

Abstract
Cystic fibrosis (CF) is a multiorgan recessive genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Gene therapy efforts have focused on treating the lung, since it manifests the most significant life-threatening disease. Over two decades have past since the first CF lung gene therapy trials and significant advances in the therapeutic implementation of pharmacologic CFTR modulators have renewed the field's focus on developing gene therapies for the 10% of CF patients these modulators cannot help. This review summarizes recent progress made in developing vectors for airway transduction and CF animal models required for understanding the relevant cellular targets in the lung and testing the efficacy of gene therapy approaches. We also highlight future opportunities in emerging gene editing strategies that may offer advantages for treating diseases like CF where the gene target is highly regulated at the cellular level. The outcomes of CF lung gene therapy trials will likely inform productive paths toward gene therapy for other complex genetic disorders, while also advancing treatments for all CF patients.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease that affects over 70 000 people in the United States and Europe. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (1), which encodes a chloride and bicarbonate channel expressed in epithelial cells of the many organs affected in this disease. Nearly 2000 distinct CFTR variants have been identified; however, further research is needed to understand whether many of these cause disease (http://www.genet.sickkids.on.ca/app). CFTR plays an important role in conducting anions across epithelia to regulate secretory volume, pH, and mucus viscosity of liquid secretions (2,3). CFTR also interacts with other ion channels, such as epithelial sodium channel (ENaC), to regulate fluid movement across epithelia (4,5), and thus can have cell-intrinsic functions based on the composition of other channels coexpressed with CFTR (6). Lung disease is the primary cause morbidity and mortality of CF where the loss of CFTR-mediated chloride and bicarbonate transport leads to progressive chronic bacterial infection and inflammation (7).
The discovery of CFTR modulators that target gating and trafficking and conductance defects of the mutant CFTR protein have shown tremendous promise for the majority of CF patients (8–10). However, approximately 10% of CF patients are refractory to CFTR modulators because they either produce no or too little CFTR protein due to nonsense and splicing mutations or because they cannot tolerate the modulator therapies. Thus, gene therapy remains an attractive approach for these patients because it is mutation agnostic. Since CFTR modulators are a systemic therapy for all diseased organs, gene therapy in CF patients with efficacious CFTR modulator options will likely not occur until the field advances its ability to genetically treat multiple affected organs in CF (i.e. lung, pancreas, and intestine). For these reasons, current gene therapy development efforts target life-threatening lung disease in a subset of CF patients with no therapeutic options.
In vivo gene therapy has shown promise in treating certain genetics disorders, with the most notable success in treating blindness caused by a defect in the RPE65 gene (11,12). By contrast, gene therapy for CF lung disease has had a more protracted journey with the first clinical trials initiated in the early 1990s shortly after the cloning of the CFTR gene (13–19). Subsequently, 27 clinical gene therapy trials involving ~600 CF patients have yet to achieve their desired outcomes. The largest of these was a Phase IIb multiple-dose trial completed by the UK Cystic Fibrosis Gene Therapy Consortium (UK CFGTC) in 2015. This trial of liposome-mediated delivery of a CFTR expression plasmid administered on a monthly basis over the course of a year demonstrated for the first time that gene therapy could provide a modest stabilization in the rate of decline of lung function in CF patients. However, the approach was not sufficient to improve the lung disease (20). For the first two decades following cloning of the CFTR gene, the field of CF gene therapy has worked to identify efficient vector systems for delivery to the lung. Additionally, the lack of suitable CF animal models to test efficacy of new vector systems had handicapped the field. Enhanced technologies for engineering new viral vector systems, increased understanding of the virology of these vector systems, and improved larger animal models of CF have led to a resurgence in gene therapy efforts for CF over the past decade. In this review, we focus on recent progress in the field that is anticipated to advance the CF gene therapy efforts.
Viral vectors for airway transduction
The conducting airways are considered the primary target for gene therapy of CF lung disease. As an organ exposed to the external environment, the lung has evolved considerable innate defense mechanisms that protect the airways from infections and these mechanisms also create barriers to gene transfer agents. In the CF airway, these barriers are enhanced due to the increased mucus within the airways. While non-viral vectors have the advantage of being less immunogenic than viral vectors, viral vectors have historically been more efficient at gene delivery to the airway than non-viral vectors. However, the failure of previous CF lung trials using recombinant adenovirus (rAd) and adeno-associated virus (rAAV2) vectors has emphasized the need for an appropriate understanding of the biology of viral transduction of the airway from the apical surface and the appropriate models in which to test them. This review will limit discussion to viral gene transfer vectors that show promise as therapeutic tools for CF lung disease.
Lentiviral vectors
This class of vectors offers the advantage of integration. If cells with progenitor capacity are transduced, the therapeutic gene will be expressed in daughter cells (21). While self-inactivating lentiviral vectors have been widely used for ex vivo gene therapy of hematopoetic disorders such as adenosine deaminase and X-linked severe combined immunodeficiency, there are relatively few examples of in vivo use. One recent example is ProSavin, a lentiviral vector-based gene therapy for Parkinson's disease, which delivers three dopamine biosynthetic enzymes to convert the transduced non-dopaminergic striatal neurons into dopamine-producing cells in brain (22). Lentiviruses, such as of human, simian and feline immunodeficiency virus (FIV), as well as equine infectious anemia virus, do not naturally infect the airways. However, advancements in pseudotyping lentiviral envelope proteins to adopt broad tissue tropism or specific tropisms for the airway epithelium have created vectors with significantly improved transduction from the apical surface of differentiated human airway epithelia grown at an air-liquid interface (HAE-ALI) and the airways of experimental animals in vivo. Sinn et al. (23) screened a wide variety of envelope proteins and found that the glycoprotein GP64 of baculovirus (24) conferred apical entry of a FIV vector into HAE-ALI and a broad tropism in transducing mouse, ferret, and porcine airways in vivo (25–27). Recently, directed evolution of a library of GP64 envelope mutants through repetitive infections in HAE-ALI led to the identification of a novel GP64 variant (E45K/T259A), which demonstrated 8-fold greater efficiency than that of the wild-type GP64 (28).
The UK CFGTC had assessed the transduction efficiency of a lentiviral vector pseudotyped with F and HN (fusion and hemagglutinin-neuraminidase) proteins from Sendai virus (29), in HAE-ALI in vitro and mouse lung in vivo. They demonstrated such a vector could produce persistent reporter expression in the lung and nose of mice. Transgene expression from this lentiviral transduction was 100-fold higher than that from the optimal formulation of plasmid/liposome that was used in their clinical trial (30). Currently, the Consortium is pursuing a phase I/IIa lentiviral vector CF gene therapy trial, using the nasal epithelium as a surrogate organ to allow easy monitoring of safety and gene expression. The vector rSIV.F/HN-hCEF-CFTR uses a chimeric promoter of CpG-free CMV enhancer and human EF1α promoter to drive a codon-optimized and CpG depleted CFTR cDNA (31,32). Also of note, following lentiviral transduction, transgene expression persists for the life of the animal in mice (23,30,33). Lentiviral vectors have also been successfully readministered to respiratory epithelia in mice without eliciting a blocking immune response (30,34,35).
Helper-dependent adenovirus and piggyBac/Adenovirus vectors
Recombinant adenoviral vectors were the first used for CFTR delivery in the earliest attempts of CF gene therapy. Although those clinical trials demonstrated partial correction of the Cl− transport defect in CF nasal epithelium by transepithelial potential difference (13,36), the effect was only observed when the nasal epithelium was damaged during delivery. Subsequently, it was found that the coxsackie-adenovirus receptor (CAR) localizes to the basolateral membrane of the human airway epithelium (37), explaining these clinical trial findings. Furthermore, intrapulmonary delivery of a first-generation recombinant adenovirus vector was found to promote an immune response that limited transgene persistence (38) and at high doses caused a transient systemic and pulmonary syndrome that may have involved an innate immune response (17).
An improved rAd vector platform includes helper-dependent adenovirus (HD-Ad) for which all viral encoded genes are removed. Thus, T-cell responses to cryptic viral protein expression observed in first-generation rAd vectors are obviated in HD-Ad. However, the large capsid of Ad still induces adaptive immune response by CD8+ T-cells through the presentation of HD-Ad-derived epitopes by dendritic cells (39). The applications of HD-Ad to the lung have used a lysophosphatidylcholine (LPC) formulation to disrupt tight junctions and facilitate access to CAR at the basolateral cell surface required for infection. This strategy has achieved much longer transgene expression in vivo than first-generation rAd and demonstrates highly efficient gene transfer to the airways of mouse, pig, and ferrets (27,40).
The TTAA-specific DNA transposon piggyBac can promote persistent gene transfer via a “cut and paste” transposition mechanism without leaving a footprint. Incorporation of the transposase-mediated integration approach into recombinant adenovirus has led to the development of the hybrid piggyBac/Adenovirus vector. Cooney et al. (41) demonstrated efficient lung-directed transgene expression in pigs using piggyBac/Adenovirus transduction. Notably, HD-Ad formulated with LPC efficiently transduces basal cells in the mouse and pig airways (40). Given that basal cells in the airways are multipotent and self-renewing, incorporation of a piggyBac transposon into HD-Ad could be used to permanently insert a CFTR expression cassette into basal stem cells and thus also correct the gene defect in their differentiated progeny.
AAV-based vectors
Despite the failure of initial rAAV CF gene therapy clinical trials, these studies established a favorable safety profile of aerosol delivery of rAAV2 in the lung (42). These trials failed for several reasons that are pertinent to developing improved rAAV vector systems. First, the efficiency of rAAV2 transduction following apical infection of differentiated airway epithelia was 100-fold greater in the preclinical model chosen (rhesus monkey) than for human (43). Second, apical transduction of HAE-ALI with rAAV2 encounters a postentry block in nuclear transport of the virion that can be overcome using proteasome inhibitors (44). Lastly, the packaging limits of the rAAV genome necessitated the use of the AAV2 inverted terminal repeat as a cryptic promoter to drive expression of the full-length CFTR cDNA, giving rise to extremely low expression. Although no additional rAAV clinical trials have been performed since 2005, research developing better AAV-based vectors for airway transduction has led to significant advances.
Issues regarding the packaging size of the rAAV2 genome and its ability to effectively deliver a CFTR expression cassette were solved by creating a synthetic promoter of 183 bp in length (45) and a shortened CFTR minigene with a 156-bp deletion in the regulatory (R) domain (46). Improved viral capsids for efficient transduction of HAE-ALI from the apical membrane were also generated by directed evolution from a library of AAV2 and AAV5 capsids following multiple rounds of infection in HAE-ALI and generated the AAV2.5T vector (47). Another novel parvovirus vector with high tropism for the apical surface of HAE-ALI is human bocavirus type 1 (HBoV1). HBoV1 is a human respiratory virus that naturally infects the human airways early in life and is a relative of rAAV. Cross genera packaging of the rAAV2 genome into the HBoV1 capsid has created the rAAV2/HBoV1 hybrid vector that is highly tropic for human airway epithelia (48). rAAV2/HBoV1 vectors also have an increased packaging capacity of up to 5.8 kb, allowing for a strong promoter and the full length CFTR cDNA. Packaging systems for rAAV2/HBoV1 have significantly improved since the vector was first created (49). rAAV2/HBoV1 demonstrates apical tropism equivalent to rAAV2/2.5 T in transducing HAE-ALI. Both rAAV2/2.5 T and rAAV2/HBoV1 can transduce the ferret lung in vivo (50), enabling the preclinical studies in CF ferret models.
Animal models for CF gene therapy
Animal models are indispensible when developing gene therapies. For gene therapy of CF lung disease, many questions can be addressed in animal models. These include identifying the necessary cellular targets, doses required to complement CFTR defects, and establishing the efficacy of gene delivery approaches in the hostile environment of the CF lung. To date, CF animals have been produced in mouse, rat, rabbit, zebrafish, ferret, pig, and sheep (51). Characterization of lung disease in CF rabbits and sheep has lagged behind the other species. While CFTR-null mice and rats do not acquire spontaneous airway infection, CFTRKO ferret and pig models recapitulate many features of the lung disease phenotype observed in humans with CF (51–53). However, these models also exhibit severe gastrointestinal and pancreatic disease at birth (54,55), which has limited their broad use in developing gene therapies. Despite these limitations, CF pigs have been successfully used to demonstrate efficacy of both lentivirus and rAAV-mediated in vivo CFTR gene therapy approaches (26,56).
Several approaches have been used to create better pig and ferret CF models that will better enable their use in testing gene therapies. For example, the creation of gut-corrected CF pigs has reduced the incidence of meconium ileus at birth (57). Using a different strategy, CF ferrets have been engineered to harbor the CFTR-G551D mutation (CFTRG551D), which is responsive to the CFTR modulator VX-770 (58). Importantly, delivery of VX-770 to pregnant jills protects the pancreas and intestine of CFTRG551D/G551D kits, allowing for normal growth and survival after birth with continued VX-770. Cessation of VX-770 at any age reinitiates disease in the pancreas, gut, and lung. Thus, the use of VX-770 in this model will allow for the testing of gene therapies that either prevent the emergence lung disease or reverse lung disease (Fig. 1).
Fig. 1.
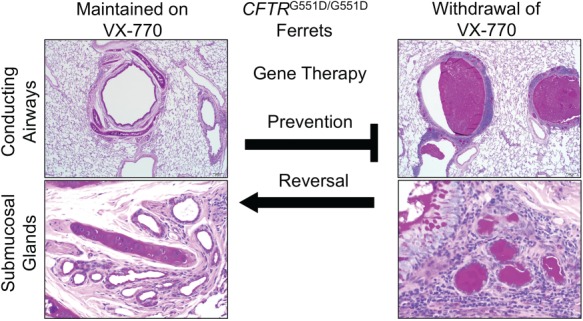
Utilizing CFTRG551D ferrets for testing gene therapies to the CF lung. CFTRG551D/G551D ferrets are protected from lung disease while treated with VX-770. Following termination of VX-770, these CF animals develop mucus accumulation in the airways and submucosal glands and eventually develop lethal bacterial infections of the lung (58). Testing gene therapies in this model can utilize two approaches. Prophylactic gene therapies administered to the lungs of CF ferrets prior to cessation of VX-770 can be used to test whether the gene therapy approach prevents the development of lung disease. This approach circumvents a potential barrier (thick mucus) in the CF airway that may impede gene transfer to the airways and addresses whether the gene therapy approach would be successful in a pristine airway. The more clinically relevant use of this CF ferret model is to ask whether the gene therapy approach can reverse lung disease. In this approach, VX-770 treatment would be terminated prior to gene delivery to the lung. In this scenario, varying extents of lung disease and mucus accumulation could be controlled by the time animals are removed from VX-770 to understand components of disease that are reversible and irreversible. Irreversible components might include structural changes to the lung such as bronchiectasis and bacteria that have adapted to the damaged lung. Comparing the efficacy of both approaches to either prevent or reverse CF lung disease will inform disease specific barriers to gene delivery to the airways and therapeutic obstacles to the approaches tested. Histology panels were reformatted from Supplementary Figure 4 (panels N, P, F and G) in Ref. (58). In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis, Sci. Transl. Med., 11, eaau7531 (Reprinted with permission from AAAS).
Heterogeneity of CFTR expressing cell types in lung
Gene therapies for CF lung disease will likely need to navigate complexities of CFTR biology and function in the lung. CFTR expression is highly regulated in specific cell types of the lung, each having its own potentially unique composition of channels and functions that coordinate clearance and innate immunity in the lung (6). In the proximal airways, CFTR protein and mRNA expression are very abundant in a small subset of columnar non-ciliated cells in the surface airway epithelium and submucosal gland duct and serous cells (59). In the more distal airways, CFTR expression is found at very high levels in respiratory bronchioles and a small subset of columnar non-ciliated cells in the terminal bronchioles (60). Recent studies using single-cell RNAseq (sc-RNAseq) are helping to better specify the CFTR expressing cell types in the airways and identified the ionocyte as the columnar non-ciliated cell that expresses approximately 50% of CFTR transcripts in the surface airway epithelium. Interestingly, little to no CFTR mRNA transcripts were detected in secretory and ciliated cell types (61,62). While human ionocytes may be a major source of CFTR expression in the proximal airway epithelium, they comprise only 0.5% to 1.5% of epithelial cells along the conducting airways.
The functions of CFTR expressing ionocytes in the airways remain unclear, but these findings raise several important questions relevant to gene therapy for CF. For example, will it be necessary to express CFTR in ionocytes due to cell-autonomous CFTR function controlled by the unique composition of channels in this cell type? Can CFTR expression in other columnar cell types compensate for the function of ionocytes in the airways? How are these CFTR-expressing cell types, including ionocytes, regionally distributed in the lung? Answers to these questions require an improved understanding of the pathophysiology of CF lung disease and how CFTR functions at the cellular level to control airway clearance and innate immunity. However, the implications for gene therapy of CF lung disease will only be fully appreciated when new gene delivery approaches are tested in clinical trials.
The complexities of CFTR regulation at the cellular level in the lung theoretically become less of an obstacle if an effective gene editing approach that targets airway stem cells can be generated. The discovery of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) provides specific programmable nucleases (63–65) and has greatly accelerated the development of practical RNA-guided genome editing approaches. These tools provide an opportunity to advance CF gene therapy from gene addition to precise gene correction (66,67). Theoretically, gene editing enables precise correction of any CFTR mutation or the rebuilding of a CFTR minigene that expresses a corrected mRNA without altering endogenous patterns of expression. Although the machinery required for homology-directed repair (HDR) is inactivated in non-dividing airway epithelial cells (68), a new non-homologous end joining (NHEJ)-based approach called homology-independent targeted integration (HITI) has been shown to efficiently insert a sequence into a desired genomic locus in post-mitotic cells in vitro and in vivo (69). Thus, HITI is compatible with an exon trap CFTR minigene gene editing approach in postmitotic airway epithelia or non-proliferating stem cells, whereas CRISPR/Cas9 HDR approaches could be used to target CFTR gene correction to proliferating airway stem cells.
Future considerations
Significant advances in the development of efficient vectors for airway gene delivery, coupled with an improved understanding of CF pathophysiology and new animals models that recapitulate the human CF phenotypes, have renewed enthusiasm for advancing effective genetic therapies for CF. Restoration of CF lung function relies on rebuilding normal homeostatic mechanisms that regulate effective airway clearance and innate immunity. Open questions remain regarding the cellular targets in the lung that are required or sufficient to prevent or reverse disease. Additionally, the most abundant cell types accessible to a CFTR gene transfer vector on the airway surfaces include terminally differentiated ciliated cells and secretory cells with the capacity to divide, and the lifespan of these cell types in the human airway is undefined. However, the average half-life of airway ciliated cells epithelial cells in mouse is estimated to be 6 months in the trachea and 17 months in lung (70); thus, effective CFTR gene delivery using a non-integrating viral-mediated approach might last for several years in the human lung in the absence of disease. Repeat dosing of a gene therapy agent to the CF lung will likely be required as the field develops, and for certain viral vectors, this will necessitate clearer understanding of methods to modulate the immune response in the CF lung. However, as the field advances to more sophisticated gene editing approaches targeting stem cell compartment in the lung, questions about whether CFTR regulation is required for complementation of disease and the need for repeat dosing of a gene transfer agent would become less important. Regardless of the strategy, CF animal models will play an important role in helping to guide the development of effective gene therapies to treat or prevent CF lung disease. The utility of any gene therapy approach will ultimately only be clearly defined by testing in CF patients using well-designed clinical trials with strong molecular and physiologic endpoints for efficacy. This will likely be an iterative process, and the opportunities for continued advancement are great.
Conflict of Interest statement
J.F.E. and P.B.M. are founders and hold equity in Talee Bio and have sponsored research with this company. Z.Y. receives income for consulting with Talee Bio. J.F.E. has sponsored research with Vertex Pharmaceuticals.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL051670, HL91842, HL123482), Cystic Fibrosis Foundation, the University of Iowa Center for Gene Therapy (DK54759), and the Roy J. Carver Chair in Molecular Medicine (to J.F.E.) and the Roy J. Carver Chair in Pulmonary Research (to P.B.M.).
References
- 1. Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science, 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- 2. Pezzulo A.A., Tang X.X., Hoegger M.J., Abou Alaiwa M.H., Ramachandran S., Moninger T.O., Karp P.H., Wohlford-Lenane C.L., Haagsman H.P., Eijk M. et al. (2012) Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature, 487, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wine J.J., Hansson G.C., Konig P., Joo N.S., Ermund A. and Pieper M. (2018) Progress in understanding mucus abnormalities in cystic fibrosis airways. J. Cyst. Fibros., 17, S35–S39. [DOI] [PubMed] [Google Scholar]
- 4. Shei R.J., Peabody J.E., Kaza N. and Rowe S.M. (2018) The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol., 43, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu D., Boucher R.C., Button B., Elston T. and Lin C.L. (2018) An integrated mathematical epithelial cell model for airway surface liquid regulation by mechanical forces. J. Theor. Biol., 438, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang Q. and Engelhardt J.F. (1998) Cellular heterogeneity of CFTR expression and function in the lung: implications for gene therapy of cystic fibrosis. Eur. J. Hum. Genet., 6, 12–31. [DOI] [PubMed] [Google Scholar]
- 7. Stoltz D.A., Meyerholz D.K. and Welsh M.J. (2015) Origins of cystic fibrosis lung disease. N. Engl. J. Med., 372, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clancy J.P., Cotton C.U., Donaldson S.H., Solomon G.M., VanDevanter D.R., Boyle M.P., Gentzsch M., Nick J.A., Illek B., Wallenburg J.C. et al. (2019) CFTR modulator theratyping: current status, gaps and future directions. J. Cyst. Fibros., 18, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyle M.P., Bell S.C., Konstan M.W., McColley S.A., Rowe S.M., Rietschel E., Huang X., Waltz D., Patel N.R., Rodman D. et al. (2014) A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir. Med., 2, 527–538. [DOI] [PubMed] [Google Scholar]
- 10. Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A. et al. (2015) Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med., 373, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. et al. (2017) Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet, 390, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morrison C. (2018) Fresh from the biotech pipeline-2017. Nat. Biotechnol., 36, 131–136. [DOI] [PubMed] [Google Scholar]
- 13. Zabner J., Couture L.A., Gregory R.J., Graham S.M., Smith A.E. and Welsh M.J. (1993) Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell, 75, 207–216. [DOI] [PubMed] [Google Scholar]
- 14. Wilson J.M., Engelhardt J.F., Grossman M., Simon R.H. and Yang Y. (1994) Gene therapy of cystic fibrosis lung disease using E1 deleted adenoviruses: a phase I trial. Hum. Gene Ther., 5, 501–519. [DOI] [PubMed] [Google Scholar]
- 15. Welsh M.J., Zabner J., Graham S.M., Smith A.E., Moscicki R. and Wadsworth S. (1995) Adenovirus-mediated gene transfer for cystic fibrosis: part a. Safety of dose and repeat administration in the nasal epithelium. Part B. Clinical efficacy in the maxillary sinus. Hum. Gene Ther., 6, 205–218. [DOI] [PubMed] [Google Scholar]
- 16. Knowles M.R., Hohneker K.W., Zhou Z., Olsen J.C., Noah T.L., Hu P.C., Leigh M.W., Engelhardt J.F., Edwards L.J., Jones K.R. et al. (1995) A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med., 333, 823–831. [DOI] [PubMed] [Google Scholar]
- 17. Crystal R.G., McElvaney N.G., Rosenfeld M.A., Chu C.S., Mastrangeli A., Hay J.G., Brody S.L., Jaffe H.A., Eissa N.T. and Danel C. (1994) Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet., 8, 42–51. [DOI] [PubMed] [Google Scholar]
- 18. Crystal R.G., Jaffe A., Brody S., Mastrangeli A., McElvaney N.G., Rosenfeld M., Chu C.S., Danel C., Hay J. and Eissa T. (1995) A phase 1 study, in cystic fibrosis patients, of the safety, toxicity, and biological efficacy of a single administration of a replication deficient, recombinant adenovirus carrying the cDNA of the normal cystic fibrosis transmembrane conductance regulator gene in the lung. Hum. Gene Ther., 6, 643–666. [DOI] [PubMed] [Google Scholar]
- 19. Boucher R.C., Knowles M.R., Johnson L.G., Olsen J.C., Pickles R., Wilson J.M., Engelhardt J., Yang Y. and Grossman M. (1994) Gene therapy for cystic fibrosis using E1-deleted adenovirus: a phase I trial in the nasal cavity. The University of North Carolina at Chapel Hill. Hum. Gene Ther., 5, 615–639. [DOI] [PubMed] [Google Scholar]
- 20. Alton E., Armstrong D.K., Ashby D., Bayfield K.J., Bilton D., Bloomfield E.V., Boyd A.C., Brand J., Buchan R., Calcedo R. et al. (2015) Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med., 3, 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marquez Loza L.I., Yuen E.C. and McCray P.B. Jr. (2019) Lentiviral vectors for the treatment and prevention of cystic fibrosis lung disease. Genes (Basel), 10, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palfi S., Gurruchaga J.M., Lepetit H., Howard K., Ralph G.S., Mason S., Gouello G., Domenech P., Buttery P.C., Hantraye P. et al. (2018) Long-term follow-up of a phase I/II study of ProSavin, a lentiviral vector gene therapy for Parkinson's disease. Hum. Gene Ther. Clin. Dev, 29, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sinn P.L., Burnight E.R., Hickey M.A., Blissard G.W. and McCray P.B. Jr. (2005) Persistent gene expression in mouse nasal epithelia following feline immunodeficiency virus-based vector gene transfer. J. Virol., 79, 12818–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blissard G.W. and Wenz J.R. (1992) Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J. Virol., 66, 6829–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oakland M., Maury W., McCray P.B. Jr. and Sinn P.L. (2013) Intrapulmonary versus nasal transduction of murine airways with GP64-pseudotyped viral vectors. Mol. Ther. Nucleic Acids, 2, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cooney A.L., Abou Alaiwa M.H., Shah V.S., Bouzek D.C., Stroik M.R., Powers L.S., Gansemer N.D., Meyerholz D.K., Welsh M.J., Stoltz D.A. et al. (2016) Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight, 1, e88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan Z., Stewart Z.A., Sinn P.L., Olsen J.C., Hu J., McCray P.B. Jr. and Engelhardt J.F. (2015) Ferret and pig models of cystic fibrosis: prospects and promise for gene therapy. Hum. Gene Ther. Clin. Dev., 26, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinn P.L., Hwang B.Y., Li N., Ortiz J.L.S., Shirazi E., Parekh K.R., Cooney A.L., Schaffer D.V. and McCray P.B. Jr. (2017) Novel GP64 envelope variants for improved delivery to human airway epithelial cells. Gene Ther., 24, 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitomo K., Griesenbach U., Inoue M., Somerton L., Meng C., Akiba E., Tabata T., Ueda Y., Frankel G.M., Farley R. et al. (2010) Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol. Ther., 18, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griesenbach U., Inoue M., Meng C., Farley R., Chan M., Newman N.K., Brum A., You J., Kerton A., Shoemark A. et al. (2012) Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am. J. Respir. Crit. Care Med., 186, 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Griesenbach U., Davies J.C. and Alton E. (2016) Cystic fibrosis gene therapy: a mutation-independent treatment. Curr. Opin. Pulm. Med., 22, 602–609. [DOI] [PubMed] [Google Scholar]
- 32. Alton E.W., Beekman J.M., Boyd A.C., Brand J., Carlon M.S., Connolly M.M., Chan M., Conlon S., Davidson H.E., Davies J.C. et al. (2017) Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax, 72, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stocker A.G., Kremer K.L., Koldej R., Miller D.S., Anson D.S. and Parsons D.W. (2009) Single-dose lentiviral gene transfer for lifetime airway gene expression. J. Gene Med., 11, 861–867. [DOI] [PubMed] [Google Scholar]
- 34. Sinn P.L., Arias A.C., Brogden K.A. and McCray P.B. Jr. (2008) Lentivirus vector can be readministered to nasal epithelia without blocking immune responses. J. Virol., 82, 10684–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patel M., Giddings A.M., Sechelski J. and Olsen J.C. (2013) High efficiency gene transfer to airways of mice using influenza hemagglutinin pseudotyped lentiviral vectors. J. Gene Med., 15, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zabner J., Ramsey B.W., Meeker D.P., Aitken M.L., Balfour R.P., Gibson R.L., Launspach J., Moscicki R.A., Richards S.M., Standaert T.A. et al. (1996) Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J Clin. Invest., 97, 1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walters R.W., Grunst T., Bergelson J.M., Finberg R.W., Welsh M.J. and Zabner J. (1999) Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem., 274, 10219–10226. [DOI] [PubMed] [Google Scholar]
- 38. Harvey B.G., Hackett N.R., Ely S. and Crystal R.G. (2001) Host responses and persistence of vector genome following intrabronchial administration of an E1(−)E3(−) adenovirus gene transfer vector to normal individuals. Mol. Ther., 3, 206–215. [DOI] [PubMed] [Google Scholar]
- 39. Kushwah R., Cao H. and Hu J. (2008) Characterization of pulmonary T cell response to helper-dependent adenoviral vectors following intranasal delivery. J Immunol, 180, 4098–4108. [DOI] [PubMed] [Google Scholar]
- 40. Cao H., Ouyang H., Grasemann H., Bartlett C., Du K., Duan R., Shi F., Estrada M., Seigel K.E., Coates A.L. et al. (2018) Transducing airway basal cells with a helper-dependent adenoviral vector for lung gene therapy. Hum. Gene Ther., 29, 643–652. [DOI] [PubMed] [Google Scholar]
- 41. Cooney A.L., Singh B.K., Loza L.M., Thornell I.M., Hippee C.E., Powers L.S., Ostedgaard L.S., Meyerholz D.K., Wohlford-Lenane C., Stoltz D.A. et al. (2018) Widespread airway distribution and short-term phenotypic correction of cystic fibrosis pigs following aerosol delivery of piggyBac/adenovirus. Nucleic Acids Res., 46, 9591–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moss R.B., Milla C., Colombo J., Accurso F., Zeitlin P.L., Clancy J.P., Spencer L.T., Pilewski J., Waltz D.A., Dorkin H.L. et al. (2007) Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther., 18, 726–732. [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Luo M., Trygg C., Yan Z., Lei-Butters D.C., Smith C.I., Fischer A.C., Munson K., Guggino W.B., Bunnell B.A. et al. (2007) Biological differences in rAAV transduction of airway epithelia in humans and in Old World non-human primates. Mol. Ther., 15, 2114–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duan D., Yue Y., Yan Z., Yang J. and Engelhardt J.F. (2000) Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J. Clin. Invest., 105, 1573–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yan Z., Sun X., Feng Z., Li G., Fisher J.T., Stewart Z.A. and Engelhardt J.F. (2015) Optimization of recombinant adeno-associated virus-mediated expression for large transgenes using a synthetic promoter and tandem array enhancers. Hum. Gene Ther., 26, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ostedgaard L.S., Rokhlina T., Karp P.H., Lashmit P., Afione S., Schmidt M., Zabner J., Stinski M.F., Chiorini J.A. and Welsh M.J. (2005) A shortened adeno-associated virus expression cassette for CFTR gene transfer to cystic fibrosis airway epithelia. Proc. Natl. Acad. Sci. U. S. A., 102, 2952–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Excoffon K.J., Koerber J.T., Dickey D.D., Murtha M., Keshavjee S., Kaspar B.K., Zabner J. and Schaffer D.V. (2009) Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. U. S. A., 106, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan Z., Keiser N.W., Song Y., Deng X., Cheng F., Qiu J. and Engelhardt J.F. (2013) A novel chimeric adenoassociated virus 2/human bocavirus 1 parvovirus vector efficiently transduces human airway epithelia. Mol. Ther., 21, 2181–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan Z., Zou W., Feng Z., Shen W., Park S.Y., Deng X., Qiu J. and Engelhardt J.F. (2019) Establishment of a high-yield recombinant adeno-associated virus/human bocavirus vector production system independent of bocavirus nonstructural proteins. Hum. Gene Ther., 30, 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yan Z., Feng Z., Sun X., Zhang Y., Zou W., Wang Z., Jensen-Cody C., Liang B., Park S.Y., Qiu J. et al. (2017) Human bocavirus type-1 capsid facilitates the transduction of ferret airways by adeno-associated virus genomes. Hum. Gene Ther., 28, 612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rosen B.H., Chanson M., Gawenis L.R., Liu J., Sofoluwe A., Zoso A. and Engelhardt J.F. (2018) Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros., 17, S28–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun X., Olivier A.K., Liang B., Yi Y., Sui H., Evans T.I., Zhang Y., Zhou W., Tyler S.R., Fisher J.T. et al. (2014) Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am. J. Respir. Cell Mol. Biol., 50, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stoltz D.A., Meyerholz D.K., Pezzulo A.A., Ramachandran S., Rogan M.P., Davis G.J., Hanfland R.A., Wohlford-Lenane C., Dohrn C.L., Bartlett J.A. et al. (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med., 2, 29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun X., Olivier A.K., Yi Y., Pope C.E., Hayden H.S., Liang B., Sui H., Zhou W., Hager K.R., Zhang Y. et al. (2014) Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am. J. Pathol., 184, 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rogers C.S., Stoltz D.A., Meyerholz D.K., Ostedgaard L.S., Rokhlina T., Taft P.J., Rogan M.P., Pezzulo A.A., Karp P.H., Itani O.A. et al. (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science, 321, 1837–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steines B., Dickey D.D., Bergen J., Excoffon K.J., Weinstein J.R., Li X., Yan Z., Abou Alaiwa M.H., Shah V.S., Bouzek D.C. et al. (2016) CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight, 1, e88728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stoltz D.A., Rokhlina T., Ernst S.E., Pezzulo A.A., Ostedgaard L.S., Karp P.H., Samuel M.S., Reznikov L.R., Rector M.V., Gansemer N.D. et al. (2013) Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J. Clin. Invest., 123, 2685–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun X., Yi Y., Yan Z., Rosen B.H., Liang B., Winter M.C., Evans T.I.A., Rotti P.G., Yang Y., Gray J.S. et al. (2019) In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med., 11, eaau7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Engelhardt J.F., Yankaskas J.R., Ernst S.A., Yang Y., Marino C.R., Boucher R.C., Cohn J.A. and Wilson J.M. (1992) Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet., 2, 240–248. [DOI] [PubMed] [Google Scholar]
- 60. Engelhardt J.F., Zepeda M., Cohn J.A., Yankaskas J.R. and Wilson J.M. (1994) Expression of the cystic fibrosis gene in adult human lung. J. Clin. Invest., 93, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Montoro D.T., Haber A.L., Biton M., Vinarsky V., Lin B., Birket S.E., Yuan F., Chen S., Leung H.M., Villoria J. et al. (2018) A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature, 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Plasschaert L.W., Zilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A.M. and Jaffe A.B. (2018) A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature, 560, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A. and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y. et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cox D.B., Platt R.J. and Zhang F. (2015) Therapeutic genome editing: prospects and challenges. Nat. Med., 21, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Suzuki K. and Izpisua Belmonte J.C. (2018) In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet., 63, 157–164. [DOI] [PubMed] [Google Scholar]
- 67. Nami F., Basiri M., Satarian L., Curtiss C., Baharvand H. and Verfaillie C. (2018) Strategies for in vivo genome editing in nondividing cells. Trends Biotechnol., 36, 770–786. [DOI] [PubMed] [Google Scholar]
- 68. Hustedt N. and Durocher D. (2016) The control of DNA repair by the cell cycle. Nat. Cell Biol., 19, 1–9. [DOI] [PubMed] [Google Scholar]
- 69. Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. et al. (2016) In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature, 540, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rawlins E.L. and Hogan B.L. (2008) Ciliated epithelial cell lifespan in the mouse trachea and lung. Am. J. Physiol. Lung Cell Mol. Physiol., 295, L231–L234. [DOI] [PMC free article] [PubMed] [Google Scholar]