

Abstract
Muscular dystrophy (MD) is a group of progressive genetic diseases affecting the musculature that are characterized by inflammatory infiltrates, necrosis and connective tissue and fat replacement of the affected muscles. Unfortunately, treatments do not exist for the vast majority of MD patients. Adeno-associated viral vector (AAV)-based gene therapy is thus emerging as a potential treatment for many types of MD. Treatments strategies based on AAV are being adapted for replacement of mutant disease-causing genes, knockdown of dominant disease-causing genes using antisense oligonucleotides or inhibitory RNAs, delivery of gene editing tools such as clustered regularly interspaced short palindromic repeats/Cas9 and effecting alterations in pre-mRNA splicing and by manipulating expression levels of modifier genes. Translational and clinical trial work focused on these types of AAV treatments for Duchenne MD, various limb girdle MDs, myotonic dystrophy 1, facioscapulohumeral MD, dysferlinopathies and congenital MDs are discussed here, with a focus on recent studies, pre-clinical large animal work and many promising ongoing and upcoming AAV clinical trials.
Introduction
Muscular dystrophy (MD) is a class of heterogeneous genetic myopathies characterized primarily by progressive, inflammatory, skeletal- and/or cardiac-muscle disease. Some dystrophies can also affect smooth muscles, while others can impact cognitive function. Their most unifying feature is histological: affected muscle has macrophage infiltrates, varying myofiber size, necrosis and muscle fibers replaced with connective tissue and fat (1). However, the severity and distribution of affected muscles can vary greatly from MD to MD and patient to patient. This is in part due to the variety of genetic mutations that can cause the different MDs (Fig. 1). Many of the proteins associated with MD are involved in the formation of the cytoskeleton–sarcolemma complex known as the dystrophin–glycoprotein complex (DGC), which protects muscle fibers from contraction-induced damage, stabilizes post-synaptic machinery at neuromuscular junctions (NMJs) and contributes to signal transduction (2). However, there are also nuclear proteins, nuclear membrane proteins and enzymes that result in MD, some due to unknown pathways. Generally, MDs have few treatments available, and most of the treatments that are available address quality of life and symptom management rather than correction of underlying disease pathology. Gene therapy has thus emerged as a promising form of treatment for many MDs, and we have entered an era of rapid clinical development.
Figure 1.
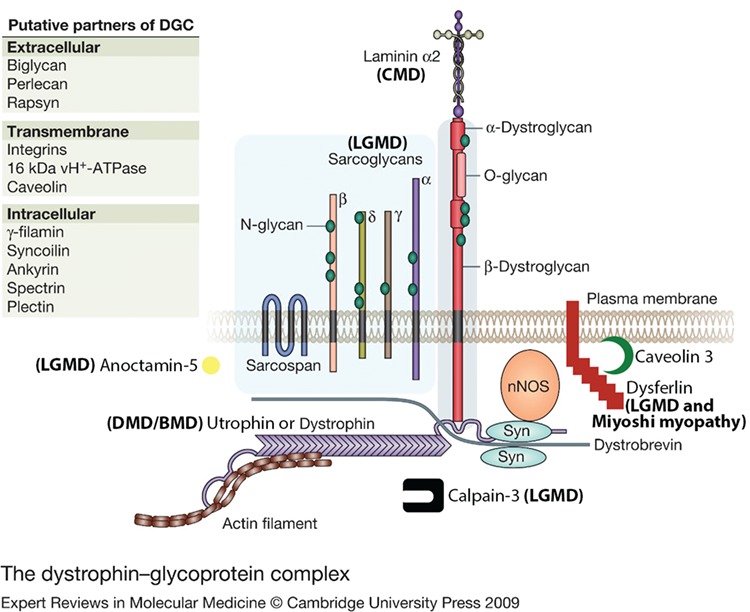
DGC and associated proteins and the MDs they cause. Modified from Dorianna Sandonà and Romeo Betto (55) under Creative Commons license BY-NC-SA 4.0. BMD means Becker MD.
Much of the current MD gene therapy pre-clinical and clinical research—like much of the gene therapy field in general—is utilizing adeno-associated viral vectors (AAVs), which were discussed in detail in another article in this issue. Briefly, these vectors can transduce a wide variety of tissues depending on serotype tropism and have small (~5 kb) genomes that exist as extrachromosomal concatemers. While body-wide, skeletal- and cardiac-muscle expression of AAV-delivered transgenes has been achieved in various animal models, transduction of satellite cells—the muscle stem cells—has proven to be difficult. However, adult cardiomyocyte turnover is < 1% per year (3); muscle fibers are multinucleated; and skeletal muscle growth and repair typically consist of satellite cells fusing and/or myotubes splitting without myonuclei mitosis (4) that could dilute vector genomes; together this suggests that expression will be relatively long-lasting in muscle, even without stem cell transduction or chromosomal integration. Indeed, muscle expression of factor IX in a patient with hemophilia B was still seen a decade after transduction (5). As long as gene therapy expression levels stabilize MD disease progression in the transduced fibers and an immune response is avoided, it is probable that diseased muscle will also have expression lasting for a least a decade.
There are several gene therapy approaches for treatment of various MDs. This review will focus on four main categories of AAV-delivered gene therapy—disease-causing gene replacement, modifier gene expression, gene editing and gene knockdown—while highlighting the exciting level of ongoing commercialization and clinical trial developments.
Gene replacement
Gene replacement is perhaps the most straightforward method of treating amenable MDs with gene therapy. It is ideal for monogenetic, recessive diseases with a known genetic cause where the gene of interest can fit into the delivery vector. Duchenne MD (DMD) is a severe X-linked recessive MD characterized by loss of ambulation around ages 8–13 and death usually in the third decade of life (1,6). DMD is caused by mutations in the gene encoding dystrophin, a protein responsible for nucleating assembly of the DGC, and also protecting against contraction-induced injury, likely through its many repetitive rod domains. Unfortunately, the cDNA of dystrophin is 14 kb, well over the carrying capacity of AAV. Interestingly, Becker MD is likewise caused by dystrophin mutations, but BMD patients can have a much milder phenotype. One such family of BMD patients, which included an ambulatory 61-year-old, had a mutation leading to a dystrophin mRNA of only 8.8 kb (7), illustrating that much of the dystrophin protein can be omitted with minimal impact on function. Based on these findings, our laboratory developed a number of truncated forms of dystrophin (microdystrophins) that can be packaged into AAV (8,9). The most recent, μDys5 (8), incorporates a recently discovered syntrophin-binding domain (10), which localizes neuronal nitric oxide synthase to the DGC (11). AAV microdystrophin has been shown to reduce the disease phenotype in canine models of DMD (12–15). Currently there are three ongoing clinical trials utilizing microdystrophins in the USA (Pfizer ClinicalTrials.gov identifier NCT03362502, Sarepta Therapeutics NCT03375164 and Solid Biosciences NCT03368742; Fig. 2), with another by Genethon in partnership with Sarepta planned to start in Europe this year (16). At the time of this writing, Solid (17) and Sarepta (18) have released interim data, with Sarepta’s higher initial dose of 2 × 1014 vector genomes (vg)/kg reflecting higher efficacy, including positive dystrophin immunofluorescence staining of muscle biopsies in all four patients biopsied to date, with an average of 81% fiber positivity, 90 days post-treatment. Patients also saw improvements in serum levels of creatine kinase (CK; an intracellular protein released into the blood with muscle damage), North Star Ambulatory Assessment and times to rise, climb four stairs and walk 100 m. Solid Biosciences reported lower levels of gene expression, in line with their lower initial vector dose. Pfizer is slated to release preliminary data in mid-2019.
Figure 2.
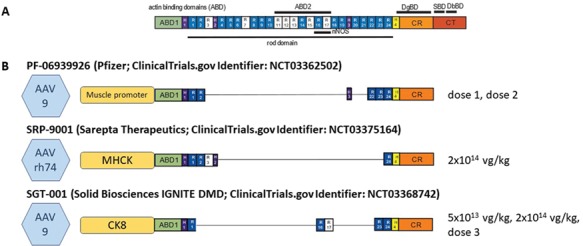
Microdystrophins in clinical trial. (A) Full-length dystrophin versus (B) the delivery vectors, promoters, microdystrophins and doses being used in current clinical trials by Pfizer, Sarepta Therapeutics and Solid Biosciences.
Sarepta has also released interim data (18) from another gene replacement clinical trial (ClinicalTrials.gov identifier NCT03652259) for limb girdle MD (LGMD) 2E, a β-sarcoglycanopathy. The sarcoglycans form a complex which is necessary to stabilize the DGC (see Figure 1) and without which muscle is subjected to contraction-induced damage. With doses of 5 × 1013 vg/kg patients had an average of 51% β-sarcoglycan positive fibers along with restoration of the sarcoglycan complex at the membrane in a 3 month post-treatment muscle biopsy accompanied with a 90% decrease in CK (18). Additional Sarepta MD gene replacement therapies are currently in clinical trials for LGMD 2D (α-sarcoglycan; ClinicalTrials.gov identifier NCT01976091) and 2B (dysferlin; ClinicalTrials.gov identifier NCT02710500) or in pre-clinical development for LGMD 2C (γ-sarcoglycan), 2L (anoctamin 5) and 2A (calpain 3).
Modifier gene expression
For some MDs, gene therapy may not be as simple as gene replacement for the disease-causing gene. Dominant disease traits, large gene sizes and immune rejection are a few of the challenges that face gene replacement. In these instances, utilizing AAV gene therapy to deliver a disease-modifying gene may be beneficial for some patients or types of MD, such as myotonic dystrophy and DMD.
In myotonic dystrophy type 1 (DM1), patients have an expansion of a CTG trinucleotide repeat found in the 3′ UTR of the dystrophia myotonica protein kinase (DMPK) gene (19). These expansions—which are dominant negative mutations—sequester the muscleblind-like protein 1 (Mbnl1) splicing regulator, leading to splicing misregulation of numerous genes necessary for normal neuromuscular physiology (20). While some groups are developing techniques to directly address the mutation through knockdown of the DMPK mRNA (see Gene Knockdown section), others are attempting to overcome the splicing bottleneck by adding more Mbnl1 to the system with AAV-mediated gene therapy (21). While an original AAV-Mbnl1 publication showed rescue of myotonia and splicing in a DM mouse model (21), a more recent publication unfortunately showed no rescue of disease phenotype with Mbnl1 overexpression that rescued splicing (22). These conflicting results warrant further studies, but in the meantime commercialization efforts for DM1 are beginning to focus on gene knockdown.
While clinical trials with microdystrophins are showing promising results, the limited transgene size and threat of an anti-dystrophin immune response (23) have led some groups and companies to pursue alternative strategies. Utrophin is a component of the cytoskeleton with high homology with dystrophin. In adults, it is typically found at myotendinous and NMJs, while it localizes to the sarcolemma during fetal development prior to dystrophin expression (24). Interestingly, it is upregulated in skeletal muscles of patients with DMD (25), and recent studies have shown that AAV-microutrophin modulates skeletal (26) and cardiac muscle disease pathology in a DMD mouse model (27). Any theoretical risk of anti-transgene immune responses is avoided since DMD patients have central tolerance to utrophin, though utrophin’s large size still requires micro-utrophins to be utilized in AAV gene therapies.
Using an alternative strategy to target the same pathway, Sarepta Therapeutics has an ongoing clinical trial (ClinicalTrials.gov identifier NCT03333590) at Nationwide Children’s Hospital utilizing AAV delivery of beta 1,4-n-acetylgalactosamine galactosyltransferase (GALGT2), a GalNac glycosyltransferase, to the legs of DMD patients. Like utrophin, GALGT2 is normally localized at the NMJ, but with gene therapy can be distributed throughout the membrane, leading to widespread membrane distribution of endogenous, full-length utrophin as well as other important members of the basement membrane including laminins and integrins. In a mouse model of DMD, AAV-GALGT2 leads to improvements in skeletal (28) and cardiac (29) muscle phenotypes, even in utrophin-deficient mice (30). GALGT2 overexpression has also been shown to improve disease phenotypes of congenital MD (CMD) MDC1A (31) and LGMD2D (32) and 2I (33) by increasing laminin and dystrophin levels and alpha-dystroglycan glycosylation (34, 35), perhaps expanding the potential commercialization market.
Gene knockdown
Gene knockdown, such as with antisense oligonucleotides (ASOs), shRNAs or miRNAs, is one method to treat dominant negative and gain-of-function mutations such as those seen in DM1 and facioscapulohumeral MD (FSHD), respectively.
As previously mentioned, one avenue for treating DM1 is through knockdown of DMPK mRNA in order to prevent the sequestration of Mbnl1 and restore normal splicing regulation. Based on preclinical successes showing infused (36, 37) and vector-delivered (38) anti-DMPK ASOs can improve the DM1 phenotype in mouse models, a phase I/II clinical trial was initiated by Ionic Pharmaceuticals and Biogen with an infused ASO. Unfortunately, the trial reported poor efficacy due to limited penetration of the ASO into muscle (39). A possible work around is to deliver the ASOs with AAV, as is currently being developed by Audentes Therapeutics (AT466, along with exon skipping for DM1) (40) and the laboratory of Joel Chamberlain at the University of Washington, who has previously shown that AAV-delivered RNAi can ameliorate disease phenotype in a DM1 mouse model (41).
FSHD type 1 is caused by contractions in the macrosatellite repeat D4Z4 that leads to chromatin relaxation and de-repressed expression of Dux4, a fetal transcription factor (42). As with DM1, this leads to a dominant phenotype, requiring Dux4 knockdown. AAV delivery of anti-Dux4 miRNAs has been shown to prevent the development of FSHD in a vector-induced mouse model (43); while disease-reversal has not been demonstrated due to drawbacks with the model, it is possible that due to the focal nature of FSHD, preventative measures may be adequate.
Gene editing
Two major approaches to AAV-based gene editing are currently developing in the pre-clinical pipeline: clustered regularly interspaced short palindromic repeats (CRISPR)-based direct genome editing and ASO-based exon skipping at the RNA level.
CRISPR
Cas (CRISPR-associated) proteins are a class of enzymes that utilize CRISPR sequences to direct genome cleavage. Such cleavage events can be utilized to remove sections of DNA through non-homologous end joining or add/replace sections of DNA through homology-directed repair. DMD is an enticing target for CRISPR-based genome editing since minidystrophin and microdystrophin are truncated versions of the protein with the potential to simply turn DMD into mild BMD. However, the extent to which CRISPR can be fully curative will still depend on the underlying disease mutation and the gene editing strategy employed. Unlike microdystrophins, a series of CRISPR-based treatments would need to be developed based on patients’ underlying mutations, since complete gene restoration is not possible with AAV-CRISPR strategies as the technology currently stands.
One of the most easily achieved strategies is to utilize CRISPR editing for exon skipping with a single, destructive cut at a splicing acceptor site, as has been successfully done following AAV gene therapy in a canine (44) model of DMD caused by exon 50 deletion. However, many mutations will require the removal of more than a single exon, and so an AAV-based, double-cutting strategy to remove exons 6–8 has also been employed by our laboratory (manuscript in preparation) in an alternative canine model of DMD. While we did demonstrated limited genome editing following intramuscular injections (45), strategies that require two simultaneous cuts, the removal of a large piece of DNA and subsequent end joining, rather than simply the destruction of a single cut site, have unsurprisingly proven to be more difficult. Still, a number of companies have DMD CRISPR treatments in their pre-clinical pipeline—undoubtedly with an eye toward improving the efficacy and exploring the immune challenges (46) associated with in vivo CRISPR—including Exonics Therapeutics (utilizing the aforementioned SingleCut CRISPR), CRISPR Therapeutics, Sarepta Therapeutics and Editas Medicine. Additionally, trans-splicing AAV was used to deliver a modified Cas protein known as an adenine base editor for successful single-nucleotide editing correction of a DMD-causing nonsense mutation in mice (47) and may be a future avenue for commercialization of CRISPR–Cas-based treatments.
Exon skipping
The only treatment for DMD currently on the market that directly addresses the underlying disease mechanism is Sarepta Therapeutics’ Exondys 51 (eteplirsen), a morpholino ASO that causes exon 51 to be spliced out in pre-mRNA, restoring the reading frame in the 13% of patients with amenable frame-shifting mutations (48). Like CRISPR, such frame restoration may lead to fuller proteins than microdystrophins, depending on the underlying mutation and the exon that is skipped. Exondys 51 and other ASOs following it up the pipeline are infused intravenously once weekly and must make it out of the circulation, through the muscle cells’ cell membranes and into the cytosol and nucleus. Some studies suggest that ASOs like Exondys 51 may be best able to enter cells that are developing, damaged or in a state of repair (49)—as would be the case in diseased MD muscle—but this suggests that an equilibrium would be reached between ASO efficacy and ASO cytosol access. These considerations may relate to the inefficiency of dystrophin production seen in patients treated with ASOs. While Exondys 51 has been shown to be exceedingly safe, there is always a theoretical risk of accumulation in the kidneys and liver, the development of antibodies against infused treatments or of off-target splicing in other tissues, and weekly, hour-long infusion sessions can be constricting to patients and their families while also resulting in drug peaks and troughs.
While Sarepta is developing advanced ASOs meant to increase cell penetration, studies utilizing AAV to deliver muscle-specific ASO-expressing cassettes are also underway in order to increase efficacy (current formulations do not target cardiac muscle) while maintaining safety. Pre-clinical work in DMD mice (50) and dogs (51) has shown that AAV-delivered ASOs are able to improve disease phenotype, although there was some loss of efficacy over time (51). Audentes Therapeutics recently announced a partnership with Nationwide Children’s Hospital to develop AT702, an ASO-delivering AAV for exon 2 skipping for DMD patients with mutations in exons 1–5, including exon 2 duplications (52). A phase I/II study is currently planned for the end of 2019. Audentes is also in the pre-clinical development stages for AAV-based ASO exons 51 and 53 skipping for DMD (AT751 and AT753, respectively). AT702, 751 and 753 together would target >25% of the DMD patient population, and the company has declared their intention to ultimately expand the platform to the 80% of patients that might benefit from exon skipping. Should this platform prove successful, many other MDs, including dysferlinopathies such as Miyoshi myopathy and LGMD2B (53, 54), could be amenable to exon skipping, expanding the patients that may ultimately be treated with infused or AAV-based exon skipping therapies.
Conclusions
Patients with MDs have been an underserved group in terms of treatment availability and commercial research and development. However, with the AAV gene therapy boom, a number of these neglected rare diseases are seeing rapid pre-clinical and clinical developments. Ongoing clinical trials for Duchenne and LGMDs, which are already showing promising results, will hopefully lead to commercial products, and additional treatments fill the pipeline behind them. It is a promising time for MD gene therapy.
Funding
National Institutes of Health Grants (AR40864 to J.S.C.); Muscular Dystrophy Association (USA).
Conflict of Interest statement
J.S.C. is a member of Solid Biosciences’ scientific advisory board. J.M.C. and J.S.C. hold equity in Solid Biosciences.
References
- 1. Emery A.E. (2002) The muscular dystrophies. Lancet, 359, 687–695. [DOI] [PubMed] [Google Scholar]
- 2. Mercuri E. and Muntoni F. (2013) Muscular dystrophies. Lancet, 381, 845–860. [DOI] [PubMed] [Google Scholar]
- 3. Bergmann O., Zdunek S., Felker A., Salehpour M., Alkass K., Bernard S., Sjostrom S.L., Szewczykowska M., Jackowska T., Dos Remedios C. et al. (2015) Dynamics of cell generation and turnover in the human heart. Cell, 161, 1566–1575. [DOI] [PubMed] [Google Scholar]
- 4. Dayanidhi S. and Lieber R.L. (2014) Skeletal muscle satellite cells: mediators of muscle growth during development and implications for developmental disorders. Muscle Nerve, 50, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchlis G., Podsakoff G.M., Radu A., Hawk S.M., Flake A.W., Mingozzi F. and High K.A. (2012) Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood, 119, 3038–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eagle M., Baudouin S.V., Chandler C., Giddings D.R., Bullock R. and Bushby K. (2002) Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord., 12, 926–929. [DOI] [PubMed] [Google Scholar]
- 7. England S.B., Nicholson L.V., Johnson M.A., Forrest S.M., Love D.R., Zubrzycka-Gaarn E.E., Bulman D.E., Harris J.B. and Davies K.E. (1990) Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature, 343, 180–182. [DOI] [PubMed] [Google Scholar]
- 8. Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D. and Chamberlain J.S. (2019) Development of novel micro-dystrophins with enhanced functionality. Mol. Ther., 27, 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Banks G.B., Judge L.M., Allen J.M. and Chamberlain J.S. (2010) The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet., 6, e1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams M.E., Odom G.L., Kim M.J., Chamberlain J.S. and Froehner S.C. (2018) Syntrophin binds directly to multiple spectrin-like repeats in dystrophin and mediates binding of nNOS to repeats 16–17. Hum. Mol. Genet., 27, 2978–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L. et al. (2009) Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest., 119, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Guiner C., Servais L., Montus M., Larcher T., Fraysse B., Moullec S., Allais M., Francois V., Dutilleul M., Malerba A. et al. (2017) Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun., 8, 16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birch S., Lawlor M.W., Guo L.-J., Crudelle J., Hawkins E.C., Nghiem P.P., Styner M.A., Struharik M.J., Brown K.J., Golebiowski D., et al. (2017) A blinded, placebo-controlled systemic gene therapy efficacy study in the GRMD model of Duchenne muscular dystrophy. Mol. Ther., 25 (5S1), 193 (abstract). [Google Scholar]
- 14. Hakim C.H., Kodipilli, K., Jenkins G., Hsiao T.Y., Pan X., Lessa T.B., Leach S.B., Emter C., Yue Y., Zhang K. et al. (2018) AAV micro-dystrophin therapy ameliorates muscular dystrophy in young adult Duchenne muscular dystrophy dogs for up to thirty months following injection. Mol. Ther., 26 (5S1), 5 (abstract). [Google Scholar]
- 15. Yue Y., Pan X., Hakim C.H., Kodippili K., Zhang K., Shin J.H., Yang H.T., McDonald T. and Duan D. (2015) Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet., 24, 5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Genethon 2019Duchenne muscular dystrophy. http://www.genethon.fr/en/products/duchenne-muscular-dystrophy/ (accessed May 15, 2019)..
- 17. Solid Biosciences (2019) Solid biosciences announces preliminary SGT-001 data and intention to dose escalate in IGNITE DMD clinical trial for duchenne muscular dystrophy. Globe Newswire.
- 18. Rodino-Klapac L.R. (2019) Presented at the annual meeting of The American Society of Gene and Cell Therapy meeting. Washington, DC.
- 19. Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T. et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- 20. Jiang H., Mankodi A., Swanson M.S., Moxley R.T. and Thornton C.A. (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet., 13, 3079–3088. [DOI] [PubMed] [Google Scholar]
- 21. Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A. and Swanson M.S. (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly (CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yadava R.S., Kim Y.K., Mandal M., Mahadevan K., Gladman J.T., Yu Q. and Mahadevan M.S. (2019) MBNL1 overexpression is not sufficient to rescue the phenotypes in a mouse model of RNA toxicity. Hum. Mol. Genet., 28, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G. et al. (2010) Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med., 363, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clerk A., Morris G.E., Dubowitz V., Davies K.E. and Sewry C.A. (1993) Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J., 25, 554–561. [PubMed] [Google Scholar]
- 25. Helliwell T.R., Man N.T., Morris G.E. and Davies K.E. (1992) The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscul. Disord., 2, 177–184. [DOI] [PubMed] [Google Scholar]
- 26. Odom G.L., Gregorevic P., Allen J.M., Finn E. and Chamberlain J.S. (2008) Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther., 16, 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kennedy T.L., Guiraud S., Edwards B., Squire S., Moir L., Babbs A., Odom G., Golebiowski D., Schneider J., Chamberlain J.S. et al. (2018) Micro-utrophin improves cardiac and skeletal muscle function of severely affected D2/mdx mice. Mol. Ther. Methods Clin. Dev., 11, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nguyen H.H., Jayasinha V., Xia B., Hoyte K. and Martin P.T. (2002) Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl. Acad. Sci. U. S. A., 99, 5616–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu R., Jia Y., Zygmunt D.A. and Martin P.T. (2019) rAAVrh74.MCK.GALGT2 protects against loss of hemodynamic function in the aging mdx mouse heart. Mol. Ther., 27, 636–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu R., Camboni M. and Martin P.T. (2007) Postnatal overexpression of the CT GalNAc transferase inhibits muscular dystrophy in mdx mice without altering muscle growth or neuromuscular development: evidence for a utrophin-independent mechanism. Neuromuscul. Disord., 17, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu R., Chandrasekharan K., Yoon J.H., Camboni M. and Martin P.T. (2007) Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am. J. Pathol., 171, 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu R., DeVries S., Camboni M. and Martin P.T. (2009) Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am. J. Pathol., 175, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thomas P.J., Xu R. and Martin P.T. (2016) B4GALNT2 (GALGT2) gene therapy reduces skeletal muscle pathology in the FKRP P448L mouse model of limb girdle muscular dystrophy 2I. Am. J. Pathol., 186, 2429–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia B., Hoyte K., Kammesheidt A., Deerinck T., Ellisman M. and Martin P.T. (2002) Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev. Biol., 242, 58–73. [DOI] [PubMed] [Google Scholar]
- 35. Yoon J.H., Johnson E., Xu R., Martin L.T., Martin P.T. and Montanaro F. (2012) Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res., 11, 4413–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mulders S.A., Broek W.J., Wheeler T.M., Croes H.J., Kuik-Romeijn P., Kimpe S.J., Furling D., Platenburg G.J., Gourdon G., Thornton C.A. et al. (2009) Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 106, 13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F. and Thornton C.A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Furling D., Doucet G., Langlois M.A., Timchenko L., Belanger E., Cossette L. and Puymirat J. (2003) Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther., 10, 795–802. [DOI] [PubMed] [Google Scholar]
- 39. Ionis Pharmaceuticals and Biogen (2017) Letter from Ionis Pharmaceuticals & Biogen to the MDF Community, Myotonic Dystrophy Foundation. Myotonic Dystrophy Foundation.
- 40. Audentes Therapeutics Audentes is developing AT466 for the treatment of myotonic dystrophy type 1. https://www.audentestx.com/myotonic-dystrophy-type-1/ (accessed May 15, 2019).
- 41. Bisset D.R., Stepniak-Konieczna E.A., Zavaljevski M., Wei J., Carter G.T., Weiss M.D. and Chamberlain J.R. (2015) Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum. Mol. Genet., 24, 4971–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeSimone A.M., Pakula A., Lek A. and Emerson C.P. Jr. (2017) Facioscapulohumeral muscular dystrophy. Compr. Physiol., 7, 1229–1279. [DOI] [PubMed] [Google Scholar]
- 43. Wallace L.M., Saad N.Y., Pyne N.K., Fowler A.M., Eidahl J.O., Domire J.S., Griffin D.A., Herman A.C., Sahenk Z., Rodino-Klapac L.R. et al. (2018) Pre-clinical safety and off-target studies to support translation of AAV-mediated RNAi therapy for FSHD. Mol. Ther. Methods Clin. Dev., 8, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. et al. (2018) Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science, 362, 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bengtsson N.E., Crudele J., Klaiman J.M., Snyder J.M. and Chamberlain J.S (2019) In vivocorrection of dystrophin expression in old dystrophic dogs. Mol. Ther., 27 (4S1), 22.(abstract). [Google Scholar]
- 46. Crudele J.M. and Chamberlain J.S. (2018) Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun., 9, 3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E. et al. (2018) Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol., 36, 536–539. [DOI] [PubMed] [Google Scholar]
- 48. Lim K.R., Maruyama R. and Yokota T. (2017) Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther., 11, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Novak J.S., Hogarth M.W., Boehler J.F., Nearing M., Vila M.C., Heredia R., Fiorillo A.A., Zhang A., Hathout Y., Hoffman E.P. et al. (2017) Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun., 8, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goyenvalle A., Babbs A., Wright J., Wilkins V., Powell D., Garcia L. and Davies K.E. (2012) Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum. Mol. Genet., 21, 2559–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vulin A., Barthelemy I., Goyenvalle A., Thibaud J.L., Beley C., Griffith G., Benchaouir R., Hir M., Unterfinger Y., Lorain S. et al. (2012) Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther., 20, 2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Therapeutics A. (2019) Audentes Therapeutics announces expansion of AAV technology platform and pipeline with new development programs for Duchenne muscular dystrophy and myotonic dystrophy. PR Newswire.
- 53. Barthelemy F., Blouin C., Wein N., Mouly V., Courrier S., Dionnet E., Kergourlay V., Mathieu Y., Garcia L., Butler-Browne G. et al. (2015) Exon 32 skipping of dysferlin rescues membrane repair in patients’ cells. J. Neuromuscul. Dis., 2, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J.J.A., Maruyama R., Duddy W., Sakurai H. and Yokota T. (2018) Identification of novel antisense-mediated exon skipping targets in DYSF for therapeutic treatment of dysferlinopathy. Mol. Ther. Nucleic Acids, 13, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sandona D. and Betto R. (2009) Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev. Mol. Med., 11, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]