

Abstract
Triplet repeat diseases (TRDs) are caused by pathogenic expansions of trinucleotide sequence repeats within coding and non-coding regions of different genes. They are typically progressive, very disabling and frequently involve the nervous system. Currently available symptomatic therapies provide modest benefit at best. The development of interventions that interfere with the natural history of these diseases is a priority. A common pathogenic process shared by most TRDs is the presence of toxicity from the messenger RNA or protein encoded by the gene harboring the abnormal expansion. Strategies to interfere with the expression of these genes using different molecular approaches are being pursued and have reached the clinical stage. This review will summarize the significant progress made in this field in the last few years, focusing on three main areas: the discovery of biomarkers of disease progression and target engagement, advances in preclinical studies for the polyglutamine ataxias and the initial clinical application in myotonic dystrophy type 1 and Huntington’s disease.
Introduction
Triplet repeat diseases
Microsatellites or short tandem DNA repeats are polymorphic repetitive sequences of 1–6 nucleotides that account for about 3% of the genomic DNA (1) and play a key regulatory role on gene expression (2). Around a million microsatellites are dispersed throughout the human genome, with over 40 linked to human disease to date that preferentially involve the nervous system (3, 4).
Triplet repeat diseases (TRD) are caused by three nucleotide repeats that expand beyond a pathological threshold (4). They are often categorized according to their nucleotide sequence or location within coding or non-coding regions. Near half of the known TRDs are linked to codons encoding for the amino acid glutamine, known as the polyglutamine (polyQ) diseases. From a translational viewpoint, a classification between polyQ and non-polyQ TRDs is particularly helpful (Table 1), as common pathogenic features among polyQ diseases facilitate joint efforts to overcome common obstacles along the therapeutic development path. There are currently no disease-modifying therapies available for any TRDs.
Table 1.
Triplet repeat diseases listed as polyglutamine and non-polyglutamine disorders. Black bolded font indicates disease, for which recent progress has been made as referenced in the manuscript
| PolyQ | Non PolyQ |
|---|---|
| Huntington’s disease | Myotonic dystrophy type 1 |
| SCA1 | Oculopharyngeal muscular dystrophy |
| SCA2 | Friedreich’s ataxia |
| SCA3 | Fragile X Syndrome |
| SCA6 | Fragile X-associated tremor/ataxia syndrome |
| SCA7 | Fragile XE mental retardation |
| SCA17 | SCA8 |
| Spinal and bulbar muscular atrophy | SCA12 |
| Dentatorubropallidoluysian atrophy | Huntington disease-like 2 |
| Fuchs corneal dystrophy |
A feature shared by many TRDs is the occurrence of protein or RNA toxicity that triggers neuronal dysfunction and death, underlying the emergence of gradual neurological impairment. The most advanced therapeutic molecular strategies for TRDs aim to lower expression of the mutated gene by targeting messenger RNA (mRNA) with antisense oligonucleotides (ASO) or RNA interference (RNAi).
The therapeutic development path: from proof of concept to commercialization
To understand where we stand in the therapeutic pipeline, it is a helpful exercise to plot the full path toward clinical application (Fig. 1). Visualizing the entire process before its execution helps prevent inadequate experimental design that would ultimately increase time to human application and costs. Owing to the substantial biological and clinical overlap among TRDs, only a fraction of the steps in this path are private to each specific therapeutic program. The knowledge acquired by a given translational program is very informative to others, catalyzing progress.
Figure 1.
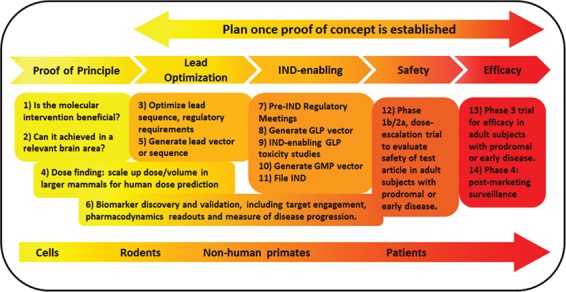
The process of therapeutic development for triplet repeat diseases. A general overview of the entire path from proof of concept to commercialization, including model systems typically used at each step. The boxes indicate key questions that need to be addressed. The full process should be carefully planned before initiating its execution, once proof of concept is established.
This review will summarize recent advances on therapeutic ASO and RNAi for TRDs. Three specific areas of focus include the identification of biomarkers, the progress in the preclinical path for in the polyQ ataxias and initial human trials in myotonic dystrophy and Huntington’s disease (HD).
Biomarker Discovery
Advances on gene-targeting therapies led translational teams to anticipate the need for reliable biomarkers of disease progression and target engagement before launching human trials. The most exciting progress on this area has materialized on fluid biomarkers for HD.
Pharmacodynamic readouts and target engagement
Most ongoing therapeutic programs for TRDs use nucleic acids to reduce expression of the target gene at the mRNA level in a non-specific (targeting both the mutant and normal alleles) or allele-specific manner (selectively targeting the mutant form). The engagement of the ligand (mRNA) by the therapeutic agent [such as a microRNA (miRNA) or ASO] occurs in neurons, a physical interaction that cannot be visualized in vivo. Target identification, specificity and engagement have been thus far established in preclinical models. However, a novel ultrasensitive immunoassay reliably quantifies levels of mutant huntingtin (mHTT), the toxic protein responsible for HD, in cerebrospinal fluid [CSF (5)] following US and European regulatory guidelines (6). This assay demonstrated that levels of mHTT correlate with disease stage and with other biomarkers of disease progression even in pre-manifesting HD mutation carriers (5, 7). For huntingtin-targeting therapies, it provides an excellent pharmacodynamic readout and an indirect measure of target engagement. For therapeutic approaches not targeting mHTT, it also serves as a biomarker of disease progression.
Disease progression
The most promising fluid biomarker of disease progression is the concentration of neurofilament light chain (NfL) in CSF and blood. NfL levels correlate with mHTT (5) exhibiting the same or superior correlation with clinical, magnetic resonance imaging and other biofluid measures (including tau) in pre-manifest and manifest HD. Remarkably, NfL in CSF and plasma discriminates pre-manifest from manifest HD and correlates with severity in manifest HD better than mHTT, thereby effectively reducing the sample size required for clinical trials (7–9). A combination of mHTT and NfL levels have emerged as optimal fluid biomarkers to be used in HD trials, complementing neuroimaging tools. This knowledge should accelerate biomarker discovery for other TRDs, such as the spinocerebellar ataxias (SCAs). Sensitive assays, private to each disease, need to be developed to detect the mutant protein in CSF. On the other hand, NfL in CSF and serum is a promising common biomarker of disease progression for all polyQ diseases that is being evaluated in several SCAs.
Advancing Beyond Proof of Concept on the Ataxias
RNAi
The first proof of concept study demonstrating the therapeutic potential of RNAi for brain disease was completed in a mouse model of SCA1, with adeno-associated virus (AAV) vectors expressing short hairpin RNAs that efficiently reduced expression levels of the mutant protein, ataxin-1 (10). Ensuing reports over the following decade confirmed the therapeutic potential of RNAi for SCA1 and extended it to SCA3, 6 and 7, including demonstration of successful allele-specific and splice-isoform specific targeting (11–20). Recent progress on RNAi for TRDs includes the development of a lentiviral vector to deliver short hairpin RNAs into SCA3 mouse striatum (21) and a novel silencing-replacement strategy for SCA7 in cultured cells (22).
Progress beyond proof of concept has been reported for SCA1, by refining the therapeutic window for an optimized lead vector in SCA1 mice (23) and scalability to primate brain (24). Infusion of the test article into the cerebellar nuclei of rhesus macaque led to widespread transduction and efficient target suppression not only to the cerebellar nuclei and cortex but also in brainstem nuclei implicated in ataxia, such as the inferior olive. This delivery approach, currently undergoing IND-enabling GLP toxicology studies for SCA1, should be applicable to other SCAs.
Antisense oligonucleotides
Target cleavage
ASO are short sequences of nucleic acids designed to bind a specific target RNA and alter its normal behavior through different mechanisms (25), including RNase H-mediated mRNA cleavage. This approach is moving forward as a therapeutic option for several SCAs and, although started a few years after RNAi programs were launched, will likely reach the clinic first.
SCA1 cleared the proof of concept milestone, with the demonstration of efficient downregulation of Atxn1 in SCA1 knockin mice after intraventricular injection of three lead ASOs. The phenotypical rescue was achieved when applied at an early stage, including improvement on motor function, transcriptional deregulation and magnetic resonance spectroscopy-based neurochemical abnormalities in affected tissue (26). In SCA2, a screen of 152 ASOs against the human gene identified a lead sequence that, when applied through intracerebroventricular (icv) injection to two different symptomatic SCA2 mice, led to a significant reduction of the target transcript in the right tissue with benefit on motor function and no toxicity (27). Because the product of the gene harboring the repeat expansion, ATXN2, is also implicated in the pathogenesis of amyotrophic lateral sclerosis, this sequence is first being developed for that devastating disease (28). An initial study for SCA3, the most common dominant ataxia worldwide, screened hundreds of ASOs targeting the human ATXN3 in cultured cells, yielding five candidates for an in vivo screen in two transgenic mouse models of SCA3. Widespread delivery to target regions in SCA3 efficiently reduced target protein levels without evidence of toxicity in one of the models used but failed to achieve similar silencing in the other (29). After this initial screening, the top ASO was evaluated in early symptomatic transgenic SCA3 mice. Significant target suppression lasted 2 months after infusion, preventing oligomerization and nuclear accumulation of ataxin-3 while improving Purkinje neurons physiology and motor function (28). SCA7 is unique among polyQ ataxias in that, in addition to a cerebellar syndrome, it also causes retinal degeneration leading to blindness. After selecting a lead sequence from a screening of 150 ASOs targeting mouse ATXN7, SCA7 knock in received ASOs targeting ataxin-7 through injections into the vitreous humor with improvement in many phenotypical features even when applied after onset of retinal disease (30). Treatment of the cerebellar syndrome in SCA7 would require adding a similar intervention targeting the cerebellum as in the SCA1, 2 and 3 programs.
For SCA7, next steps include the identification of ASOs targeting human ATXN7 and efficacy and safety screening in retinal and brain tissue in rodents. Initial human trials will likely target retinal disease. For SCA1, larger screening of sequences against human ATXN1 is likely the next step to identify a lead vector. In SCA2 and 3, the focus should be on biomarker identification (assays to measure the mutant protein in CSF and evaluation of NfL in CSF and serum) and dosing studies to define the therapeutic window before moving on to IND-enabling GLP evaluation. SCA2 will likely follow initial trials in patients with amyotrophic lateral sclerosis. Scalability to a larger mammal brain can be informed by other ASO therapeutic programs using the same route of administration (31), including post-mortem evaluation in children who received ASO for spinal muscular atrophy [SMA (32)].
Modifying splicing
ASO can also be designed to alter splicing of the targeted gene product, an approach that reached commercialization for another neurogenetic disorder, SMA (32). Therapeutic splicing modulation is being evaluated for SCA3. Using ASOs that remove the polyQ-encoding sequence from the mutated gene product, ataxin-3, through exon skipping does not interfere with key biochemical functions of the normal protein. A single infusion of this ASO modifies splicing for over 2 months in SCA3 mice, lowering levels of insoluble ataxin-3 and correcting cytological features (33, 34). Demonstration of clinical efficacy in SCA3 models should be completed before investing in a full-fledged therapeutic program.
Reaching Clinical Application: Myotonic Dystrophy and HD
Myotonic dystrophy
A CUG expansion in DMPK (dystrophia myotonica protein kinase) generates toxic transcripts that are retained in the nucleus, triggering transcriptional deregulation and splicing defects that cause myotonic dystrophy type 1 [DM1 (35)]. The use of ASOs to downregulate DMPK faces the added challenge of required delivery to multiple tissues, including skeletal and cardiac muscle. After initial cell- and animal-based studies following this approach were successful (36–40), 3000 sequences were screened to yield an ASO that was successfully tested in rodents and non-human primates and achieved efficient (>50%) and well-tolerated inhibition of DMPK in skeletal and cardiac muscle for both species (41). This led to the design and execution of a phase 1/2 dose escalation clinical trial in subjects with DM1, evaluating safety and biomarkers for target engagement in muscle biopsies. Although not published in peer-reviewed form, Ionis Pharmaceuticals and Biogen reported the results of the DMPKRx trial in early 2017. The distribution of the ASO (IONIS-DMPK-2.5Rx) was not sufficient to reach the required RNA splicing rescue in skeletal muscle, highlighting the critical importance of biomarkers. This human trial has provided very valuable information and serves as a cautionary tale for the future development of molecular therapies for DM1 and other therapeutic programs.
Huntington’s disease
RNAi
After an initial demonstration of efficacy in a mouse model of HD, a wealth of publications followed supporting the development of therapeutic RNAi for HD (reviewed in 42–44) and optimization of the first-generation therapeutic vectors ensued (45, 46). The triad of motor, cognitive and psychiatric symptoms in HD initially arises from striatal pathology that subsequently extends to other cerebral areas (47). The most common RNAi approach has been the infusion of AAV encoding sequences targeting HTT into the striatum. The main advances in RNAi for HD in the last few years cluster on the refinement of the therapeutic vector and scalability studies in large mammals.
In efforts to improve efficacy and safety, sequences have been engineered to target preferentially the mutant allele (48–50), and different AAV serotypes (49, 51, 52) and lentiviral vectors (53) have been tested. Moreover, different promoters driving expression of miRNAs have been compared. The higher expression levels of the miRNA targeting HTT achieved by a pol-3 (human U6) than a pol-2 (chicken β-actin) promoter were associated with toxicity in HD mice (52) but not in sheep (54). As a result, different AAV serotype-promoter-miRNA combinations are under development by different groups.
Regarding scalability to larger mammalian brains, the efficacy and short-term safety of striatal infusion of AAV miRNA targeting HTT were previously shown in non-human primates, with evidence of spread beyond the putamen to cortical areas (55, 56). More recently, the striatum of HD transgenic sheep was targeted with AAV9 miRNA targeting human HTT. This intervention achieved sustained HTT mRNA and protein suppression in the caudate and putamen for at least 6 months post-injection with no safety concerns (54). A recent clinical trial in patients with Parkinson’s disease (PD) provides very valuable information for AAV-based HD programs. Patients with PD underwent an intraputamenal infusion of AAV encoding a potentially beneficial enzyme. Despite infusing significantly larger volumes and doses than in previous trials, the highest dose cohort did not surpass 50% of putamenal coverage, which might be insufficient for gene silencing in HD (57). Nevertheless, this approach is actively being pursued by different companies that reportedly completed IND-enabling studies and are close to launching early phase human trials (42).
Antisense oligonucleotides
Following initial preclinical proof of concept, optimization and scalability studies in different rodent HD models and non-human primates (31, 58–60), Ionis Pharmaceuticals launched a Phase 1/2 clinical trial evaluating IONIS-HTTRx (or RG6042) in 46 patients with early-stage HD. Subjects were randomized to receive four different doses of IONIS-HTTRx or placebo through monthly intrathecal applications (lumbar puncture). The results of this trial were recently published (61), reporting safety and tolerability of all tested doses. There was a remarkable dose-dependent reduction in levels of mutant HTT in CSF of ~60%. An open-label extension trial (NCT03342053) for those who completed the Phase 1/2 trial and a new Phase 3 randomized double-blind placebo-controlled trial involving 660 subjects (GENERATION-HD1) has been launched.
Similar to RNAi vectors, ASOs can be designed to selectively target mutant HTT without affecting levels of the wild-type allele (62–64). WAVE Life Sciences designed two different SNP-targeting ASOs for allele-preferential suppression of mutant HTT expression. A Phase 1/2 trial has been initiated for each ASO (PRECISION-HD1 and PRECISION-HD2), although no preclinical information or reports of trial progress are available (42, 65).
Other progress
For HD, AAV-based expression of zinc finger proteins successfully silenced transcription at the DNA level in vivo (66). No recent progress has been reported on this program. The use of therapeutic ASOs has been explored for other TRDs, such as Friedreich’s Ataxia (67) or spinal and bulbar muscular atrophy (68, 69). AAV-based treatments are in early stages of development for fragile X mental retardation protein-related disorders (70). Finally, silencing-replacement strategies are being pursued for oculopharyngeal muscular dystrophy (71, 72).
An extremely exciting development to target disease genes at the DNA level is through gene editing. The therapeutic application of CRISPR/Cas9 technologies is rapidly advancing (73). In HD, evidence of efficacy in cells and in vivo allele-specific targeting has been reported (74–80), and this approach is also being pursued for other TRDs (81). Once hurdles related to delivery and safety are cleared, this field is likely to take off and readily move toward IND-enabling studies.
Conclusion
As the preclinical gap narrows and human application is reached in a growing number of programs (Fig. 2), we advanced from an era of ‘cautious optimism’ to a state of remarkable excitement. Although high expectations are often counterproductive, a growing number of translational scientists expect a demonstration of clinical efficacy in a neurodegenerative disease in the next 5 years. Other precision-based therapeutics, biologicals and small molecules are being developed in parallel for TRDs (65, 82). In the future, it is reasonable to envision the use of several of these therapies in combination, perhaps driven by phenotypical features and disease stage.
Figure 2.
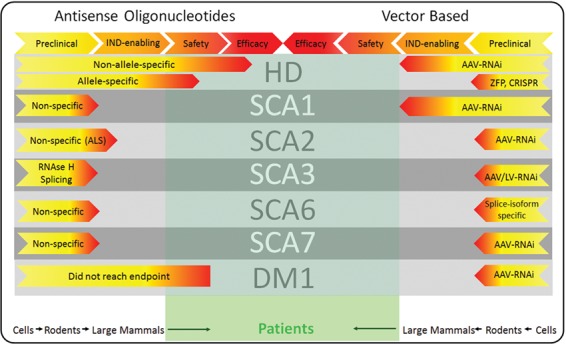
Current status of molecular therapies for selected triplet repeat disorders. The information included in this figure is inferred from published or publicly available information. Therapeutic ASOs are taking the lead in the ‘race’ to commercialization for TRDs.
Even if approved for clinical use, new challenges will appear. For instance, the repeated administration of Spinraza for SMA has led to the appearance of meningitis and hydrocephalus in some patients, information now included in the product label. New delivery methods might need to be developed. The involvement of other organs unmasked by improvement on brain disease or unwanted extension of end stages of the disease is also a possible concern. Issues of cost and reimbursement will have to be addressed. These are challenges the field is willing to embrace and will be ready to tackle. Slowing or halting the progression of the disease process in TRDs in humans is within reach.
Funding
National Institutes of Health/National Institute of Neurological Disorders and Stroke (grant number UH3NS094355).
References
- 1. Subramanian S., Mishra R.K. and Singh L. (2003) Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol., 4, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gymrek M., Willems T., Guilmatre A., Zeng H., Markus B., Georgiev S., Daly M.J., Price A.L., Pritchard J.K., Sharp A.J. et al. (2016) Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet., 48, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hannan A.J. (2018) Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet., 19, 286–298. [DOI] [PubMed] [Google Scholar]
- 4. Paulson H. (2018) Repeat expansion diseases. Handb. Clin. Neurol., 147, 105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wild E.J., Boggio R., Langbehn D., Robertson N., Haider S., Miller J.R., Zetterberg H., Leavitt B.R., Kuhn R., Tabrizi S.J. et al. (2015) Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J. Clin. Invest., 125, 1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fodale V., Boggio R., Daldin M., Cariulo C., Spiezia M.C., Byrne L.M., Leavitt B.R., Wild E.J., Macdonald D., Weiss A. et al. (2017) Validation of ultrasensitive mutant Huntingtin detection in human cerebrospinal fluid by single molecule counting immunoassay. J. Huntingtons Dis., 6, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Byrne L.M., Rodrigues F.B., Johnson E.B., Wijeratne P.A.,De Vita E., Alexander D.C., Palermo G., Czech C., Schobel S., Scahill R.I. et al. (2018) Evaluation of mutant huntingtin and neurofilament proteins as potential markers in Huntington's disease. Sci. Transl. Med., 10. [DOI] [PubMed] [Google Scholar]
- 8. Byrne L.M., Rodrigues F.B., Blennow K., Durr A., Leavitt B.R., Roos R.A.C., Scahill R.I., Tabrizi S.J., Zetterberg H., Langbehn D. et al. (2017) Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol., 16, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson E.B., Byrne L.M., Gregory S., Rodrigues F.B., Blennow K., Durr A., Leavitt B.R., Roos R.A., Zetterberg H., Tabrizi S.J. et al. (2018) Neurofilament light protein in blood predicts regional atrophy in Huntington disease. Neurology, 90, e717–e723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia H., Mao Q., Eliason S.L., Harper S.Q., Martins I.H., Orr H.T., Paulson H.L., Yang L., Kotin R.M. and Davidson B.L. (2004) RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med., 10, 816–820. [DOI] [PubMed] [Google Scholar]
- 11. Alves S., Nascimento-Ferreira I., Auregan G., Hassig R., Dufour N., Brouillet E., Pedroso de Lima M.C., Hantraye P., Pereira de Almeida L. and Deglon N. (2008) Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS One, 3, e3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alves S., Nascimento-Ferreira I., Dufour N., Hassig R., Auregan G., Nobrega C., Brouillet E., Hantraye P., Pedroso de Lima M.C., Deglon N. et al. (2010) Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3? Hum. Mol. Genet., 19, 2380–2394. [DOI] [PubMed] [Google Scholar]
- 13. Costa Mdo C., Luna-Cancalon K., Fischer S., Ashraf N.S., Ouyang M., Dharia R.M., Martin-Fishman L., Yang Y., Shakkottai V.G., Davidson B.L. et al. (2013) Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther., 21, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keiser M.S., Geoghegan J.C., Boudreau R.L., Lennox K.A. and Davidson B.L. (2013) RNAi or overexpression: alternative therapies for Spinocerebellar ataxia type 1. Neurobiol. Dis., 56, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramachandran P.S., Boudreau R.L., Schaefer K.A., La Spada A.R. and Davidson B.L. (2014) Nonallele specific silencing of ataxin-7 improves disease phenotypes in a mouse model of SCA7. Mol. Ther., 22, 1635–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scholefield J., Greenberg L.J., Weinberg M.S., Arbuthnot P.B., Abdelgany A. and Wood M.J. (2009) Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One, 4, e7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholefield J., Watson L., Smith D., Greenberg J. and Wood M.J. (2014) Allele-specific silencing of mutant Ataxin-7 in SCA7 patient-derived fibroblasts. Eur. J. Hum. Genet., 22, 1369–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsou W.L., Soong B.W., Paulson H.L. and Rodriguez-Lebron E. (2011) Splice isoform-specific suppression of the Cav2.1 variant underlying spinocerebellar ataxia type 6. Neurobiol. Dis., 43, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J., Pendergraff H., Narayanannair K.J., Lackey J.G., Kuchimanchi S., Rajeev K.G., Manoharan M., Hu J. and Corey D.R. (2013) RNA duplexes with abasic substitutions are potent and allele-selective inhibitors of huntingtin and ataxin-3 expression. Nucleic Acids Res., 41, 8788–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J., Yu D., Aiba Y., Pendergraff H., Swayze E.E., Lima W.F., Hu J., Prakash T.P. and Corey D.R. (2013) Ss-siRNAs allele selectively inhibit ataxin-3 expression: multiple mechanisms for an alternative gene silencing strategy. Nucleic Acids Res., 41, 9570–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nóbrega C., Codêsso J.M., Mendonça L. and Pereira de Almeida L. (2019) RNA interference therapy for Machado-Joseph disease: long-term safety profile of lentiviral vectors encoding short hairpin RNAs targeting mutant Ataxin-3. Hum Gene Ther. doi: 10.1089/hum.2018.157. [DOI] [PubMed]
- 22. Curtis H.J., Seow Y., Wood M.J.A. and Varela M.A. (2017) Knockdown and replacement therapy mediated by artificial mirtrons in spinocerebellar ataxia 7. Nucleic Acids Res., 45, 7870–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keiser M.S., Monteys A.M., Corbau R., Gonzalez-Alegre P. and Davidson B.L. (2016) RNAi prevents and reverses phenotypes induced by mutant human ataxin-1. Ann. Neurol., 80, 754–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keiser M.S., Kordower J.H., Gonzalez-Alegre P. and Davidson B.L. (2015) Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain, 138, 3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schoch K.M. and Miller T.M. (2017) Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron, 94, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friedrich J., Kordasiewicz H.B., O'Callaghan B., Handler H.P., Wagener C., Duvick L., Swayze E.E., Rainwater O., Hofstra B., Benneyworth M. et al. (2018) Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight, 3, DOI: 10.1172/jci.insight.123193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scoles D.R., Meera P., Schneider M.D., Paul S., Dansithong W., Figueroa K.P., Hung G., Rigo F., Bennett C.F., Otis T.S. et al. (2017) Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature, 544, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McLoughlin H.S., Moore L.R., Chopra R., Komlo R., McKenzie M., Blumenstein K.G., Zhao H., Kordasiewicz H.B., Shakkottai V.G. and Paulson H.L. (2018) Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol., 84, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore L.R., Rajpal G., Dillingham I.T., Qutob M., Blumenstein K.G., Gattis D., Hung G., Kordasiewicz H.B., Paulson H.L. and McLoughlin H.S. (2017) Evaluation of antisense oligonucleotides targeting ATXN3 in SCA3 mouse models. Mol. Ther. Nucleic Acids, 7, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niu C., Prakash T.P., Kim A., Quach J.L., Huryn L.A., Yang Y., Lopez E., Jazayeri A., Hung G., Sopher B.L. et al. (2018) Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med., 10, DOI: 10.1126/scitranslmed.aap8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., Mazur C., McAlonis M.M., Pytel K.A., Artates J.W., Weiss A., Cheng S.H., Shihabuddin L.S. et al. (2012) Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron, 74, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De Vivo D.C., Yamashita M., Rigo F., Hung G., Schneider E. et al. (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet, 388, 3017–3026. [DOI] [PubMed] [Google Scholar]
- 33. Evers M.M., Tran H.D., Zalachoras I., Pepers B.A., Meijer O.C., Dunnen J.T., Ommen G.J., Aartsma-Rus A. and Roon-Mom W.M. (2013) Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: removal of the CAG containing exon. Neurobiol. Dis., 58, 49–56. [DOI] [PubMed] [Google Scholar]
- 34. Toonen L.J.A., Rigo F., Attikum H. and Roon-Mom W.M.C. (2017) Antisense oligonucleotide-mediated removal of the Polyglutamine repeat in Spinocerebellar ataxia type 3 mice. Mol. Ther. Nucleic Acids, 8, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pettersson O.J., Aagaard L., Jensen T.G. and Damgaard C.K. (2015) Molecular mechanisms in DM1 - a focus on foci. Nucleic Acids Res., 43, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao Z. and Cooper T.A. (2013) Antisense oligonucleotides: rising stars in eliminating RNA toxicity in myotonic dystrophy. Hum. Gene Ther., 24, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F. and Thornton C.A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wojtkowiak-Szlachcic A., Taylor K., Stepniak-Konieczna E., Sznajder L.J., Mykowska A., Sroka J., Thornton C.A. and Sobczak K. (2015) Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res., 43, 3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carrell S.T., Carrell E.M., Auerbach D., Pandey S.K., Bennett C.F., Dirksen R.T. and Thornton C.A. (2016) Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet., 25, 4328–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jauvin D., Chretien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M. et al. (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in Myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids, 7, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pandey S.K., Wheeler T.M., Justice S.L., Kim A., Younis H.S., Gattis D., Jauvin D., Puymirat J., Swayze E.E., Freier S.M. et al. (2015) Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther., 355, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wild E.J. and Tabrizi S.J. (2017) Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol., 16, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnson C.D. and Davidson B.L. (2010) Huntington's disease: progress toward effective disease-modifying treatments and a cure. Hum. Mol. Genet., 19, R98–R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gonzalez-Alegre P. and Paulson H.L. (2007) Technology insight: therapeutic RNA interference--how far from the neurology clinic? Nat. Clin. Pract. Neurol., 3, 394–404. [DOI] [PubMed] [Google Scholar]
- 45. McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B. et al. (2008) Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U. S. A., 105, 5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monteys A.M., Spengler R.M., Dufour B.D., Wilson M.S., Oakley C.K., Sowada M.J., McBride J.L. and Davidson B.L. (2014) Single nucleotide seed modification restores in vivo tolerability of a toxic artificial miRNA sequence in the mouse brain. Nucleic Acids Res., 42, 13315–13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bates G.P., Dorsey R., Gusella J.F., Hayden M.R., Kay C., Leavitt B.R., Nance M., Ross C.A., Scahill R.I., Wetzel R. et al. (2015) Huntington disease. Nat. Rev. Dis. Primers., 1, 15005. [DOI] [PubMed] [Google Scholar]
- 48. Monteys A.M., Wilson M.J., Boudreau R.L., Spengler R.M. and Davidson B.L. (2015) Artificial miRNAs targeting mutant Huntingtin show preferential silencing in vitro and in Vivo. Mol. Ther. Nucleic Acids, 4, e234. [DOI] [PubMed] [Google Scholar]
- 49. Miniarikova J., Zimmer V., Martier R., Brouwers C.C., Pythoud C., Richetin K., Rey M., Lubelski J., Evers M.M., Deventer S.J. et al. (2017) AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington's disease. Gene Ther., 24, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller J.R.C., Pfister E.L., Liu W., Andre R., Trager U., Kennington L.A., Lo K., Dijkstra S., Macdonald D., Ostroff G. et al. (2017) Allele-selective suppression of mutant Huntingtin in primary human blood cells. Sci. Rep., 7, 46740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keeler A.M., Sapp E., Chase K., Sottosanti E., Danielson E., Pfister E., Stoica L., DiFiglia M., Aronin N. and Sena-Esteves M. (2016) Cellular analysis of silencing the Huntington's disease gene using AAV9 mediated delivery of artificial micro RNA into the striatum of Q140/Q140 mice. J. Huntingtons Dis., 5, 239–248. [DOI] [PubMed] [Google Scholar]
- 52. Pfister E.L., Chase K.O., Sun H., Kennington L.A., Conroy F., Johnson E., Miller R., Borel F., Aronin N. and Mueller C. (2017) Safe and efficient silencing with a pol II, but not a pol lII, promoter expressing an artificial miRNA targeting human Huntingtin. Mol. Ther. Nucleic Acids, 7, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cambon K., Zimmer V., Martineau S., Gaillard M.C., Jarrige M., Bugi A., Miniarikova J., Rey M., Hassig R., Dufour N. et al. (2017) Preclinical evaluation of a Lentiviral vector for Huntingtin silencing. Mol. Ther. Methods Clin. Dev., 5, 259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pfister E.L., DiNardo N., Mondo E., Borel F., Conroy F., Fraser C., Gernoux G., Han X., Hu D., Johnson E. et al. (2018) Artificial miRNAs reduce human mutant Huntingtin throughout the striatum in a transgenic sheep model of Huntington's disease. Hum. Gene Ther., 29, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McBride J.L., Pitzer M.R., Boudreau R.L., Dufour B., Hobbs T., Ojeda S.R. and Davidson B.L. (2011) Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol. Ther., 19, 2152–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hadaczek P., Stanek L., Ciesielska A., Sudhakar V., Samaranch L., Pivirotto P., Bringas J., O'Riordan C., Mastis B., San Sebastian W. et al. (2016) Widespread AAV1- and AAV2-mediated transgene expression in the nonhuman primate brain: implications for Huntington's disease. Mol. Ther. Methods Clin. Dev., 3, 16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Christine C.W., Bankiewicz K.S., Van Laar A.D., Richardson R.M., Ravina B., Kells A.P., Boot B., Martin A.J., Nutt J., Thompson M.E. and Larson P.S. (2019) Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson's disease. Ann. Neurol., 85, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu X.H. and Yang X.W. (2012) "Huntingtin holiday": progress toward an antisense therapy for Huntington's disease. Neuron, 74, 964–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stanek L.M., Yang W., Angus S., Sardi P.S., Hayden M.R., Hung G.H., Bennett C.F., Cheng S.H. and Shihabuddin L.S. (2013) Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington's disease. J. Huntingtons Dis., 2, 217–228. [DOI] [PubMed] [Google Scholar]
- 60. Roon-Mom W.M.C., Roos R.A.C. and Bot S.T. (2018) Dose-dependent lowering of mutant Huntingtin using antisense oligonucleotides in Huntington disease patients. Nucleic Acid Ther., 28, 59–62. [DOI] [PubMed] [Google Scholar]
- 61. Tabrizi S.J., Leavitt B.R., Landwehrmeyer G.B., Wild E.J., Saft C., Barker R.A., Blair N.F., Craufurd D., Priller J., Rickards H. et al. (2019) Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med., 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- 62. Carroll J.B., Warby S.C., Southwell A.L., Doty C.N., Greenlee S., Skotte N., Hung G., Bennett C.F., Freier S.M. and Hayden M.R. (2011) Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther., 19, 2178–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gagnon K.T., Pendergraff H.M., Deleavey G.F., Swayze E.E., Potier P., Randolph J., Roesch E.B., Chattopadhyaya J., Damha M.J., Bennett C.F. et al. (2010) Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry, 49, 10166–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Skotte N.H., Southwell A.L., Ostergaard M.E., Carroll J.B., Warby S.C., Doty C.N., Petoukhov E., Vaid K., Kordasiewicz H., Watt A.T. et al. (2014) Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS One, 9, e107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rodrigues F.B., Quinn L. and Wild E.J. (2019) Huntington's disease clinical trials corner: January 2019. J. Huntingtons Dis., 8, 115–125. [DOI] [PubMed] [Google Scholar]
- 66. Garriga-Canut M., Agustin-Pavon C., Herrmann F., Sanchez A., Dierssen M., Fillat C. and Isalan M. (2012) Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. U. S. A., 109, E3136–E3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li L., Shen X., Liu Z., Norrbom M., Prakash T.P., O'Reilly D., Sharma V.K., Damha M.J., Watts J.K., Rigo F. et al. (2018) Activation of Frataxin protein expression by antisense oligonucleotides targeting the mutant expanded repeat. Nucleic Acid Ther., 28, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lieberman A.P., Yu Z., Murray S., Peralta R., Low A., Guo S., Yu X.X., Cortes C.J., Bennett C.F., Monia B.P. et al. (2014) Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep., 7, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sahashi K., Katsuno M., Hung G., Adachi H., Kondo N., Nakatsuji H., Tohnai G., Iida M., Bennett C.F. and Sobue G. (2015) Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet., 24, 5985–5994. [DOI] [PubMed] [Google Scholar]
- 70. Hampson D.R., Hooper A.W.M. and Niibori Y. (2019) The application of Adeno-associated viral vector gene therapy to the treatment of fragile X syndrome. Brain Sci., 9, DOI: 10.3390/brainsci902003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Abu-Baker A., Kharma N., Perreault J., Grant A., Shekarabi M., Maios C., Dona M., Neri C., Dion P.A., Parker A. et al. (2019) RNA-based therapy utilizing Oculopharyngeal muscular dystrophy transcript knockdown and replacement. Mol. Ther. Nucleic Acids, 15, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Malerba A., Klein P., Bachtarzi H., Jarmin S.A., Cordova G., Ferry A., Strings V., Espinoza M.P., Mamchaoui K., Blumen S.C. et al. (2017) PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun., 8, 14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hsu P.D., Lander E.S. and Zhang F. (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Batra R., Nelles D.A., Pirie E., Blue S.M., Marina R.J., Wang H., Chaim I.A., Thomas J.D., Zhang N., Nguyen V. et al. (2017) Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell, 170, 899–912e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dabrowska M., Juzwa W., Krzyzosiak W.J. and Olejniczak M. (2018) Precise excision of the CAG tract from the Huntingtin gene by Cas9 Nickases. Front. Neurosci., 12, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kolli N., Lu M., Maiti P., Rossignol J. and Dunbar G.L. (2017) CRISPR-Cas9 mediated gene-silencing of the mutant Huntingtin gene in an in vitro model of Huntington's disease. Int. J. Mol. Sci., 18, 10.3390/ijms18040754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Monteys A.M., Ebanks S.A., Keiser M.S. and Davidson B.L. (2017) CRISPR/Cas9 editing of the mutant Huntingtin allele in vitro and in vivo. Mol. Ther., 25, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shin J.W., Kim K.H., Chao M.J., Atwal R.S., Gillis T., MacDonald M.E., Gusella J.F. and Lee J.M. (2016) Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet., 25, 4566–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vachey G. and Deglon N. (2018) CRISPR/Cas9-mediated genome editing for Huntington's disease. Methods Mol. Biol., 1780, 463–481. [DOI] [PubMed] [Google Scholar]
- 80. Yang S., Chang R., Yang H., Zhao T., Hong Y., Kong H.E., Sun X., Qin Z., Jin P., Li S. et al. (2017) CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J. Clin. Invest., 127, 2719–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yrigollen C.M. and Davidson B.L. (2019) CRISPR to the rescue: advances in gene editing for the FMR1 gene. Brain Sci., 9, 10.3390/brainsci9010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bushart D.D., Murphy G.G. and Shakkottai V.G. (2016) Precision medicine in spinocerebellar ataxias: treatment based on common mechanisms of disease. Ann. Transl. Med., 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]