

We report the engineering and characterization of designed ankyrin proteins as potent neutralizers of TcdB toxin secreted by a hypervirulent ribotype 027 strain of Clostridium difficile. We further show that although TcdB toxins from both ribotype 027 and VPI 10461 interact efficiently with TcdB receptors CSPG4 and Pvrl3, TcdB027 lacks significant ability to bind the only known physiologically relevant TcdB receptor, Frizzled 1/2/7.
KEYWORDS: toxin, therapeutic, infection, protein, antibody, hypervirulent, enterotoxins
ABSTRACT
Clostridium difficile infection (CDI) is a leading cause of hospital-acquired diarrhea. In recent decades, the emergence of the “hypervirulent” BI/NAP1/027 strains of C. difficile significantly increased the morbidity and mortality of CDI. The pathogenesis of CDI is primarily mediated by the action of two toxins, TcdA and TcdB, with TcdB being the major virulent factor in humans. In this report, we describe the engineering of a panel of designed ankyrin repeat proteins (DARPins) that potently neutralize TcdB from the BI/NAP1/027 strains (e.g., TcdBUK1). The most effective DARPin, D16, inhibits TcdBUK1 with a 50% effective concentration (EC50) of 0.5 nM, which is >66-fold lower than that of the FDA-approved anti-TcdB antibody bezlotoxumab (EC50, ∼33 nM). Competitive enzyme-linked immunosorbent assays (ELISAs) showed that D16 blocks interactions between TcdB and its receptor, chondroitin sulfate proteoglycan 4 (CSPG4). The dimeric DARPin U3D16, which pairs D16 with DARPin U3, a disrupter of the interaction of TcdB with Frizzled 1/2/7 receptor, exhibits 10-fold-to-20-fold-enhanced neutralization potency against TcdB from C. difficile strains VPI 10463 (laboratory strain) and M68 (CF/NAP9/017) but identical activity against TcdBUK1 relative to D16. Subsequent ELISAs revealed that TcdBUK1 did not significantly interact with Frizzled 1/2/7. Computation modeling revealed 4 key differences at the Frizzled 1/2/7 binding interface which are likely responsible for the significantly reduced binding affinity.
IMPORTANCE We report the engineering and characterization of designed ankyrin proteins as potent neutralizers of TcdB toxin secreted by a hypervirulent ribotype 027 strain of Clostridium difficile. We further show that although TcdB toxins from both ribotype 027 and VPI 10461 interact efficiently with TcdB receptors CSPG4 and Pvrl3, TcdB027 lacks significant ability to bind the only known physiologically relevant TcdB receptor, Frizzled 1/2/7.
INTRODUCTION
Clostridium difficile is a bacterial pathogen that causes a variety of intestinal diseases collectively referred to as C. difficile infection (CDI). The symptoms of CDI range from mild diarrhea to life-threatening pseudomembranous colitis and toxic megacolon (1–3). Each year, C. difficile causes half a million infections and ∼15,000 deaths and results in over $1 billion in treatment-associated costs in the United States, leading the CDC to declare C. difficile an urgent threat to public health (4). A major culprit responsible for CDI is believed to be the administration of broad-spectrum antibiotics, which disrupts the natural gut microflora that would otherwise suppress C. difficile proliferation (5). It is estimated that ∼7% of hospitalized patients succumb to CDI, making C. difficile among the most dangerous nosocomial pathogens (5).
Prior to 2000, CDI could be effectively controlled by treatment with additional antibiotics such as metronidazole and vancomycin (6, 7). However, disturbing trends of increased morbidity and mortality due to relapse of C. difficile-infected patients after antibiotic treatment have since emerged (8–12). These trends correlated with the emergence of the “hypervirulent” BI/NAP1/027 strains of C. difficile (11, 13, 14) (C. difficile 027), which at one point were responsible for ∼1/3 of the CDI in the United States (15). Infection with C. difficile 027 is associated with more-severe disease and a higher death rate (15). The exact reason for the increased virulence of C. difficile 027 remains enigmatic, although many factors such as antibiotic resistance, sporulation ability, and toxin production have been proposed to contribute to its virulence (12, 16–19).
The pathology of CDI is primarily due to the action of two bacterial secreted exotoxins, toxin A (TcdA) and toxin B (TcdB) (20), that target small GTPases within the host cells, leading to disruption of tight junctions, loss of colonic epithelial barrier function, and bloody diarrhea (21). Administration of spores from nontoxigenic C. difficile strain M3 was found to significantly reduce CDI recurrence (22), highlighting a pivotal role of the toxins in CDI pathology. Vaccines against C. difficile toxins are currently being actively pursued and have enjoyed some preliminary successes (23–27). However, since CDI most often afflicts elderly hospitalized patients, the efficacy of vaccine in this unique population may be less than ideal (28, 29). The TcdB-neutralizing monoclonal antibody (MAb) bezlotoxumab was found to reduce the CDI recurrence rate from 28% to 16% in a phase III clinical trial (30) and was approved by the FDA in 2016 (31). Curiously, despite its toxin neutralization ability, bezlotoxumab did not improve the initial cure rate of CDI in patients (31) and is not approved by the FDA as a treatment for CDI. Bezlotoxumab neutralizes TcdB by directly blocking its carbohydrate binding pocket and thus preventing its attachment to the colonic mucosal cells (32). Although bezlotoxumab exhibits potent neutralization activity against TcdB from a broad range of C. difficile strains, its potency is significantly (∼185-fold) weaker against toxin from C. difficile 027 than against that from laboratory strain VPI 10463 (33).
Recently, our laboratory successfully engineered several designed ankyrin repeat proteins (DARPins) with ultrapotent neutralization activity against TcdB using phage display coupled with functional screening (34). DARPin is a small non-antibody-binding scaffold that exhibits very high thermostability and low immunogenicity and has the potential to be produced at a low cost in microbial cells (35, 36). The best dimer DARPin from our previous study—DLD4—exhibited 50% effective concentration (EC50) values of 4 pM and 16 pM against TcdB from C. difficile strains VPI 10463 (TcdBVPI, ribotype 087) and M68 (TcdBM68, ribotype 017), respectively, representing ∼330-fold-higher and ∼30-fold-higher in vitro potency than bezlotoxumab (34). DLD4 consists of two monomeric DARPins, 1.4E and U3. Cryo-electron microscopy (Cryo-EM) structural studies combined with competitive enzyme-linked immunosorbent assays (ELISAs) revealed that 1.4E and U3 interfere with the interaction between TcdB and its receptors chondroitin sulfate proteoglycan 4 (CSPG4) (37) and Frizzled receptor 1/2/7 (FZD1/2/7) (38), respectively. Unfortunately, like bezlotoxumab, all of these DARPins showed significantly weaker activity against TcdB from C. difficile 027 (TcdBUK1). TcdBUK1 and TcdBVPI share a high level of sequence homology (92% identical). However, critical sequence differences at the CSPG4 and FZD1/2/7 interacting regions render anti-TcdBVPI DARPins powerless against TcdBUK1.
In this study, we performed phage panning and functional screening to identify a panel of new DARPins with significantly improved TcdBUK1 neutralization activity. The best DARPin, D16, neutralized TcdBUK1 with an EC50 of 0.5 nM, making it >66-fold more potent than bezlotoxumab (EC50 of ∼33 nM) in vitro. Importantly, D16 also potently neutralizes TcdBVPI (EC50 of 5 nM) and TcdBM68 (EC50 of 1.6 nM). Competitive ELISAs showed that all our anti-TcdBUK1 DARPins block the toxin interaction with CSPG4. Since DARPin U3 from our previous study impedes TcdB interaction with FZD1/2/7 receptor, which binds an epitope distinct from that of CSPG4, we constructed multiple-dimer DARPins composed of D16 and U3 with different linker sizes and topologies with a view to creating synergistic blocking of toxin-receptor interactions. These dimers were expected to exhibit enhanced neutralization activity through the avidity effect. All the dimer DARPins exhibited (10-fold-to-20-fold) enhanced neutralization potency against TcdBVPI and TcdBM68, pointing to their potential as new antitoxin biologics for treating CDI and/or preventing its recurrence.
Intriguingly, none of the constructed dimer DARPins showed enhanced neutralization activity against TcdBUK1. Subsequent ELISAs revealed that, unlike TcdBVPI, which binds strongly to both purified ectodomains of CSPG4 and FZD2, TcdBUK1 lacks significant ability to interact with FZD2 despite strong ability to associate with CSPG4. Consistent with this result, TcdBUK1 was found to be minimally toxic to Caco-2 colon epithelium cells, which express multiple Frizzled proteins, including FZD2 and FZD7 (38, 39), but lack CSPG4 (38, 40). A closer analysis of the crystal structure of TcdB Frizzled binding domain (FBD) (TcdB-FBD; PDB code: 6C0B) revealed multiple differences between TcdBUK1 and TcdBVPI at the FBD binding interface which likely abolish or significantly weaken the TcdBUK1-FZD1/2/7 interaction.
RESULTS
Selection of TcdB-neutralizing monomer DARPins.
DARPins were designed based on repeat modules of natural ankyrin proteins and consist of an N-terminal capping repeat (N-cap), three (N3C) internal ankyrin repeats (ARs), and a C-terminal capping repeat (C-cap) (41). In a DARPin library, each internal repeat contains six randomized positions on the flexible surface-exposed loop and one partially randomized position on the hinge region, yielding a total of 18 plus 3 randomized positions in each N3C DARPin. Additional mutations at the framework positions can emerge during repeated PCR amplification. A previously constructed DARPin library comprising a complex of ∼2 × 109 variants was used in phage panning against biotinylated TcdBUK1. DARPin variants from the 4th round of panning, which showed significantly elevated levels of TcdBUK1 binding ability (see Fig. S1 in the supplemental material), were cloned into the pET28a vector and expressed in BL21(DE3) Escherichia coli cells and underwent functional screening. Among the 760 individual clones screened, 57 clones rescued Vero cell viability from TcdBUK1 toxicity by >70%. All these clones were sequenced, and 16 unique clones were identified (see Table S1 in the supplemental material). Interestingly, these 16 unique DARPins were found to be composed of only 6 distinct ARs in 4 different configurations (Fig. 1), suggesting that these DARPins likely bind to overlapping epitopes on TcdBUK1. Four DARPins (one from each unique configuration [D2, D3, D8, and D16]) that lacked any additional framework mutations were selected for further characterization.
FIG 1.
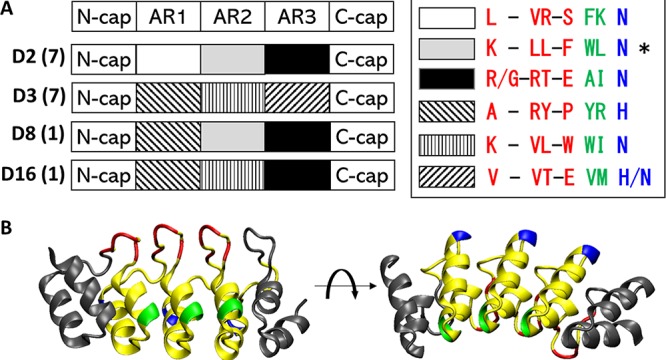
(A) Schematics of the different DARPins. The identity of the randomized residues in each repeat module is indicated in the legend on the right. The different colors represent the different positions on the ankyrin repeat module and are pictorially represented in panel B. The number of clones with the same AR configuration (with or without additional framework mutations) is shown in parentheses. The asterisk (*) indicates a DARPin that contains one framework mutation. (B) A crystal structure illustrating the structure of DARPin. Residues colored in red, green, and blue are randomized in the library.
Purification of DARPins. Each DARPin was expressed in 5 ml of E. coli culture and purified by Ni-NTA affinity chromatography. Purified DARPins were analyzed by SDS-PAGE. Download FIG S1, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Amino acid sequences of the unique clones of anti-TcdBUK1 DARPins. Download Table S1, DOCX file, 0.02 MB (19.1KB, docx) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In addition to neutralizing TcdBUK1, all four DARPins also inhibited TcdBVPI and TcdBM68 (Fig. 2A). The best DARPin, D16, exhibited an EC50 of 0.5 nM against TcdBUK1, representing a >66-fold-higher in vitro potency than bezlotoxumab against the same toxin. D16 also exhibited low nanomolar neutralization potency against TcdBVPI (ribotype 087, EC50 of 5.2 nM) and TcdBM68 (ribotype 017, EC50 of 1.6 nM), representing lower potencies for these toxins relative to bezlotoxumab. The ability of the DARPins to bind the different toxins generally matches their toxin neutralization potency (Fig. 2B), with D16 being both the strongest binder and the most potent neutralizer of all the toxins.
FIG 2.

(A and B) DARPins strongly exhibited the ability to neutralize (A) and bind (B) the different TcdB toxins. conc., concentration. (C) TcdB neutralization potency of different DARPins and bezlotoxumab. For neutralization assays, serially diluted immobilized-metal affinity chromatography (IMAC)-purified DARPins were mixed with the appropriate toxin and then added to Vero cells seeded the night before in 96-well plates. The cell viability was quantified by the CellTiterGlo assay 72 h later and normalized to naive Vero cells. For ELISAs, the MaxiSorp plates were coated with the appropriate toxin followed by treatment with serially diluted DARPins. The amounts of plate-bound DARPins (containing Myc tags) were quantified using an anti-c-Myc antibody. Data in panel A represent averages of results from at least 2 independent experiments. Data presented in panel B are representative of results from two independent experiments performed in duplicate.
Mechanism of TcdBUK1 neutralization by monomeric DARPins.
There are three known TcdB receptors: chondroitin sulfate proteoglycan 4 (CSPG4), poliovirus receptor-like 3 (PVRL3 or NECTIN3), and members of the Frizzled protein family FZD1/2/7, which share identical sequences in the TcdB-binding region (37, 38, 42). In our previous study performed with TcdBVPI, most of the identified antitoxin DARPins interfered with the TcdB-CSPG4 interaction based on Cryo-EM structural studies and competitive ELISA results (34). The high frequency of antitoxin DARPin hits that block CSPG4 binding seen in our previous study likely stemmed from the use of Vero cells, which express a high level of CSPG4 (40), in our functional screening.
We repeated the competitive ELISA for the four unique DARPins (i.e., D2, D3, D8, and D16). The wells of the ELISA plate were first coated with TcdBUK1 (4 μg/ml) overnight at 4°C. The next day, a green fluorescent protein (GFP)-tagged extracellular domain of CSPG4 (CSPG4-EC-GFP) was added in the presence or absence of the different DARPins. The plate was incubated at room temperature for 2 h, and the amounts of bound CSPG4-EC-GFP were detected using anti-GFP antibody. As shown in Fig. 3, all four DARPins significantly reduced the binding signal from CSPG4-EC-GFP, with D16 producing the most signal reduction, indicating that all these monomer DARPins interfere with the TcdBUK1-CSPG4 interaction. Thus, consistent with our previous finding, inhibition of CSPG4 interaction emerged as a dominant mechanism used by antitoxin DARPins in Vero cell-based functional screening.
FIG 3.
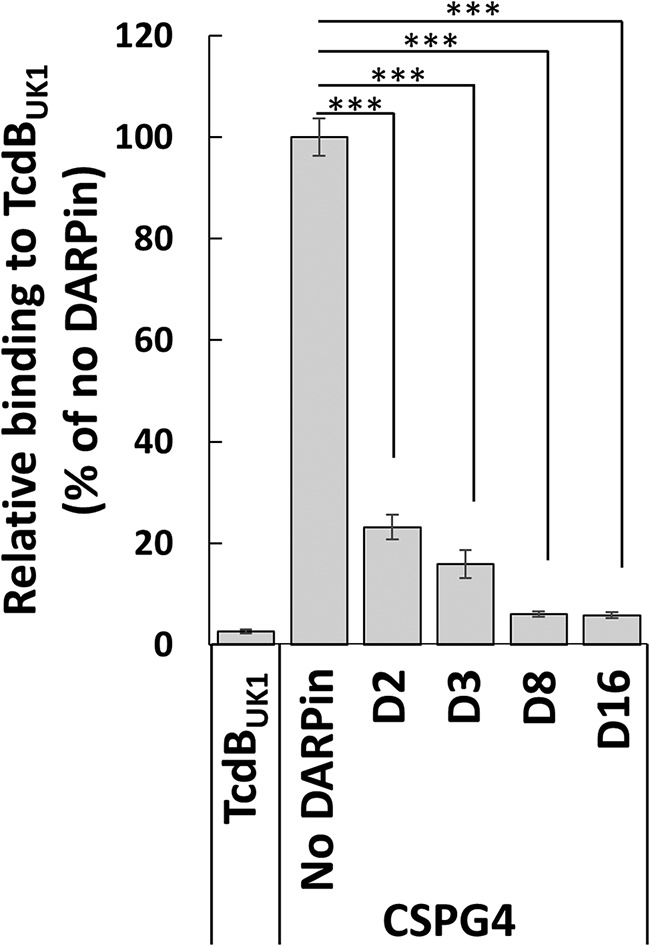
Anti-TcdBUK1 DARPins block the interaction between TcdB and its receptor CSPG4. ***, P < 0.001 (t test). The wells of an ELISA plate were coated with TcdBUK1 followed by treatment with a 1 nM concentration of a CSPG4 extracellular domain-GFP fusion protein alone or in a mixture with a 250 nM concentration of the indicated DARPins. The amounts of plate-bound CSPG4 were detected using an anti-GFP antibody. The data are representative of results from two independent experiments and of averages of results from quadruplicate samples.
Dimeric DARPins with enhanced potency against TcdBVPI and TcdBM68.
Fusion of multiple binders to nonoverlapping epitopes has been reported to significantly enhance the overall target-binding affinity via the avidity effect (43, 44). Previously, we identified DARPin U3, which interferes with the interaction between TcdB and its receptor FZD1/2/7 (34). Dimer DARPin—DLD4—consisting of U3 and 1.4E joined by a 3× GGGGS linker exhibited >100-fold-higher neutralization potency against TcdBVPI than either constituent monomer. Since both D16 and 1.4E interfere with the TcdB-CSPG4 interaction, we reasoned that a dimeric DARPin (Fig. S2B) comprising U3 and D16 joined by the same linker should exhibit stronger toxin neutralization potency than D16 alone.
Schematic of monomeric and dimeric DARPins. (A) Monomeric DARPins contain an N-terminal 6×His tag and a Myc tag. The sequence from D16 is presented as an example. (B) The constituent DARPins in a dimeric DARPin are separated by a (GGGGS) × 3 linker sequence. The sequence from D16U3 is presented as an example. Download FIG S2, TIF file, 1.1 MB (1.2MB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Indeed, dimeric DARPin U3D16 showed 10-fold-to-20-fold-higher activity toward TcdBVPI and TcdBM68 than D16 alone (Fig. 4A and B). However, surprisingly, the TcdBUK1 neutralization ability shown by U3D16 was weaker than that seen with D16 alone (Fig. S3A). We subsequently prepared additional dimeric DARPins with a reverse configuration (i.e., D16U3) and/or different linker lengths (4×, 5×, and 6× GGGGS), but to no avail (Fig. 3 and 5). Furthermore, a mixture containing both D16 and U3 showed activity identical to that seen with D16 alone (Fig. S3C). These results suggest that, although U3 efficiently neutralizes TcdBVPI and TcdBM68, it is powerless against TcdBUK1. Subsequent ELISAs confirmed that U3 lacks the ability to bind TcdBUK1 (Fig. 4C).
FIG 4.
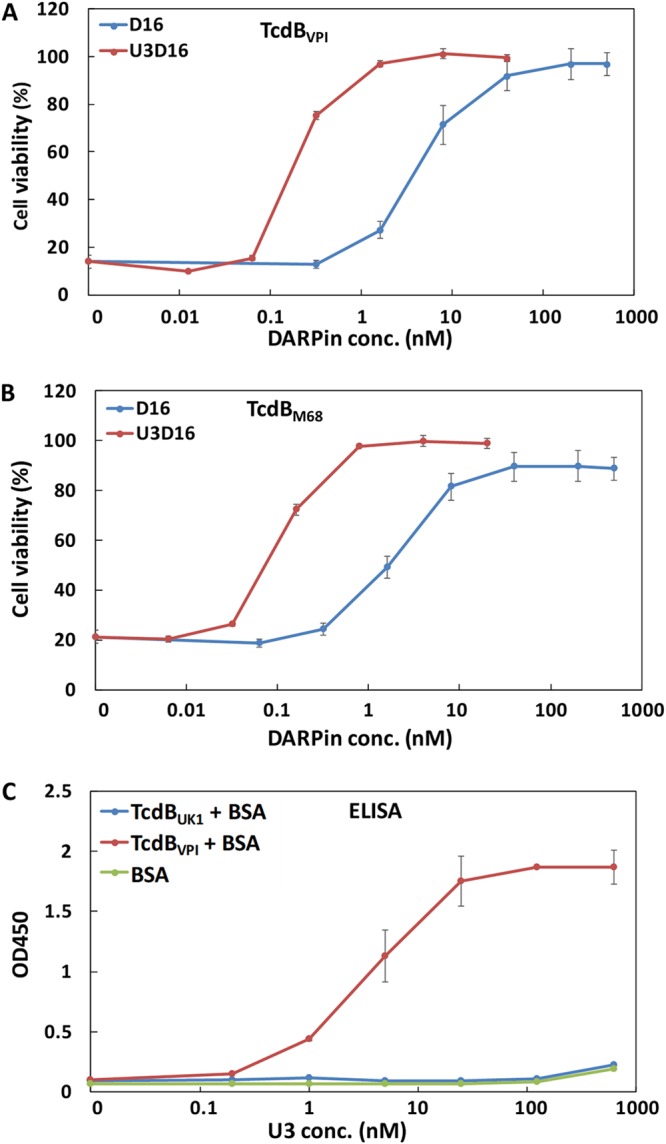
DARPin dimer U3D16 showed enhanced neutralization ability against TcdBVPI (A) and TcdBM68 (B). Serially diluted DARPins were mixed with the appropriate toxins and then added to Vero cells that had been seeded the night before. Cell viability was quantified by the CellTiterGlo assay 72 h later and normalized to naive Vero cells. The error bars represent mean deviations of results from two independent experiments. (C) U3 lacks the ability to bind to TcdBUK1 as determined by ELISA. The ELISA plates were coated with the appropriate toxin and then blocked with BSA prior to the addition of serially diluted DARPin U3. The amounts of plate-bound DARPin were quantified using an anti-c-Myc antibody. The data are representative of results from two independent experiments performed in duplicate. OD450, optical density at 450 nm.
FIG 5.

TcdBUK1 lacks significant ability to interact with FZD2. The wells of an ELISA plate were coated with TcdBVPI or TcdBUK1 followed by treatment with CSPG4-EC-GFP (1 nM) (A), FZD2-Fc (4 nM) (B), or PVRL3 (100 nM) (C). The amount of plate-bound CSPG4, FZD2, or PVRL3 was detected using each of the respective antibodies. Results are representative of at least two independent experiments. Error bars represent the mean deviations of results from duplicate samples.
Neutralization potency of engineered dimeric DARPins against UK1 TcdB. Purified DARPins were added to Vero cells together with TcdBUK1 (1 pg/ml). Cell viability was quantified by the CellTiter Glo assay 72 h later and normalized to naïve Vero cells. Experiments were conducted in triplicate. Error bars represent the standard deviation. (A) D16 versus U3D16. (B) D16 versus D16U3. (C) D16 versus mixture of D16 and U3. (D) D16 versus U3D16 and D16U3 with longer linkers. Download FIG S3, TIF file, 0.8 MB (804.5KB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
TcdBUK1 lacks significant ability to interact with FZD2.
The inability of U3 to bind/neutralize TcdBUK1 was surprising. DARPin U3 neutralizes TcdB by interfering with the interaction between TcdB and the FZD1/2/7 receptor (34). The members of the Frizzled family of receptors are important for Wnt signaling, a key signaling pathway that regulates cell proliferation and self-renewal (45). Unlike CSPG4, which is not present in the colon epithelium but abundant in the subepithelium layer, FZD2 and FZD7 are highly expressed in mouse and human colonic epithelium, making them the most physiologically relevant receptors for TcdB (38).
The lack of binding of U3 to TcdBUK1 prompted us to examine the interaction between TcdBUK1 and FZD2. ELISA plates were coated with TcdBVPI or TcdBUK1 prior to the addition of the extracellular domain of CSPG4 or FZD2. After thorough washing, the amounts of plate-bound CSPG4 and FZD2 were detected using the respective antibodies. As shown in Fig. 5, CSPG4 can bind efficiently to both TcdBVPI and TcdBUK1, with the TcdBUK1 interaction being the stronger. In contrast, only TcdBVPI was able to significantly associate with FZD2 at the test concentration, indicating that the binding affinity of TcdBUK1 to FZD2 is much weaker than that shown by TcdBVPI. The extracellular domain of PVRL3 appeared to bind TcdBVPI and TcdBUK1 with similar levels of affinity (Fig. 5C).
To interrogate the inability of TcdBUK1 to interact with FZD1/2/7 from a different angle, we compared the levels of toxicity of TcdBUK1 and TcdBVPI in Caco-2 and Vero cells (Fig. 6). Caco-2 cells lack CSPG4 but express multiple Frizzled proteins, including FZD2 and FZD7 (38, 39). On the other hand, Vero cells are abundant with respect to CSPG4, FZD2, and PVRL3 (40). In Vero cells, TcdBUK1 (50% lethal concentration [LC50], 0.15 ± 0.01 pg/ml) appeared to be 6.5-fold more toxic than TcdBVPI (LC50, 0.98 ± 0.04 pg/ml). However, in Caco-2 cells, the order was reversed and TcdBUK1 (LC50, >125 pg/ml) appeared to be far less toxic than TcdBVPI (LC50, ∼10 pg/ml). These data indicate that TcdBUK1 lacks significant ability to enter cells through the FZD1/2/7 receptor and are consistent with our finding that TcdBUK1 exhibits poor binding affinity for FZD2 (Fig. 5).
FIG 6.
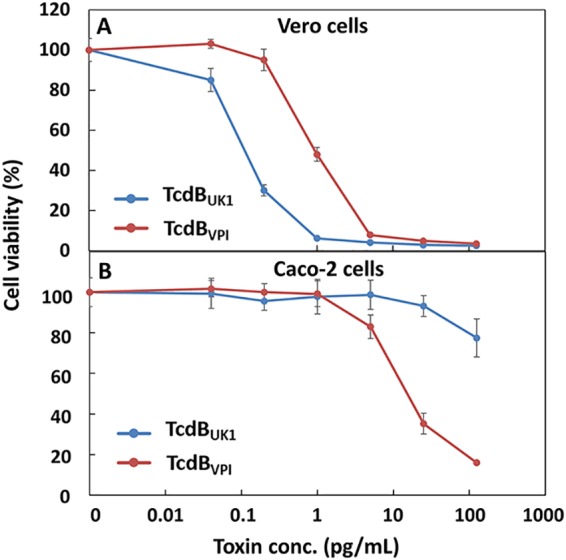
Vero and Caco-2 cells exhibit different levels of sensitivity to TcdBUK1 and TcdBVPI. Vero cells (A) or Caco-2 cells (B) were incubated with serial dilutions of TcdBUK1 or TcdBVPI. The cell viability was quantified using CellTiter-Glo reagent 72 h later and normalized to naive cells. The error bars represent standard deviations of results from two independent experiments performed in triplicate.
DISCUSSION
C. difficile infection (CDI) is the most common cause of antibiotic-associated diarrhea and gastroenteritis-associated death in developed countries. The prevalence, mortality, and costs associated with CDI make C. difficile a major threat to public health. The pathology of CDI stems primarily from the two exotoxins secreted by C. difficile bacteria, TcdA and TcdB, of which TcdB is considered the primarily virulence factor in human (46). C. difficile was first reported to cause human disease in 1978 (47, 48). In the past, CDI has been routinely treated with supportive therapy and regimens of antibiotics. However, the cure rate has been steadily decreasing over the last decades largely due to the emergence of a hypervirulent (NAP1/BI/027) strain of C. difficile (9–11).
Previously, using phage display coupled with functional screening, we successfully isolated a panel of DARPins with potent neutralization activity against TcdB from the laboratory strain of C. difficile VPI 10463 (ribotype 087) and the clinical strain M68 (NAP9/CF/017). The goal of this study was to identify strong binders/neutralizers of TcdB from hypervirulent but intractable C. difficile strain UK1 (NAP1/BI/027). The sequence of TcdBUK1 shares 92% and 88% similarity with those of TcdBVPI and TcdBM68, respectively. In comparison, TcdBVPI shares 93.7% sequence similarity with TcdBM68. TcdB from a NAP1/BI/027 strain was reported to induce a greater cytopathic effect on a variety of cell types (49) and to exhibit a substantially lower lethal dose and more-extensive brain hemorrhaging in mice than that were seen with the laboratory strain (50).
To investigate these factors, we performed four rounds of panning of phage displaying a randomized library of designed ankyrin repeat proteins (DARPins) against TcdB from the UK1 strain of C. difficile. TcdBUK1 binders that emerged from this approach were subjected to an in vitro potency screen carried out on Vero cells, resulting in the identification of a panel of DARPins with potent neutralization activity against TcdBUK1. DARPins are synthetic ankyrin repeat proteins composed of three internal ankyrin repeat (AR) domains sandwiched between N-capping and C-capping domains. Interestingly, the top 57 anti-TcdBUK1 DARPins that emerged from this study share the same 6 ARs in 4 different configurations, suggesting that these DARPins likely target regions surrounding a common epitope (Fig. 1). The four DARPins representing each unique repeat configuration and without any framework mutations were further characterized. The most effective DARPin, D16, neutralized TcdB from C. difficile strains UK1, VPI 10463, and M68 with EC50 values of 0.5 nM, 5.2 nM, and 1.6 nM, respectively. The in vitro potency of D16 toward TcdBUK1 is >66-fold higher than that of the toxin-neutralizing therapeutic antibody bezlotoxumab (EC50 of >33 nM) (Fig. 2).
All four unique anti-TcdBUK1 DARPins from our screen were found to block the interaction of TcdB with the receptor CSPG4 (Fig. 3), much like the antitoxin DARPins. This finding is consistent with results from our previous study in which the vast majority of the isolated anti-TcdBVPI DARPins neutralized the toxin by interfering with the TcdB-CSPG4 interaction (34). There are currently three known receptors for TcdB: CSPG4, Frizzled 1/2/7, and PVRL3. Although Vero cells express high levels of all these receptors (40), the recurrent emergence of CSPG4-interfering DARPins from functional screens using Vero cells points to a dominant role of CSPG4 in mediating TcdB entry in these cells.
Previously, we identified DARPin 1.4E, which inhibited the CSPG4-TcdBVPI interaction but showed neither activity toward nor binding to TcdBUK1 (34). Since both TcdBVPI and TcdBUK1 bind CSPG4 (Fig. 5A), the ability of the antitoxin DARPins reported in this study to neutralize both TcdBUK1 and TcdBVPI, albeit with different potencies, indicates that these DARPins bind epitopes on TcdB that are distinct from those bound by DARPin 1.4E and that these epitopes partially overlap the footprint of CSPG4, which in part overlaps the epitope of DARPin 1.4E. Unfortunately, the data representing the binding interface of CSPG4 and DARPin 1.4E lack sufficient resolution to support detailed mutagenesis studies to elucidate the exact epitopes for these DARPins (34).
With a view to creating higher-potency antitoxin molecules, the dimeric DARPin U3D16 was created by fusing monomeric D16 to DARPin U3, which was earlier found to disrupt the interaction of TcdB from C. difficile VPI with the Frizzled 1/2/7 receptor. U3D16 exhibits 10-fold-to-20-fold-enhanced neutralization potency against TcdBVPI and TcdBM68 relative to the D16 monomer, likely through an avidity effect (Fig. 4). However, unexpectedly, all the tested dimeric DARPins composed of U3 and D6 (which included variations in configuration and linker length) not only did not show enhanced activity but showed ∼10-fold-reduced activity (see Fig. S3 in the supplemental material). U3 targets an adjacent epitope on TcdB and blocks its interaction with FZD1/2/7 (34). Further studies showed that neither U3 nor FZD2 bound TcdBUK1 efficiently (Fig. 4C and 5). To corroborate this finding, we compared the levels of toxicity of TcdBUK1 and TcdBVPI in Caco-2 cells and Vero cells. The Caco-2 cells are derived from the colon epithelium and lack detectable CSPG4 expression (38). On the other hand, Vero cells, derived from the kidney epithelium cells of an African green monkey, express all three known TcdB receptors (CSPG4, FZD2/7, and PVRL3) (40). At the same molar concentration, TcdBUK1 is more toxic to Vero cells than TcdBVPI but is far less toxic to Caco-2 cells than TcdBVPI (Fig. 6), indicating that TcdBUK1 lacks significant ability to enter cells via the FZD1/2/7 receptor. Our ELISA results indicated that, while TcdBUK1 and TcdBVPI bound PVRL3 with similar affinities, TcdBUK1 associated more strongly with CSPG4 than TcdBVPI and lacked significant ability to bind to FZD1/2/7 (Fig. 5). The higher affinity of TcdBUK1 than TcdBVPI for CSPG4 is likely responsible for its greater toxicity in Vero cells, whereas the weaker affinity of TcdBUK1 for FZD1/2/7 may explain its reduced toxicity in Caco-2 cells.
Sequence alignment of TcdBUK1 with TcdBVPI revealed six residue differences at the FZD1/2/7 binding interface (Fig. S4). Among these, we posit that four differences are most likely responsible for the weaker affinity between TcdBUK1 and FZD1/2/7, namely, E1468K, D1501N, Y1509C, and F1597S. The E1468 and Y1509 residues in TcdBVPI form charge interactions/hydrogen bonds with Q83 and H74, respectively, in FZD2 (Fig. 7A). The presence of a positively charged Lys in position 1468 of TcdBUK1 instead of a negatively charged glutamic acid likely abolishes this hydrogen bond interaction. The same applies to the presence of a relatively small cysteine residue in position 1509 of TcdBUK1 in place of tyrosine. Residue D1501 in TcdBUK1 was previously shown to be critical for binding to FZD2, as the D1501A mutation abolished the ability of TcdBVPI to interact with FZD2 (51). In TcdBUK1, position 1501 is occupied by polar residue Asn (Fig. 7B), which reduces the original two hydrogen bond interactions with K127 in FZD2 to only one. Finally, the F1597S substitution may reduce the hydrophobic interaction between Phe and the nearby F130 on FZD2. In fact, an earlier study found that the F1597G mutation in TcdBVPI abolished its ability to associate with FZD2 in a pulldown assay (51). Collectively, the data indicate that there appears to be a strong structural basis for the lack of interaction between TcdBUK1 and FZD1/2/7.
FIG 7.

Overlay of homology model of FBD from TcdBUK1 with the crystal structure of TcdBVPI (PDB code: 6C0B) in two different views (A, side view; B, front view). The toxin and the cysteine-rich domain 2 (CRD2) from FZD2 are represented in brown and green, respectively. Key positive allosteric modulator (PAM) (silver) binding residues from the toxin (brown) and CRD2 (green) are shown as stick models. The FBD sequence from TcdBM68 is identical to that from TcdBVPI.
Sequence alignment of the FBD from TcdBUK1 and TcdBVPI. Key residues at the FZD binding interface are shown in blue and red. Download FIG S4, TIF file, 0.7 MB (729.3KB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The weakened ability of TcdBUK1 to use FZD1/2/7 for cell entry is surprising, as FZD7 is abundant in the colon epithelium. On the other hand, CSPG4 is absent from the colon epithelium but is predominantly expressed in multinucleated intestinal subepithelial myofibroblasts (52). Independently, the same phenomenon was recently reported by Chung et al., who noted a lack of interaction between TcdBR20291 (with the same amino acid sequence as TcdBUK1) and FZD2-Fc by Western blotting (53), and by Lopez-Urena et al., whose results demonstrated that the uptake of TcdBVPI by HeLa cells (expressing both CSPG4 and Frizzled receptors [38]) was not blocked by TcdBNPI (with the same protein sequence as TcdBUK1) (57).
The explanation for the reduced activity of the dimeric DARPin composed of U3 and D16 toward TcdBUK1 compared to D16 is not immediately clear. It may due to a nonspecific interaction between U3 and D16 which partially obscures the target binding interface on D16. The presence of a binding partner of U3 on TcdBVPI and TcdBM68 draws U3 away from D16, enabling the two DARPins to simultaneously bind the toxin and enhancing the neutralization potency.
In summary, we identified a panel of DARPins with potent neutralization potency toward TcdB from the hypervirulent UK1 strain of C. difficile (NAP1/BI/027). These DARPins neutralize TcdB by blocking its interaction with the receptor CSPG4. We further showed that TcdBUK1 does not strongly associate with FZD1/2/7, bringing into question the significance of this receptor in CDI caused by ribotype 027 hypervirulent strains of C. difficile.
MATERIALS AND METHODS
Protein expression and purification.
TcdBUK1 was recombinantly expressed in Bacillus megaterium cells and purified via the use of a nickel-nitrilotriacetic acid (Ni-NTA) column essentially as described previously (54). The fractions containing TcdB were combined and concentrated and subjected to buffer exchange using phosphate-buffered saline (PBS) (10-fold dilution of 10× PBS [Fisher catalog no. BP3991]) and ultrafiltration units (Amicon; molecular-weight cutoff [MWCO], 100 kDa). Protein purity was confirmed using SDS-PAGE. The concentration of purified protein was determined by calculating the absorbance at 280 nm with a theoretical extinction coefficient of 293,620 M−1 cm−1. We typically obtain ∼2 mg of purified toxin per liter of culture. The purified protein was stored at −20°C in 50% glycerol. TcdBVPI, TcdBM68, CSPG4-EC-GFP, and bezlotoxumab were recombinantly expressed and purified as described previously (34, 54).
Phage panning and functional screening.
An in-house N3C DARPin library with a diversity level of ∼109 was used in the phage panning essentially as described previously (34, 55). Purified TcdBUK1 was biotinylated via the use of EZ-Link-Sulfo NHS-LC [succinimidyl 6-(biotinamido)hexanoate] biotin (Pierce) and used as the target protein in four rounds of sequential phage panning. A significant level of TcdBUK1 binding enrichment was observed after 4 rounds of selection using phage ELISA (55), indicative of successful phage panning.
DARPin variants from the fourth-round phage library were cloned into pET28a vector (containing an N-terminal His tag and a Myc tag; see Fig. S2A in the supplemental material) via the use of BamHI and HindIII restriction sites. A total of 760 individual E. coli BL21(DE3) clones were picked and grown in v-bottom 96-well plates (200 μl/well) in Luria broth (LB) supplemented with kanamycin (50 μg/ml) at 37°C with shaking for ∼18 h. Cells were harvested by centrifugation (1,048 × g for 10 min at 4°C). Each cell pellet was resuspended in 200 μl lysis buffer (PBS supplemented with 1 mM CaCl2, 0.5 mM EDTA, and 200 μg/ml lysozyme) and incubated at 37°C for 30 min. These cells then underwent 2 cycles of freeze-thaw between −80°C and 37°C and centrifugation at 1,048 × g for 20 min at 4°C. The soluble cell lysates (2 μl/well) were added to Vero cells that had been seeded the previous day at 1,500 cells/well together with TcdBUK1 (1 pg/ml final concentration) in 200 μl complete growth medium (Dulbecco’s modified Eagle’s medium [DMEM] supplemented with 10% fetal bovine serum, 1× nonessential amino acids, and 1× antibiotic antimycotic (Life Technologies catalog no. 15240062). The plates were incubated at 37°C and 5% CO2 for 72 h, and the viability of these Vero cells was quantified by the use of CellTiter-Glo reagent (Promega) and normalized to that of naive Vero cells.
Candidate DARPin clones were grown in 5 ml autoinduction medium (6 g/liter Na2HPO4, 3 g/liter KH2PO4, 20 g/liter tryptone, 5 g/liter yeast extract, 5 g/liter NaCl, 0.6% [vol/vol] glycerol, 0.05% [wt/vol] glucose, 0.2% [wt/vol] lactose) supplemented with 50 μg/ml kanamycin at 37°C with shaking for ∼18 h. These DARPins were purified by the use of Ni-NTA beads and subjected to buffer exchange using PBS and Zeba desalting columns (Thermo Scientific catalog no. 89877). Typically, 5 ml of bacterial culture yields ∼100 μg of purified DARPin. Protein purity was estimated to be >90% based on SDS-PAGE results (Fig. S1).
In vitro toxicity and neutralization assay.
To compare the levels of toxicity of the different toxins (i.e., TcdBUK1, TcdBVPI, and TcdBM68), purified toxins were serially diluted and added to Vero cells or Caco-2 cells that had been seeded in 96-well plates the day before at 1,500 cells/well. The plates were incubated at 37°C and 5% CO2, and the viability of these cells was quantified 72 h later using CellTiter-Glo reagent. The minimum concentrations of the toxin that led to <20% Vero cell viability were determined to be 1 pg/ml for TcdBUK1, 5 pg/ml for TcdBVPI, and 5 pg/ml for TcdBM68.
To determine the neutralization potencies of the different DARPins, serially diluted DARPins were added to Vero cells (seeded at 1,500 cells/well the day before) together with the minimum dose of the appropriate toxin that led to <20% cell viability.
Enzyme-linked immunosorbent assay (ELISA).
To compare the abilities of the different DARPins to bind TcdBUK1, the wells of MaxiSorp immunoplates (Nunc) were coated with 100 μl of TcdBUK1 (4 μg/ml) at 4°C overnight. The next day, the wells were washed and blocked with PBSTB buffer (PBS supplemented with 0.1% Tween 20 and 0.2% bovine serum albumin [BSA]) and were then incubated with serially diluted DARPins for 2 h at room temperature. After thorough washing, the amount of bound DARPin was quantified using mouse anti-c-Myc antibody (Invitrogen catalog no. 13-2500) (0.5 μg/ml) and horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Jackson Immuno Research catalog no. 115-035-146) (0.13 μg/ml) followed by color development using 3,3′,5,5′-tetramethylbenzidine (TMB).
To determine the interaction between CSPG4/FZD2/PVRL3 and TcdBUK1/VPI, the immunoplates were first coated with the appropriate toxin as described above. After blocking and thorough washing, CSPG4-EC-GFP (1 nM), a GFP-tagged extracellular domain of CSPG4 (34); FZD2-Fc (R&D Systems catalog no. 1307-FZ-050) (4 nM); or PVRL3 (Sino Biological catalog no. 10852-H08H) (100 nM) was added to each well alone or in the presence of the relevant DARPin (250 nM) followed by incubation at room temperature for 2 h. After thorough washing, the amount of bound CSPG4-EC-GFP was determined using rabbit anti-GFP antibody (Proteintech catalog no. 50430-2-AP) (0.08 μg/ml) plus HRP-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology catalog no. SC-2004) (0.8 μg/ml), and the bound FZD2-Fc amount was determined using HRP-conjugated goat anti-human antibody (Jackson Immuno Research catalog no. 109-035-088) (0.2 μg/ml), while the bound PVRL3 was detected using a goat anti-PVRL3 antibody (R&D Systems catalog no. AF3064) plus HRP-conjugated donkey anti-goat antibody (Santa Cruz Biotechnology catalog no. SC-2020) followed by color development using TMB.
Modeling study.
A homology model of TcdBUK1 Frizzled binding domain (FBD; amino acids 1284 to 1803) was constructed using SWISS-MODEL and was overlaid onto the FBD of TcdBVPI (PDB code: 6C0B). The complex was visualized using Visual Molecular Dynamics (VMD) (56).
Data availability.
The sequences of our engineered proteins are presented in Table S1.
ACKNOWLEDGMENTS
The funding for this work was graciously provided by the National Institutes of Health through grants R21AI126025, DP2OD024146, and R21AI137803 (to Z.P., R.S., and Z.C.).
Z.C., Z.P., and R.S. designed the experiments. Z.P., R.S., and S.B.M. performed the experiments. Z.P., R.S., and Z.C. wrote the manuscript. J.Z. and H.F. contributed critical reagents, analyzed the results, and edited the manuscript.
REFERENCES
- 1.Carter GP, Rood JI, Lyras D. 2012. The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol 20:21–29. doi: 10.1016/j.tim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Aktories K, Schwan C, Jank T. 2017. Clostridium difficile toxin biology. Annu Rev Microbiol 71:281–307. doi: 10.1146/annurev-micro-090816-093458. [DOI] [PubMed] [Google Scholar]
- 3.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–2370. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CDC. 2013. Antibiotic resistance threats in the United States, 2013. https://www.cdc.gov/drugresistance/biggest_threats.html.
- 5.McFarland LV, Mulligan ME, Kwok RYY, Stamm WE. 1989. Nosocomial acquisition of Clostridium difficile infection. N Engl J Med 320:204–210. doi: 10.1056/NEJM198901263200402. [DOI] [PubMed] [Google Scholar]
- 6.Fekety R, Shah AB. 1993. Diagnosis and treatment of Clostridium difficile colitis. JAMA 269:71–75. doi: 10.1001/jama.1993.03500010081036. [DOI] [PubMed] [Google Scholar]
- 7.Bartlett JG. 2008. The case for vancomycin as the preferred drug for treatment of Clostridium difficile infection. Clin Infect Dis 46:1489–1492. doi: 10.1086/587654. [DOI] [PubMed] [Google Scholar]
- 8.Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, Godin D, Bourassa C. 2005. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin Infect Dis 40:1591–1597. doi: 10.1086/430315. [DOI] [PubMed] [Google Scholar]
- 9.Muto CA, Pokrywka M, Shutt K, Mendelsohn AB, Nouri K, Posey K, Roberts T, Croyle K, Krystoflak S, Patel-Brown S, Pasculle AW, Paterson DL, Saul M, Harrison LH. 2005. A large outbreak of Clostridium difficile-associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infect Control Hosp Epidemiol 26:273–280. doi: 10.1086/502539. [DOI] [PubMed] [Google Scholar]
- 10.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 11.McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 12.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. 2005. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 13.Hubert B, Loo VG, Bourgault AM, Poirier L, Dascal A, Fortin E, Dionne M, Lorange M. 2007. A portrait of the geographic dissemination of the Clostridium difficile North American pulsed-field type 1 strain and the epidemiology of C. difficile-associated disease in Quebec. Clin Infect Dis 44:238–244. doi: 10.1086/510391. [DOI] [PubMed] [Google Scholar]
- 14.Karas JA, Enoch DA, Aliyu SH. 2010. A review of mortality due to Clostridium difficile infection. J Infect 61:1–8. doi: 10.1016/j.jinf.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 15.See I, Mu Y, Cohen J, Beldavs ZG, Winston LG, Dumyati G, Holzbauer S, Dunn J, Farley MM, Lyons C, Johnston H, Phipps E, Perlmutter R, Anderson L, Gerding DN, Lessa FC. 2014. NAP1 strain type predicts outcomes from Clostridium difficile infection. Clin Infect Dis 58:1394–1400. doi: 10.1093/cid/ciu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akerlund T, Persson I, Unemo M, Noren T, Svenungsson B, Wullt M, Burman LG. 2008. Increased sporulation rate of epidemic Clostridium difficile type 027/NAP1. J Clin Microbiol 46:1530–1533. doi: 10.1128/JCM.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourgault AM, Lamothe F, Loo VG, Poirier L, CDAD-CSI Study Group. 2006. In vitro susceptibility of Clostridium difficile clinical isolates from a multi-institutional outbreak in Southern Quebec, Canada. Antimicrob Agents Chemother 50:3473–3475. doi: 10.1128/AAC.00479-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drudy D, Quinn T, O'Mahony R, Kyne L, O'Gaora P, Fanning S. 2006. High-level resistance to moxifloxacin and gatifloxacin associated with a novel mutation in gyrB in toxin-A-negative, toxin-B-positive Clostridium difficile. J Antimicrob Chemother 58:1264–1267. doi: 10.1093/jac/dkl398. [DOI] [PubMed] [Google Scholar]
- 19.Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan VK, Johnson S, Gerding DN, Vedantam G. 2010. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol 192:4904–4911. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 21.Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ. 2016. Clostridium difficile infection. Nat Rev Dis Primers 2:16020. doi: 10.1038/nrdp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerding DN, Meyer T, Lee C, Cohen SH, Murthy UK, Poirier A, Van Schooneveld TC, Pardi DS, Ramos A, Barron MA, Chen H, Villano S. 2015. Administration of spores of nontoxigenic Clostridium difficile strain M3 for prevention of recurrent C. difficile infection: a randomized clinical trial. JAMA 313:1719–1727. doi: 10.1001/jama.2015.3725. [DOI] [PubMed] [Google Scholar]
- 23.Gardiner DF, Rosenberg T, Zaharatos J, Franco D, Ho DD. 2009. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine 27:3598–3604. doi: 10.1016/j.vaccine.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Sun X, Zhang Y, Li S, Chen K, Shi L, Nie W, Kumar R, Tzipori S, Wang J, Savidge T, Feng H. 2012. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun 80:2678–2688. doi: 10.1128/IAI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin K, Wang S, Zhang C, Xiao Y, Lu S, Huang Z. 2013. Protective antibody responses against Clostridium difficile elicited by a DNA vaccine expressing the enzymatic domain of toxin B. Hum Vaccin Immunother 9:63–73. doi: 10.4161/hv.22434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leuzzi R, Spencer J, Buckley A, Brettoni C, Martinelli M, Tulli L, Marchi S, Luzzi E, Irvine J, Candlish D, Veggi D, Pansegrau W, Fiaschi L, Savino S, Swennen E, Cakici O, Oviedo-Orta E, Giraldi M, Baudner B, D'Urzo N, Maione D, Soriani M, Rappuoli R, Pizza M, Douce GR, Scarselli M. 2013. Protective efficacy induced by recombinant Clostridium difficile toxin fragments. Infect Immun 81:2851–2860. doi: 10.1128/IAI.01341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steele J, Mukherjee J, Parry N, Tzipori S. 2013. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 207:323–330. doi: 10.1093/infdis/jis669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tucker M. 2013. Influenza vaccine efficacy significantly lower in elderly. In Medscape Medical News. http://www.medscape.com/viewarticle/779816.
- 29.Schmader KE, Levin MJ, Gnann JW Jr, McNeil SA, Vesikari T, Betts RF, Keay S, Stek JE, Bundick ND, Su SC, Zhao Y, Li X, Chan IS, Annunziato PW, Parrino J. 2012. Efficacy, safety, and tolerability of herpes zoster vaccine in persons aged 50–59 years. Clin Infect Dis 54:922–928. doi: 10.1093/cid/cir970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilcox MH, Gerding DN, Poxton IR, Kelly C, Nathan R, Birch T, Cornely OA, Rahav G, Bouza E, Lee C, Jenkin G, Jensen W, Kim YS, Yoshida J, Gabryelski L, Pedley A, Eves K, Tipping R, Guris D, Kartsonis N, Dorr MB; MODIFY I and MODIFY II Investigators. 2017. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med 376:305–317. doi: 10.1056/NEJMoa1602615. [DOI] [PubMed] [Google Scholar]
- 31.Anonymous. 2016. FDA approves Zinplava for recurrent C. difficile. http://www.pharmacypracticenews.com/Web-Only/Article/10-16/FDA-Approves-Zinplava-for-Recurrent-em-C-difficile-em-/38344/ses=ogst?enl=true.
- 32.Orth P, Xiao L, Hernandez LD, Reichert P, Sheth PR, Beaumont M, Yang X, Murgolo N, Ermakov G, DiNunzio E, Racine F, Karczewski J, Secore S, Ingram RN, Mayhood T, Strickland C, Therien AG. 2014. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem 289:18008–18021. doi: 10.1074/jbc.M114.560748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez LD, Racine F, Xiao L, DiNunzio E, Hairston N, Sheth PR, Murgolo NJ, Therien AG. 2015. Broad coverage of genetically diverse strains of Clostridium difficile by actoxumab and bezlotoxumab predicted by in vitro neutralization and epitope modeling. Antimicrob Agents Chemother 59:1052–1060. doi: 10.1128/AAC.04433-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simeon R, Jiang M, Chamoun-Emanuelli AM, Yu H, Zhang Y, Meng R, Peng Z, Jakana J, Zhang J, Feng H, Chen Z. 2019. Selection and characterization of ultrahigh potency designed ankyrin repeat protein inhibitors of C. difficile toxin B. PLoS Biol 17:e3000311. doi: 10.1371/journal.pbio.3000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamaskovic R, Simon M, Stefan N, Schwill M, Pluckthun A. 2012. Designed ankyrin repeat proteins (Darpins): from research to therapy. Methods Enzymol 203:101–134. [DOI] [PubMed] [Google Scholar]
- 36.Binz HK, Bakker TR, Phillips DJ, Cornelius A, Zitt C, Gottler T, Sigrist G, Fiedler U, Ekawardhani S, Dolado I, Saliba JA, Tresch G, Proba K, Stumpp MT. 2017. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin (R) drug candidate. MAbs 9:1262. doi: 10.1080/19420862.2017.1305529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, He R, Li C, Guo S, Li S, Huang T, Perez-Cordon G, Feng H, Wei W. 2015. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25:157–168. doi: 10.1038/cr.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X, Hurdle JG, Breault DT, Brass AL, Dong M. 2016. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538:350–355. doi: 10.1038/nature19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueno K, Hiura M, Suehiro Y, Hazama S, Hirata H, Oka M, Imai K, Dahiya R, Hinoda Y. 2008. Frizzled-7 as a potential therapeutic target in colorectal cancer. Neoplasia 10:697–705. doi: 10.1593/neo.08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta P, Zhang Z, Sugiman-Marangos SN, Tam J, Raman S, Julien JP, Kroh HK, Lacy DB, Murgolo N, Bekkari K, Therien AG, Hernandez LD, Melnyk RA. 2017. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor-binding determinants. J Biol Chem 292:17290–17301. doi: 10.1074/jbc.M117.806687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A. 2003. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 42.LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB. 2015. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc Natl Acad Sci U S A 112:7073–7078. doi: 10.1073/pnas.1500791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boersma YL, Chao G, Steiner D, Wittrup KD, Pluckthun A. 2011. Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J Biol Chem 286:41273–41285. doi: 10.1074/jbc.M111.293266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hausammann S, Vogel M, Kremer Hovinga JA, Lacroix-Desmazes S, Stadler BM, Horn MP. 2013. Designed ankyrin repeat proteins: a new approach to mimic complex antigens for diagnostic purposes? PLoS One 8:e60688. doi: 10.1371/journal.pone.0060688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gregorieff A, Clevers H. 2005. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 19:877–890. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 46.Chandrasekaran R, Lacy DB. 2017. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 41:723–750. doi: 10.1093/femsre/fux048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang TW, Bartlett JG, Gorbach SL, Onderdonk AB. 1978. Clindamycin-induced enterocolitis in hamsters as a model of pseudomembranous colitis in patients. Infect Immun 20:526–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, Burdon DW. 1978. Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J 1:695. doi: 10.1136/bmj.1.6114.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanis JM, Heinlen LD, James JA, Ballard JD. 2013. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog 9:e1003523. doi: 10.1371/journal.ppat.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen P, Tao L, Wang T, Zhang J, He A, Lam KH, Liu Z, He X, Perry K, Dong M, Jin R. 2018. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360:664–669. doi: 10.1126/science.aar1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. 2006. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol 17:2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 53.Chung SY, Schottelndreier D, Tatge H, Fuhner V, Hust M, Beer LA, Gerhard R. 2018. The Conserved Cys-2232 in Clostridioides difficile Toxin B Modulates Receptor Binding. Front Microbiol 9:2314. doi: 10.3389/fmicb.2018.02314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. 2008. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8:192. doi: 10.1186/1471-2180-8-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steiner D, Forrer P, Pluckthun A. 2008. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol 382:1211–1227. doi: 10.1016/j.jmb.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:27–28, 33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 57.López-Ureña D, Orozco-Aguilar J, Chaves-Madrigal Y, Ramírez-Mata A, Villalobos-Jimenez A, Ost S, Quesada-Gómez C, Rodríguez C, Papatheodorou P, Chaves-Olarte E. 2019. Toxin B variants from Clostridium difficile strains VPI 10463 and NAP1/027 share similar substrate profile and cellular intoxication kinetics but use different host cell entry factors. Toxins 11:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Purification of DARPins. Each DARPin was expressed in 5 ml of E. coli culture and purified by Ni-NTA affinity chromatography. Purified DARPins were analyzed by SDS-PAGE. Download FIG S1, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Amino acid sequences of the unique clones of anti-TcdBUK1 DARPins. Download Table S1, DOCX file, 0.02 MB (19.1KB, docx) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Schematic of monomeric and dimeric DARPins. (A) Monomeric DARPins contain an N-terminal 6×His tag and a Myc tag. The sequence from D16 is presented as an example. (B) The constituent DARPins in a dimeric DARPin are separated by a (GGGGS) × 3 linker sequence. The sequence from D16U3 is presented as an example. Download FIG S2, TIF file, 1.1 MB (1.2MB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Neutralization potency of engineered dimeric DARPins against UK1 TcdB. Purified DARPins were added to Vero cells together with TcdBUK1 (1 pg/ml). Cell viability was quantified by the CellTiter Glo assay 72 h later and normalized to naïve Vero cells. Experiments were conducted in triplicate. Error bars represent the standard deviation. (A) D16 versus U3D16. (B) D16 versus D16U3. (C) D16 versus mixture of D16 and U3. (D) D16 versus U3D16 and D16U3 with longer linkers. Download FIG S3, TIF file, 0.8 MB (804.5KB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequence alignment of the FBD from TcdBUK1 and TcdBVPI. Key residues at the FZD binding interface are shown in blue and red. Download FIG S4, TIF file, 0.7 MB (729.3KB, tif) .
Copyright © 2019 Peng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The sequences of our engineered proteins are presented in Table S1.