal fragment of PR39, PR11 (11), was sufficient to convey allosteric actions on the CP,35,36 we set out to explore peptide regulators based on the scaffold of 11 and constructed from Arg-rich, Pro-rich, and HbYX-inspired motifs. Modifications introduced to the sequence of 11 allowed us to discover inhibitors more potent than the parent peptide but also compounds able to efficiently stimulate human 20S proteasome (h20S). Our studies have shed light on allosteric interactions that may be critical for the positive and negative regulation of the proteasome.
Results and Discussion
Design of the Peptides
Compound 11 has been described before as a noncompetitive and allosteric inhibitor of human and yeast proteasomes.35 As a basis for our design, we distinguished three modules in the canonical structure of 11: the N-terminal triple-Arg, the Pro-rich module (PPPP or PRPP), and the variable C-terminal fragment (Table 1). The RRR module, which has been proved essential for proteasome targeting by 11, was present in all peptides. Some of the peptides (3, 4, 10, and 12) were devoid of the Pro-rich module but had instead an extended C-terminal part. In 6, 7, and 8 both the extended C-terminus and Pro-rich module were included. The extension of the original sequence of 11 was inspired by the fact that an aromatic residue at the ultimate or penultimate position has been indicated as crucial for binding of allosteric modulators to proteasome.37 Another rationale for such a modification was reported by Anbanandam et al., better inhibitory capacity of 11 with a Trp residue appended at the C-terminus.36 In 1 and 2 the C-terminus was modified by incorporation of the full HbYX motif (Table 1), which has been proved to be a critical element of proteasome activators.13
Table 1. Amino Acid Sequence and Corresponding IC50 of the Studied PR Modulators.
| MW [Da] |
||||
|---|---|---|---|---|
| compd | sequence | calculated | found | IC50 ± SEM [μM] |
| 11 | RRR-PRPP-YLPR-OH | 1462.8746 | 1462.7694 | 0.095 ± 0.009 |
| 1 | RRR-PPPP-LYA-OH | 1221.7095 | 1221.6209 | N/Aa |
| 2 | RRR-PPPP-YYA-OH | 1271.6888 | 1271.5974 | N/Aa |
| 3 | RRR-YLPR-WG-OH | 1258.7160 | 1258.6235 | 0.111 ± 0.013 |
| 4 | RRR-YLPR-WG-NH2 | 1257.7160 | 1257.6425 | 0.082 ± 0.013 |
| 6 | RRR-PRPP-YLPR-FG-OH | 1666.9645 | 1666.8566 | 0.099 ± 0.009 |
| 7 | RRR-PRPP-YLPR-WG-OH | 1705.9754 | 1705.8556 | 0.050 ± 0.006 |
| 8 | RRR-PRPP-YLPR-WG-NH2 | 1704.9754 | 1704.8704 | 0.113 ± 0.010 |
| 10 | RRR-YPR-WG-OH | 1145.6319 | 1145.5477 | 0.166 ± 0.017 |
| 12 | RRR-YLPR-YA-NH2 | 1248.7156 | 1248.6411 | 0.154 ± 0.003 |
Not applicable.
PR Peptides as Inhibitors of Human 20S Proteasome
All PR peptides described so far were inhibitors of human 20S proteasome.35,36 Accordingly, we found that the ChT-L peptidase activity of SDS-activated 20S was inhibited by all but two of our modulators (Figures 1 and S1). The IC50 (the concentration causing 50% inhibition of activity, compared to the vehicle-treated control) was respectably below 200 nM for all inhibitors, with the lowest values obtained for 7 and 4 (50 and 82 nM, respectively; Table 1). The common motif in the sequences of 7 and 4 was a WG moiety, added to the C-terminal module of 11. Since glycine is devoid of any productive side chain, it may be speculated that the indole ring of tryptophan is solely responsible for this improvement in the inhibitory capacity. This conclusion is in line with the results of Anbanandam et al.36 and proves the positive effect that may be exerted by tryptophan incorporated as an ultimate/penultimate residue into the sequence of PR-type inhibitors. Further analysis of the structure–activity relationships in 3–8 indicates that other elements that distinguish their sequences do not affect their inhibitory propensity in a consistent manner. While the lack of the Pro-rich module diminished the inhibitory capacity of the analog with a C-terminal carboxylate (3 vs 7), the same modification was rather advantageous for the peptide terminated with an amide group (8 vs 4). Compound 10, which has almost the same sequence as 3 except for the lack of Leu residue in the YLPR segment, was the weakest inhibitor in the tested set of compounds (Table 1). The importance of the Leu residue may result either from its ability to furnish hydrophobic interactions necessary for efficient proteasome inhibition or from its ability to position other moieties in such a way as to enable their interactions with the 20S proteasome. The most striking result of the kinetic assays utilizing the activated 20S was a complete lack of inhibitory capacity of 1 and 2 (Figure 1). These two compounds were equipped with the HbYX motif, with a tyrosine residue occupying the penultimate position and preceded by either Leu or another Tyr. On the other hand, when the penultimate aromatic residue Trp (3, 4, 7, 10), Phe (6), or Tyr (12) was flanked by Arg from the YLPR/YPR segment, the peptides retained their inhibitory propensity.
Figure 1.
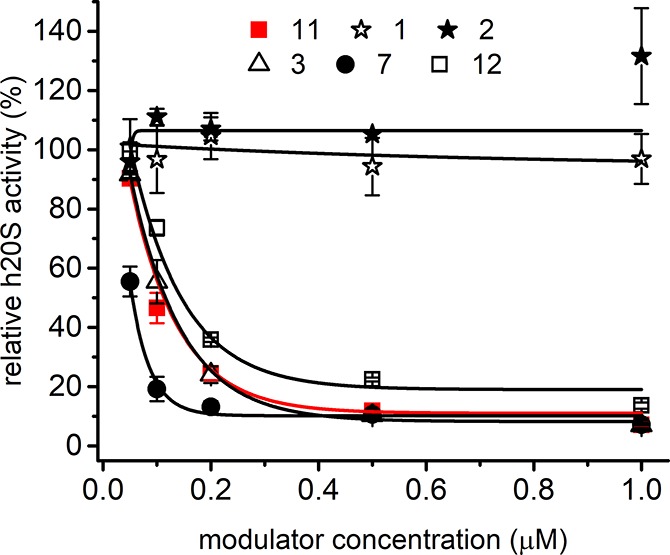
ChT-L peptidase activity of SDS-activated human 20S proteasome was inhibited by selected PR peptides in a dose dependent manner, with the exception of 1 and 2. 4 (Figure S1) and 7 were better inhibitors than 11 (red line), whereas 10 (Figure S1) and 12 were less efficient. The enzyme activity was probed with the substrate Suc-LLVY-AMC. Each titration was performed in three independent replicates. The results are presented as the mean ± SEM.
Apart from the ChT-L, we also tested the trypsin- and caspase-like activities to detect if there are any selectivity in the influence of our PR-analogs on the SDS-activated h20S proteasome. We observed that compounds 3–12 inhibited both the C-L and T-L activities but the caspase-like peptidase responded to much lower concentrations of PRs (Figure S2). This response generally resembled the sensitivity of the ChT-L peptidase (Figures 1 and S1). The T-L activity diminished significantly only when the modulators were applied at their highest tested concentration (10 μM). 1 and 2 displayed very weak inhibitory propensity against the C-L and the complete lack of capability to inhibit the T-L peptidase (Figure S2).
Mechanism of Inhibition
We attempted to determine the mechanism of inhibition for the best two inhibitors, 4 and 7, using Suc-LLVY-AMC as a substrate for ChT-L active centers. We tested three concentrations of each inhibitor and noticed that with increasing concentration the dose–response curves adopt a more pronounced sigmoidal shape, indicating a cooperative digest of the substrate (Figure 2). Such an effect is usually observed for an allosteric mode of inhibition.38
Figure 2.

Inhibition of the ChT-L peptidase activity of h20S by 4 (a) or 7 (b) followed the mechanism of noncompetitive mixed inhibition. In this mechanism, proteasome preserves partial catalytic activity, while the inhibitor binding affinity also depends on occupancy of the active centers. Each titration was performed in three independent replicates. The results are presented as the mean ± SEM.
Numerical analysis of the data, performed with the enzyme kinetics applications of OriginPro, pointed at the partial noncompetitive mixed inhibition model as the most probable mechanism to explain the PR peptide interactions with proteasome (Table 2). Calculated Ki values were 0.0101 ± 0.0055 μM and 0.0048 ± 0.0026 μM for 4 and 7, respectively, which closely matched the calculated IC50 values (Table 1).
Table 2. Kinetic Parameters of Inhibition of SDS-Activated h20S Proteasome.
| compound 4 |
compound 7 |
|||
|---|---|---|---|---|
| kinetic parameter | value | SEM | value | SEM |
| Vmax [s–1] | 13.46 | 0.91 | 13.64 | 0.88 |
| KM [μM] | 14.02 | 5.55 | 17.42 | 5.79 |
| Ki [μM] | 0.0101 | 0.0055 | 0.0048 | 0.0026 |
| β | 4.98 × 106 | 4.20 × 108 | 1347 | 1370 |
| α | 180878 | 7.65 × 106 | 4620 | 2963 |
The mixed inhibition indicates that affinity of binding of a substrate to the active center and of an inhibitor to its regulatory pocket is mutually cross-dependent. Furthermore, partial noncompetitive inhibition may explain the substantial remaining catalytic activity of proteasome in the presence of PR peptides. Apparently, these inhibitors can significantly slow formation of the substrate–enzyme complex or dissociation of the product but cannot completely block the catalytic cycle. It is worth noting that this partial inhibition, without total abolishment of proteasome activity even at high concentrations of an inhibitor, is typical for allosteric modulators and may be very beneficial when the cells treated with the drug are meant to survive rather than die from apoptosis. Both tested peptides presented similar kinetic parameters with a notable difference in Ki value, which designated 7 as the better inhibitor of the two (Tables 2 and 1). The proposed mechanism of inhibition is in contrast to the previously studied case of PR39, which was classified as consistent with a pure noncompetitive mode of action.35 A putative penetration of the catalytic chamber by a 39-residue poly-Pro-rich peptide is less plausible than the similar action performed by the short peptides considered here. The lack of a competitive component in the case of PR39 could be rationalized in this way.
PR Inhibitors Impair Activity of h20S Stimulated with Rpt5 C-Terminal Peptide
Activation of the core proteasome with SDS is an established intervention for studying inhibitor efficiency;39 however it can be considered an artificial way of enzyme stimulation. Therefore, we decided to also test our PR peptides using h20S activated with the 10-aa C-terminal peptide of the Rpt5 subunit of the 19S regulatory particle (KKK-ANLQ-YYA-OH, named here “Rpt5”). Rpt5 activated proteasome with an AC50 of 3.14 μM and can be considered a “minimal RP” model for activity tests with proteasome regulators.24 We used two concentrations of Rpt5: 1 μM, which stimulated the ChT-L activity nearly 4-fold, and a saturating 10 μM concentration (Figure S3), which left the proteasome activity about 8-fold higher than the latent control.
As demonstrated in Figure 3, both 7 and the original peptide 11 inhibited the Rpt5-activated proteasome. 7 was a more potent inhibitor than 11, with an IC50 of about 0.5 μM (0.56 μM with 1 μM Rpt5 and 0.50 μM with 10 μM Rpt5, respectively). The IC50 of 11 was approximated as 1.60 μM (1 μM Rpt5) and 1.90 μM (10 μM Rpt5). The effect of 2 was quite interesting: it did not affect the performance of CP activated by 10 μM Rpt5 (Figure 3b) but was able to stimulate 2-fold the activity of CP treated with 1 μM Rpt5 (Figure 3a). As a result, the CP activity rose 8-fold (4-fold activation by Rpt5 × 2-fold activation by 2), which is similar to the activation achieved with the saturating concentration of Rpt5. This newly observed activity of 2 prompted us to test the effects of our PR peptides on the latent h20S.
Figure 3.

Compounds 7 and 11 inhibited 20S proteasome activated with 1 μM (a) or 10 μM Rpt5 (b). IC50 calculated for 7 did not depend on Rpt5 concentration, whereas for 11 it was slightly increased at the higher concentration of Rpt5. In contrast, in the presence of 1 μM Rpt5, 2 additively activated proteasome (a). This effect was abolished at 10 μM Rpt5 (b).
There Are Efficient Proteasome Activators among PR-Type Modulators
PR analogs that displayed distinct inhibitory capacity against SDS-activated h20S (Figures 1, S1, and S2) differed in their influence on the latent enzyme. At low concentrations, clustering around 1 μM, 6, 11, and 12 activated the ChT-L peptidase of proteasome, with the best-performing 12 achieving nearly 5-fold activation at 5 μM concentration (Figure 4). The activating capacity of 4 was weaker (maximum 2-fold at 5 μM concentration), while 7 practically did not activate h20S at all. The transient nature of the activation observed for compounds 12, 11, and 6 may be explained by presence of a low affinity secondary binding site that exercises the inhibitory actions on ChT-L activity. An alternative explanation would call for a negative cooperativity between binding sites where the occupation of the first binding site would allosterically modify the second binding site to turn it into the inhibitory site upon binding of the same compound. So far our analyses of enzyme kinetics performed in the presence of the compounds exclude the possibility that the digest products may play a role of inhibitors leading to the bell shaped dose–response.
Figure 4.

ChT-L activity of the latent 20S proteasome, probed with the substrate Suc-LLVY-AMC, was strongly and diversely regulated by PR peptides. At low concentrations 6, 11, and 12 transiently activated the proteasome. 1 and 2 proved to be strong activators of the latent CP and induced typical sigmoidal dose response stimulation. The results are presented as the mean ± SEM.
Interestingly, 1 and 2 that did not inhibit the SDS-activated 20S proved to be strong activators of the latent CP. When probed with the substrate Suc-LLVY-AMC (ChT-L activity) they stimulated the enzyme up to 7-fold at 25 μM concentration (Figure 4). The caspase-like peptidase was even more sensitive to activation by the compounds; they stimulated it 5-fold to 7-fold at 10 μM concentration (Figure S4).
Proteasome has a dynamic structure, and even in the latent state about 25% of its molecules adopt a partially open conformation, which enables small molecules to penetrate the catalytic channel and be degraded.38 Therefore, using only small fluorogenic substrates as probes can produce misleading results, especially in the case of identification of an activator.40 To verify the results obtained with Suc-LLVY-AMC, we included in our kinetic studies the internally quenched LFP nonapeptide, which has been described as a substrate that is very slowly hydrolyzed by the nonactivated 20S proteasome.41 Using this substrate, we did not detect any, even transient, stimulating propensity of 3, 4, 6, 7, 8, 10, 11, and 12 (Figures 5b and S5b). The only PR analogs capable of efficiently, and in a dose dependent manner, stimulating ChT-L peptidase activity of the latent h20S were 1 and 2. At the highest tested concentration (25 μM) these two peptides accelerated LFP hydrolysis 8- and 14-fold, respectively (Figure 5b). In kinetic tests with the SDS-activated h20S 1 and 2 also behaved differently from other PRs (Figure 1). While almost all analogs strongly inhibited degradation of the LFP substrate, 1 and 2 did not exert any inhibitory capacity, consistent with the results obtained with Suc-LLVY-AMC substrate. 7 and 8 were better inhibitors when compared to 11, whereas 3 and 10 were much less efficient (Figures 5a and S5a).
Figure 5.

(a) ChT-L activity of the SDS-activated proteasome, probed with LFP substrate. Almost all analogs, except for 1 and 2, inhibited h20S, with 8 (Figure S5a) and 7 being more efficient and 10 (Figure S5a) and 3 significantly less efficient than 11 (red line). (b) 1 and 2 were the only compounds among the studied set of PR analogs able to stimulate the activity of the latent h20S. The maximum activation effect reached 8-fold for 1 and 14-fold for 2.
The observed diverse effects of PR peptides on proteasome activity cannot be explained based on conformational differences between the positive and negative modulators. Although some structural diversity was detected when comparing the modulators’ CD spectra, it was not helpful in identification of activators and inhibitors (Figure S6). We thus turned our attention to the primary structure differences. There are two regions that differentiate the positive and negative modulators’ sequences: the proline-rich module and the C-terminal sequence. To check the influence of the first one, we have synthesized an analog of inhibitor 7 with the PRPP segment substituted with the module PPPP, present in the sequence of 1 and 2 activators. The peptide 7-4P inhibited the chymotrypsin-like activity of the SDS-activated proteasome with similar effectiveness as 7 and was not able to activate this peptidase in the latent h20S (Figure S7). The possible explanations of the observed diverse effects of PR peptides on h20S activity should be thus sought within the C-terminal sequence. 1 and 2 are furnished with the HbYX module (Table 1), which they share with protein activators such as PA200/Blm10, PAN, or Rpt2, 3, and 5 subunits of 19S. This motif has been proved to be a key factor in the mechanism of proteasome activation, due to the contacts delivered by the hydroxyl group of the penultimate Tyr with Gly19. This interaction causes a shifting of the Pro17 reverse turn in one or more α subunits, which results in a partially or fully opened entrance leading to the catalytic chamber.7,13 The stimulating propensity of 1 and 2 may result from similar interactions. It is also tempting to speculate that the limited activating potency of the remaining PR analogs may originate from shortening their C-terminal motif to only HbX (with “Hb” denoting a hydrophobic Trp or Phe residue), which precludes the canonic contacts with Gly19. Compound 12, which displayed quite significant (although transient) activating capacity when probed with Suc-LLVY-AMC (Figure 4), comprises the penultimate Tyr in its sequence. However, in contrast to 1 and 2, not the hydrophobic but basic arginine residue precedes the penultimate Tyr in 12, forming an alternative RYX motif, similar to RHbX present in all PR analogs which displayed inhibitory capacity (Table 1). Arginine possesses a long side chain grafted with the highly basic guanidyl group and thus may provide interactions not available to 1 and 2. The interactions of both Arg and the penultimate Tyr may be responsible for the ambivalent properties of 12, i.e., the concomitant demonstration of considerable inhibitory and activating capacity against the latent h20S.
PR Peptides Affect Degradation of Disorganized/Unfolded Proteins
The model peptide substrates provide a convenient way to probe proteasome activity. However, utilization of protein substrates has the advantage of a better approximation of the kinetic challenges encountered in the cellular context. Therefore, we tested degradation of α-synuclein (α-syn) which, as a natively unfolded protein, can be degraded by 20S alone, without the assistance of the 19S regulatory complex.42,43Figure 6a shows that PR modulators influence α-syn proteolysis differently. At 1 μM concentration the most efficient inhibitors were 6 and 7, but at higher concentration 4, 11, and 12 impeded α-syn degradation to similar extent. 1 and 2 did not display any inhibitory capacity at 1 μM concentration. On the contrary, at 10 μM concentration, both compounds increased the level of α-syn hydrolysis. In the presence of 2, h20S was able to almost completely degrade this protein (Figure 6a).
Figure 6.

Degradation of protein substrates by human 20S proteasome was strongly affected by PR peptides. Levels of the remaining substrates were determined based on SDS–PAGE electrophoretic separation of proteins incubated with 20S proteasome. Almost 70% of α-synuclein (a) and 50% of enolase (b) were degraded by 20S proteasome under the applied experimental conditions. Representative SDS–PAGE electrophoregrams with Coomassie-stained substrate bands are presented below the columns. At 10 μM, 1 and 2 accelerated the digest to completely degrade enolase. Other PR peptides, especially at 10 μM concentration, efficiently blocked degradation of the model proteins, with 6 and 7 being the best inhibitors. NS = not statistically significant; all other cases are statistically significant (p < 0.05).
To further survey the catalytic response of 20S to PRs, we expanded our tests to enolase. This 436-residue thermolabile protein is an established substrate of 20S proteasome,44 although we found that it was less efficiently degraded by CP than α-syn (Figure 6b).
The enolase degradation assay confirmed that 6 and 7 are efficient inhibitors of proteolysis, whereas 11, 4, and 12 influence this process slightly less effectively. In contrast to α-syn, there were no distinct differences in the level of inhibition at 1 and 10 μM concentration of the inhibitor. Importantly, 1 and 2 stimulated degradation of enolase, a poor substrate, much better than of α-syn. Indeed, stimulation with 1 μM concentration of 2 led to more than 2-fold acceleration of enolase digestion. Surprisingly, 20S proteasome activated with 10 μM of either peptide completely degraded enolase under the employed reaction conditions. HPLC analysis of the products showed short fragments of the hydrolyzed protein. The detection of such products strongly indicates that the proteolytic activity of h20S was responsible for the disappearance of the enolase band at the electropherograms.
Stimulation of Proteasome Activity in Cell Culture
Intrigued by the strong potency of 2 as an activator of the peptidase and proteinase activity of the proteasome, we decided to test the performance of this peptide in cell culture. We chose human primary fibroblasts, which provide an advantageous system for investigating dynamic molecular regulatory processes without the confounding effects of a disease state. The cells were treated with 2 at concentrations ranging from 0.1 μM to 20 μM. Even the highest concentration of 2 did not exert any cytotoxic effects (Figure 7a). Importantly, the total activity of proteasomes was significantly elevated in total cell lysates prepared from cells treated with 2, as compared with the vehicle-treated cells. The activation was nearly 3-fold when 10 μM concentration of 2 was present in the cell culture medium (Figure 7b). This important result indicates that the activating PR derivatives can be safely used in cellulo to stimulate UPS.
Figure 7.

Treatment of human cultured fibroblasts with compound 2 (24 h) did not affect viability of the cells, even at high peptide concentrations (a). ChT-L activity of proteasome in cell extracts prepared from human fibroblasts treated with 2 was activated up to 3-fold in a dose dependent manner (b).
Dynamics of the Gate Probed by Atomic Force Microscopy
We had established earlier that the latent 20S proteasome can switch freely between two major conformational states: a prevailing “closed” state with no detectable gate in the α-face and a less populous state with AFM-detectable indentation in the gate area (the “indented” or “open” state).45 Interaction of control proteasomes with a substrate was followed by a switch to a majority of “open” particles (about 75%). Consequently, we proposed an allosteric model of the gate movements with a positive feedback loop running between the gate and the active centers and enabling efficient passage of substrates and products to and from the catalytic chamber.46 Treatment of proteasomes with the canonical PR peptides, 11 and PR39, induced a “shaky” conformation, with multiple shallow indentations indicating destabilization of the α-face.35 Moreover, we noticed that while the images of “closed” particles were very uniform, the conformers with detectable indentation presented two distinct morphologies of the α-face: with a deep indentation surrounded by a symmetrical rim and with a shallow dip and irregular rims. Computational analysis of sections through the α-face of imaged control proteasome particles allowed distinction between three conformers, “closed” (as previously) as well as “intermediate” and “open”, replacing the previously described state with the α-face indentation. We found the control proteasome particles in a conformational equilibrium of nearly three-quarters of closed (71% ± 2%), 22% ± 3% of intermediate, and 7% ± 3% of open particles (Figure 8). Interestingly, this refined partition of conformers in the control sample closely resembles the recently proposed cryo-EM based model of conformational states of the gate in 26S proteasome, where only about 8% of particles assumed the fully open conformation.47
Figure 8.

PR peptides influenced the abundance of the α-face conformations of 20S proteasome, as detected by AFM. (a) In control, untreated samples, about 70% of molecules had their central channel covered and classified as a closed gate conformation. About 7% of molecules had their gate completely open, and the remaining 22% were in the process of switching between these conformations and were classified as intermediates. In the presence of an activating 1 μM concentration of 2, the abundance of open and intermediate conformations increased to 54% (18% and 36%, respectively). In contrast, a 1 μM concentration of 7 decreased the number of open conformers to less than 3%. The higher concentration of 7 pushed proteasome to open the gate in 18% of molecules, with a slightly higher contribution of intermediates (25%). (b) Representative AFM images of the three conformational forms of the core proteasome. The top-view images with the α-face exposed were zoomed-in from 1 μm × 1 μm fields. The images are raw and have been subjected only to planefitting/flattening, linear adjustments of brightness and contrast, and linear interpolation for viewing clarity.
Treatment with 1 μM 2 increased the contribution of both open and intermediate conformers, which together amounted to half of the analyzed standing proteasomes (Figure 8). In contrast, treatment with 1 μM 7 resulted in a slightly decreased partition of open proteasomes and a high contribution of closed particles (73% ± 2%). Interestingly, treatment with a high concentration of 7 (10 μM) resulted in apparent destabilization of the α-face, with the content of the closed conformers decreasing to only 57% ± 6% (Figure 8). The destabilization of the α-face by high concentrations of 7 may seem an unusual effect for an inhibitor; however it follows our previous observations with PR39 and 11, when closed conformers were poorly detected in the peptide-treated sets of particles.34 We speculate that at higher concentrations PR inhibitors may occupy additional binding sites that include pockets between the α subunits. The pocket binding is likely to be critical for activation by the HbYX-containing 1 and 2; however engagement of other sites may contribute to the effects displayed by the PR inhibitors.
Binding Site of PR Inhibitor in the Proteasome Molecule
Molecular modeling and yeast two-hybrid studies have indicated that PR peptides bind to the outer rim of the yeast proteasome α-ring.34,48 To elucidate the structural basis of the interaction, we performed trials to crystallize our modulators in complexes with both human and yeast 20S proteasomes. Crystals were obtained for both orthologous enzymes; however a complex was only successfully formed in the case of 6 and the yeast 20S. Since 6 is also an efficient inhibitor of the yeast enzyme (Figure S8), the structural information provided by this complex may be extended to the human counterpart. The structure of the complex was determined at 2.7 Å resolution (PDB code 4X6Z) and confirmed that PR peptides bind within the proteasome α-ring. The structure solved based on a single data collection showed 6 bound in the pocket between the α5 and α6 subunits, and the unidentified electron density was localized symmetrically between the α5′ and α6′ subunits. Merging the three data sets collected for the same complex reinforced the electron density which allows us to identify the peptide electron density also in the α5′/α6′ pocket. The three C-terminal amino acid residues of 6 that were visible in the crystal structure created the main-chain to main-chain hydrogen bonds with α6Ser33 and α6Gly76, and the salt bridge with the α6Lys62 side chain (Figure 9). The rest of the peptide sequence was not defined by the electron density probably because of its flexibility and the lack of stable interactions with the proteasome. Degradation of 6 by 20S, as other possible explanation, was excluded by the peptide stability tests (Figure S9).
Figure 9.

Interaction of 6 with yeast 20S proteasome (PDB code 4X6Z). (a) General (left) and detailed (blow-up) localization of the inhibitor binding site between α5 (wheat color) and α6 (gray) subunits. It is visible in the crystal structure that the C-terminal fragment of 6 (green) binds to the ε-amine group of the conserved α6Lys62 through its carboxylate group. (b) Electron density defining the fragment of 6 included in the model (2Fo – Fc omit map contoured at 1σ level). The cartoon of general proteasome structure in (a), left, has also been used in our earlier work.30
Binding in the α5/α6 pocket and similar network of interactions utilized to anchor the modulator within the pocket were observed by us in the crystal structure of the Blm10-based proteasome activator, Blm-pep.30 This compound possesses the C-terminal HbYX motif (YYA) and was able to stimulate h20S 8-fold when probed with the LFP substrate. The same site of binding of two modulators, displaying the opposite influence on the proteasome activity, may indicate that the pockets between the α subunits may be universal binding places for allosteric modulators. The allosteric signal that transduces either inhibition or stimulation effects to the active sites may thus result from transient interactions, which due to their nature could be observed in the crystal structure of neither compound 6 nor Blm-pep. The importance of transient interactions in proteasome allosteric activation is supported by the cryo-EM structures of yeast and human 26S proteasomes, in which the opened entrance to the catalytic chamber was observed in only one of the detected few conformational states.7,47,49 It remains to be established which allosteric routes contribute to the effects of ligands of the α-face pockets. The nonexclusive options include direct allosteric signaling from the pockets to the active sites, the pocket-gate signaling resulting in conformational shifts favoring distinct states of the gate and influencing substrate uptake, as well as the complex pocket-gate-antechamber/catalytic chamber/active sites network. PR peptides constitute excellent tools for the exploration of proteasome allostery.
Conclusions
We demonstrated that manipulations of the structural modules of Pro- and Arg-rich peptides could provide compounds that exhibit diverse effects on the activity of the core proteasome particle, both in vitro and in cellulo. Mixing-and-matching of Pro-rich and HbYX or RHbX motifs produces strong activators or strong inhibitors. Studies of interactions of PR39 and 11 with yeast proteasome suggested binding to the outer rim of the α ring.34,48 While we cannot exclude and should still consider such binding site(s) for our PR derivatives, their binding to the intersubunit pockets, used by the HbYX-containing activators, is an intriguing opportunity. The plausible common binding sites for allosteric inhibitors and activators imply that the type of the signal that is transmitted to the binding pockets and allosterically transduced to the gate and/or the active sites may depend on small but precisely adjusted changes in the modulator design. Further exploration of the diverse effects of PR peptides on performance of the core proteasome will undoubtedly provide more information on the mechanism of proteasome degradation, the significance of the α-face pockets in modulating proteasome activity, and the usefulness of small peptides as universal proteasome regulators.
Experimental Section
General Information
20S proteasome, isolated from human erythrocytes, was purchased from Enzo Life Sciences Inc. (Farmingdale, NY). All reagents used in tests with proteasome were of molecular biology grade. The pH of all buffers was determined at 20 °C.
Peptide Synthesis
Syntheses of all peptides were carried out on a solid support (TentaGel R PHB or TentaGel R RAM resin), using a Liberty Blue microwave peptide synthesizer (CEM). Coupling of orthogonally protected Fmoc-amino acid residues was carried out using, as a coupling agent, 1 M solution of N,N′-diisopropylcarbodiimide in dimethylformamide with 0.5 M ethyl cyano(hydroxyimino)acetate as an antiracemization additive. Crude peptides were purified by reverse phase high performance liquid chromatography (RP-HPLC), using a C12 semipreparative Jupiter Proteo column (21.2 mm × 250 mm, 4 μm, Phenomenex) and H2O/acetonitrile gradients. The purity of the peptides was assessed by HPLC analysis performed using a Luna C18 column (4.6 mm × 250 mm, 5 μm, 100 Å; Phenomenex) and a LC-20A HPLC system (Shimadzu). 60 min gradients of 100% A → 100% B (A, H2O + 0.1% TFA; B, 80% acetonitrile/H2O + 0.1% TFA) and a detection wavelength of λ = 223 nm were applied. The purity of the peptides has been determined based on the integration of the area under the peaks, using the Lab Solution software provided by the HPLC manufacturer (Shimadzu), The purity of all PRs was at least 95%. The identity of the pure products was confirmed based on m/z signals detected by a LCMS-ESI-IT-TOF Prominence mass spectrometer (Shimadzu). The MS spectra and the chromatograms, alongside with their quantitative analysis, are available in Supporting Information.
Enzymatic Activity Tests
Boc-LRR-AMC and Z-LLE-AMC were used as a probes in the trypsin-like and caspase-like activity assays, respectively. Two peptide substrates were employed in the assessment of the proteasome chymotrypsin-like activity: the classic fluorogenic succinyl-Leu-Leu-Val-Tyr-4-methylcoumarin-7-amide (Suc-LLVY-AMC, Enzo Life Sciences Inc.) and a homemade FRET-type nonapeptide (LFP; mca-AKVYPYPME-dap(dnp)-amide) as an alternative which is more reliable for assessment of the activating propensities of the studied compounds.41 Latent h20S proteasome was activated with 0.005% SDS. Stock solutions of the peptides (10 mM) were prepared in dimethyl sulfoxide (DMSO). The activity assays were performed in a 96-well plate in 50 mM TrisHCl, (pH 8.0) at a 100 μL final volume. The CP was used at a final concentration of 0.001 mg/mL (1.4 nM). Suc-LLVY-AMC, Boc-LRR-AMC, and Z-LLE-AMC were added at 100 μM and LFP at 20 μM final concentration. The release of aminomethylcoumarin (AMC) was followed by monitoring the fluorescence emission in the range 380–460 nm. LFP hydrolysis was detected by measuring the emission in the range 322–398 nm (Infinite M200 Pro, Tecan). Fluorescence was measured continuously every 2 min for up to 60 min, at 37 °C. All activity assays were performed in at least three independent replicates. The relative activity was calculated in relation to the catalytic activity of the vehicle (DMSO) treated proteasome.
Activity tests with Rpt5 peptide and Rpt5 combined with PRs were performed in 45 mM Tris/HCl (pH 8.0) buffer containing 100 mM KCl and 1 mM EDTA, to ensure latency of the h20S proteasome. The Fluoroskan Ascent plate reader (Thermo Fisher Scientific Inc., Waltham, MA) was used in these assays, with fluorescence measured every 1 min for 60 min at 37 °C.
Determination of the Type of Inhibition
To determine the type of inhibition induced by PRs, the ChT-L activity of h20S proteasome was tested in the presence of three different concentrations of 7 and 4 (0.05, 0.1, and 0.2 μM). The proteasome concentration was 1.4 nM. ChT-L activity was probed with Suc-LLVY-AMC in a concentration range 20–200 μM. The kinetic parameters were calculated with the kinetic module of OriginPro 2017 software (OriginLab, Northampton, MA).
Protein Substrate Degradation Assay
Human α-synuclein (rPeptide) or yeast enolase (Sigma-Aldrich) was incubated with human 20S proteasome activated with 0.01% SDS. The h20S/protein substrate ratio was 1:100 pmol and 1:10 pmol for α-synuclein and enolase, respectively. Degradation experiments were carried out at 37 °C in 20 mM HEPES, pH 7.4, at a 10 μL total sample volume for 1 h (α-synuclein) or 4 h (enolase). Either DMSO (control) or compounds dissolved in DMSO were added to evaluate the influence of PR peptides on the degradation process. The DMSO concentration never exceeded 0.05%. The reaction was stopped with 4× Laemmli buffer, and then samples were boiled for 5 min at 95 °C and loaded (8 μL) onto a 12% (α-synuclein) or 10% (enolase) SDS–PAGE gel. The protein bands were detected with Coomassie Blue-based reagent (InstantBlue, Sigma-Aldrich). Quantitative image analysis was carried out with Quantity One 1-D analysis software (Bio-Rad). The amount of the nondegraded protein was calculated after reduction of the background intensity and expressed as a percentage of the control. Each value represents an average of at least three experiments. All results are presented as a mean ± SEM. Statistical analysis was performed using SigmaPlot 12.3 and one-way ANOVA followed by a Bonferroni post hoc test for pairwise comparison. A P value of <0.05 was considered statistically significant.
Fibroblast Culture
Primary human fibroblasts (GM04390, Coriell cell repositories) were cultured in complete media made of Dulbecco’s modified Eagle medium (high-glucose variant, Gibco-Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B; Gibco-Invitrogen). Cells were incubated at 3% O2, hypoxic with respect to atmospheric O2 concentration, to mimic their normal physiological environment; the incubators were also maintained at 5% CO2 and 37 °C. The medium was replaced every 3–4 days. For experimental end points, cells were seeded at 100 000 cells/mL in either 6- or 96-well plates. Cells were washed with PBS and medium replaced with Optimem (Gibco-Invitrogen) 24 h after seeding. Cells were treated with 0.1–20 μM 2 (or an equal volume of DMSO diluent) 48 h after seeding. Experimental end points were typically performed 24 h after initiation of treatment with 2.
Proteolytic Activity Assay in Cell Lysates
Cells were harvested through scraping in PBS and then lysed in proteolysis buffer (50 mM Tris/HCl, pH 7.4, 5 mM MgCl2, 1 mM DTT) through mechanical lysis followed by freeze fractionation. Protein content was measured using a Bradford assay, after which 1 μg of cell lysate was diluted to 100 μL and incubated with 50 μM Suc-LLVY-AMC (Sigma-Aldrich). Fluorescence emission was measured at 460 nm with excitation at 370 nm.
WST-1 Assay
Cell viability was evaluated using a WST-1 assay following the Sigma recommended protocol. Cells were incubated with 10% WST-1 reagent (Sigma-Aldrich) for 4 h after which absorbance was measured at 450 nm.
Atomic Force Microscopy (AFM) Imaging
We used our established procedures to image proteasome particles in AFM oscillating (tapping) mode in liquid using a MultiMode Nanoscope IIIa microscope (Bruker Corp., Billerica, MA).46,50 In short, about 50 ng of h20S proteasomes in 3 μL of 5 mM Tris/HCl buffer (pH 7.0) was deposited on freshly cleaved muscovite mica, which provides a flat, clean, and mildly negatively charged surface that electrostatically binds a majority of proteins. After 2 min of incubation at room temperature the enzyme molecules were overlaid with 30 μL of 5 mM Tris/HCl buffer, pH 8.0. Scanning was performed with SNL (Sharp Nitride Lever) probes with a nominal spring constant of 0.35 N/m, mounted in a liquid-mode chamber (Bruker Corp.) and tuned to 9–10 kHz. Fields of 1 μm2 containing multiple proteasome particles, the majority of them standing on the α ring and not touching their neighbors, were scanned at a rate of 3.05 Hz, with a drive voltage of 200–600 mV and a setpoint ranging from 1.5 to 2.0 V. The relatively high setpoint ensured very gentle imaging conditions. The trace and retrace images were collected with a digital resolution of 512 × 512 pixels. Numerical values of the height of particles were collected in SPIP software. The analyzed data remained “raw”, since the images were subjected only to the standard order 1 flattening and planefitting in the Nanoscope software. To determine the conformational status of the α face, height values of the six-pixel scan across the center of the α face were harvested. When a plot of these values (a section through the center of the α face) revealed a local minimum, the particle was classified as “open”. When a plot presented a concave function devoid of a local minimum, the particle was considered to be an “intermediate” conformer. The remaining “closed” particles displayed convex functions in their section plots.
Crystallization
The proteasome crystals were grown at 20 °C using the hanging drop vapor diffusion method. Drops contained a 1:1 mixture of the protein (2.5 mg/mL) and the reservoir solution (30 mM magnesium acetate, 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 7.2, and 12% (v/v) 2-methyl-2,4-pentanediol (MPD)). The proteasome–peptide complex was obtained by soaking the crystals with a solution of 6 at a final concentration of 1 μM for 24 h. Crystals were cryoprotected in the mother liquor and flash-frozen in liquid nitrogen.
Structure Determination and Refinement
Diffraction data were collected at the beam 19 ID at Advanced Photon Source, Argonne National Laboratory, USA (λ = 0.979 Å). The images were indexed, integrated, and scaled using the HKL3000 program package.51 The crystal structure was determined by molecular replacement using MOLREP52 and the coordinates of the yeast 20S proteasome (PDB code 1RYP) as a search model. The model was refined using Refmac53 from the CCP4 package,54 with stereochemical restraints and, in the later stages, TLS parameters for rigid-body segments established by the TLSMD server.55 The model was constructed in Coot.56 Progress of the refinement was monitored, and the model was validated using Rfree.57 The quality of the final structure was assessed using a MolProbity server.58 Data collection and refinement statistics are summarized in Table 1S.
Acknowledgments
This work was supported by National Science Center (Grants UMO-2014/15/B/NZ7/01014 (E.J.), UMO-2015/17/N/NZ1/01772 (J.W.), and UMO-2016/23/N/ST5/02812 (M.Gi.)), the William and Ella Owens Medical Research Foundation award (M.Ga. and P.O.), the San Antonio Nathan Shock Center of Excellence in the Biology of Aging Pilot Grants Program NIH/P30 (A.M.P.), and the Voelcker Fund New Investigator Award (A.M.P.). We thank Dr. Dominika Borek from the Department of Biochemistry, University of Texas Southwestern Medical Center (U.S.), for her assistance in the X-ray data refinement.
Glossary
Abbreviations Used
- AC50
the concentration causing 50% of maximal activation
- AFM
atomic force microscopy
- AMC
aminomethylcoumarin
- ChT-L
chymotrypsin-like activity
- CP
core particle
- h20S
human 20S proteasome
- Hb
hydrophobic
- IC50
the concentration causing 50% inhibition of activity
- LFP
mca-AKVYPYPME-dap(dnp)-amide
- MW
molecular weight
- PR peptide
proline- and arginine-rich peptide
- RP
regulatory particle
- Rpt5
regulatory particle ATPase 5 and its C-terminal fragment
- SEM
standard error of the mean
- UPS
ubiquitin–proteasome system
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01025.
HPLC and HR MS analysis of the designed analogs, supplementary figures displaying the ChT-L, T-L, and C-L activity of human proteasome in the presence of the modulators, comparison of 7 and 7-4P influence on the latent and SDS-activated h20S, CD spectra of the modulators, inhibitory capacity of 6 against yeast 20S proteasome, HPLC chromatogram confirming stability of 6 against proteasomal proteolytic activity, and crystallographic data statistics (PDF)
Accession Codes
PDB code for the yeast 20S proteasome in the complex with 6 modulator is 4X6Z. The atomic coordinates and experimental data have been already released.
Author Present Address
∥ R.R.: Polpharma SA Pharmaceutical Works, Pelplińska 19, Starogard Gdański 83-200, Poland.
Author Contributions
Author contributions were the following: M.Gi., synthesis and kinetic studies of modulators, proteolytic stability tests; J.W., synthesis and kinetic studies of modulators, X-ray structure refinement; P.K., protein substrates degradation tests; A.M.P. and E.S.C., cellular tests; P.O. and M.Ga., AFM and kinetic studies with Rpt5 peptide, design of the experiments, and data analysis; R.R., crystallization of 20S-6 complex; E.J., design of the experiments and data analysis. All authors contributed to the manuscript preparation.
The authors declare no competing financial interest.
Supplementary Material
References
- Ciechanover A.; Orian A.; Schwartz A. L. The Ubiquitin-Mediated Proteolytic Pathway: Mode of Action and Clinical Implications. J. Cell. Biochem. 2000, 77 (Suppl. 34), 40–51. . [DOI] [PubMed] [Google Scholar]
- Jankowska E.; Stoj J.; Karpowicz P.; Osmulski P. A.; Gaczynska M. The Proteasome in Health and Disease. Curr. Pharm. Des. 2013, 19, 1010–1028. 10.2174/138161281906141012124358. [DOI] [PubMed] [Google Scholar]
- Crawford L. J.; Walker B.; Irvine A. E. Proteasome Inhibitors in Cancer Therapy. J. Cell Commun. Signal. 2011, 5 (2), 101–110. 10.1007/s12079-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford L. J.; Irvine A. E. Targeting the Ubiquitin Proteasome System in Haematological Malignancies. Blood Rev. 2013, 27 (6), 297–304. 10.1016/j.blre.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Cohen S.; Nathan J. A.; Goldberg A. L. Muscle Wasting in Disease: Molecular Mechanisms and Promising Therapies. Nat. Rev. Drug Discovery 2015, 14 (1), 58–74. 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- Li X.-J.; Li S. Proteasomal Dysfunction in Aging and Huntington Disease. Neurobiol. Dis. 2011, 43 (1), 4–8. 10.1016/j.nbd.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Wu J.; Lu Y.; Ma Y.-B.; Lee B.-H.; Yu Z.; Ouyang Q.; Finley D. J.; Kirschner M. W.; Mao Y. Structural Basis for Dynamic Regulation of the Human 26S Proteasome. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (46), 12991–12996. 10.1073/pnas.1614614113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwe J.; Stock D.; Jap B.; Zwickl P.; Baumeister W.; Huber R. Crystal Structure of the 20S Proteasome from the Archaeon T. Acidophilum at 3.4 Å Resolution. Science 1995, 268 (5210), 533–539. 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Groll M.; Ditzel L.; Löwe J.; Stock D.; Bochtler M.; Bartunik H. D.; Huber R. Structure of 20S Proteasome from Yeast at 2.4Å Resolution. Nature 1997, 386 (6624), 463–471. 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Unno M.; Mizushima T.; Morimoto Y.; Tomisugi Y.; Tanaka K.; Yasuoka N.; Tsukihara T. The Structure of the Mammalian 20S Proteasome at 2.75 Å Resolution. Structure 2002, 10 (5), 609–618. 10.1016/S0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- Fabre B.; Lambour T.; Delobel J.; Amalric F.; Monsarrat B.; Burlet-Schiltz O.; Bousquet-Dubouch M.-P. Subcellular Distribution and Dynamics of Active Proteasome Complexes Unraveled by a Workflow Combining in Vivo Complex Cross-Linking and Quantitative Proteomics. Mol. Cell. Proteomics 2013, 12 (3), 687–699. 10.1074/mcp.M112.023317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre B.; Lambour T.; Garrigues L.; Ducoux-Petit M.; Amalric F.; Monsarrat B.; Burlet-Schiltz O.; Bousquet-Dubouch M. P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13 (6), 3027–3037. 10.1021/pr500193k. [DOI] [PubMed] [Google Scholar]
- Sadre-Bazzaz K.; Whitby F. G.; Robinson H.; Formosa T.; Hill C. P. Structure of a Blm10 Complex Reveals Common Mechanisms for Proteasome Binding and Gate Opening. Mol. Cell 2010, 37 (5), 728–735. 10.1016/j.molcel.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby F. G.; Masters E. I.; Kramer L.; Knowlton J. R.; Yao Y.; Wang C. C.; Hill C. P. Structural Basis for the Activation of 20S Proteasomes by 11S Regulators. Nature 2000, 408 (6808), 115–120. 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- Baugh J. M.; Viktorova E. G.; Pilipenko E. V. Proteasomes Can Degrade a Significant Proportion of Cellular Proteins Independent of Ubiquitination. J. Mol. Biol. 2009, 386 (3), 814–827. 10.1016/j.jmb.2008.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Lanzas R.; Castaño J. G. Proteins Directly Interacting with Mammalian 20S Proteasomal Subunits and Ubiquitin-Independent Proteasomal Degradation. Biomolecules 2014, 4 (4), 1140–1154. 10.3390/biom4041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynes R.; Pomatto L. C. D.; Davies K. J. A. Degradation of Oxidized Proteins by the Proteasome: Distinguishing between the 20S, 26S, and Immunoproteasome Proteolytic Pathways. Mol. Aspects Med. 2016, 50, 41–55. 10.1016/j.mam.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A.; Kwon Y. T. Degradation of Misfolded Proteins in Neurodegenerative Diseases: Therapeutic Targets and Strategies. Exp. Mol. Med. 2015, 47 (3), e147. 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D.; Saez I.; Dillin A. The Role of Protein Clearance Mechanisms in Organismal Ageing and Age-Related Diseases. Nat. Commun. 2014, 5, 5659. 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- Ruschak A. M.; Slassi M.; Kay L. E.; Schimmer A. D. Novel Proteasome Inhibitors to Overcome Bortezomib Resistance. J. Natl. Cancer Inst. 2011, 103 (13), 1007–1017. 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- McDaniel T. J.; Lansdell T. A.; Dissanayake A. A.; Azevedo L. M.; Claes J.; Odom A. L.; Tepe J. J. Substituted Quinolines as Noncovalent Proteasome Inhibitors. Bioorg. Med. Chem. 2016, 24 (11), 2441–2450. 10.1016/j.bmc.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell T. A.; Hurchla M. A.; Xiang J.; Hovde S.; Weilbaecher K. N.; Henry R. W.; Tepe J. J. Noncompetitive Modulation of the Proteasome by Imidazoline Scaffolds Overcomes Bortezomib Resistance and Delays MM Tumor Growth in Vivo. ACS Chem. Biol. 2013, 8 (3), 578–587. 10.1021/cb300568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. L.; Njomen E.; Sjögren B.; Dexheimer T. S.; Tepe J. J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12 (9), 2240–2247. 10.1021/acschembio.7b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmulski P. A.; Gaczynska M. Rapamycin Allosterically Inhibits the Proteasome. Mol. Pharmacol. 2013, 84 (1), 104–113. 10.1124/mol.112.083873. [DOI] [PubMed] [Google Scholar]
- Njomen E.; Osmulski P. A.; Jones C. L.; Gaczynska M.; Tepe J. J. Small Molecule Modulation of Proteasome Assembly. Biochemistry 2018, 57 (28), 4214–4224. 10.1021/acs.biochem.8b00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Ho P.; Chen C.-H. Activation and Inhibition of Proteasomes by Betulinic Acid and Its Derivatives. FEBS Lett. 2007, 581 (25), 4955–4959. 10.1016/j.febslet.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk S.; Chen W.-E. Synthetic Peptide-Based Activators of the Proteasome. Mol. Biol. Rep. 1997, 24 (1), 119–124. 10.1023/A:1006851428691. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 2015, 20 (1), 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Chang S. C.; Park S.; Finley D.; Cheng Y.; Goldberg A. L. Docking of the Proteasomal ATPases’ Carboxyl Termini in the 20S Proteasome’s Ring Opens the Gate for Substrate Entry. Mol. Cell 2007, 27 (5), 731–744. 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkowska J.; Gizyńska M.; Grudnik P.; Golik P.; Karpowicz P.; Giełdoń A.; Dubin G.; Jankowska E. Crystal Structure of a Low Molecular Weight Activator Blm-Pep with Yeast 20S Proteasome - Insights into the Enzyme Activation Mechanism. Sci. Rep. 2017, 7 (1), 6177. 10.1038/s41598-017-05997-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Vechio F. H.; Cerqueira F.; Augusto O.; Lopes R.; Demasi M. Peptides That Activate the 20S Proteasome by Gate Opening Increased Oxidized Protein Removal and Reduced Protein Aggregation. Free Radical Biol. Med. 2014, 67, 304–313. 10.1016/j.freeradbiomed.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Karpowicz P.; Osmulski P. A.; Witkowska J.; Sikorska E.; Giżyńska M.; Belczyk-Ciesielska A.; Gaczynska M. E.; Jankowska E. Interplay between Structure and Charge as a Key to Allosteric Modulation of Human 20S Proteasome by the Basic Fragment of HIV-1 Tat Protein. PLoS One 2015, 10 (11), e0143038. 10.1371/journal.pone.0143038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Post M.; Volk R.; Gao Y.; Li M.; Metais C.; Sato K.; Tsai J.; Aird W.; Rosenberg R. D.; Hampton T. G.; Li J.; Sellke F.; Carmeliet P.; Simons M. PR39, a Peptide Regulator of Angiogenesis. Nat. Med. 2000, 6 (1), 49–55. 10.1038/71527. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Lecker S.; Post M. J.; Hietaranta A. J.; Li J.; Volk R.; Li M.; Sato K.; Saluja A. K.; Steer M. L.; Goldberg A. L.; Simons M. Inhibition of Ubiquitin-Proteasome Pathway-mediated IκBα Degradation by a Naturally Occurring Antibacterial Peptide. J. Clin. Invest. 2000, 106 (3), 439–448. 10.1172/JCI9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaczynska M.; Osmulski P. A.; Gao Y.; Post M. J.; Simons M. Proline- and Arginine-Rich Peptides Constitute a Novel Class of Allosteric Inhibitors of Proteasome Activity. Biochemistry 2003, 42 (29), 8663–8670. 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- Anbanandam A.; Albarado D. C.; Tirziu D. C.; Simons M.; Veeraraghavan S. Molecular Basis for Proline- and Arginine-Rich Peptide Inhibition of Proteasome. J. Mol. Biol. 2008, 384 (1), 219–227. 10.1016/j.jmb.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmueller B. M.; Ferrell K.; Whitby F. G.; Heroux A.; Robinson H.; Myszka D. G.; Hill C. P. Structural Models for Interactions between the 20S Proteasome and Its PAN/19S Activators. J. Biol. Chem. 2010, 285 (1), 13–17. 10.1074/jbc.C109.070425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut T. W.Allosteric Regulatory Enzymes; Springer US, 2008. [Google Scholar]
- Stein R. L.; Melandri F.; Dick L. Kinetic Characterization of the Chymotryptic Activity of the 20S Proteasome. Biochemistry 1996, 35 (13), 3899–3908. 10.1021/bi952262x. [DOI] [PubMed] [Google Scholar]
- Coleman R. A.; Trader D. J. Development and Application of a Sensitive Peptide Reporter to Discover 20S Proteasome Stimulators. ACS Comb. Sci. 2018, 20 (5), 269–276. 10.1021/acscombsci.7b00193. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Kafri G.; Cheng Y.; Ng D.; Walz T.; Goldberg A. L. ATP Binding to PAN or the 26S ATPases Causes Association with the 20S Proteasome, Gate Opening, and Translocation of Unfolded Proteins. Mol. Cell 2005, 20 (5), 687–698. 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Alvarez-Castelao B.; Goethals M.; Vandekerckhove J.; Castaño J. G. Mechanism of Cleavage of Alpha-Synuclein by the 20S Proteasome and Modulation of Its Degradation by the RedOx State of the N-Terminal Methionines. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843 (2), 352–365. 10.1016/j.bbamcr.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Trader D. J.; Simanski S.; Dickson P.; Kodadek T. Establishment of a Suite of Assays That Support the Discovery of Proteasome Stimulators. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861 (4), 892–899. 10.1016/j.bbagen.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum A. K.; Dick T. P.; Keilholz W.; Schirle M.; Stevanovic S.; Dietz K.; Heinemeyer W.; Groll M.; Wolf D. H.; Huber R.; Rammensee H.-G.; Schild H. Cleavage Motifs of the Yeast 20S Proteasome Subunits Deduced from Digests of Enolase 1. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (21), 12504–12509. 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmulski P. A.; Gaczynska M. Atomic Force Microscopy Reveals Two Conformations of the 20 S Proteasome from Fission Yeast. J. Biol. Chem. 2000, 275 (18), 13171–13174. 10.1074/jbc.C901035199. [DOI] [PubMed] [Google Scholar]
- Osmulski P. A.; Hochstrasser M.; Gaczynska M. A Tetrahedral Transition State at the Active Sites of the 20S Proteasome Is Coupled to Opening of the α-Ring Channel. Structure 2009, 17 (8), 1137–1147. 10.1016/j.str.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Wu J.; Dong Y.; Chen S.; Sun S.; Ma Y. B.; Ouyang Q.; Finley D.; Kirschner M. W.; Mao Y. Conformational Landscape of the p28-Bound Human Proteasome Regulatory Particle. Mol. Cell 2017, 67 (2), 322–333. 10.1016/j.molcel.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Osmulski P. A. A.; Gaczynska M. Allosteric Regulators of the Proteasome: Potential Drugs and a Novel Approach for Drug Design. Curr. Med. Chem. 2006, 13 (2), 155–165. 10.2174/092986706775197926. [DOI] [PubMed] [Google Scholar]
- Wehmer M.; Rudack T.; Beck F.; Aufderheide A.; Pfeifer G.; Plitzko J. M.; Förster F.; Schulten K.; Baumeister W.; Sakata E. Structural Insights into the Functional Cycle of the ATPase Module of the 26S Proteasome. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (6), 1305–1310. 10.1073/pnas.1621129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaczynska M.; Osmulski P. A.. Atomic Force Microscopy of Proteasome Assemblies. In Atomic Force Microscopy in Biomedical Research; Braga P., Ricci D., Eds.; Methods in Molecular Biology, Vol. 736; Humana Press: New York, 2011; pp 117–132, 10.1007/978-1-61779-105-5_9. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. IUCr. Molecular Replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (1), 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. IUCr. REFMAC 5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (Part 4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. IUCr. TLSMD Web Server for the Generation of Multi-Group TLS Models. J. Appl. Crystallogr. 2006, 39 (1), 109–111. 10.1107/S0021889805038987. [DOI] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A. T. Free R Value: A Novel Statistical Quantity for Assessing the Accuracy of Crystal Structures. Nature 1992, 355 (6359), 472–475. 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (1), 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.