

Abstract
Inherited neuromuscular diseases are a heterogeneous group of developmental and degenerative disorders that affect motor unit function. Major challenges toward developing therapies for these diseases include heterogeneity with respect to clinical severity, age of onset and the primary cell type that is affected (e.g. motor neurons, skeletal muscle and Schwann cells). Here, we review recent progress toward the establishment of genetic therapies to treat inherited neuromuscular disorders that affect both children and adults with a focus on spinal muscular atrophy, Charcot–Marie–Tooth disease and spinal and bulbar muscular atrophy. We discuss clinical features, causative mutations and emerging approaches that are undergoing testing in preclinical models and in patients or that have received recent approval for clinical use. Many of these efforts employ antisense oligonucleotides to alter pre-mRNA splicing or diminish target gene expression and use viral vectors to replace expression of mutant genes. Finally, we discuss remaining challenges for optimizing the delivery and effectiveness of these approaches. In sum, therapeutic strategies for neuromuscular diseases have shown encouraging results, raising hope that recent strides will translate into significant clinical benefits for patients with these disorders.
Introduction
Remarkable progress during the past several decades has defined causative mutations that drive pathogenesis in a large number of inherited neuromuscular diseases. Mechanistic studies have uncovered diverse consequences of disease-causing mutations, including both loss-of-function and toxic gain-of-function effects that often occur concurrently to contribute to pathogenesis. This complexity reverberates as dysfunction in an unexpectedly large number of pathways. As a result, therapeutically targeting any of one of these downstream pathways most often leads to incomplete or inadequate clinical responses. To address this challenge, recent therapeutic efforts for inherited neuromuscular diseases have focused on the mutant protein or gene as the proximal mediator of pathogenesis, often using emerging technologies to drug these targets.
We review recent progress in developing genetic therapies for three types of neuromuscular disease. We highlight efforts toward treating disorders that affect children [spinal muscular atrophy (SMA) and some cases of SMA] and adults [Charcot–Marie–Tooth (CMT) disease and spinal and bulbar muscular atrophy (SBMA)]. Foundational work on these disorders is aimed at identifying disease-specific clinical outcome measures that are both sensitive and reliable. These quantitative metrics are essential for evaluating efficacy in the clinic. Additional significant challenges are notable for therapy development. For adult onset diseases, a slowly worsening clinical course can make it difficult to measure rescue of disease progression during clinical trials that may last a year or less. Moreover, experiences in model systems suggest that beneficial effects may be most robust when interventions occur early in disease course. However, this can be challenging to achieve, as treatment of asymptomatic adults is not yet standard practice and age of onset is often poorly defined. Limitations of current genetic approaches are also important to consider. Poor uptake of therapeutic oligonucleotides and limited accessibility to or durability of viral vectors in critical target cells raises the likelihood of partial treatment responses. Moreover, gene therapy approaches are presumably irreversible, potentially providing sustained benefits but also raising the specter of long-term untoward effects.
While these issues present notable hurdles to translating approaches to the clinic, there has also been striking progress. We start by reviewing recent successes in the treatment of SMA, as this progress may provide a framework that is applicable to other neuromuscular diseases.
SMA
SMA is an inherited neuromuscular disease characterized by motor neuron degeneration causing progressive muscle atrophy and weakness. SMA occurs in approximately 1 in 10, 000 live births and is the leading inherited cause of infant mortality (1). Disease progression is often most rapid at onset and slower at later stages (2,3). Clinical symptoms are highly variable in terms of age of onset and motor milestones reached by patients. Type 0 (congenital) and type I (0–6 months) are the most severe forms with affected individuals failing to reach any major motor milestones and dying by 2 years without intervention (4–6). Although type I SMA is the most common form of SMA, there is vast phenotypic heterogeneity in SMA with type II SMA patients achieving the ability to sit but not stand whereas type III SMA patients are able to stand. Patients with type IV SMA show adult onset (second or third decade of life) and only mild to moderate muscle weakness (4,5). SMA arises from recessive loss-of-function mutations (often large deletions) of the survival motor neuron 1 (SMN1) gene leading to reduced expression of the SMN protein. However, disease severity scales inversely with copy number of the paralogous SMN2 gene (7–9). Although SMN2 is ubiquitously transcribed, a single base change (C→T) in exon 7 leads to its frequent exclusion from its mature mRNA and the resulting protein is truncated and unstable (Fig. 1A; 10–13). A small proportion (~20%) of SMN2 transcripts retain exon 7 and produce some functional protein. While this is sufficient to prevent the embryonic lethality that results from the complete absence of SMN, it is insufficient to prevent motor neuron degeneration (14). Why motor neurons are particularly vulnerable to SMN protein deficiency remains unknown, but disease pathogeness is a direct result of loss of the SMN protein and early SMN replacement by various methods prevents the SMA phenotype in preclinical models (21, 23, 31–33). The specific genetics of SMA has enabled rapid therapeutic developments aimed at increasing SMN levels by targeting either SMN1 and SMN2 as described below.
Figure 1.
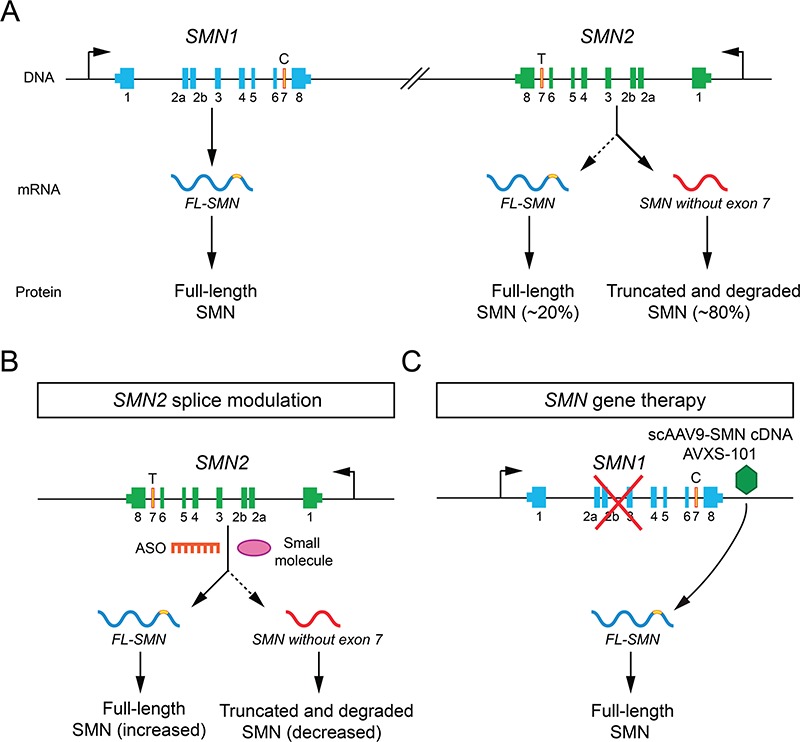
Genetics and therapeutic strategies in SMA. (A) Diagram of SMN genes, mRNA and protein products. (B)SMN2 splice modulation. (C)SMN gene therapy.
Gene targeting therapeutics
Antisense oligonucleotides (ASOs) are short, synthetic single-stranded nucleic acids that bind complementary sense sequences in targeted RNAs in order to either promote RNase-mediated degradation or modify splicing by acting as a steric hindrance to the binding of RNA splicing factors (15). Development of splice-switching oligonucleotide platforms to target SMN2 pre-mRNAs has resulted in the first FDA-approved treatment for SMA—nusinersen/Spinraza [Ionis (Carlsbad, CA)/Biogen (Cambridge, MA); 16–18]. This ASO with a modified nucleotide backbone hybridizes with the intronic splice silencer N1 in SMN2 pre-mRNAs to promote exon 7 inclusion, leading to an increase in full-length SMN protein (Fig. 1B; 19,20). Nusinersen is administered by intrathecal injections as it is unable to cross the blood brain barrier. It has been shown to increase SMN protein levels both in vitro and in pre-clinical models of SMA (21–23). Several clinical trials have been conducted to test the therapeutic potential of nusinersen in type I and type II/III SMA patients (14,24–26).
Another approach to target the SMN2 gene is by using small molecules. Bioavailable small molecules that also function to promote exon 7 inclusion in SMN2 mRNA have the advantage that they can be orally administered and have widespread tissue bioavailability. Two SMN-inducing small molecule therapies, Risdiplam (developed by Roche, Basel, Switzerland) and Branaplam (developed by Novartis, Basel, Switzerland), have proven to be as effective as ASOs and gene replacement in SMA mouse models and are being tested in ongoing clinical trials (27,28).
The third approach that has shown success in treating SMA is gene replacement therapy. In this case, a wild-type copy of the SMN cDNA packaged in adeno-associated virus 9 vector (AAV9) is delivered intravenously (Fig. 1C). AAVs are non-pathogenic, capable of crossing the blood brain barrier, and can efficiently transduce neurons (29,30). Following viral transduction, the SMN cDNA is maintained as a constitutively expressed episome, which offers long-term durable expression in terminally differentiated cells such as neurons after a single dose, but is diluted in mitotically active cells that might limit beneficial effects in non-neuronal tissues (14). The low rate of integration into the host genome reduces risk of oncogenesis. Avexis-101(scAAV9-SMN) (Avexis, Chicago, IL) uses double-stranded, self-complementary recombinant AAV vector (scAAV9) to deliver SMN cDNA leading to a rapid induction of SMN protein. This drug has shown marked improvements of survival and motor function in SMA mice in preclinical studies (31–33) and efficacy in infantile SMA patients (34). The FDA recently granted the approval of Zolgensma (onasemnogene abeparvovec-xioi) to Avexis, Inc (Avexis, Inc, Chicago, IL) for the treatment of SMA patients less than two years of age.
Optimizing SMA therapeutics
Challenges remain for SMN-inducing approaches in terms of optimizing drug timing, dosage, delivery route, distribution and uptake in vulnerable neuronal populations. The importance of targeting cells and tissues other than motor neurons remains unclear. In addition, long-term durability and potential toxicities remain to be defined. Table 1 summarizes the key pharmacological features of gene targeting therapeutics in SMA and the challenges that need to be addressed. It is clear that while patients treated very early in disease can show marked improvements, many patients have only modest responses. The institution of neonatal testing for SMA will increase the number of patients who receive therapy soon after birth. In addition, identification of biomarkers of disease progression and methods to quantify SMN levels in target tissues are keys for progress. Studies of combinatorial treatment that includes more than one SMN-inducing drug as well as SMN induction with SMN-independent strategies such as drugs to improve muscle growth and contractility are ongoing (35). Lessons learned from the success of gene targeting platforms in SMA can be applied to other diseases including CMT disease and SBMA as discussed in the following sections. These therapeutic approaches are summarized in Fig. 2.
Table 1.
Overview of SMN-inducing therapies and their pharmacological characteristics
| Drug (manufacturer) | Nusinersen (Biogen/Ionis) | scAAV9-SMN (Avexis/Novartis) | Risdiplam (Roche/PTC/SMA Foundation) | Branaplam (Novartis) |
|---|---|---|---|---|
| Category | ASO | Gene therapy | Small molecule | Small molecule |
| Mechanism | SMN2 splice switching | Viral-mediated SMN replacement | SMN2 splice switching | SMN2 splice switching |
| Delivery route | Intrathecal | Intravenous | Oral | Oral |
| Dosage frequency | Four loading doses over 2 months, followed by single dose every 4 months | Once | Daily | Once weekly |
| Tissue distribution | ||||
| -Cell targets | CNS | Systemic (lost from mitotically active cells) | Systemic | Systemic |
| -Topographical distribution | Possibility of high drug levels in caudal and low levels in rostral regions of the spinal cord (humans) (86). CSF flow dynamics could potentially affect drug distribution. | IV and CSF delivery shows transduction of motor neurons in the spinal cord (pre-clinical work in mice and non-human primates) (31,87). | Distributes to a similar extent in the brain, plasma and muscles (pre-clinical study in mice, rats and monkeys) (89) | Not reported |
| -Neuronal tropism | ASOs are taken up well by neurons compared to other cell types. | Efficient transduction of motor neurons (88) | Likely shows similar uptake levels in all tissue types | Likely shows similar uptake levels in all tissue types |
| Pharmacokinetics | Median time to max (Tm) in plasma, 1.7–6 h; Mean terminal elimination half-life in CSF, 135–177 days and in plasma, 63–87 days (61) | Not reported | Not reported | Not reported |
| Pharmacodynamics | Exon 7 inclusion increased by 20–40% across the spinal cord. SMN levels increased in motor neurons (86); SMN induction is likely limited by amount of SMN2 pre-mRNA; Speed of SMN induction and thresholds needed for efficacy are unknown. |
Possibly results in a relatively rapid induction of SMN compared to other approaches. How long increased SMN levels are sustained over the long-term is not known. | Likely causes a dose-dependent induction of SMN in CNS and other tissues; SMN induction is likely limited by amount of SMN2 pre-mRNA. |
Likely causes a dose-dependent induction of SMN in CNS and other tissues; SMN induction is likely limited by amount of SMN2 pre-mRNA. |
| Long-term durability | Requires monthly dosing (following the initial four loading doses) to sustain SMN levels | Possible stable long-term expression in terminally differentiated cells with single administration | Requires daily dosing to sustain SMN levels | Requires daily dosing to sustain SMN levels |
| Potential toxicities | • Mild renal toxicity• mild thrombocytopenia• complications arising from repeated lumbar punctures | • Potential inflammatory reaction during administration• exclusion of subjects with antibodies for AAV9 in serum• insertional genotoxicity leading to oncogenesis | • Potential off-target effects• possible build-up of toxic byproducts after prolonged daily dosage | • Potential off-target effects• possible toxic byproduct build-up after prolonged daily dosage |
| Stage in clinical development | FDA-approved | FDA-approved for SMA patients aged less than 2 years | FDA submission expected | Phase II |
| Current SMA target patient population(s) | FDA-approved for all types | Type I; trial involving intrathecal injections for Type II SMA is ongoing | Types I–III | Type I |
Figure 2.

Therapeutic approaches for SMA, CMT disease and SBMA. Diagram of therapeutic approaches and target tissues for SMA, CMT disease and SBMA discussed in this review. Shown is a lower motor neuron (green), myelinated axon (yellow) and skeletal muscle (red).
CMT disease
Charcot-Marie-Tooth (CMT) disease—named after the three physicians who reported the phenotype in 1886—represents a broad class of inherited peripheral neuropathies. Patients with CMT disease experience muscle weakness in the hands and feet that results in impaired mobility and fine motor function as well as reduced sensation in distal extremities (36). Over the past three decades significant advances have been made in mapping genes responsible for CMT disease. These data now facilitate precise diagnoses. While management strategies have been developed to improve quality of life, no effective molecular therapies are available for CMT disease.
A major challenge in developing therapies for CMT disease is the vast heterogeneity. First, CMT disease is divided into two major subtypes based on the primary site of pathogenesis (37). In CMT1, genetic lesions have a primary effect on the myelinating Schwann cells of the peripheral nerve, in part indicated by reduced motor nerve conduction velocities (MNCVs). In CMT2, the primary effect is on peripheral nerve axons, in part indicated by preserved MNCVs but reduced amplitudes of evoked nerve responses. This ‘cellular heterogeneity’ poses challenges for the delivery of therapeutics to motor neurons, sensory neurons and Schwann cells. Second, CMT disease exhibits all inheritance patterns. Over 100 loci have been implicated in disease (38), and each locus can be associated with dozens of disease-causing alleles acting through diverse pathogenic mechanisms (e.g. loss- and gain-of-function), resulting in the need to develop gene-specific and even allele-specific therapeutics. Third, the clinical heterogeneity of CMT disease is such that even patients with molecularly indistinguishable disease-causing variants can display diverse clinical severities. For example, patients with a 1.4 Mb duplication surrounding the peripheral myelin protein 22 locus (PMP22, which encodes an integral myelin transmembrane protein) have wide variability in age of onset (39), which raises questions about the appropriate timing for therapeutic interventions.
While there are currently no effective therapies for patients with CMT disease, much has been learned from pre-clinical studies in animal models (40). Recessive CMT phenotypes are largely due to a loss (or severe reduction) of gene function, suggesting that gene replacement therapies will be effective once timing and cell delivery issues are solved. In contrast, dominant CMT phenotypes often involve a mutant allele that is toxic to the peripheral nerve, suggesting that gene knockdown may be required to alleviate phenotypes. Below, we highlight recent advances in therapeutic strategies aimed at the more common forms of dominant CMT disease.
Dominant, demyelinating CMT disease (CMT1)
The majority of genes implicated in CMT1 encode proteins that are critical for peripheral nerve myelination (38). A common phenotypic characteristic is impaired development and progressive thinning of the myelin sheath. In terms of generalized therapeutic strategies for CMT1, improving lipid biosynthesis may be a reasonable approach since lipids are important for myelin formation and maintenance. In support of this notion, RNA sequencing analysis of sciatic nerve from a transgenic rat model of PMP22-related neuropathy revealed reduced transcript expression of genes involved in lipid biosynthesis (41). To test the ability of phospholipid therapy to rescue neuropathy in this model, (1) animals received fluorescently labeled phosphatidylcholine in the tail vain during myelination (postnatal day 15) and (2) mothers and weened animals were provided a lipid-rich diet. These studies revealed incorporation of lipids into peripheral nerve myelin and rescue of motor behavioral deficits in mutation-positive animals. Encouragingly, lipid supplementation was also effective at advanced disease stages, suggesting that it may be relevant for symptomatic patients (41). Future studies should assess the relevance of this strategy in other genetic models of CMT1 (e.g. those associated with myelin protein zero gene mutations) and the efficacy of the approach in patients with PMP22-related CMT1.
Because duplication of PMP22 is the most common cause of CMT1 (42), therapeutic development has focused on down-regulating PMP22 transcripts. Initial attempts focused on ascorbic acid to reduce cAMP levels in Schwann cells; this was based on the structure of the PMP22 promoter, which harbors a repressor that functions in the absence of cyclic adenosine monophosphate (cAMP) (43). While ascorbic acid was effective at reducing PMP22 expression, increasing myelination and alleviating neuropathy in transgenic mice (44), clinical trials in CMT1 patients were unsuccessful (45,46). Current approaches focus on reducing PMP22 expression via ASO. This strategy was successful in transgenic mice harboring multiple copies of a yeast artificial chromosome spanning human PMP22 (47). The authors designed an ASO to target the 3′ untranslated region (UTR) of human PMP22 and administered it via subcutaneous (s.c.) injections in a range of concentrations. They noted a concentration-dependent reduction of both human and mouse PMP22, which was associated with improved motor function, enhanced myelination and a rescue of transcriptional changes. This strategy was similarly successful in PMP22 transgenic rats. Notably, PMP22 haploinsufficiency causes hereditary neuropathy with liability to pressure palsies (48), indicating that translation of this strategy to humans will require careful titration and monitoring.
Dominant, axonal CMT disease (CMT2)
In contrast to CMT1, loci associated with CMT2 encode proteins with myriad functions. A potential common, downstream effect of CMT2-associated mutations is impaired axonal transport (49). If therapeutics were developed to improve axonal transport, this might be applicable to multiple forms of CMT disease. Supporting this notion, inhibitors of histone deacetylase 6 (HDAC6) improved axonal transport deficits and rescued associated behavioral phenotypes in two mouse models of CMT2 [heat shock protein B1 and glycyl-tRNA synthetase (Gars); 50,51]. The positive effect was attributed to increased acetylated α-tubulin, which may stabilize axonal microtubules and improve transport. While these findings are promising, the mechanism of action with respect to the gene mutations is controversial (51,52); the approach has not been tested on other models of CMT2, and the safety of HDAC6 inhibitors in humans is undetermined.
Mutations in the gene encoding mitofusin 2 (MFN2) are the most common cause of CMT2 (53). MFN2 is a mitochondrial GTPase required for fusion and axonal transport, which are compromised in models of disease. Interestingly, a separate locus encodes the mitochondrial GTPase MFN1, which complements MFN2 dysfunction (54). While MFN1 and MFN2 are ubiquitously expressed, they are not equally expressed in all tissues and, of note, MFN1 is expressed at lower levels in the nervous system (54). Using a neuron-specific transgenic mouse model of a CMT2-associated MFN2 mutation (R94Q), it was recently shown that transgenic expression of MFN1 (and separately MFN2) rescues behavioral and histopathological phenotypes (55). These findings give hope that if appropriate delivery systems can be developed (e.g. modulating endogenous gene expression or employing AAV-mediated delivery as in SMA), increasing the function of MFN1 and/or MFN2 will improve neuropathy.
Aminoacyl-tRNA synthetases (ARSs) are a ubiquitously expressed class of enzymes responsible for charging tRNA molecules with cognate amino acids (56). To date, five loci encoding an ARS have been implicated in CMT2: GARS, YARS, AARS, HARS and WARS (57). The overlapping function of these enzymes suggests that impaired tRNA charging plays a role in disease pathogenesis and that additional ARS loci will be implicated in CMT disease. However, while the majority of CMT2-associated ARS variants reduce or ablate enzyme function (57), it has been established that haploinsufficiency is not the mode of pathogenesis (58). There are currently two reasonable therapeutic designs for ARS-associated CMT2. First, restoring the wild-type enzyme may rescue loss-of-function effects. While over-expression of wild-type Gars did not rescue the phenotype of a spontaneous mouse model (i.e. a mutation that has not been identified in humans; 58,59), this approach should be tested on human pathogenic GARS alleles and on other CMT2-associated ARS loci. Second, regardless of the molecular pathology, it is clear that pathogenic ARS alleles are dominantly toxic to peripheral nerve axons (57). To alleviate the phenotype, it would be appropriate to design allele-specific knockdown, taking care to not dramatically reduce expression of these essential genes, as ARS loci are also associated with more severe recessive phenotypes (57). Promising allele-specific knockdown approaches for ARS-related CMT2 disease using AAV-delivered RNA interference are currently being developed in pre-clinical studies.
SBMA
SBMA is a progressive neuromuscular disorder that affects only men and is characterized by proximal limb and bulbar muscle weakness, atrophy and fasciculations (60). In addition to neuromuscular impairment patients may exhibit sensory involvement, particularly affecting vibration in the distal legs (61). These clinical features correlate with loss of lower motor neurons in the brainstem and spinal cord and with both myopathic and neurogenic changes in skeletal muscle (60,62). More mild loss of myelinated axons in the posterior column of the spinal cord also occurs (62). The causative mutation in SBMA is an expansion of a CAG/polyglutamine (polyQ) repeat in the first exon of the androgen receptor (AR) gene (63). The expanded polyQ tract promotes polyQ AR unfolding and oligomerization, steps that are critical to toxicity (61,64). That this protein aggregation is hormone dependent accounts for the striking sex bias of this disease, a feature that distinguishes SBMA from eight other neurodegenerative disorders similarly caused by CAG repeat expansions in coding regions of unrelated genes. As a group, the polyQ disorders are currently untreatable, with symptom onset typically in midlife and death several decades later (65).
Although it was long considered that lower motor neurons are the primary targets of degeneration in SBMA, recent studies have established the importance of peripheral polyQ AR expression in disease, particularly in skeletal muscle. SBMA patients show myopathic features on muscle biopsy as well as serum creatine kinase levels that are higher than those normally found in purely denervating diseases (66–68). SBMA knock-in mice develop both muscle atrophy and myopathy before the occurrence of spinal cord pathology, consistent with the idea that skeletal muscle is an early target of disease (69). Moreover, transgenic mice that over-express either wild-type or polyQ AR only in skeletal muscle show hormone-dependent myopathy and motor axon loss (70,71). Disease severity in SBMA transgenic mice is ameliorated by over-expression of insulin-like growth factor 1 (IGF-1) in skeletal muscle (72). That skeletal muscle contributes to the SBMA phenotype and provides a therapeutic target is further supported by studies showing that peripheral polyQ AR gene suppression by s.c. administration of AR-targeted ASOs or conditional gene deletion in skeletal muscle rescues disease in mice (73,74). The beneficial effects of ASOs are noteworthy, as s.c. administration diminishes peripheral but not central nervous system (CNS) expression of polyQ AR and rescues skeletal muscle weight and fiber size, improves grip strength and extends lifespan in both knock-in and bacterial artificial chromosome transgenic models of SBMA. Based on this body of work, SBMA can be considered a degenerative disorder of the neuromuscular system, with skeletal muscle pathology predominating early and motor neuron loss becoming more significant at late stages of disease (64).
More recent characterization of the phenotype of SBMA patients and mouse models has revealed unexpected metabolic abnormalities. These include the occurrence of dyslipidemia, hyperglycemia, altered skeletal muscle energy metabolism and the development of non-alcoholic steatohepatitis despite normal body mass index (75–80). These findings support the emerging concept that SBMA subjects develop marked peripheral pathologies that are a component of the disease phenotype and are important targets for therapeutic intervention. Studies in SBMA knock-in mice show that diminishing peripheral polyQ AR expression by s.c. administration of ASOs rescues metabolic abnormalities (77) and age-dependent impairment of the ubiquitin-proteasome pathway in skeletal muscle (81), supporting the concept that peripheral polyQ AR is a potent target for therapeutic intervention.
Based on studies in these preclinical models, SBMA is well positioned for the translation of therapeutic approaches to clinical trial. Beneficial effects of peripheral gene targeting are clearly established in mouse models; the extent to which CNS targeting provides added benefits in mice is model system-dependent and may reflect variable transgene expression levels (73,82). Given the disease’s slow rate of progression and peripheral manifestations, the possibility of clinical benefit from targeting peripheral polyQ AR is worthy of testing in patients. Challenges of such an approach include obtaining robust targeting of human skeletal muscle by ASOs such that knockdown is sufficient to mediate benefits. Direct targeting of lower motor neurons by ASOs would require intrathecal administration. Alternative therapeutic strategies, including approaches to achieve anti-androgenic effects (83,84) or treatment with IGF-1 mimetics (85), are under active study in SBMA patients and provide complementary approaches to ameliorate toxicity of the polyQ AR. Notably, both ASOs and other strategies that diminish AR signaling face the challenge that loss of AR action in muscle will result in diminished anabolic effects, a factor that may complicate measurements of clinical benefit.
As highlighted here, SMA, CMT disease and SBMA are well positioned for the development of genetic therapies. Patient populations with defined targetable mutations and significant unmet clinical needs characterize these inherited disorders. Natural history studies and experiences from recent clinical trials provide invaluable data for planning future studies. While significant challenges remain, the path forward for drug discovery and development to treat these disorders is remarkably encouraging.
Funding
National Institutes of Health (NS055746 to A.P.L.); Muscular Dystrophy Association (513702 to A.P.L.); National Institute of General Medical Sciences (GM118647 to A.A.); National Institutes of Health (NS062869 and NS096770 to C.J.S.)
Conflicts of Interest. C.J.S. is a consultant to Avexis, Biogen, Genentech, Roche, PTC Therapeutics, and Ionis Pharmaceuticals.
References
- 1. Ogino S., Leonard D.G., Rennert H., Ewens W.J. and Wilson R.B. (2002) Genetic risk assessment in carrier testing for spinal muscular atrophy. Am. J. Med. Genet., 110, 301–307. [DOI] [PubMed] [Google Scholar]
- 2. Finkel R.S., McDermott M.P., Kaufmann P., Darras B.T., Chung W.K., Sproule D.M., Kang P.B., Foley A.R., Yang M.L., Martens W.B. et al. (2014) Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology, 83, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolb S.J., Coffey C.S., Yankey J.W., Krosschell K., Arnold W.D., Rutkove S.B., Swoboda K.J., Reyna S.P., Sakonju A., Darras B.T. et al. (2017) Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol., 82, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lunn M.R. and Wang C.H. (2008) Spinal muscular atrophy. Lancet, 371, 2120–2133. [DOI] [PubMed] [Google Scholar]
- 5. Mercuri E., Bertini E. and Iannaccone S.T. (2012) Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol., 11, 443–452. [DOI] [PubMed] [Google Scholar]
- 6. Wadman R.I., Stam M., Gijzen M., Lemmink H.H., Snoeck I.N., Wijngaarde C.A., Braun K.P., Schoenmakers M.A., van den Berg L.H., Dooijes D. et al. (2017) Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0–4. J. Neurol. Neurosurg. Psychiatry, 88(4), 365–367. [DOI] [PubMed] [Google Scholar]
- 7. Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 8. Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G. and Melki J. (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet., 16, 265–269. [DOI] [PubMed] [Google Scholar]
- 9. Parsons D.W., McAndrew P.E., Iannaccone S.T., Mendell J.R., Burghes A.H. and Prior T.W. (1998) Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet., 63, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lorson C.L., Hahnen E., Androphy E.J. and Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A., 96, 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H. and McPherson J.D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet., 8, 1177–1183. [DOI] [PubMed] [Google Scholar]
- 12. Kashima T. and Manley J.L. (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet., 34, 460–463. [DOI] [PubMed] [Google Scholar]
- 13. Cartegni L. and Krainer A.R. (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet., 30, 377–384. [DOI] [PubMed] [Google Scholar]
- 14. Sumner C.J. and Crawford T.O. (2018) Two breakthrough gene-targeted treatments for spinal muscular atrophy: challenges remain. J. Clin. Invest., 128, 3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schoch K.M. and Miller T.M. (2017) Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron, 94, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rigo F., Hua Y., Krainer A.R. and Bennett C.F. (2012) Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol., 199, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodkey K., Aslesh T., Maruyama R. and Yokota T. (2018) Nusinersen in the treatment of spinal muscular atrophy. Methods Mol. Biol., 1828, 69–76. [DOI] [PubMed] [Google Scholar]
- 18. Claborn M.K., Stevens D.L., Walker C.K. and Gildon B.L. (2019) Nusinersen: a treatment for spinal muscular atrophy. Ann. Pharmacother., 53, 61–69. [DOI] [PubMed] [Google Scholar]
- 19. Singh N.K., Singh N.N., Androphy E.J. and Singh R.N. (2006) Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol., 26, 1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh N.N., Howell M.D., Androphy E.J. and Singh R.N. (2017) How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther., 24, 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hua Y., Sahashi K., Hung G., Rigo F., Passini M.A., Bennett C.F. and Krainer A.R. (2010) Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev., 24, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F. and Krainer A.R. (2011) Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature, 478, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G. et al. (2011) Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med., 3, 72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E. et al. (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med., 377, 1723–1732. [DOI] [PubMed] [Google Scholar]
- 25. Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K. et al. (2018) Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med., 378, 625–635. [DOI] [PubMed] [Google Scholar]
- 26. d’Ydewalle C. and Sumner C.J. (2015) Spinal muscular atrophy therapeutics: where do we stand? Neurotherapeutics, 12, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naryshkin N.A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K.K., Karp G.M., Qi H., Woll M.G. et al. (2014) Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science, 345, 688–693. [DOI] [PubMed] [Google Scholar]
- 28. Palacino J., Swalley S.E., Song C., Cheung A.K., Shu L., Zhang X., Van Hoosear M., Shin Y., Chin D.N., Keller C.G. et al. (2015) SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol., 11, 511–517. [DOI] [PubMed] [Google Scholar]
- 29. Serguera C. and Bemelmans A.P. (2014) Gene therapy of the central nervous system: general considerations on viral vectors for gene transfer into the brain. Rev Neurol (Paris), 170, 727–738. [DOI] [PubMed] [Google Scholar]
- 30. Saraiva J., Nobre R.J. and Pereira de Almeida L. (2016) Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J. Control. Release, 241, 94–109. [DOI] [PubMed] [Google Scholar]
- 31. Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H. et al. (2010) Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol., 28, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Valori C.F., Ning K., Wyles M., Mead R.J., Grierson A.J., Shaw P.J. and Azzouz M. (2010) Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med., 2, 35ra42. [DOI] [PubMed] [Google Scholar]
- 33. Dominguez E., Marais T., Chatauret N., Benkhelifa-Ziyyat S., Duque S., Ravassard P., Carcenac R., Astord S., Pereira de Moura A., Voit T. et al. (2011) Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet., 20, 681–693. [DOI] [PubMed] [Google Scholar]
- 34. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. et al. (2017) Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med., 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- 35. d’Ydewalle C., Ramos D.M., Pyles N.J., Ng S.Y., Gorz M., Pilato C.M., Ling K., Kong L., Ward A.J., Rubin L.L. et al. (2017) The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron, 93, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skre H. (1974) Genetic and clinical aspects of Charcot–Marie–Tooth’s disease. Clin. Genet., 6, 98–118. [DOI] [PubMed] [Google Scholar]
- 37. Dyck P.J. and Lambert E.H. (1968) Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch. Neurol., 18, 619–625. [DOI] [PubMed] [Google Scholar]
- 38. Timmerman V., Strickland A.V. and Zuchner S. (2014) Genetics of Charcot–Marie–Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel), 5, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murphy S.M., Herrmann D.N., McDermott M.P., Scherer S.S., Shy M.E., Reilly M.M. and Pareyson D. (2011) Reliability of the CMT neuropathy score (second version) in Charcot–Marie–Tooth disease. J. Peripher. Nerv. Syst., 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Juneja M., Burns J., Saporta M.A. and Timmerman V. (2019) Challenges in modelling the Charcot–Marie–Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry, 90, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fledrich R., Abdelaal T., Rasch L., Bansal V., Schutza V., Brugger B., Luchtenborg C., Prukop T., Stenzel J., Rahman R.U. et al. (2018) Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat. Commun., 9, 3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patel P.I., Roa B.B., Welcher A.A., Schoener-Scott R., Trask B.J., Pentao L., Snipes G.J., Garcia C.A., Francke U., Shooter E.M. et al. (1992) The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat. Genet., 1, 159–165. [DOI] [PubMed] [Google Scholar]
- 43. Saberan-Djoneidi D., Sanguedolce V., Assouline Z., Levy N., Passage E. and Fontes M. (2000) Molecular dissection of the Schwann cell specific promoter of the PMP22 gene. Gene, 248, 223–231. [DOI] [PubMed] [Google Scholar]
- 44. Passage E., Norreel J.C., Noack-Fraissignes P., Sanguedolce V., Pizant J., Thirion X., Robaglia-Schlupp A., Pellissier J.F. and Fontes M. (2004) Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot–Marie–Tooth disease. Nat. Med., 10, 396–401. [DOI] [PubMed] [Google Scholar]
- 45. Pareyson D., Reilly M.M., Schenone A., Fabrizi G.M., Cavallaro T., Santoro L., Vita G., Quattrone A., Padua L., Gemignani F. et al. (2011) Ascorbic acid in Charcot–Marie–Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol., 10, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lewis R.A., McDermott M.P., Herrmann D.N., Hoke A., Clawson L.L., Siskind C., Feely S.M., Miller L.J., Barohn R.J., Smith P. et al. (2013) High-dosage ascorbic acid treatment in Charcot–Marie–Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol., 70, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao H.T., Damle S., Ikeda-Lee K., Kuntz S., Li J., Mohan A., Kim A., Hung G., Scheideler M.A., Scherer S.S. et al. (2018) PMP22 antisense oligonucleotides reverse Charcot–Marie–Tooth disease type 1A features in rodent models. J. Clin. Invest., 128, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M. and Bird T.D. (1993) DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell, 72, 143–151. [DOI] [PubMed] [Google Scholar]
- 49. Prior R., Van Helleputte L., Benoy V. and Van Den Bosch L. (2017) Defective axonal transport: a common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis., 105, 300–320. [DOI] [PubMed] [Google Scholar]
- 50. Benoy V., Vanden Berghe P., Jarpe M., Van Damme P., Robberecht W. and Van Den Bosch L. (2017) Development of improved HDAC6 inhibitors as pharmacological therapy for axonal Charcot–Marie–Tooth disease. Neurotherapeutics, 14, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benoy V., Van Helleputte L., Prior R., d’Ydewalle C., Haeck W., Geens N., Scheveneels W., Schevenels B., Cader M.Z., Talbot K. et al. (2018) HDAC6 is a therapeutic target in mutant GARS-induced Charcot–Marie–Tooth disease. Brain, 141, 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mo Z., Zhao X., Liu H., Hu Q., Chen X.Q., Pham J., Wei N., Liu Z., Zhou J., Burgess R.W. et al. (2018) Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot–Marie–Tooth neuropathy. Nat. Commun., 9, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zuchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J. et al. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat. Genet., 36, 449–451. [DOI] [PubMed] [Google Scholar]
- 54. Detmer S.A. and Chan D.C. (2007) Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol., 176, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Y., Carmona S., Muhammad A., Bell S., Landeros J., Vazquez M., Ho R., Franco A., Lu B., Dorn G.W. II et al. (2019) Restoring mitofusin balance prevents axonal degeneration in a Charcot–Marie–Tooth type 2A model. J. Clin. Invest., 130, 1756–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Antonellis A. and Green E.D. (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet., 9, 87–107. [DOI] [PubMed] [Google Scholar]
- 57. Meyer-Schuman R. and Antonellis A. (2017) Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet., 26, R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seburn K.L., Nangle L.A., Cox G.A., Schimmel P. and Burgess R.W. (2006) An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot–Marie–Tooth 2D mouse model. Neuron, 51, 715–726. [DOI] [PubMed] [Google Scholar]
- 59. Motley W.W., Seburn K.L., Nawaz M.H., Miers K.E., Cheng J., Antonellis A., Green E.D., Talbot K., Yang X.L., Fischbeck K.H. et al. (2011) Charcot–Marie–Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet., 7, e1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kennedy W.R., Alter M. and Sung J.H. (1968) Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology, 18, 671–680. [DOI] [PubMed] [Google Scholar]
- 61. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209531s003s004lbl.pdf, reference ID: 4262307.
- 62. Sobue G., Hashizume Y., Mukai E., Hirayama M., Mitsuma T. and Takahashi A. (1989) X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain, 112, 209–232. [DOI] [PubMed] [Google Scholar]
- 63. La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E. and Fischbeck K.H. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature, 352, 77–79. [DOI] [PubMed] [Google Scholar]
- 64. Giorgetti E. and Lieberman A.P. (2016) Polyglutamine androgen receptor-mediated neuromuscular disease. Cell. Mol. Life Sci., 73, 3991–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lieberman A.P., Shakkottai V.G. and Albin R.L. (2019) Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol., 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Atsuta N., Watanabe H., Ito M., Banno H., Suzuki K., Katsuno M., Tanaka F., Tamakoshi A. and Sobue G. (2006) Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain, 129, 1446–1455. [DOI] [PubMed] [Google Scholar]
- 67. Rhodes L.E., Freeman B.K., Auh S., Kokkinis A.D., La Pean A., Chen C., Lehky T.J., Shrader J.A., Levy E.W., Harris-Love M. et al. (2009) Clinical features of spinal and bulbar muscular atrophy. Brain, 132, 3242–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Soraru G., D’Ascenzo C., Polo A., Palmieri A., Baggio L., Vergani L., Gellera C., Moretto G., Pegoraro E. and Angelini C. (2008) Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J. Neurol. Sci., 264, 100–105. [DOI] [PubMed] [Google Scholar]
- 69. Yu Z., Dadgar N., Albertelli M., Gruis K., Jordan C., Robins D.M. and Lieberman A.P. (2006) Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest., 116, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Monks D.A., Johansen J.A., Mo K., Rao P., Eagleson B., Yu Z., Lieberman A.P., Breedlove S.M. and Jordan C.L. (2007) Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. U. S. A., 104, 18259–18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ramzan F., McPhail M., Rao P., Mo K., Halievski K., Swift-Gallant A., Mendoza-Viveros L., Cheng H.Y. and Monks D.A. (2015) Distinct etiological roles for myocytes and motor neurons in a mouse model of Kennedy’s disease/spinobulbar muscular atrophy. J. Neurosci., 35, 6444–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Palazzolo I., Stack C., Kong L., Musaro A., Adachi H., Katsuno M., Sobue G., Taylor J.P., Sumner C.J., Fischbeck K.H. et al. (2009) Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron, 63, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lieberman A.P., Yu Z., Murray S., Peralta R., Low A., Guo S., Yu X.X., Cortes C.J., Bennett C.F., Monia B.P. et al. (2014) Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep., 7, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cortes C.J., Ling S.C., Guo L.T., Hung G., Tsunemi T., Ly L., Tokunaga S., Lopez E., Sopher B.L., Bennett C.F. et al. (2014) Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron, 82, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Querin G., Bertolin C., Da Re E., Volpe M., Zara G., Pegoraro E., Caretta N., Foresta C., Silvano M., Corrado D. et al. (2016) Non-neural phenotype of spinal and bulbar muscular atrophy: results from a large cohort of Italian patients. J. Neurol. Neurosurg. Psychiatry, 87, 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rocchi A., Milioto C., Parodi S., Armirotti A., Borgia D., Pellegrini M., Urciuolo A., Molon S., Morbidoni V., Marabita M. et al. (2016) Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol., 132, 127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Giorgetti E., Yu Z., Chua J.P., Shimamura R., Zhao L., Zhu F., Venneti S., Pennuto M., Guan Y., Hung G. et al. (2016) Rescue of metabolic alterations in AR113Q skeletal muscle by peripheral androgen receptor gene silencing. Cell Rep., 17, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guber R.D., Takyar V., Kokkinis A., Fox D.A., Alao H., Kats I., Bakar D., Remaley A.T., Hewitt S.M., Kleiner D.E. et al. (2017) Nonalcoholic fatty liver disease in spinal and bulbar muscular atrophy. Neurology, 89, 2481–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nakatsuji H., Araki A., Hashizume A., Hijikata Y., Yamada S., Inagaki T., Suzuki K., Banno H., Suga N., Okada Y. et al. (2017) Correlation of insulin resistance and motor function in spinal and bulbar muscular atrophy. J. Neurol., 264, 839–847. [DOI] [PubMed] [Google Scholar]
- 80. Rosenbohm A., Hirsch S., Volk A.E., Grehl T., Grosskreutz J., Hanisch F., Herrmann A., Kollewe K., Kress W., Meyer T. et al. (2018) The metabolic and endocrine characteristics in spinal and bulbar muscular atrophy. J. Neurol., 265, 1026–1036. [DOI] [PubMed] [Google Scholar]
- 81. Nath S.R., Yu Z., Gipson T.A., Marsh G.B., Yoshidome E., Robins D.M., Todi S.V., Housman D.E. and Lieberman A.P. (2018) Androgen receptor polyglutamine expansion drives age-dependent quality control defects and muscle dysfunction. J. Clin. Invest., 128, 3630–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sahashi K., Katsuno M., Hung G., Adachi H., Kondo N., Nakatsuji H., Tohnai G., Iida M., Bennett C.F. and Sobue G. (2015) Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet., 24, 5985–5994. [DOI] [PubMed] [Google Scholar]
- 83. Hashizume A., Katsuno M., Suzuki K., Banno H., Takeuchi Y., Kawashima M., Suga N., Mano T., Araki A., Hijikata Y. et al. (2019) Efficacy and safety of leuprorelin acetate for subjects with spinal and bulbar muscular atrophy: pooled analyses of two randomized-controlled trials. J. Neurol., 266, 1211–1221. [DOI] [PubMed] [Google Scholar]
- 84. Katsuno M., Banno H., Suzuki K., Takeuchi Y., Kawashima M., Yabe I., Sasaki H., Aoki M., Morita M., Nakano I. et al. (2010) Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol., 9, 875–884. [DOI] [PubMed] [Google Scholar]
- 85. Grunseich C., Miller R., Swan T., Glass D.J., El Mouelhi M., Fornaro M., Petricoul O., Vostiar I., Roubenoff R., Meriggioli M.N. et al. (2018) Safety, tolerability, and preliminary efficacy of an IGF-1 mimetic in patients with spinal and bulbar muscular atrophy: a randomised, placebo-controlled trial. Lancet Neurol., 17, 1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De D.C., Yamashita M., Rigo F., Hung G., Schneider E. et al. (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet, 388, 3017–3026. [DOI] [PubMed] [Google Scholar]
- 87. Meyer K., Ferraiuolo L., Schmelzer L., Braun L., McGovern V., Likhite S., Michels O., Govoni A., Fitzgerald J., Morales P. et al. (2015) Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol. Ther., 23, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R. and Samulski R.J. (2011) Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther., 19, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Poirier A., Weetall M., Heinig K., Bucheli F., Schoenlein K., Alsenz J., Bassett S., Ullah M., Senn C., Ratni H. et al. (2018) Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect., 6, e00447. [DOI] [PMC free article] [PubMed] [Google Scholar]