

Summary
The human microbiota composition is associated with a number of diseases including obesity, inflammatory bowel disease, and bacterial vaginosis. Thus, microbiome research has the potential to reshape clinical and therapeutic approaches. However, raw microbiome count data require careful pre-processing steps that take into account both the sparsity of counts and the large number of taxa that are being measured. Filtering is defined as removing taxa that are present in a small number of samples and have small counts in the samples where they are observed. Despite progress in the number and quality of filtering approaches, there is no consensus on filtering standards and quality assessment. This can adversely affect downstream analyses and reproducibility of results across platforms and software. We introduce PERFect, a novel permutation filtering approach designed to address two unsolved problems in microbiome data processing: (i) define and quantify loss due to filtering by implementing thresholds and (ii) introduce and evaluate a permutation test for filtering loss to provide a measure of excessive filtering. Methods are assessed on three “mock experiment” data sets, where the true taxa compositions are known, and are applied to two publicly available real microbiome data sets. The method correctly removes contaminant taxa in “mock” data sets, quantifies and visualizes the corresponding filtering loss, providing a uniform data-driven filtering criteria for real microbiome data sets. In real data analyses PERFect tends to remove more taxa than existing approaches; this likely happens because the method is based on an explicit loss function, uses statistically principled testing, and takes into account correlation between taxa. The PERFect software is freely available at https://github.com/katiasmirn/PERFect.
Keywords: 16S rRNA, Filtering, Microbiome, Normalization, Permutation test
1. Introduction
Microbiome studies yield data as counts of microbes from the 16S rRNA marker gene using next generation sequencing (NGS) technology. Specifically, a sample gives counts of DNA fragments which are then grouped into species level operational taxonomic units (OTUs), also referred to as taxa; in statistical terminology, these are random variables. The resulting data, usually referred to as the “OTU table” is typically high dimensional; for example, human gut samples provide counts on 1000 to 1500 taxa, while vaginal samples yield 200 to 400 taxa. In contrast to gene expression data, microbial data are sparse as many taxa are rare and often have zero counts in most samples.
The role of the microbiome in human health and disease has received increased attention over the last decade (Human Microbiome Project Consortium, 2012) with the gut and vaginal body sites being among the best-studied. Studies on the gut microbiome have explored the role of microbiota in the immune system, inflammatory bowel disease, and development of the infant gut (Greenblum and others, 2012; Lozupone and others, 2012; Maynard and others, 2012). Vaginal microbiome studies are important for understanding conditions such as bacterial vaginosis (BV), a disruption of the microbiome that is associated with increased risk of sexually transmitted infections and preterm births (Ma and others, 2012; Romero and others, 2014). Given the clinical and translational implications of microbiome research, it is crucial to identify and agree on high data quality standards and statistical methodology.
Stulberg and others (2016) assessed the current state of microbiome research in the USA, identifying standardized protocols for data processing as the highest priority technical need. Every aspect of the process from sample collection to DNA extraction to data analysis can contribute different sources of errors and variability. Herein, we concentrate on filtering or removing spurious taxa from the 16S data set, which are observed mainly because of contamination and/or sequencing errors. Contamination occurs during the sample preparation, DNA extraction and polymerase chain reaction (PCR) amplification. Potential sources of contamination are bacteria that are frequently handled in the lab, those that reside on the skin of lab workers, or in the extraction kits (Salter and others, 2014). Several studies have been conducted using “mock” samples curated so that they consist of known microbial species in prescribed proportions and, after cultivation, the samples are sequenced using NGS technology to identify the taxa and evaluate the effects of such contamination on the observed taxa counts (Brooks and others, 2015). Errors, especially due to misclassification, arise as the sequencing technology employs a combination of statistical and computational algorithms that make assumptions about identifying nucleotide bases (Cacho and others, 2016) and for assembling the DNA fragments during the alignment process (Li and Homer, 2010). Overall, contamination and sequencing errors lead to either falsely identifying taxa that were not in the sample or misclassifying the taxa of DNA fragment reads.
In practice, filtering is a variation of an ad hoc, albeit simple, procedure. One of the most widely used techniques for filtering in microbiome studies selects taxa that have a number of counts above 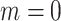 in at least
in at least 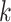 samples. This approach is borrowed from the RNA-seq gene expression literature and is implemented in the R package genefilter (Gentleman and others, 2016) and in QIIME bioinformatics pipeline function filter_otus_from_otu_table.py (Caporaso and others, 2010). Another popular approach is to remove taxa that are observed in fewer than
samples. This approach is borrowed from the RNA-seq gene expression literature and is implemented in the R package genefilter (Gentleman and others, 2016) and in QIIME bioinformatics pipeline function filter_otus_from_otu_table.py (Caporaso and others, 2010). Another popular approach is to remove taxa that are observed in fewer than 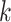 % of the samples. The advantage of these methods is that they are simple, intuitive, and easy to communicate with collaborators. However, they do not have an explicit loss function and objective criteria for choosing the tuning parameters
% of the samples. The advantage of these methods is that they are simple, intuitive, and easy to communicate with collaborators. However, they do not have an explicit loss function and objective criteria for choosing the tuning parameters 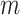 and
and 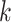 .
.
Recently, several techniques have been proposed to detect contaminant taxa. One approach, developed by Knights and others (2011) and implemented in R package sourcetracker, relies on microbial source tracking to identify the proportion of contaminant taxa in each sample by matching the taxa table against the database of known contaminants. However, this method does not detect individual contaminant taxa that should be removed from the data set. Davis and others (2017) addressed this problem by introducing decontam R package that identifies contaminants by: (i) inversely correlating taxa frequencies with sample DNA concentration; and (ii) using the prevalence of sequenced negative controls (Salter and others, 2014). A major practical limitation of this method is that the auxiliary data from DNA quantitation that is in most cases intrinsic to sample preparation or negative controls data that is intrinsic to sequencing protocol might not be available.
We propose a filtering loss measure and a principled filtering test, PERFect, for deciding which taxa to remove. In contrast to the standard procedures, which assume that taxa in a biological network are isolated, PERFect filters out taxa with insignificant contribution to the total covariance. Our proposal relies on ranking taxa importance, measuring their contribution to the total covariance, and quantifying the chance that the loss increase for a set of filtered taxa is due to randomness. We choose the contribution to covariance as the measure of filtering loss because it provides a measure of taxa contribution to the biological network. We introduce two principled filtering methods: simultaneous and permutation PERFect, that rely on estimating the null distribution for the increase in filtering loss due to each taxon. We compare our proposal to traditional filtering on two data sets acquired from mock community experiments carried out at Virginia Commonwealth University (VCU) (Fettweis and others, 2012; Brooks and others, 2015) and a reagent and laboratory contamination data set (Salter and others, 2014). We also illustrate our methods using a publicly available vaginal microbiome data set published in Ravel and others (2011) and Bacterial Diversity in Neonatal Intensive Care Units data (Knights and others, 2011). Methods are described for relative OTU abundance (proportions data), but can be used on other OTU table representations including raw OTU counts or presence-absence.
The main goal of PERFect is to extend traditional filtering approaches to find the best subset of taxa to retain for further analysis by implementing statistical data-driven significance cut-off thresholds. This method is remotely related to the sparse covariance and precision matrix estimation techniques which are pairwise methods and, in the context of microbiome data, identify pairs of marginally or conditionally uncorrelated taxa, respectively. In contrast, PERFect removes columns of low-signal taxa as opposed to individual covariance pairs. The goals of PERFect are closely related to the idea of sure screening method introduced by Fan and Lv (2008), however PERFect is an unsupervised method, in which the response information, such as health outcomes, is not used in identifying signal taxa.
Results show that in the high signal-to-noise ratio scenarios, PERFect is consistent with standard filtering and outperforms it on one of the mock data sets. In low signal-to-noise ratio scenario, PERFect permutation approach significantly outperforms other microbiome filtering methods. Most taxa in these samples are uncorrelated, and 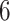 out of
out of 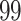 ,
, 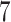 out
out 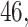 and
and 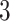 out of
out of 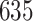 are signal taxa respectively. In the real correlated data scenario with low to moderate signal, PERFect removes the same taxa as the traditional approaches, but removes many additional taxa that are found not to contribute to the overall signal. Taxa removed by PERFect are consistent with expectations based on biological knowledge of these organisms. In summary, PERFect has several practical and theoretical advantages over standard approaches. First, PERFect allows dimension reduction consistent with minimal total covariance loss. It retains a smaller subset of taxa that provide highest contribution to the total covariance. Second, in contrast to recently developed decontam method, PERFect can be used in any data set, where additional information required by decontam might not be available. Third, PERFect is implemented in R and provides an easy-to-use, data-driven approach for choosing a filtering cut-off combined with the visualization of the relationship between taxa P-values and filtering loss.
are signal taxa respectively. In the real correlated data scenario with low to moderate signal, PERFect removes the same taxa as the traditional approaches, but removes many additional taxa that are found not to contribute to the overall signal. Taxa removed by PERFect are consistent with expectations based on biological knowledge of these organisms. In summary, PERFect has several practical and theoretical advantages over standard approaches. First, PERFect allows dimension reduction consistent with minimal total covariance loss. It retains a smaller subset of taxa that provide highest contribution to the total covariance. Second, in contrast to recently developed decontam method, PERFect can be used in any data set, where additional information required by decontam might not be available. Third, PERFect is implemented in R and provides an easy-to-use, data-driven approach for choosing a filtering cut-off combined with the visualization of the relationship between taxa P-values and filtering loss.
We introduce criteria for measuring filtering loss and develop the PERFect methodology in Section 2. In Section 3, we evaluate traditional filtering approaches, simultaneous and permutation PERFect on three mock community data sets, a data set with known taxa biology. We test our method in Section 4 and one vaginal microbiome data set. In Section 5, we present the concluding remarks and the directions for future work. PERFect software features follow in Section 6.
2. Methods
The microbiome studies data structure is an 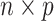 matrix of OTU counts
matrix of OTU counts 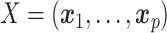 , where each column
, where each column 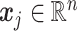 contains the
contains the 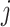 th taxon counts observed across
th taxon counts observed across 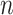 samples. Filtering is the process of identifying and removing a subset of taxa
samples. Filtering is the process of identifying and removing a subset of taxa 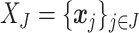 , where
, where 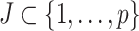 , according to a particular criterion. Let
, according to a particular criterion. Let 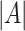 denote the cardinality of the set of indices
denote the cardinality of the set of indices 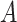 . The original data matrix
. The original data matrix 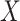 can be written (after re-arranging some columns) as
can be written (after re-arranging some columns) as 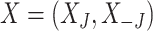 , where
, where 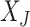 is the
is the 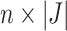 dimensional matrix containing the taxa that are removed and
dimensional matrix containing the taxa that are removed and 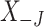 is the
is the 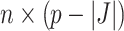 dimensional matrix containing the taxa that are retained for further analysis.
dimensional matrix containing the taxa that are retained for further analysis.
2.1. Filtering loss
We base the filtering loss on the Frobenius norm since it measures the total covariance of the data. Specifically, we define the loss due to filtering out the 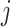 th taxon as,
th taxon as,
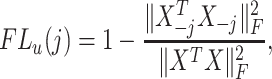 |
(2.1) |
where 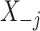 is the
is the 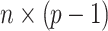 dimensional matrix obtained by removing the
dimensional matrix obtained by removing the 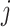 th column from the data matrix
th column from the data matrix 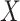 . Here,
. Here, 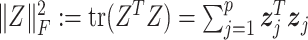 is the square of the Frobenius norm of matrix
is the square of the Frobenius norm of matrix 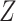 . The covariance matrix of column-wise centered data
. The covariance matrix of column-wise centered data 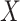 is estimated as
is estimated as 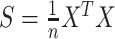 , so that the filtering loss can be viewed as the ratio of filtered and full covariance matrix magnitudes. Thus, the quantity
, so that the filtering loss can be viewed as the ratio of filtered and full covariance matrix magnitudes. Thus, the quantity 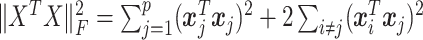 measures total covariance of the data, and the filtering loss criterion accounts both for the contribution of the
measures total covariance of the data, and the filtering loss criterion accounts both for the contribution of the 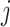 th taxon and its co-occurrence with other taxa. Similarly, we define the filtering loss due to removing a group of taxa,
th taxon and its co-occurrence with other taxa. Similarly, we define the filtering loss due to removing a group of taxa, 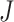 , as
, as
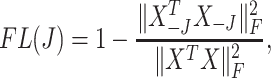 |
(2.2) |
where 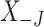 is the
is the 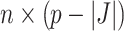 dimensional matrix obtained by removing the columns indexed by the set
dimensional matrix obtained by removing the columns indexed by the set 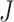 from the data matrix
from the data matrix 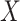 .
.
The filtering loss 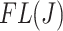 is a number between
is a number between 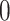 and
and 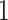 , with values close to
, with values close to 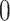 if the set of taxa
if the set of taxa 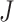 has small contribution to the total covariance and
has small contribution to the total covariance and 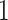 otherwise. The methods presented here are based on the Frobenius norm, but other filtering losses can be considered without changes in the methodology. An small subset of mock data example presented in Section 1 in the supplementary material available at Biostatistics online and PERFect software vignette (https://github.com/katiasmirn/PERFect) illustrates how these measures detect the differences between signal and noise taxa.
otherwise. The methods presented here are based on the Frobenius norm, but other filtering losses can be considered without changes in the methodology. An small subset of mock data example presented in Section 1 in the supplementary material available at Biostatistics online and PERFect software vignette (https://github.com/katiasmirn/PERFect) illustrates how these measures detect the differences between signal and noise taxa.
We start by re-arranging the columns of the matrix 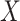 with respect to the number of occurrences of the taxa in the
with respect to the number of occurrences of the taxa in the 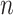 samples. More precisely, we define
samples. More precisely, we define
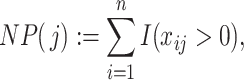 |
(2.3) |
where 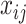 is the
is the 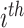 element in
element in 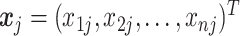 , the
, the 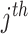 column of
column of 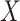 , and
, and 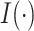 is the indicator function. Taxa with smaller values of
is the indicator function. Taxa with smaller values of 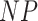 are more likely candidates to be filtered and the columns of
are more likely candidates to be filtered and the columns of 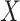 are re-ordered to ensure that
are re-ordered to ensure that 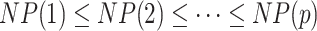 . This ordering will be shown to have a very good performance in applications, though alternative or more refined orderings could also be considered; we provide and discuss some alternatives and their effect on the choice of filtering cut-off in Section 2 in supplementary material available at Biostatistics online.
. This ordering will be shown to have a very good performance in applications, though alternative or more refined orderings could also be considered; we provide and discuss some alternatives and their effect on the choice of filtering cut-off in Section 2 in supplementary material available at Biostatistics online.
Once taxa are ordered, we propose calculating the filtering loss sequentially by removing the taxa in increasing order of 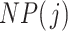 . If
. If 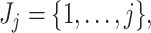 then we define the filtering loss for removing the first
then we define the filtering loss for removing the first 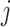 taxa as
taxa as 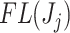 and the difference in filtering loss as
and the difference in filtering loss as
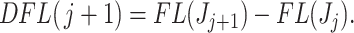 |
(2.4) |
To better understand the two measures in Figure 1, we display the results for a mock community data set (Fettweis and others, 2012), where only six taxa to the right of the vertical dashed line correspond to the true signal. The left panel displays the filtering loss (relative to the total covariance), while the right panel displays the difference in filtering loss (relative to the sample variance after removing 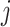 taxa). Both panels provide the intuition that many taxa can be removed from the OTU matrix based on our chosen loss function. However, around
taxa). Both panels provide the intuition that many taxa can be removed from the OTU matrix based on our chosen loss function. However, around 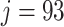 , the filtering loss starts to increase dramatically. In the following section, we provide a principled approach on deciding which increases in filtering loss can be attributed to randomness and which increases correspond to true signal in the data.
, the filtering loss starts to increase dramatically. In the following section, we provide a principled approach on deciding which increases in filtering loss can be attributed to randomness and which increases correspond to true signal in the data.
Fig. 1.

Filtering loss plots for Fettweis and others (2012) data. For each plot, taxa on the x-axis are arranged in the order of increasing values of 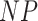 and dashed vertical lines indicate taxa for which
and dashed vertical lines indicate taxa for which 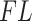 have faster increase rates. Left panel: cumulative filtering loss
have faster increase rates. Left panel: cumulative filtering loss 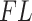 . Right panel: values of
. Right panel: values of 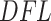 that approximate filtering loss slope at each taxon.
that approximate filtering loss slope at each taxon.
2.2. PERFect
Our main goal is to decide whether the set of first 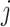 taxa,
taxa, 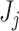 , is needed to explain the microbiome structure observed in the OTU table, or if a smaller set of
, is needed to explain the microbiome structure observed in the OTU table, or if a smaller set of 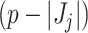 taxa suffices. We define
taxa suffices. We define
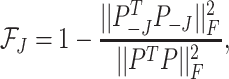 |
where 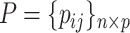 is a matrix of the true relative abundance of microbe
is a matrix of the true relative abundance of microbe 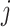 in sample
in sample 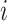 . The theoretical quantity
. The theoretical quantity 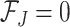 if the
if the 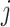 th taxon is included erroneously. Then
th taxon is included erroneously. Then 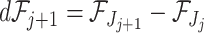 is the theoretical improvement to the signal from adding the taxon
is the theoretical improvement to the signal from adding the taxon  . We propose estimating
. We propose estimating 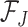 and
and 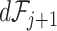 using the filtering loss
using the filtering loss 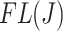 (2.2) and corresponding differences in filtering loss
(2.2) and corresponding differences in filtering loss 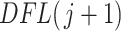 (2.4) statistics. Therefore, we test
(2.4) statistics. Therefore, we test
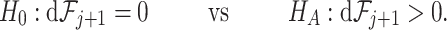 |
To test this hypothesis we need to estimate a filtering cut-off threshold. To achieve this, we introduce two approaches: (i) fitting a distribution to the differences in filtering loss for 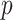 taxa simultaneously; and (ii) fitting a null distribution for each set
taxa simultaneously; and (ii) fitting a null distribution for each set 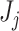 of first
of first 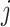 taxa by permuting the order of taxa. Both methods depend on the assumption that a substantial percentage of the taxa have
taxa by permuting the order of taxa. Both methods depend on the assumption that a substantial percentage of the taxa have 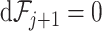 . In our case, we assume that at least half the taxa will need to be removed, but other probabilities can be used. Although the theoretical underlying hypothesis is stated in terms of relative abundances, counts can be used interchangeably in calculations because of the definition of the loss function.
. In our case, we assume that at least half the taxa will need to be removed, but other probabilities can be used. Although the theoretical underlying hypothesis is stated in terms of relative abundances, counts can be used interchangeably in calculations because of the definition of the loss function.
2.2.1. Simultaneous filtering.
The simultaneous filtering approach is very fast and requires only fitting a distribution to the filtering loss differences 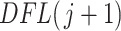 shown in Figure 1 (right panel). Figure 2 (left panel) provides the histogram of the log-transformed
shown in Figure 1 (right panel). Figure 2 (left panel) provides the histogram of the log-transformed 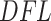 values for the Fettweis and others (2012) data. Under the assumption that at least
values for the Fettweis and others (2012) data. Under the assumption that at least 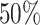 of the taxa are not informative, the left part of the distribution can be approximated by a Skew-Normal distribution (Azzalini, 2005) using quantile matching. We suggest using
of the taxa are not informative, the left part of the distribution can be approximated by a Skew-Normal distribution (Azzalini, 2005) using quantile matching. We suggest using 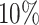 ,
, 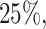 and
and 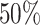 quantiles for matching, though these quantiles can be adjusted in specific scenarios when one expects a larger or smaller percentage of taxa that are not informative. This part of the approach does not depend on the choice of filtering loss. A random variable
quantiles for matching, though these quantiles can be adjusted in specific scenarios when one expects a larger or smaller percentage of taxa that are not informative. This part of the approach does not depend on the choice of filtering loss. A random variable 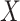 is said to have a Skew-Normal distribution with location parameter
is said to have a Skew-Normal distribution with location parameter 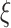 , scale parameter
, scale parameter 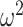 , and shape parameter
, and shape parameter 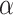 denoted by
denoted by 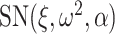 if its probability density function (pdf) is
if its probability density function (pdf) is
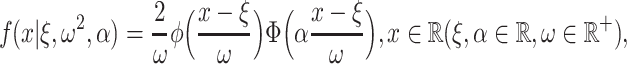 |
where 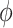 and
and 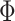 denote the probability density and the cumulative distribution function of the standard normal distribution, respectively. Because there are three parameters, we used three quantiles for matching the distributions. The estimated distribution for the Fettweis and others (2012) data is shown in Figure 2 (left panel) as a blue line superimposed over the histogram of the log-transformed
denote the probability density and the cumulative distribution function of the standard normal distribution, respectively. Because there are three parameters, we used three quantiles for matching the distributions. The estimated distribution for the Fettweis and others (2012) data is shown in Figure 2 (left panel) as a blue line superimposed over the histogram of the log-transformed 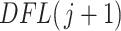 values. The estimated distribution approximates the empirical distribution of sequential losses. The Skew-Normal fits the left part of the distribution reasonably well, while the right tail of the distribution is assumed to be generated from an unspecified alternative distribution and is not expected to fit the same distribution as the left part. It is important to understand that our Skew-Normal distribution is designed to capture the null distribution of
values. The estimated distribution approximates the empirical distribution of sequential losses. The Skew-Normal fits the left part of the distribution reasonably well, while the right tail of the distribution is assumed to be generated from an unspecified alternative distribution and is not expected to fit the same distribution as the left part. It is important to understand that our Skew-Normal distribution is designed to capture the null distribution of 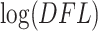 values, whereas the alternative distribution remains unspecified. This is crucial for making decisions about which taxa do not contribute to the signal (provide
values, whereas the alternative distribution remains unspecified. This is crucial for making decisions about which taxa do not contribute to the signal (provide 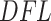 values corresponding to the null component) and which taxa contribute to the signal (provide
values corresponding to the null component) and which taxa contribute to the signal (provide 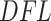 values that correspond to the alternative component). The log Skew-Normal was necessary in our case because of the nature of the
values that correspond to the alternative component). The log Skew-Normal was necessary in our case because of the nature of the 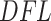 measures. Once we have an estimate of the null distribution, we define the significance of the set of first
measures. Once we have an estimate of the null distribution, we define the significance of the set of first 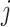 taxa
taxa 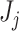 as the P-value
as the P-value
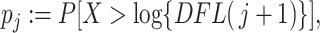 |
(2.5) |
where the random variable 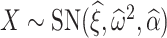 and
and 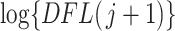 is the log-transformed value of filtering loss difference due to removing the
is the log-transformed value of filtering loss difference due to removing the 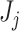 taxa. Here
taxa. Here 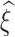 ,
, 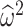 , and
, and 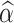 are the estimated parameters of the Skew-Normal distribution on the log-transformed losses. We further suggest using smoothed P-values, obtained by averaging
are the estimated parameters of the Skew-Normal distribution on the log-transformed losses. We further suggest using smoothed P-values, obtained by averaging  (or more) subsequent P-values (2.5). This practice allows the PERFect procedure to be robust to the choice of quantiles used to fit the reference distribution in rare cases when a marginally significant taxon has a low ordering rank and thus appears very early in the data. Finally, we filter out the set
(or more) subsequent P-values (2.5). This practice allows the PERFect procedure to be robust to the choice of quantiles used to fit the reference distribution in rare cases when a marginally significant taxon has a low ordering rank and thus appears very early in the data. Finally, we filter out the set 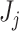 of taxa whose P-values are larger than a given significance level
of taxa whose P-values are larger than a given significance level 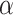 . The simultaneous filtering procedure method is outlined in Algorithm 1.
. The simultaneous filtering procedure method is outlined in Algorithm 1.
Fig. 2.

Left panel: histogram of log-transformed 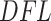 values for the Fettweis and others (2012) data. The blue line indicates
values for the Fettweis and others (2012) data. The blue line indicates 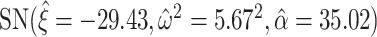 density fitted to the log-transformed data using quantile matching method. Note that the Skew-Normal fits well the left part of the distribution (the null P-values), while the right tail of the distribution is assumed to be generated by an unspecified alternative distribution. Right panel: histogram of log-transformed
density fitted to the log-transformed data using quantile matching method. Note that the Skew-Normal fits well the left part of the distribution (the null P-values), while the right tail of the distribution is assumed to be generated by an unspecified alternative distribution. Right panel: histogram of log-transformed  values for the Fettweis and others (2012) data obtained by permuting the labels for
values for the Fettweis and others (2012) data obtained by permuting the labels for  and
and  taxa. The red and green lines indicate Skew-Normal density fitted to the log-transformed data to taxon
taxa. The red and green lines indicate Skew-Normal density fitted to the log-transformed data to taxon 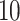 and
and 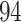 respectively using quantile matching method.
respectively using quantile matching method.
Algorithm 1
PERFect: simultaneous filtering
Input: OTU table X, test critical value
Output: Filtered OTU table X, p-value for each taxon
1: Order columns of
such that
end
3: Using quantile matching fit the Skew-Normal distribution to the logarithm of the sample
to obtain
the null distribution
4: Calculate the p-value
for
as
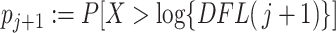
5: Average
subsequent P-values
6: Filter out the set of taxa
with the first P-value such that
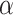
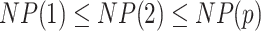
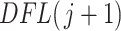
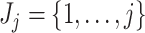
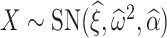
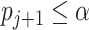
2.2.2. Permutation filtering.
In the previous section, we assumed that all sequential increments in filtering loss can be attributed to the same distribution. This assumption allows us to quickly calculate the mixture of the null and alternative distributions. However, there is no a priori reason to assume that all distributions for each step  are identical and equal to the distribution of simultaneous filtering. To address this issue, we propose a simple alternative we call permutation filtering. More precisely, we randomly permute the labels of the taxa and calculate
are identical and equal to the distribution of simultaneous filtering. To address this issue, we propose a simple alternative we call permutation filtering. More precisely, we randomly permute the labels of the taxa and calculate 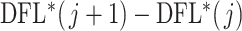 for every permutation. Once we draw this sample to evaluate
for every permutation. Once we draw this sample to evaluate 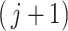 th taxon significance, the permutation distribution for each set of taxa is a mixture distribution between the null distribution corresponding to noise and signal taxa. In some cases that taxon will have weak signal, the
th taxon significance, the permutation distribution for each set of taxa is a mixture distribution between the null distribution corresponding to noise and signal taxa. In some cases that taxon will have weak signal, the 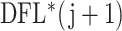 loss will be small and contribute to the null part of the distribution. In other cases that taxon will have strong signal and will have a larger contribution, which will become a component of the alternative distribution. The underlying assumption is that if the
loss will be small and contribute to the null part of the distribution. In other cases that taxon will have strong signal and will have a larger contribution, which will become a component of the alternative distribution. The underlying assumption is that if the 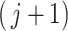 th taxon is unimportant (weak signal) then it will contribute very little above and beyond any combination of other
th taxon is unimportant (weak signal) then it will contribute very little above and beyond any combination of other 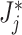 taxa and will provide a small value, corresponding to the null distribution. } Assuming that at least
taxa and will provide a small value, corresponding to the null distribution. } Assuming that at least 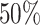 of taxa are not informative, we suggest fitting the log Skew-Normal distribution by matching the
of taxa are not informative, we suggest fitting the log Skew-Normal distribution by matching the 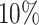 ,
, 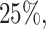 and
and 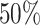 percentiles of the log-transformed samples to the Skew-Normal distribution. Thus, we estimate the location
percentiles of the log-transformed samples to the Skew-Normal distribution. Thus, we estimate the location 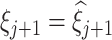 , scale
, scale 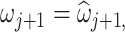 and shape
and shape 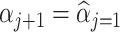 of the
of the 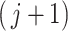 th taxon null distribution. If the
th taxon null distribution. If the 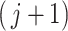 th taxon is not important then the difference is drawn from the null distribution, otherwise it is drawn from the alternative.
th taxon is not important then the difference is drawn from the null distribution, otherwise it is drawn from the alternative.
The difference between simultaneous and permutation filtering is as follows. In permutation filtering the assumption is that a taxon with weak signal remains unimportant to any combination of other 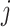 taxa. In simultaneous filtering the assumption is that the weak signal taxon is unimportant in the particular ordering imposed by Step
taxa. In simultaneous filtering the assumption is that the weak signal taxon is unimportant in the particular ordering imposed by Step  of Algorithm
of Algorithm  . In validation studies, we have found that in most cases the methods perform similarly, though the permutation filtering is more robust to the choice of tuning parameters.
. In validation studies, we have found that in most cases the methods perform similarly, though the permutation filtering is more robust to the choice of tuning parameters.
Figure 2 (right panel) illustrates the histogram of log-transformed 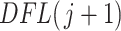 values for
values for 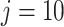 (red) and
(red) and 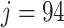 (green) filtered taxa for the Fettweis and others (2012) data set, where only
(green) filtered taxa for the Fettweis and others (2012) data set, where only 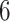 out of
out of 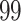 taxa correspond to the true signal. Both PERFect approaches correctly identify the true signal, though the null distributions are quite different, especially in the left tail of the distribution. We chose these examples because they are quite extreme. Indeed, in practice, we propose stopping much earlier with removing taxa. While most null distributions agree quite closely for
taxa correspond to the true signal. Both PERFect approaches correctly identify the true signal, though the null distributions are quite different, especially in the left tail of the distribution. We chose these examples because they are quite extreme. Indeed, in practice, we propose stopping much earlier with removing taxa. While most null distributions agree quite closely for 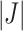 up to around
up to around 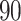 , differences start to increase for
, differences start to increase for 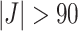 .
.
The method then proceeds just as simultaneous filtering and the significance of the 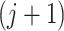 th taxon is the P-value
th taxon is the P-value 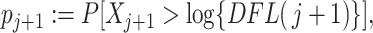 where the random variable
where the random variable 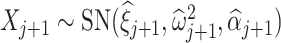 and
and 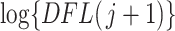 is the log-transformed value of filtering loss difference due to removing the
is the log-transformed value of filtering loss difference due to removing the 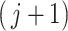 th taxon. Similar to the simultaneous testing approach, we filter out taxa sequentially based on large smoothed P-values. The permutation filtering is outlined in Algorithm 2.
th taxon. Similar to the simultaneous testing approach, we filter out taxa sequentially based on large smoothed P-values. The permutation filtering is outlined in Algorithm 2.
In contrast to simultaneous filtering, permutation filtering does not require a particular ordering of taxa, and the 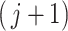 th taxa significance can be evaluated using any ordering of the taxa, not necessarily the number of occurrences in
th taxa significance can be evaluated using any ordering of the taxa, not necessarily the number of occurrences in 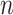 samples given by (2.3). While the distribution of
samples given by (2.3). While the distribution of 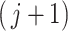 th taxa does not depend on ordering, the value of
th taxa does not depend on ordering, the value of 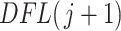 used in Algorithm 2 to evaluate the significance of the
used in Algorithm 2 to evaluate the significance of the 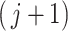 th taxon depends on the choice of taxa in the cutoff set
th taxon depends on the choice of taxa in the cutoff set 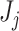 . Therefore, we suggest using taxa ordering according to (2.3) in simultaneous PERFect approach as a preliminary measure of taxa importance. Thus, we propose using the decreasing order of simultaneous PERFect p-values (2.5) in permutation PERFect. The permutation PERFect is computationally more expensive than the simultaneous PERFect. It requires
. Therefore, we suggest using taxa ordering according to (2.3) in simultaneous PERFect approach as a preliminary measure of taxa importance. Thus, we propose using the decreasing order of simultaneous PERFect p-values (2.5) in permutation PERFect. The permutation PERFect is computationally more expensive than the simultaneous PERFect. It requires 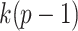 permutations, where the number of permutations for each taxon
permutations, where the number of permutations for each taxon 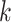 is large (we use
is large (we use 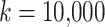 ) leading to longer computational time when the number of observed taxa is large.
) leading to longer computational time when the number of observed taxa is large.

Algorithm 2 PERFect: permutation filtering
3. Methods validation
We apply traditional, decontamination (Davis and others, 2017) (where appropriate) and PERFect filtering methods to:
1. Mock community data 1: positive controls data. These data (Fettweis and others, 2012) were generated as a part of a sequencing protocol where two control samples (one positive and one negative) were placed on a sequencing plate at each run. The positive control samples consisted of six species combined in prescribed proportions where the proportions of each microbial community was held the same in all samples. Negative controls samples were comprised of distilled water; ideally, no bacteria should be detected in these samples as a result of sequencing. In this article, we consider the positive controls data, where 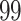 taxa were observed as a result of sequencing. The negative controls samples were used to test the decontam package performance.
taxa were observed as a result of sequencing. The negative controls samples were used to test the decontam package performance.
2. Mock community data 2: bias experiment data. These publicly available data (Brooks and others, 2015) were generated as a part of a study designed to evaluate the bias at each step of the VCU sequencing protocol, namely, DNA extraction, PCR amplification, sequencing, and taxonomic classification. Mock community samples were created out of seven vaginally relevant bacteria by mixing prescribed quantities of cells, with quantities varying across samples according to an experimental design described in Brooks and others (2015). As opposed to the positive controls data, bacteria appear in different proportions across samples. The number of taxa identified by the sequencing and bioinformatics pipeline was 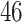 .
.
3. Mock community data 3: reagent and laboratory contamination data. These 16S Amplicon Data (Salter and others, 2014) was generated as part of the effect of contaminants present in DNA extraction kits and other laboratory reagents on sequencing results study. Mock samples of a pure Salmonella bongori culture had undergone five rounds of serial ten-fold dilutions to generate a series of high to low biomass samples. The taxa counts table was not reported in the study. We use samples for the Salmonella bongori culture 16S rRNA gene profiling deposited as FASTQ files deposited under ENA project accession EMBL: ERP006808 and processed using the dada2 R package. We follow data processing steps described in Davis and others (2017). The final data set contained 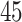 samples and
samples and 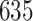 taxa, out of which
taxa, out of which 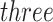 taxa corresponded to Salmonella bongori culture.
taxa corresponded to Salmonella bongori culture.
4. Bacterial diversity in neonatal intensive care units (NICUs) data. These data (Knights and others, 2011), were collected to investigate the sources of bacteria found on surfaces and equipment in NICU. The data contains 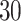 samples and
samples and 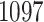 taxa and was previously analyzed using the sourcetracker software to identify the proportion of bacteria from each environment using published data sets from environments likely to be sources of indoor contaminants, namely human skin, oral cavities, feces (Costello and others, 2009), and soils (Lauber and others, 2009).
taxa and was previously analyzed using the sourcetracker software to identify the proportion of bacteria from each environment using published data sets from environments likely to be sources of indoor contaminants, namely human skin, oral cavities, feces (Costello and others, 2009), and soils (Lauber and others, 2009).
We now evaluate and compare 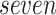 different approaches: (i) simultaneous PERFect with abundance ordering; (ii) permutation PERFect with abundance ordering; (iii) permutation PERFect P-values ordering from simultaneous PERFect; (iv) traditional filtering; (v) decontam prevalence method in which the proportions of features in signal samples are compared with their occurrence proportions in negative control samples; (vi) decontam frequency pooled, where batches of samples that were processed separately are pooled together; and (vii) decontam frequency batched method, which accounts for batches of samples that were processed separately. For each approach, we validate several settings. Namely, we evaluate the robustness of PERFect methods 1–3 to the choice of taxa ordering and quantiles used to fit the Skew-Normal for the null distribution using the following combination of quantiles: (i) 5%, 10%, 25%; (ii) 10%, 25%, 40%; (iii) 10%, 25%, 50%; and (iv) 20%, 30%, 60% quantiles. We also investigate the effect of the size of the test by varying
different approaches: (i) simultaneous PERFect with abundance ordering; (ii) permutation PERFect with abundance ordering; (iii) permutation PERFect P-values ordering from simultaneous PERFect; (iv) traditional filtering; (v) decontam prevalence method in which the proportions of features in signal samples are compared with their occurrence proportions in negative control samples; (vi) decontam frequency pooled, where batches of samples that were processed separately are pooled together; and (vii) decontam frequency batched method, which accounts for batches of samples that were processed separately. For each approach, we validate several settings. Namely, we evaluate the robustness of PERFect methods 1–3 to the choice of taxa ordering and quantiles used to fit the Skew-Normal for the null distribution using the following combination of quantiles: (i) 5%, 10%, 25%; (ii) 10%, 25%, 40%; (iii) 10%, 25%, 50%; and (iv) 20%, 30%, 60% quantiles. We also investigate the effect of the size of the test by varying 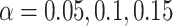 . We consider two rules for traditional filtering methods: (i) Rule
. We consider two rules for traditional filtering methods: (i) Rule 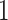 : Remove all taxa present in fewer than five samples and (ii) Rule
: Remove all taxa present in fewer than five samples and (ii) Rule 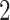 : Adopted from Milici and others (2016) that first selects taxa with abundance levels
: Adopted from Milici and others (2016) that first selects taxa with abundance levels 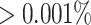 , and then further selects taxa that satisfy at least one of the following conditions: (i) Present in at least one sample at a relative abundance
, and then further selects taxa that satisfy at least one of the following conditions: (i) Present in at least one sample at a relative abundance 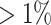 of the reads of that sample, (ii) Present in at least
of the reads of that sample, (ii) Present in at least 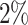 of samples at a relative abundance
of samples at a relative abundance 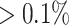 for a given sample, and (iii) Present in at least
for a given sample, and (iii) Present in at least 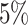 of samples at any abundance level. We compare the performance of PERFect to traditional filtering and decontam with
of samples at any abundance level. We compare the performance of PERFect to traditional filtering and decontam with 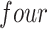 significance levels settings
significance levels settings 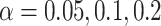 , and
, and 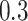 . (in data sets where decontam can be used) in terms of the total number of taxa preserved after filtering, percent of filtered taxa, and percent of preserved contaminants in the mock data sets.
. (in data sets where decontam can be used) in terms of the total number of taxa preserved after filtering, percent of filtered taxa, and percent of preserved contaminants in the mock data sets.
Validation results presented in Tables 1 and 2 indicate that our approach is highly robust to the choice of quantiles, especially in higher signal-to-noise ratio scenarios. While the PERFect simultaneous performs well in higher signal-to-noise ratio scenarios, this procedure is sensitive to the choice of quantiles used to fit the null distribution in low signal-to-noise ratio in mock data set 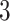 . This highlights a rare, but yet possible case when a marginally significant taxon has a low ordering rank and appears very early in the data, which can lead to large differences in the number of preserved taxa. However, this issue is alleviated by PERFect permutation procedure. PERFect permutation consistently outperforms the other approaches and can be used in situations when decontam cannot. All filtering procedures correctly select the true taxa present in the three mock data sets. Moreover, the permutation PERFect with P-values ordering provides major improvement over existing procedures and other PERFect methods in low signal-to-noise ratio mock data set
. This highlights a rare, but yet possible case when a marginally significant taxon has a low ordering rank and appears very early in the data, which can lead to large differences in the number of preserved taxa. However, this issue is alleviated by PERFect permutation procedure. PERFect permutation consistently outperforms the other approaches and can be used in situations when decontam cannot. All filtering procedures correctly select the true taxa present in the three mock data sets. Moreover, the permutation PERFect with P-values ordering provides major improvement over existing procedures and other PERFect methods in low signal-to-noise ratio mock data set 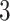 .
.
Table 1.
Comparison of traditional, decontam and PERFect filtering results for: Mock data set 1 (Fettweis and others, 2012) and Mock data set 2 (Brooks and others, 2015)
| Significance | Mock data set 1: 6 true, 99 total | Mock data set 2: 7 true, 46 total | ||||||
|---|---|---|---|---|---|---|---|---|
| Method | level 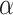
|
Setting (%) | # Taxa preserved | % Filtered | % Contaminants preserved | # Taxa preserved | % Filtered | % Contaminants preserved |
| 1. Simultaneous PERFect abundance ordering |
|
5, 10, 25 |
|
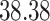
|
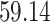
|
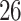
|
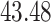
|
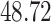
|
| 10, 25, 40 |
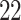
|
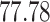
|
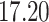
|
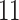
|
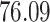
|
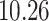
|
||
| 10, 25, 50 |
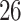
|
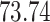
|
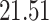
|
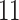
|
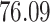
|
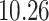
|
||
| 20, 30, 60 |
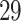
|
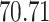
|
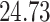
|
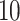
|
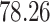
|
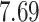
|
||
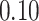
|
5, 10, 25 |
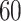
|
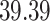
|
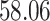
|
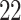
|
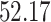
|
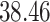
|
|
| 10, 25, 40 |
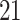
|
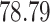
|
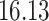
|
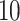
|
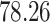
|
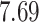
|
||
| 10, 25, 50 |
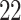
|
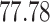
|
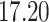
|
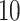
|
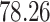
|
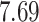
|
||
| 20, 30, 60 |
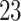
|
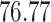
|
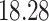
|
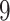
|
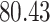
|
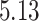
|
||
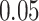
|
5, 10, 25 |
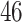
|
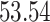
|
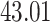
|
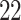
|
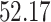
|
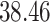
|
|
| 10, 25, 40 |
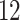
|
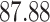
|
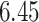
|
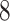
|
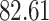
|
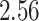
|
||
| 10, 25, 50 |
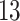
|
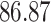
|
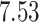
|
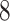
|
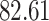
|
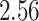
|
||
| 20, 30, 60 |
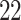
|
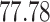
|
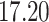
|
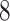
|
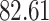
|
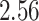
|
||
| 2. Permutation PERFect abundance ordering |
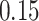
|
5, 10, 25 |
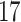
|
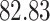
|
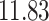
|
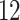
|
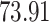
|
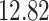
|
| 10, 25, 40 |
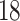
|
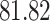
|
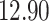
|
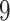
|
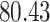
|
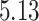
|
||
| 10, 25, 50 |
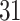
|
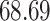
|
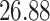
|
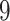
|
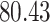
|
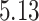
|
||
| 20, 30, 60 |
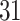
|
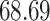
|
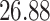
|
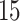
|
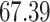
|
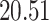
|
||
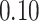
|
5, 10, 25 |
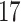
|
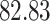
|
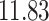
|
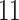
|
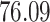
|
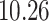
|
|
| 10, 25, 40 |
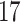
|
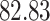
|
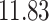
|
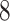
|
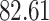
|
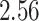
|
||
| 10, 25, 50 |
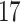
|
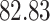
|
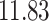
|
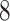
|
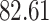
|
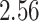
|
||
| 20, 30, 60 |
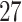
|
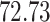
|
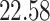
|
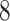
|
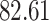
|
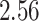
|
||
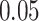
|
5, 10, 25 |
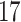
|
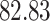
|
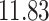
|
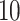
|
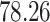
|
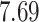
|
|
| 10, 25, 40 |
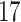
|
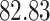
|
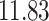
|
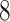
|
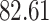
|
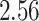
|
||
| 10, 25, 50 |
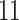
|
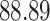
|
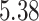
|
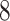
|
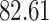
|
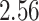
|
||
| 20, 30, 60 |
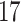
|
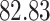
|
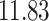
|
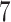
|
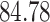
|
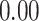
|
||
| 3. Permutation PERFect P-values ordering |
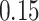
|
5, 10, 25 |
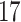
|
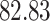
|
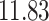
|
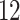
|
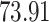
|
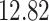
|
| 10, 25, 40 |
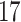
|
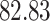
|
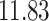
|
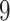
|
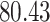
|
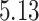
|
||
| 10, 25, 50 |
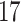
|
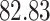
|
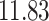
|
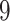
|
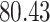
|
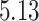
|
||
| 20, 30, 60 |
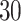
|
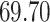
|
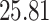
|
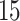
|
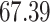
|
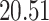
|
||
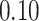
|
5, 10, 25 |
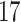
|
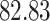
|
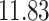
|
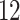
|
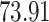
|
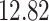
|
|
| 10, 25, 40 |
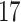
|
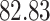
|
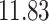
|
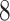
|
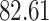
|
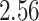
|
||
| 10, 25, 50 |
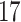
|
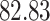
|
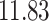
|
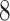
|
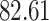
|
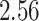
|
||
| 20, 30, 60 |
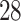
|
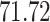
|
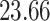
|
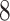
|
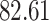
|
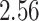
|
||
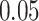
|
5, 10, 25 |
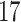
|
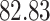
|
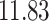
|
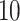
|
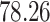
|
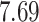
|
|
| 10, 25, 40 |
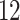
|
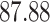
|
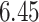
|
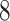
|
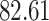
|
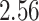
|
||
| 10, 25, 50 |
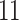
|
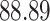
|
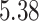
|
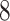
|
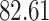
|
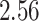
|
||
| 20, 30, 60 |
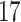
|
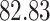
|
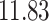
|
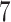
|
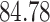
|
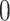
|
||
| 4. Traditional | Rule 1 |
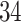
|
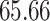
|
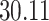
|
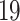
|
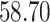
|
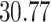
|
|
| Rule 2 |
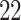
|
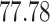
|
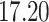
|
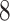
|
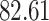
|
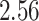
|
||
| 5. decontam prevalence |
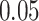
|
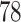
|
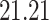
|
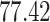
|
NA | NA | NA | |
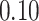
|
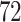
|
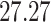
|
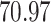
|
NA | NA | NA | ||
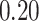
|
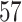
|
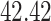
|
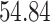
|
NA | NA | NA | ||
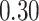
|
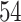
|
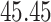
|
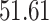
|
NA | NA | NA | ||
NA: Not available.
Table 2.
Comparison of traditional, decontam and PERFect filtering results for: (i) reagent and laboratory contamination data (Salter and others, 2014) and (ii) bacterial diversity in NICU (Knights and others, 2011) data sets
| Significance | Mock data set 3: 3 true, 635 total | NICU bacterial diversity | |||||
|---|---|---|---|---|---|---|---|
| Method | level 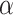
|
Setting % | # Taxa preserved | % Filtered | % Contaminants preserved | # Taxa preserved | % Filtered |
| 1. Simultaneous PERFect abundance ordering |
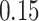
|
5, 10, 25 |
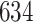
|
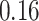
|
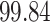
|
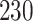
|
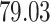
|
| 10, 25, 40 |
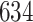
|
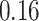
|
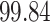
|
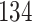
|
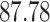
|
||
| 10, 25, 50 |
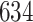
|
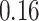
|
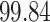
|
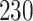
|
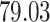
|
||
| 20, 30, 60 |
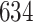
|
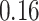
|
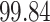
|
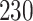
|
|
||
|
5, 10, 25 |
|
|
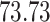
|
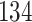
|
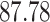
|
|
| 10, 25, 40 |
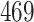
|
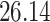
|
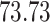
|
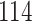
|
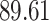
|
||
| 10, 25, 50 |
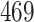
|
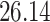
|
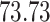
|
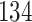
|
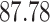
|
||
| 20, 30, 60 |
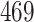
|
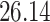
|
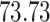
|
|
|
||
|
5, 10, 25 |
|
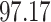
|
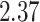
|
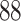
|
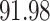
|
|
| 10, 25, 40 |
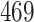
|
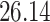
|
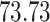
|
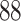
|
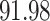
|
||
| 10, 25, 50 |
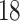
|
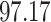
|
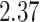
|
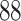
|
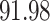
|
||
| 20, 30, 60 |
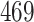
|
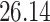
|
|
|
|
||
| 2. Permutation PERFect abundance ordering |
|
5, 10, 25 |
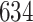
|
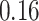
|
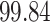
|
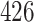
|
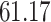
|
| 10, 25, 40 |
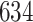
|
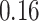
|
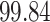
|
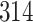
|
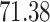
|
||
| 10, 25, 50 |
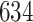
|
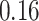
|
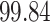
|
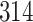
|
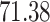
|
||
| 20, 30, 60 |
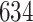
|
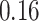
|
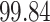
|
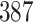
|
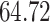
|
||
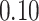
|
5, 10, 25 |
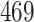
|
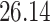
|
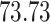
|
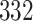
|
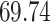
|
|
| 10, 25, 40 |
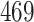
|
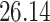
|
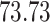
|
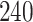
|
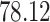
|
||
| 10, 25, 50 |
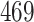
|
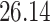
|
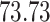
|
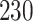
|
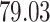
|
||
| 20, 30, 60 |
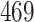
|
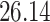
|
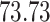
|
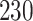
|
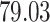
|
||
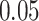
|
5, 10, 25 |
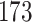
|
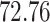
|
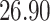
|
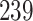
|
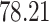
|
|
| 10, 25, 40 |
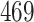
|
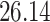
|
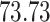
|
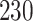
|
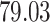
|
||
| 10, 25, 50 |
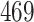
|
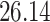
|
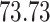
|
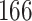
|
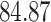
|
||
| 20, 30, 60 |
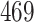
|
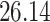
|
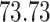
|
|
|
||
| 3. Permutation PERFect p-values ordering |
|
5, 10, 25 |
|
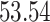
|
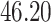
|
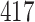
|
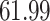
|
| 10, 25, 40 |
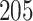
|
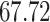
|
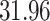
|
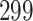
|
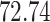
|
||
| 10, 25, 50 |
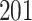
|
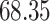
|
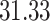
|
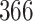
|
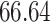
|
||
| 20, 30, 60 |
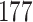
|
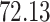
|
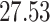
|
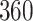
|
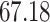
|
||
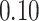
|
5, 10, 25 |
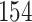
|
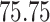
|
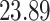
|
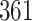
|
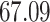
|
|
| 10, 25, 40 |
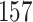
|
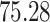
|
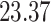
|
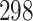
|
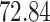
|
||
| 10, 25, 50 |
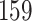
|
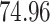
|
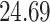
|
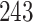
|
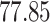
|
||
| 20, 30, 60 |
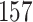
|
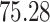
|
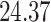
|
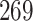
|
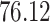
|
||
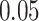
|
5, 10, 25 |
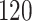
|
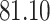
|
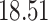
|
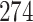
|
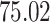
|
|
| 10, 25, 40 |
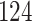
|
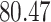
|
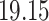
|
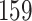
|
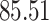
|
||
| 10, 25, 50 |
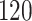
|
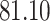
|
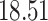
|
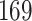
|
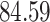
|
||
| 20, 30, 60 |
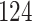
|
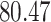
|
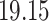
|
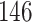
|
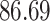
|
||
| 4. Traditional | Rule 1 |
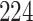
|
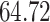
|
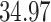
|
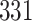
|
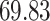
|
|
| Rule 2 |
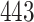
|
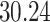
|
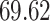
|
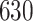
|
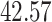
|
||
| 6. decontam frequency pooled |
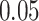
|
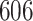
|
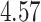
|
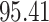
|
NA | NA | |
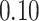
|
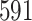
|
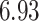
|
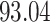
|
NA | NA | ||
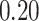
|
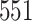
|
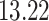
|
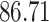
|
NA | NA | ||
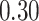
|
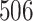
|
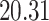
|
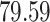
|
NA | NA | ||
| 7. decontam frequency batched |
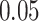
|
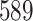
|
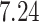
|
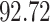
|
NA | NA | |
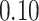
|
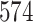
|
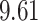
|
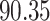
|
NA | NA | ||
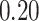
|
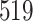
|
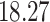
|
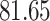
|
NA | NA | ||
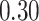
|
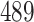
|
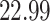
|
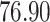
|
NA | NA | ||
NA: Not available.
We further validate PERFect methods on Knights and others (2011) data set, which contains microbial samples from NICU surface and equipment (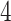 buttons,
buttons, 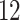 handles,
handles, 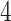 keyboards,
keyboards, 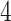 counters,
counters, 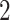 screens,
screens, 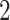 incubators, and
incubators, and 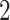 plastics), and could be used indirectly to validate the PERFect approach to filtration. Indeed, one would expect for the oral- and gut-related microbes to not truly be present in the samples but for some of the skin-related taxa to be preserved. The reason is that these NICU samples come from samples potentially touched by lab employees. Therefore, for these data, we used the known contaminants reference data sets available at sourcetracker github webpage (Knights and others, 2011) to match taxa found in skin, soil, oral, and gut environments. We focus here only on skin, soil, oral, gut, and unknown taxa and do not consider taxa mapped to more than two environments (e.g. skin and soil). We apply PERFect with a significance level
plastics), and could be used indirectly to validate the PERFect approach to filtration. Indeed, one would expect for the oral- and gut-related microbes to not truly be present in the samples but for some of the skin-related taxa to be preserved. The reason is that these NICU samples come from samples potentially touched by lab employees. Therefore, for these data, we used the known contaminants reference data sets available at sourcetracker github webpage (Knights and others, 2011) to match taxa found in skin, soil, oral, and gut environments. We focus here only on skin, soil, oral, gut, and unknown taxa and do not consider taxa mapped to more than two environments (e.g. skin and soil). We apply PERFect with a significance level 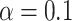 and present results in Table 3. Since these NICU samples come from the equipment touched by the lab employees, we expect that most signal taxa originate from the skin and possibly soil environments. Table 3 reveals that the data set filtered by PERFect using either method, preserves no gut (except for
and present results in Table 3. Since these NICU samples come from the equipment touched by the lab employees, we expect that most signal taxa originate from the skin and possibly soil environments. Table 3 reveals that the data set filtered by PERFect using either method, preserves no gut (except for 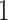 gut taxon preserved by permutation method with abundance ordering) and no oral taxa, which are almost surely not in the data. Moreover, a large proportion of preserved taxa are associated with the known skin environment: with
gut taxon preserved by permutation method with abundance ordering) and no oral taxa, which are almost surely not in the data. Moreover, a large proportion of preserved taxa are associated with the known skin environment: with 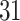 ,
, 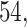 and
and 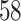 out of
out of 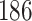 preserved by simultaneous abundance ordering, permutation abundance ordering and ordering by the P-value, respectively. We conclude that PERFect performed as expected by filtering out the taxa that almost surely could not have been in the samples (oral and gut) and preserving some of the taxa that have a reasonable likelihood of being in the sample (skin, soil, and unknown). Interestingly, PERFect also removes some of these taxa indicating that some of the skin- and soil-related taxa may truly not be in the sample. Many preserved taxa did not match any of the
preserved by simultaneous abundance ordering, permutation abundance ordering and ordering by the P-value, respectively. We conclude that PERFect performed as expected by filtering out the taxa that almost surely could not have been in the samples (oral and gut) and preserving some of the taxa that have a reasonable likelihood of being in the sample (skin, soil, and unknown). Interestingly, PERFect also removes some of these taxa indicating that some of the skin- and soil-related taxa may truly not be in the sample. Many preserved taxa did not match any of the  environments and were labeled “unknown” in Table 3. Results indicate that PERFect also removes a large number of taxa with unknown environmental provenance. We consider that this is another very encouraging characteristic of PERFect. We would like to note that decontam package could not be applied to these data because these data release does not contain the concentration of amplified DNA in each sample prior to sequencing or negative controls samples. A major advantage of PERFect over decontam is that it does not require this information. We conclude that in this data PERFect provides reasonable results using the available information.
environments and were labeled “unknown” in Table 3. Results indicate that PERFect also removes a large number of taxa with unknown environmental provenance. We consider that this is another very encouraging characteristic of PERFect. We would like to note that decontam package could not be applied to these data because these data release does not contain the concentration of amplified DNA in each sample prior to sequencing or negative controls samples. A major advantage of PERFect over decontam is that it does not require this information. We conclude that in this data PERFect provides reasonable results using the available information.
Table 3.
Percent of taxa preserved in each environment by PERFect results for the bacterial diversity in NICU data (Knights and others 2011) and taxa preserved inRavel and others (2011)vaginal microbiome data. To determine taxa to retain in the data set, we use 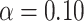 significance level.
significance level.
| NICU data | Vaginal microbiome data | |||||||
|---|---|---|---|---|---|---|---|---|
| Total number of taxa | skin 
|
soil 
|
oral 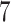
|
gut 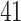
|
unknown 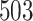
|
# Taxa preserved | % Filtered | |
| PERFect | ||||||||
| Simultaneous abundance |
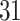
|
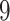
|
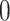
|
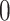
|
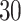
|
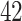
|
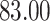
|
|
| Permutation abundance |
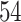
|
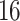
|
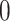
|
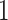
|
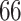
|
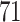
|
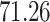
|
|
| Permutation P-values |
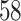
|
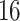
|
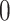
|
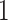
|
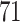
|
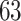
|
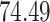
|
|
4. Vaginal microbiome data filtering
We apply traditional and PERFect filtering methods to a vaginal microbiome study of a cohort of 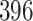 reproductive age women previously published in Ravel and others (2011), where data and details on data collection and pre-processing are also available. The goal of the study was to understand the role and ultimate function of vaginal microbiota in reducing risk of infectious diseases and to identify factors leading to disease susceptibility. Microbiome data were obtained by pyrosequencing of barcoded 16S rRNA genes; in this data set
reproductive age women previously published in Ravel and others (2011), where data and details on data collection and pre-processing are also available. The goal of the study was to understand the role and ultimate function of vaginal microbiota in reducing risk of infectious diseases and to identify factors leading to disease susceptibility. Microbiome data were obtained by pyrosequencing of barcoded 16S rRNA genes; in this data set 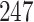 taxa were identified.
taxa were identified.
We applied PERFect simultaneous with abundance ordering and permutation with abundance and P-values ordering to the two traditional filtering methods outlined in Section 3 using the Ravel and others (2011) data set; results are summarized in Table 3 and comparison with traditional filtering methods is available in Table 5 in the supplementary material available at Biostatistics online. Table 5 in the supplementary material available at Biostatistics online reflects that traditional filtering approaches preserve a large proportion of taxa (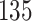 and
and 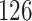 , respectively), while the PERFect rules are more aggressive in eliminating taxa (preserving
, respectively), while the PERFect rules are more aggressive in eliminating taxa (preserving 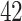 ,
, 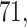 and
and 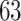 taxa, respectively). The taxa preserved by PERFect form a subset of taxa preserved by the two traditional filtering rules, and the five methods agree on
taxa, respectively). The taxa preserved by PERFect form a subset of taxa preserved by the two traditional filtering rules, and the five methods agree on 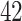 taxa retained in the filtered data set. These taxa are listed in Table 4 of supplementary material available at Biostatistics online. Because we do not have a gold standard, it is impossible to say that having fewer preserved taxa is better. However, PERFect provides an objective way of saying that given the preserved taxa the other taxa do not add much to the overall covariance. Thus, objectively, adding any of the taxa that traditional methods retain and PERFect rejects does not add much or anything to the overall observed variability.
taxa retained in the filtered data set. These taxa are listed in Table 4 of supplementary material available at Biostatistics online. Because we do not have a gold standard, it is impossible to say that having fewer preserved taxa is better. However, PERFect provides an objective way of saying that given the preserved taxa the other taxa do not add much to the overall covariance. Thus, objectively, adding any of the taxa that traditional methods retain and PERFect rejects does not add much or anything to the overall observed variability.
These differences in results are likely due to in-built differences between the traditional and PERFect filtering approaches. First, while traditional filtering approaches use rule of thumb criteria, which may be different across data sets, PERFect assigns a data driven significance value to each subset 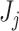 of
of 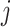 filtered taxa. Second, traditional filtering rules evaluate a taxon’s importance in isolation, while PERFect evaluates a significance value for the set of taxa, thus considering each taxon in connection with other taxa. While treating each taxon in isolation may be valid in the mock samples, in a more realistic biological environment, bacteria may, in fact, compete for finite resources and the presence of one taxon may affect the presence of other taxa. Furthermore, PERFect with the taxa ordering according to (2.3) can be viewed as an extension of the traditional Rule 1 filtering, where instead of choosing a filtering threshold arbitrarily we: (i) take into account taxa covariance; and (ii) quantify the chance that the set of taxa
filtered taxa. Second, traditional filtering rules evaluate a taxon’s importance in isolation, while PERFect evaluates a significance value for the set of taxa, thus considering each taxon in connection with other taxa. While treating each taxon in isolation may be valid in the mock samples, in a more realistic biological environment, bacteria may, in fact, compete for finite resources and the presence of one taxon may affect the presence of other taxa. Furthermore, PERFect with the taxa ordering according to (2.3) can be viewed as an extension of the traditional Rule 1 filtering, where instead of choosing a filtering threshold arbitrarily we: (i) take into account taxa covariance; and (ii) quantify the chance that the set of taxa 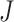 is observed due to noise. Third, since the smaller values of filtering loss criteria (2.2) are consistent with a smaller loss from the total covariance of the data, taxa retained by PERFect provide a dimension reduction approximation of taxa covariance. This reduction is crucial for further analysis, such as data visualization via principal components analysis, and inference approaches that require covariance matrix estimation.
is observed due to noise. Third, since the smaller values of filtering loss criteria (2.2) are consistent with a smaller loss from the total covariance of the data, taxa retained by PERFect provide a dimension reduction approximation of taxa covariance. This reduction is crucial for further analysis, such as data visualization via principal components analysis, and inference approaches that require covariance matrix estimation.
5. Discussion
We introduced two PERFect approaches for microbiome filtering and compared them the to the traditional filtering procedures and the recently developed R package decontam for identifying contaminants in microbiome sequencing data. For mock data sets with very strong signal traditional and PERFect filtering effectively eliminate contaminants, while PERFect permutation with PERFect simultaneous p-values ordering provides a more effective reduction when the signal-to-noise ratio is low. In NICU surfaces and equipment microbial samples, our method filters out the taxa that almost surely could not have been in the samples (oral and gut) and preserving some of the taxa that have a reasonable likelihood of being in the sample (skin, soil, and unknown). Finally, our results are in agreement with results published indicating that taxa that are important were not filtered out in the vaginal microbiome data set.
The present work is the first time that a taxa reduction method is motivated by statistical hypothesis testing. The combination of PERFect taxa P-values and filtering loss information provides a useful insight into taxa co-relationship and allows identification of related groups of taxa. The proposed method provides an intuitive and biologically meaningful classification of taxa importance based on their contribution to the total covariance. This information can be used in subsequent explanatory and inferential analysis.
One limitation of the proposed approach is that it is skewed toward retaining more dominant features. That is if a first significant taxon is observed in 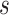 samples, then any taxon observed in less than
samples, then any taxon observed in less than 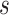 samples will also be removed by the current procedure under abundance ordering. Moreover, it might occur that a persistent contaminant feature appears in a large number of samples, has a high contribution towards covariance and is not removed from the data set. However, this is a general limitation of any filtering approach that does not take into account additional information about negative controls, or feature DNA concentrations in the samples. Another limitation is that the Skew Lognormal distribution was chosen heuristically and while its use is justified empirically on mock data sets, the finite sample, or asymptotic behavior of the test statistic are not supported theoretically. Thus, the theoretical underpinnings of this work are not yet understood.
samples will also be removed by the current procedure under abundance ordering. Moreover, it might occur that a persistent contaminant feature appears in a large number of samples, has a high contribution towards covariance and is not removed from the data set. However, this is a general limitation of any filtering approach that does not take into account additional information about negative controls, or feature DNA concentrations in the samples. Another limitation is that the Skew Lognormal distribution was chosen heuristically and while its use is justified empirically on mock data sets, the finite sample, or asymptotic behavior of the test statistic are not supported theoretically. Thus, the theoretical underpinnings of this work are not yet understood.
Finally, we would like to emphasize that PERFect can be viewed as an extension of traditional filtering approaches that provides more insight into the data and achieves better dimension reduction for correlated taxa. While both traditional and PERFect filtering methods were effective in identifying true species in mock data, permutation PERFect consistently outperformed alternative filtering rules for the mock data sets 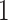 and
and 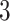 . The accompanying R software implementation and associated visualization tools makes the method easy-to-use, interpret results, and gain additional insight into taxa co-relationships. However, the vaginal microbiome data set was used to illustrate PERFect performance on real data, PERFect is designed for general sparse data, including gut and other body sites microbiome, which have similar properties and satisfy the PERFect assumptions.
. The accompanying R software implementation and associated visualization tools makes the method easy-to-use, interpret results, and gain additional insight into taxa co-relationships. However, the vaginal microbiome data set was used to illustrate PERFect performance on real data, PERFect is designed for general sparse data, including gut and other body sites microbiome, which have similar properties and satisfy the PERFect assumptions.
6. Software: PERFect
We have developed the R software package PERFect that incorporates the methods introduced in Algorithms 1 and 2 of this manuscript. The package can be found at https://github.com/katiasmirn/PERFect. PERFect takes an OTU table 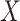 , which can be either counts, proportions, or presence-absence data, as an input and produces a filtered OTU table
, which can be either counts, proportions, or presence-absence data, as an input and produces a filtered OTU table 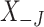 at a user-specified significance level
at a user-specified significance level 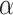 . The software has an option to center the columns of
. The software has an option to center the columns of 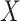 , which aids interpretability of filtering loss criterion as taxa covariance. Users can request any taxa ordering discussed in this paper, or specify an alternative ordering. We discuss the effect of alternative orderings on the vaginal microbiome data (Ravel and others, 2011) filtering in Section 2 of supplementary material available at Biostatistics online. However, we recommend fitting a Skew-Normal distribution to the log differences in filtering loss to estimate the null distribution, other distributions such as Normal may be used. We provide the list of available distributions in the software package description. PERFect permutation sampling distribution for each taxon is generated as part of the output and can be used with default, optional, or user specified taxa ordering and a collection of distributions to capture the null. Simultaneous and permutation PERFect provide
, which aids interpretability of filtering loss criterion as taxa covariance. Users can request any taxa ordering discussed in this paper, or specify an alternative ordering. We discuss the effect of alternative orderings on the vaginal microbiome data (Ravel and others, 2011) filtering in Section 2 of supplementary material available at Biostatistics online. However, we recommend fitting a Skew-Normal distribution to the log differences in filtering loss to estimate the null distribution, other distributions such as Normal may be used. We provide the list of available distributions in the software package description. PERFect permutation sampling distribution for each taxon is generated as part of the output and can be used with default, optional, or user specified taxa ordering and a collection of distributions to capture the null. Simultaneous and permutation PERFect provide 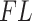 and
and 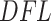 values, distribution fit details including histograms illustrated in Figure 2, P-values for the set of taxa and P-values plots illustrated in Section 2 of supplementary material available at Biostatistics online. By default, software uses smoothed P-values obtained by averaging
values, distribution fit details including histograms illustrated in Figure 2, P-values for the set of taxa and P-values plots illustrated in Section 2 of supplementary material available at Biostatistics online. By default, software uses smoothed P-values obtained by averaging 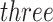 subsequent P-values obtained from the reference distribution, with an option to either use no averaging or average a different number of subsequent P-values. However, the simultaneous PERFect is faster than permutation PERFect, we recommend to verify the results using permutation PERFect. Due to the randomness in the permutation component of PERFect, results might differ across runs; in such situations a larger number of permutations should be used. Permutation PERFect can finish
subsequent P-values obtained from the reference distribution, with an option to either use no averaging or average a different number of subsequent P-values. However, the simultaneous PERFect is faster than permutation PERFect, we recommend to verify the results using permutation PERFect. Due to the randomness in the permutation component of PERFect, results might differ across runs; in such situations a larger number of permutations should be used. Permutation PERFect can finish 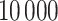 permutations within
permutations within 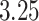 min on an Apple MacBook Pro for
min on an Apple MacBook Pro for 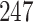 taxa.
taxa.
Supplementary Material
Acknowledgments
The authors are grateful for crucial suggestions and feedback on improving previous drafts provided by Professor Ciprian M. Crainiceanu. We thank Paul Brooks for making available access to the VCU data http://vmc.vcu.edu/ and Molecular Computation Core at the U. of Montana for its facilities. We thank two anonymous reviewers for thorough reading of this manuscript and insightful suggestions that dramatically improved the paper. Conflict of Interest: None declared.
Funding
This work was supported by National Science Foundation [DMS-1100615 to E.S. and S.H.]; and by National Institutes of Health [1U54GM104944-01A1 to S.H.] while the authors were at U. of Wyoming. The data collection at Virginia Commonwealth University was supported by National Institutes of Health [NICHD 8U54HD080784; NIAID UH3AI08326].
References
- Azzalini A. (2005). The skew-normal distribution and related multivariate families. Scandinavian Journal of Statistics 32, 159–188. [Google Scholar]
- Brooks, J. P., Edwards, D. J., Harwich, M. D., Rivera, M. C., Fettweis, J. M., Serrano, M. G., Reris, R. A., Sheth, N. U., Huang, B., Girerd, P.. and others. (2015). The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiology 15, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacho, A., Smirnova, E., Huzurbazar, S. and Cui, X. (2016). A comparison of base-calling algorithms for illumina sequencing technology. Briefings in Bioinformatics 7, 786–795. [DOI] [PubMed] [Google Scholar]
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Pena, A. G., Goodrich, J. K., Gordon, J. I.. and others (2010). Qiime allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I. and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, N. M., Proctor, D., Holmes, S. P., Relman, D. A. and Callahan, B. J. (2017). Simple statistical identification and ritoval of contaminant sequences in marker-gene and metagenomics data. bioRxiv preprint, 221499 10.1101/221499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, J. and Lv, J. (2008). Sure independence screening for ultra-high dimensional feature space. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 70, 849–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettweis, J. M., Serrano, M. G., Sheth, N. U., Mayer, C. M., Glascock, A. L., Brooks, J. P., Jefferson, K. K.; Vaginal Microbiome Consortium, and Buck, G. A. (2012). Species-level classification of the vaginal microbiome. BMC Genomics 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentlitan, R., Carey, V., Huber, W. and Hahne, F. (2016). genefilter: Methods for Filtering Genes from Microarray Experiments. R package version 1.54.2, Bioconductor.https://bioconductor.org/packages/release/bioc/html/genefilter.html. [Google Scholar]
- Greenblum, S., Turnbaugh, P. J. and Borenstein, E. (2012). Metagenomic systits biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Acadity of Sciences USA 109, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. (2012). A framework for human microbiome research. Nature 486, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., Bushman, F. D., Knight, R. and Kelley, S. T. (2011). Bayesian community-wide culture-independent microbial source tracking. Nature Methods 8, 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber, C. L., Hamady, M., Knight, R. and Fierer, N. (2009). Pyrosequencing-based assessment of soil ph as a predictor of soil bacterial community structure at the continental scale. Applied and Environmental Microbiology 75, 5111–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Homer, N. (2010). A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics 11, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, B., Forney, L. J. and Ravel, J. (2012). The vaginal microbiome: rethinking health and diseases. Annual Review of Microbiology 66, 371–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard, C. L., Elson, C. O., Hatton, R. D. and Weaver, C. T. (2012). Reciprocal interactions of the intestinal microbiota and immune systit. Nature 489, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milici, M., Tomasch, J., Wos-Oxley, M. L., Wang, H., Jáuregui, R., Camarinha-Silva, A., Deng, Z.-L., Plumeier, I., Giebel, H.-A., Wurst, M.. and others. (2016). Low diversity of planktonic bacteria in the tropical ocean. Scientific Reports 6, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S. K., McCulle, S. L., Karlebach, S., Gorle, R., Russell, J., Tacket, C. O.. and others. (2011). Vaginal microbiome of reproductive-age women. Proceedings of the National Acadity of Sciences USA 108, 4680–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, R., Dey, S. K. and Fisher, S. J. (2014). Preterm labor: one syndrome, many causes. Science 345, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., Turner, P., Parkhill, J., Loman, N. J. and Walker, A. W. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology 12, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulberg, E., Fravel, D., Proctor, L. M., Murray, D. M., LoTitpio, J., Chrisey, L., Garland, J., Goodwin, K., Graber, J., Harris, M. C.. and others. (2016). An assessment of US microbiome research. Nature Microbiology 1, 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.