

Abstract
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative condition affecting approximately 185,000 people in the UK. No drug has been proven to slow disease progression. Epidemiological and pre-clinical data support simvastatin, a widely used cholesterol-lowering drug with a well-established safety profile, having neuroprotective properties. The aim of this study (Simvastatin as a neuroprotective treatment for PD (PD STAT)) is to determine whether simvastatin has the potential to slow PD progression. The study is part of the International Linked Clinical Trials initiative coordinated by The Cure Parkinson’s Trust. This paper describes the protocol for the PD STAT study.
Methods and analysis
PD STAT is a double-blind, randomised, placebo-controlled, multi-centre, parallel group, futility trial in patients with PD of mild–moderate severity. 235 participants have been recruited and randomly allocated in a 1:1 ratio to receive either oral simvastatin or matched placebo. Treatment involves a 1-month low-dose phase (40 mg daily), followed by a 23-month high-dose phase (80 mg daily) and ends with a 2-month washout period. Participants are reviewed at clinic visits at 1 month, 6, 12, 18, 24 and 26 months post-baseline, with interim telephone follow-up to monitor for adverse events.
The primary outcome is the change in the Movement Disorder Society Unified Parkinson’s Disease Rating Scale part III motor subscale score in the practically defined OFF medication state (OFF state) between baseline and 24 months. Primary analysis will be on a modified intention to treat basis and will include only those participants who progress to the high-dose phase of the study.
Ethics and dissemination
The protocol has been approved by the North East–Newcastle and North Tyneside 2 Research Ethics Committee. The results will be disseminated via research articles in peer-reviewed journals and presentations at local, national and international scientific meetings, as well as disseminated via patient groups, websites and networks. A summary of the study findings will be posted to participants at the end of the study.
Trial Registration
ISRCTN16108482 (prospectively registered); EudraCT 2015-000148-40; ClinicalTrials.gov NCT02787590; Pre-results.
Keywords: Parkinson’s disease, statin, randomised controlled futility study, neuroprotective effect, MDS-UPDRS
Strengths and limitations of this study.
Independent, blinded outcome assessors not involved in participant treatment, reduces likelihood of bias in results.
OFF state Movement Disorder Society Unified Parkinson’s Disease Rating Scale assessments, the current gold standard for evaluating disease progression.
Standardised training for raters reduces inter-rater variability.
Embedded substudy to evaluate the participant’s trial experience and inform future trial design.
Long duration of study increases risk of drop-out/loss to follow-up.
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative condition affecting approximately one person in every 350 in the UK.1 Furthermore, with population growth and an increasingly ageing population, the estimated prevalence and incidence of PD in the UK are increasing. There are currently no known treatments that slow the rate of neuronal loss or clinical progression in PD. All currently licensed therapies are symptomatic.
Epidemiological and pre-clinical data support a possible neuroprotective role for statins in PD, with statin use being associated with lower PD incidence.2 3 Simvastatin has been shown in various toxin and genetic cell culture and rodent PD models to influence several pathways thought to be of relevance in PD etiopathogenesis, including inflammation and microglial activation, oxidative stress and α-synuclein aggregation.4 5 A beneficial effect of simvastatin on dopamine neuron survival and motor function has been observed in acute6 and chronic4 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse models. Additionally, statins may have symptomatic effects on dyskinesia and depression in PD.4 Interestingly, simvastatin has been shown to reduce the rate of brain atrophy in secondary progressive multiple sclerosis,7 and it is likely that some of the mechanisms underlying neuronal death are similar in this and other neurodegenerative diseases. This finding therefore lends support to investigating the potential long-term disease-modifying effects of simvastatin in PD. In 2012, the International PD Linked Clinical Trials initiative (LCT) was established by The Cure Parkinson’s Trust to identify potential new neuroprotective treatments for PD by repurposing drugs that have been approved, or are in current clinical development, to treat other conditions.8 On the basis that simvastatin has a well-established safety profile,9 10 it was one of the first drugs selected by the LCT committee to be tested in a clinical trial in patients with PD to determine its disease-modifying potential.
Clinical trials of potential neuroprotective agents in PD are difficult to design, given the variability in disease phenotype and rate of progression, as well as the potential confounding factor of a symptomatic response. In addition, there is no reliable biomarker for assessing disease progression.11 Initially used in oncology trials, a trial with a futility design allows for a relatively short study duration and smaller sample size in comparison with the typical phase II/III trial design.12 The futility design typically has a single treatment arm and tests whether a new treatment exceeds a pre-defined futility threshold.12 In neurological diseases such as PD, the lack of a concurrent control group has led to criticism of subsequent findings from futility trials13 but it is possible to test for futility using a randomised parallel group design. There is, therefore, increasing interest in the use of futility trials to provide an efficient method for early phase studies to ascertain whether there is sufficient evidence to justify conducting larger, longer and more expensive phase III trials. The PD STAT trial is a phase II futility study, which aims to determine whether simvastatin has potential to reduce the rate of neurodegenerative decline in patients with PD.
Aims and objectives
The aim of the study is to determine whether the cholesterol-lowering drug, simvastatin, has potential as a neuroprotective therapy in PD. The primary objective of the PD STAT study is to determine whether simvastatin is clearly ineffective (futile) in preventing the clinical decline of PD as measured by the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) motor score in patients in the OFF state.14 Secondary objectives are to confirm the safety and tolerability of simvastatin in patients with PD, to distinguish symptomatic effects of simvastatin from disease-modifying effects, and to evaluate the impact of simvastatin on activities of daily living, timed motor tests, cognitive ability, mood, behaviour, non-motor symptoms and quality of life in patients with moderate PD using standard validated tools of assessment. The results of this study will help to determine the merits of conducting a larger, definitive phase III study to assess the neuroprotective and/or disease-modifying effectiveness of simvastatin.
METHODS AND ANALYSIS
This protocol is reported in accordance with the Standard Protocol Items: Recommendations for Interventional Trials guidance for protocols of clinical trials.15
Trial design and setting
This is a double-blind, randomised, placebo-controlled, multi-centre, parallel group trial in patients with PD of moderate severity. There are three embedded substudies. Participants are individually randomised in a 1:1 ratio to receive either oral simvastatin or matched placebo for 24 months. A 1-month low-dose phase (40 mg daily) is followed by a 23-month high-dose phase (80 mg daily) and treatment ends with a 2-month washout period. Recruitment took place between March 2016 and March 2018, with a target of at least 198 participants progressing successfully to the high-dose phase of the study; 26-month follow-up of all participants is expected to be completed by May 2020. The trial design, including scheduled follow-up assessments, is summarised in figure 1.
Figure 1.
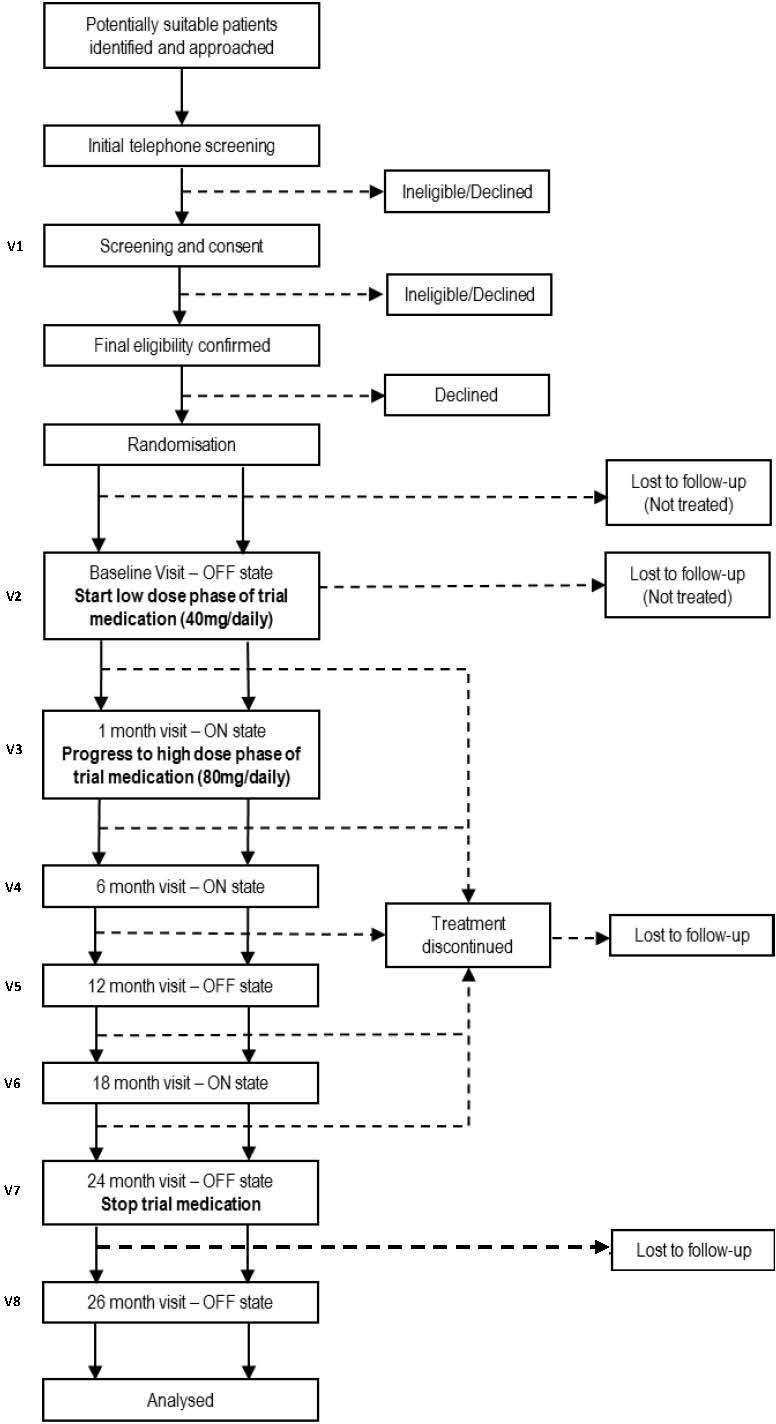
Scheduled follow-up assessments.
A 12-month treatment period was originally considered but it was felt that this might not be long enough to demonstrate any disease-modifying effect; hence, participants are each treated for 24 months. Should this futility study have positive results, the additional collection of 12-month outcome data, as well as outcome data collected at the primary endpoint of 24 months, will enable assessment of any potential benefit at 12 months to assist with design of future studies.
The trial is being conducted in 23 National Health Service (NHS) Trusts across England. A list of recruiting sites is provided in online supplementary appendix A. A local principal investigator (PI), supported by at least two other staff members (eg, research nurse or PD specialist nurse), leads the conduct of the study at each participating site. Participants are followed up on an outpatient basis at 1 month, 6, 12, 18, 24, and 26 months post-baseline (treatment start), with regular interim telephone contact.
bmjopen-2019-029740supp001.pdf (43.2KB, pdf)
Study population
The study population includes patients aged between 40 and 90 years with a diagnosis of idiopathic PD, a modified Hoehn and Yahr stage of ≤3.0 in the ON medication state, and who are on dopaminergic treatment with experience of wearing-off phenomenon (as defined by the nine-item wearing-off questionnaire (WOQ)16). Patients are excluded if they have a diagnosis (or suspicion) of another cause for their parkinsonism, or have any prior use, current use, intolerance of or requirement for, statins. A full list of patient inclusion and exclusion criteria is listed in box 1.
Box 1. Inclusion and exclusion criteria.
Inclusion criteria
Diagnosis of idiopathic PD
Modified Hoehn and Yahr stage ≤3.0 in the ON medication state
Age 40–90 years
On dopaminergic treatment with wearing-off phenomenon
Able to comply with study protocol and willing to attend necessary study visits
Exclusion criteria
Diagnosis or suspicion of other cause for parkinsonism
Known abnormality on CT or MRI brain imaging considered to be causing symptoms or signs of neurological dysfunction, or considered likely to compromise compliance with study protocol
Concurrent dementia defined by a MoCA score <21
Concurrent severe depression defined by MADRS score >31
Prior intracerebral surgical intervention for PD including deep brain stimulation, lesional surgery, growth factor administration, gene therapy or cell transplantation
Already actively participating in a research study that might conflict with this trial
Prior or current use of statins as a lipid-lowering therapy
Intolerance of statins
Untreated hypothyroidism
End-stage renal disease (creatinine clearance <30 mL/min) or history of severe cardiac disease (angina, myocardial infarction or cardiac surgery in preceding 2 years)
eGFR <30 mL/min
History of alcoholism or liver impairment
Creatine kinase >1.1 × ULN
AST or ALT >1.1 × ULN
Females who are pregnant or breast feeding or of child-bearing potential and unwilling to use appropriate contraception methods while on trial treatment
Currently taking any medication contraindicated with simvastatin use
Any requirement for statin use
Regular participation in endurance or high-impact sports
-
Unable to abstain from consumption of grapefruit-based products
ALT, alanine transaminase; AST, aspartate transaminase; eGFR, estimated glomerular filtration rate; MADRS, Montgomery and Asberg Depression Rating Scale; MoCA, Montreal Cognitive Assessment; PD, Parkinson’s disease; ULN, upper limit of normal.
Outcome measures
The primary outcome is the change in MDS-UPDRS part III motor subscale score in the OFF state between baseline and 24 months.14 Secondary outcomes at 12, 24 and 26 months include the following:
MDS-UPDRS total score in the practically defined ON state
MDS-UPDRS part II subscale score in the practically defined ON state
Timed motor tests—finger tapping and timed walk test (10 Metre Walk Test) in the OFF state, electromagnetic sensor (EMS) assessment in the OFF and ON state
Montgomery and Asberg Depression Rating Scale (MADRS)
The Addenbrooke’s Cognitive Assessment-III
Non-Motor Symptom assessment scale
Parkinson’s Disease Questionnaire-39
Changes in PD medication as measured by levodopa-equivalent dose
Cholesterol levels (total, high-density lipoprotein (HDL), total/HDL ratio)
King’s PD pain scale
EuroQoL 5D-5L health status questionnaire
Safety and tolerability of trial medication by adverse events (AEs) review
Incidence of diabetes mellitus at 24 months, using a glycated haemoglobin (HbA1c) level of 6.5% (48 mmol/mol) as diagnostic of diabetes mellitus17
Participant identification and initial telephone screening (T1)
Potentially suitable patients were identified via clinical lists, research registers and publicity/word of mouth. Patients who expressed interest in the study were sent a study invitation letter and participant information sheet. A member of the local research team subsequently telephoned the patient to discuss the study further, ascertain further interest and establish potential eligibility for the study.
Consent and screening visit (V1)
The study schedule is depicted in table 1. Interested patients deemed to be potentially eligible for the study were invited to attend a local screening appointment. After patients had any questions answered, those who were willing, and appeared to meet the study eligibility criteria, were asked to provide written informed consent before proceeding with full screening for the study. The written informed consent process was undertaken by the PI or by an appropriately trained member of the research team as delegated by the PI, depending on local arrangements. Initial screening included recording of demographic details, medical history and concomitant medication. Patients completed the WOQ-9, MoCA and MADRS with the PI (or authorised delegate) and underwent a physical examination by the PI (or authorised delegate), including assessment of modified Hoehn and Yahr stage. Blood samples for creatinine kinase (CK), aspartate transaminase (AST) or alanine transaminase (ALT), estimated glomerular filtration rate, cholesterol (HDL, total), urea, electrolytes (sodium, potassium and creatinine), thyroid stimulating hormone and glycated HbA1c were taken and analysed locally.
Table 1.
Study schedule
| Study period | ||||||||||||||||||
| Screen | Baseline | Post-allocation | Washout | |||||||||||||||
| Contacts | T1 | V1 | V2 | T2 | V3 | T3 | T4 | V4 | T5 | T6 | V5 | T7 | T8 | V6 | T9 | T10 | V7 | V8 |
| T – Telephone V – Clinic Visit |
-4 weeks | Screening | Baseline | 2 weeks | 1 month | 2 months | 4 months | 6 months | 8 months | 10 months | 12 months | 14 months | 16 months | 18 months | 20 months | 22 months | 24 months | 26 months |
| Enrolment | ||||||||||||||||||
| Eligibility screen | X | X | ||||||||||||||||
| Informed consent | X | |||||||||||||||||
| Demographics | X | |||||||||||||||||
| Randomisation |

|
|||||||||||||||||
| Treatment | ||||||||||||||||||
| Prescription | X | X | X | X | X | |||||||||||||
| Simvastatin/placebo (40 mg/day) |

|
|||||||||||||||||
| Simvastatin/placebo (80 mg/day) |

|
|||||||||||||||||
| Assessments (OFF) | ||||||||||||||||||
| MDS-UPDRS Part III | X | X | X | X | ||||||||||||||
| 10 Metre Walk Test | X | X | X | X | ||||||||||||||
| Brain (Tap) Test | X | X | X | X | ||||||||||||||
| Assessments (ON) | ||||||||||||||||||
| Complete MDS-UPDRS | X | X | X | X | ||||||||||||||
| ACE-III | X | X | X | X | ||||||||||||||
| PDQ-39 | X | X | X | X | ||||||||||||||
| KPPS | X | X | X | X | ||||||||||||||
| EQ-5D-5L | X | X | X | X | ||||||||||||||
| LED | X | X | X | X | ||||||||||||||
| NMSS | X | X | X | X | ||||||||||||||
| MADRS | X | X | X | X | ||||||||||||||
| Other | ||||||||||||||||||
| Cholesterol (HDL, total) | X | X | X | X | ||||||||||||||
| HbA1c | X | X | ||||||||||||||||
| Adverse event review |

|
|||||||||||||||||
| Concomitant medication review |

|
|||||||||||||||||
| Qualitative substudy* |

|
|||||||||||||||||
| Genetics substudy* | X | |||||||||||||||||
| EMS substudy* | X | X | ||||||||||||||||
*See embedded substudies section.
ACE-III, Addenbrooke’s Cognitive Assessment-III; EMS, electromagnetic sensor; EQ-5D-5L, EuroQoL 5D-5L health status questionnaire; HbA1c, haemoglobin A1c; HDL, high-density lipoprotein; KPPS, King’s PD pain scale; LED, levodopa-equivalent dose; MADRS, Montgomery–Åsberg Depression Rating Scale; MDS-UPDRS, Movement Disorder Society Unified Parkinson’s Disease Rating Scale; NMSS, Non-Motor Symptom assessment scale; PDQ-39, Parkinson’s disease Questionnaire-39.
Calculation of cardiovascular disease risk score
National Institute for Health and Care Excellence (NICE) guidelines recommend that people with an estimated 10-year risk of cardiovascular disease (CVD) of 10% or more should be prioritised for a full formal risk assessment for consideration of statin therapy.17 QRISK2 is a commonly used CVD risk calculator that was used in this study to assess whether there may be an underlying requirement for statin therapy.
The QRISK2 score (considering all risk factors) was calculated for each potential participant after their screening visit, by the Peninsula Clinical Trials Unit (CTU) at Plymouth University.18 A QRISK2 score <10% permitted entry to the study, assuming all other eligibility criteria were satisfied. Patients with a score ≥10% were advised to discuss the implications with their general practitioner (GP), but were able to be included in the study regardless of whether they consulted their GP or not, providing that they were not subsequently prescribed statin therapy by their GP.
Screening for type 2 diabetes
There is some evidence that long-term use of high doses of simvastatin may be associated with an increased risk of developing insulin resistance and type 2 diabetes mellitus,19 20 although in a recent analysis there was no reported evidence of a significant association at 2 years in patients taking a prescribed statin.21 To monitor this, patients were screened at baseline and month 24 using a glycated HbA1c level of 6.5% (48 mmol/mol) as diagnostic of diabetes mellitus.22
Patients with an existing diagnosis of diabetes were not excluded from study participation. Those presenting with an HbA1c≥6.5% (≥48 mmol/mol) at screening, in the absence of a diabetes diagnosis, were asked to discuss the implications with their GP before proceeding further with the study, and excluded if a statin was prescribed. These patients opting not to consult their GP were considered ineligible for the study, based on the potential requirement for statins in the future.
Final eligibility
Following the screening visit, patients who remained eligible and willing to participate in the study were invited to attend a baseline visit approximately 2 to 8 weeks after the screening visit. This interval enabled review of the screening blood results, including time for any subsequent GP discussions, in order to confirm final eligibility for the study. If more than 8 weeks had elapsed since the screening visit, all screening assessments were repeated before proceeding to the baseline visit (nine participants required re-screen on this basis, of whom one was deemed eligible).
Allocation to simvastatin or placebo
Participants were individually randomised to receive simvastatin or matched placebo in a 1:1 ratio. A 24-hour secure web-based randomisation system was created by the CTU in conjunction with an independent statistician and was accessed by research teams at local sites. Allocation used random permuted blocks, with stratification by site and modified Hoehn and Yahr stage (≤2.0 or 2.5–3) in the ‘ON’ medication state. To maintain concealment, the allocation was not displayed or otherwise accessible to the person undertaking the randomisation process. Following completion of the randomisation process (at some point between the screening and baseline visits, or at the baseline visit itself), a signed prescription was passed to the relevant hospital pharmacy so that the initial 1-month supply of trial medication could be dispensed for the baseline visit.
Trial treatment
The trial treatment is an over-encapsulated simvastatin (40 mg) tablet back-filled with microcrystalline cellulose magnesium stearate, or identically presented matched placebo containing microcrystalline cellulose magnesium stearate only. Capsules are packaged in plastic screw neck bottles with child-resistant, tamper-evident lids. Each bottle contains 100 capsules and has a unique four-digit number with an expiry date displayed on a label that meets the current regulatory requirements. Participants are provided with a 1-month supply of trial medication at baseline, a 5-month supply at the month 1 visit, and a 6-month supply at months 6, 12 and 18 visits. Participants are asked to return all empty, full or partially used medication bottles at each study visit. These are returned to the local site pharmacy for capsule count as part of the assessment of compliance with study treatment.
Baseline visit (V2)
Participants attended their baseline visit in the practically defined OFF state (see below) and underwent a series of assessments (see table 1) before being invited to take their usual PD medications. Further assessments were then undertaken in the ON state before participants were provided with a 1-month supply of trial medication (40 mg daily dose or placebo) for the initial low-dose phase. Participants were also provided with a paper-based diary in which to record any dose alterations, concomitant medications or AEs. The diary was intended to serve as an aide-memoire, with participants being asked to bring their completed diary to each study visit to aid Case Report Form completion by the local research team. Participants were advised to contact the local research team promptly should they develop unexplained muscle pain, tenderness or weakness.
Participant follow-up and data collection
Participant follow-up is via a mixture of clinic visits and telephone contacts. Clinic visits are held at 1 month (V3), then 6, 12, 18, 24 and 26 months (V4–V8) post-baseline. The 12-month, 24-month and 26-month visits require attendance in the OFF state, followed by further assessments on the same day in the ‘ON’ state after the participant has taken his/her usual PD medication—as for baseline (V2). Telephone contacts between visits are made at 2 weeks (T2), and then at 4, 8, 10, 14, 16, 20 and 22 months (T3–T9) to identify any compliance problems, AEs or changes to participants’ routine medication. Additional telephone contacts may be made, as required, at the discretion of the local research team and specifically in the event of abnormal blood results being identified at any stage during the trial.
Practically defined ‘OFF’ and ‘ON’ state
Participants are asked to attend baseline, 12-month, 24-month and 26-month study visits in the OFF state, having omitted their routine PD medication. To facilitate attendance, these visits are scheduled in the morning, and OFF assessments take approximately 30 min to complete. Short-acting PD medications are omitted from 1800 hours on the day before the clinic visit. Long-acting agents are omitted for the entire day before the clinic visit and also on the day of the visit itself. The local research team is able to make arrangements to provide the participant with a prescription for relevant supportive medications (eg, zopiclone/zolpidem for night sedation, paracetamol for pain relief and/or diazepam for treatment of anxiety) as necessary. Participants may also be prescribed dispersible Madopar as a rescue medication to be taken in the event of severe difficulty with OFF state symptoms, but this would necessitate abandonment of the study visit. The visit can be rescheduled if the patient has been unable to attend in the OFF state. If the further attempt at attending in the OFF state fails, the participant is withdrawn from the study. The delivery of OFF state assessments is challenging, but we are managing this with appropriate interaction with, and training of, study teams, encouraging them to provide support for patients, such as the use of taxis to facilitate visit attendance and the offer of home visits if necessary.
Dose adjustments
If the participant was able to tolerate the initial low-dose phase of trial medication for 4 weeks, the prescription was increased to 80 mg daily at the 1-month clinic visit. At the 24-month visit, participants stop their trial medication and a 2-month washout period follows. The final visit at 26 months will be used to differentiate whether any benefit may have been symptomatic.
Participants who were unable to tolerate the 40 mg dose during the first month due to unwanted symptoms, or who fulfilled the stopping criteria (see later), had their trial treatment permanently discontinued but were invited to continue with the study assessments.
During the higher-dose maintenance phase, participants who are unable to tolerate the 80 mg dose of study medication due to unwanted symptoms (but who do not fulfil the stopping criteria) may have their dose reduced to 40 mg daily. Participants may continue on the 40 mg dose for the remainder of the trial or, at the discretion of the local PI, may later be re-challenged with the 80 mg dose after resolution of their symptoms.
Blinding
This is a double-blind study, hence the participants, trial management team, investigator site teams and site pharmacy staff are blind to treatment allocation throughout the trial. In the event of a potential Suspected Unexpected Serious Adverse Reaction (SUSAR) to the trial medication, unblinding will be undertaken by the sponsor in accordance with the regulatory requirements. Unblinding may also be performed at the request of a senior clinician responsible for the care of a trial participant but such requests are likely to occur only in the case of a serious adverse clinical event and are expected to be rare. The Data Monitoring Committee (DMC) is able to review unblinded data as required.
Since the PI and other ‘treating’ site team members have access to participants’ blood results and review all reported AEs, a separate ‘assessing’ member of the research team undertakes the MDS-UPDRS and other outcome assessments after appropriate training. The same outcome assessor is used at all visits for an individual participant and sites are requested to identify back-up personnel to cover staff absences and avoid cross-over of ‘assessing’ and ‘treating’ team members. Telephone follow-up calls are not made by outcome assessors.
Participant monitoring
At each study visit or telephone call, participants are asked about any AEs experienced and, dependent on reported symptoms, may be asked to provide a blood sample to check CK and/or AST/ALT levels. If a raised AST/ALT is observed in the absence of a CK result, the CK should be checked. Tables 2–4 outline the possible outcomes and any action required.
Table 2.
AST/ALT monitoring outcomes and action required
| Observation | Action required | Repeat observation | Action required |
| AST/ALT>3xULN | Repeat sample within 1 week | AST/ALT>4xULN | Stop study treatment temporarily |
| AST/ALT>2 xULN but ≤4 x ULN | Repeat again within 3 weeks. If remains >2 ×ULN stop study treatment temporarily | ||
| AST/ALT>2 xULN but ≤3xULN | Repeat again within 3 weeks | AST/ALT>3xULN | Stop study treatment temporarily |
| AST/ALT>2 xULN but ≤3xULN | Repeat again within 3 weeks. If remains >2 ×ULN stop study treatment temporarily |
ALT, alanine transaminase; AST, aspartate transaminase; ULN, upper limit of normal.
Table 3.
AST/ALT monitoring outcomes and action required 6 weeks after temporary stop of study treatment
| Observation | Action required | Subsequent action required |
| AST/ALT>1.5xULN | Stop study treatment permanently | Repeat every 3 weeks until AST/ALT reverts to normal (ie, ≤1.5 xULN) |
| AST/ALT≤1.5xULN | Study treatment can be restarted | Repeat twice at 3-week intervals. AST/ALT must remain ≤2 xULN, otherwise study treatment should be stopped permanently |
ALT, alanine transaminase; AST, aspartate transaminase; ULN, upper limit of normal.
Table 4.
CK monitoring outcomes and action required
| Observation | Action required | Repeat observation | Action required |
| CK >5xULN |
Stop study treatment permanently
Repeat sample within 1 week |
Repeat every 3 weeks until CK reverts to normal (ie, ≤3 xULN) | Repeat every 3 weeks until CK reverts to normal (ie, ≤3 xULN) |
| CK >4 xULN but ≤5 xULN that cannot be explained (ie, trauma, heavy exercise, etc) | Repeat sample within 1 week | CK remains >4 xULN but ≤5 xULN | Stop study treatment temporarily Check CK again in 6 weeks
|
CK, creatine kinase; ULN, upper limit of normal.
If the participant reports jaundice or new or unusually severe nausea, malaise or lethargy, an AST/ALT level should be checked (table 2). If study treatment needs to be stopped temporarily, AST/ALT should be checked again after 6 weeks, and then action taken in accordance with table 3.
If the participant reports new or unusually severe muscle pain, tenderness or weakness, the CK level should be checked (table 4).
AEs may also be reported to the research team outside of a participant’s scheduled clinic visit, either by the participant, non-study clinician or other informant by contacting the trial centre.
Stopping criteria for discontinuation of trial treatment
The defined stopping criteria for the discontinuation of trial medication are:
Abnormalities in CK or ALT/AST fulfilling stopping criteria as outlined above, OR
New severe muscular symptoms (progressive or persistent), not attributable to other cause, which in the opinion of the PI may be related to the study medication even in the absence of abnormal CK, OR
Onset of a clinical condition for which prescription of a statin is indicated.
Pharmacovigilance
Safety and tolerability of the trial treatment are monitored throughout the study by means of regular clinic visits and interim telephone follow-up review of all participants. All serious adverse events (SAEs) are recorded and reported, whether they are deemed related to the trial treatment or not. Quarterly summaries of all SAEs are provided to the DMC and study sponsor. Any potential SUSAR will be reported immediately to the sponsor who will have the facility to unblind the treatment allocation independently of the trial team and report onwards as necessary.
Non-SAEs deemed possibly, probably or definitely related to trial treatment are also recorded, monitored by the Trial Management Group (TMG) and reported to the DMC.
Embedded sub-studies
The three embedded substudies will be described in more detail in separate publications. The ‘Experience of Trial Participation’ substudy aims to develop an understanding of the barriers and facilitators to participating in clinical trials for people living with PD. It includes a quantitative component (feedback surveys) for all participants and a qualitative component (semistructured interviews and focus groups) in a sample of participants and their carers. Part of this substudy involves an evaluation of The Cure Parkinson’s Trust’s Charter for Clinical Trials in Parkinson’s, which aims to set standards of practice for both participants and clinicians involved in clinical trials for PD. All patients approached for the PD STAT study were provided with a copy of this charter and asked to provide feedback on its usefulness.
The ‘genetic substudy’ aims to identify the genetic markers that may be associated with PD disease course, severity or variation in treatment responsiveness. PD STAT participants are asked to provide separate written informed consent, followed by collection of two 10 mL blood samples, usually at the 12-month clinic visit. One sample is sent to University College London Neurogenetics Department. The inherited materials (DNA and genes) are extracted from the sample and stored in a biobank within the Institute of Neurology. The second sample is sent to the Genetic Support Support Services, Culture Collections, Public Health England laboratory for preperation and storage of peripheral blood lymphocytes and potential future cell lines.
The ‘EMS measurement’ substudy is an exploratory study conducted in a subset of participants. It aims to evaluate the use of the EMS (Polhemus Inc.) in the measurement of bradykinesia and tremor and is completed alongside the MDS-UPDRS upper limb motor assessments at the 12-month and 24-month visits. Participants are required to wear the sensors on the index finger and thumb when performing the assessments, visual assessments are conducted as normal by the assessor.
Patient and public involvement
Patient and public involvement (PPI) representatives are members of both the TMG and Trial Steering Committee (TSC). They were involved in the design of the study and reviewed and advised on all participant-facing study documentation; they will also be closely involved in dissemination of results to participants and patient groups.
Study management
The study sponsor organisation is the University Hospitals Plymouth NHS Trust, Derriford, Plymouth PL6 5FP. Day-to-day trial management is administered through the United Kingdom Clinical Research Collaboration (UKCRC)-registered Peninsula CTU at Plymouth University. The CTU conducts central and site monitoring in accordance with a risk-based monitoring plan and the study sponsor may audit trial conduct as deemed appropriate.
The TMG, which includes two patient members, meets regularly to monitor and discuss the progress of the trial, and to address any issues that may arise. The TSC, which includes an independent chair and two PPI members, meets once or twice a year to oversee the conduct of the trial. An independent DMC, comprising two independent clinicians and a statistician, meets at similar intervals to the TSC to monitor safety and ethical issues, including any participant drop-outs and overall data completeness. The agreed roles and responsibilities of both committees are set out in written charters.
Data management and Confidentiality
Research teams at all sites ensure that participants’ anonymity is maintained for all documents.
Data are collected and stored in accordance with the Data Protection Act, 1998 and General Data Protection Regulation, 2018. Within the CTU, pseudonymised paper-based study data are stored in locked filing cabinets within a locked office. Electronic records are stored in a Structured Query Language (SQL) server database, stored on a restricted access, secure server maintained by the University of Plymouth. The study website is encrypted using Secure Sockets Layer (SSL). Study data are double-data entered on to a password-protected database within the CTU, with copies retained at the relevant study site. Double-entered data are compared for discrepancies using an established procedure to verify data entry. Discrepant data are verified using the original paper data sheets. Direct access to the trial data is overseen by the CTU, and restricted to members of the research team and the CTU, with access granted to the sponsor on request. Copies of study data retained at study sites are securely stored for the duration of the study prior to archiving.
Sample size
As this study has a futility design, the direction of the hypotheses is different from that in traditional phase II efficacy or effectiveness trials. The study sample size has been calculated based on testing the null hypothesis that simvastatin is not futile, in terms of the primary outcome. If at the end of the study there is evidence to reject the null hypothesis, then simvastatin will be considered to be futile for a phase III study.
The minimum clinically important difference in UPDRS motor score has been estimated to be 2.3–2.7 points.23 The null hypothesis (H0) in this futility study is that the mean MDS-UPDRS part III change score (between baseline and 24 months) for the simvastatin group is at least three points better (ie, smaller, as higher MDS-UPDRS scores are worse) than the corresponding mean change in the placebo group. The alternative hypothesis (HA) is that the mean MDS-UPDRS part III change score for the simvastatin group is not at least three points better. This can be written mathematically as:
H0: μs ≤ μp – 3 versus HA: μs > μp – 3
where μs is the expected mean MDS-UPDRS part III change score from baseline to 24 months for the simvastatin group and μp is the corresponding expected mean change for the placebo group. Given this hypothesis, a one-sided test (and associated significance level, alpha) is appropriate.
In futility studies, the error probabilities are interpreted differently from those in traditional efficacy/effectiveness studies. The type 1 (alpha) error pertains to recommending that an effective treatment should not be considered for a phase III study and the type 2 (beta) error pertains to recommending that an ineffective treatment should be considered for a phase III study.24 Given these different interpretations, alpha and beta are chosen relative to the futility design-based hypotheses: in this study, the one-sided alpha is set at 10% and beta at 20% (ie, 80% power).24 Under these design parameters, there is a 20% chance of failing to identify that simvastatin is ineffective.
Based on available data at the time of planning the PD STAT study, the expected mean increase in MDS-UPDRS part III from baseline to 12 months in the placebo group is 2.2 points, with SD 7.3 points.25 Assuming that this increase in MDS-UPDRS part III is linear over time, gives an expected mean increase from baseline to 24 months of 4.4 points in the placebo group, with an assumed slightly inflated SD over this period of 7.5 points.
The null hypothesis H0: μs ≤ μp – 3 can be stated equivalently as H0: μs – μp ≤ −3. To test this hypothesis, and assuming μp is 4.4 points, it is assumed that μs is 1.4 points (ie, 4.4−3). Based on a two-sample t-test with a 10% one-sided alpha, it is estimated that 24-month follow-up data are required from 57 participants per allocated group to provide 80% power to reject the null hypothesis and declare futility.
The initial calculated sample size was inflated twice. First, to allow for a small proportion of participants allocated to the simvastatin group to stop taking the trial medication during the initial 4 week low-dose phase. Assuming that this proportion is 15%, the previous sample size is inflated by a factor of (1–0.15)−2, to give 79 participants per group.26 Second, the sample size was adjusted to allow for a (non-differential) loss to follow-up rate by 24 months of 20%. Accordingly, the sample size was further inflated by a factor of (1–0.2)−1, to give a sample size of 99 participants per group and a total recruitment target of 198 participants.
Statistical analysis
The primary analyses are all pre-specified and a detailed statistical analysis plan will be drafted and agreed by the DMC and signed off by the independent statistician on the TSC, prior to commencement of analyses. The study will be reported following the principles of Consolidated Standards Of Reporting Trials guidelines.27–31 There is no planned interim analysis for this study. Primary analyses will be on a modified intention-to-treat (ITT) basis. The ITT evaluable sample will include all participants who are randomised and who commence on the higher-dose phase of the study. As this is a phase II study, no imputation of missing data is planned for the primary analysis and so the ITT sample for the primary analysis of the primary outcome will include participants with baseline and 24-month MDS-UPDRS part III scores.
The statistical analyses will be undertaken blinded to the allocated group. The primary analysis will be a between-group comparison of mean change in MDS-UPDRS part III from baseline to 24 months. Specifically, a linear regression model will be fitted to MDS-UPDRS part III scores at 24 months, with allocated treatment group, baseline MDS-UPDRS part III score, the stratification variable (modified Hoehn & Yahr stage), gender and age at baseline included as covariates. Scores will be appropriately transformed if necessary. In the primary analysis of the primary outcome, if the p value <0.1 from the regression model testing the null hypothesis that the treatment effect is <-3, then the null hypothesis that simvastatin is not futile will be rejected and simvastatin will be considered to be futile for a phase III study. For completeness, the two-sided 80% CI (confidence interval) for the estimated treatment effect will also be presented, although only the upper bound of the CI is of relevance when assessing for futility. If the upper bound of the CI is lower than −3, there will be evidence to consider simvastatin for a phase III study.
Consideration will be given to a secondary analysis of the primary outcome on a per-protocol basis. If a sufficient number of participants drop back down to the lower dose of simvastatin, consideration will be given to a subgroup analysis of the effect of dose. These, and any other secondary analyses, such as comparing participant characteristics of responders and non-responders, will be discussed with the DMC and included in the agreed statistical analysis plan.
Secondary continuous outcomes will be compared between allocated treatment groups in a similar manner, although will not be statistically tested for futility; instead the focus will be on providing appropriate summary statistics and CIs for the between-group differences. Ordered categorical outcomes will be analysed using ordinal regression analysis. Analysis of AEs will be on a per-protocol basis.
Ethics and dissemination
The protocol has been approved by the North East—Newcastle and North Tyneside 2 Research Ethics Committee (REC Reference: 15/NE/0324). The trial is conducted in accordance with the study protocol, the principles of the Declaration of Helsinki, International Conference on Harmonisation of Good Clinical Practice and the Medicines for Human Use (Clinical Trials) Regulations, 2004. The trial has been adopted by the National Institute for Health Research (NIHR) Clinical Research Network and has relevant local NHS research approvals. The study is sponsored by University Hospitals Plymouth NHS Trust and managed by the UKCRC-registered Peninsula CTU.
After the end of the study, pseudonymised information collected during the study will be made available to other researchers under an appropriate data sharing agreement, but it will not be possible to identify participants personally from any information shared.
Following analysis of the data, the results will be disseminated through publication of articles in peer-reviewed journals and presentations at local, national and international scientific meetings. A lay summary of the study results will be prepared with assistance from our patient TMG members and made available to study participants, PD charities and relevant support groups for wider dissemination among people with PD and their families.
DISCUSSION
There is currently no neuroprotective agent proven to slow or reverse the progression of PD. This phase II trial is required to inform the decision to progress to a definitive phase III randomised controlled trial evaluating the effectiveness of simvastatin as a neuroprotective agent to treat PD. In addition to this, the study will generate other important outputs related to trial delivery and how trial experience can be improved from the perspective of the participants.
This study has a number of strengths: it starts a shared resource with other studies in the LCT initiative with the pharmacogenetics substudy, and the EMS substudy provides a platform for evaluating a novel outcome measure based on wearable technology for neuroprotective studies that can be used to inform future evaluations. PD STAT importantly demonstrates that a multi-centre trial delivery platform exists within the UK to deliver a study of reasonably long duration, engaging patients with PD and clinicians, which will strengthen delivery of future similar studies.
Supplementary Material
Acknowledgments
We would like to thank all the staff and patients involved with the study at the 23 research sites, and the Trial Steering Committee and Data Monitoring Committee for their ongoing support throughout the trial. We would like to acknowledge the role of the NIHR Clinical Research Network in supporting the ongoing delivery of the trial at the sites.
Footnotes
Contributors: CBC is the Chief Investigator, provides clinical expertise and is responsible for conception and design of the study. RW, AW and JZ provide clinical and scientific expertise and helped with design of the study. JV contributed to study design and trial management. SC and KNS are the trial statisticians and provide expertise in the day-to-day running of the trial. SB and SC provided statistical expertise in the overall design of the trial. MW and AF contributed to study design and PPI input. VE and DW were the original trial managers, responsible for overseeing the day-to-day running of the trial.
Funding: This work is supported by grants from The Cure Parkinson’s Trust and JP Moulton Charitable Foundation. SB is funded by the National Institute for Health Research (NIHR) Collaboration for Leadership in Applied Health Research and Care South West Peninsula (NIHR CLAHRC South West Peninsula).
Disclaimer: The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Competing interests: RW is Director of Research and Development at The Cure Parkinson’s Trust.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. The incidence and prevalence of Parkinson's in the UK report, 2018. Available: https://www.parkinsons.org.uk/professionals/resources/incidence-and-prevalence-parkinsons-uk-report
- 2. Friedman B, Lahad A, Dresner Y, et al. Long-Term statin use and the risk of Parkinson's disease. Am J Manag Care 2013;19:626–32. [PubMed] [Google Scholar]
- 3. Undela K, Gudala K, Malla S, et al. Statin use and risk of Parkinson’s disease: a meta-analysis of observational studies. J Neurol 2013;260:158–65. 10.1007/s00415-012-6606-3 [DOI] [PubMed] [Google Scholar]
- 4. Roy A, Pahan K. Prospects of statins in Parkinson disease. Neuroscientist 2011;17:244–55. 10.1177/1073858410385006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carroll CB, Wyse RKH. Simvastatin as a Potential Disease-Modifying Therapy for Patients with Parkinson’s Disease: Rationale for Clinical Trial, and Current Progress. J Parkinsons Dis 2017;7:545–68. 10.3233/JPD-171203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghosh A, Roy A, Matras J, et al. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci 2009;29:13543–56. 10.1523/JNEUROSCI.4144-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. The Lancet 2014;383:2213–21. 10.1016/S0140-6736(13)62242-4 [DOI] [PubMed] [Google Scholar]
- 8. Brundin P, Barker RA, Conn PJ, et al. Linked clinical trials-the development of new clinical learning studies in Parkinson's disease using screening of multiple prospective new treatments. J Parkinsons Dis 2013;3:231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246 955 participants from 135 randomized, controlled trials. Circ Cardiovasc Qual Outcomes 2013;6:390–9. [DOI] [PubMed] [Google Scholar]
- 10. HL L, Jin DM, Liu M, et al. Long-Term efficacy and safety of statin treatment beyond six years: a meta-analysis of randomized controlled trials with extended follow-up. Pharmacol Res 2014;81:64–73. [DOI] [PubMed] [Google Scholar]
- 11. Lang AE, Melamed E, Poewe W, et al. Trial designs used to study neuroprotective therapy in Parkinson's disease. Mov Disord 2013;28:86–95. 10.1002/mds.24997 [DOI] [PubMed] [Google Scholar]
- 12. Schwid SR, Cutter GR. Futility studies: spending a little to save a lot. Neurology 2006;66:626–7. 10.1212/01.wnl.0000204644.81956.65 [DOI] [PubMed] [Google Scholar]
- 13. Creanor S, Vickery J, Eyre V, et al. Two-arm randomised futility trials: PD-stat - a futility trial of a potential neuroprotective treatment in people with Parkinson's disease. Trials 2015;16 10.1186/1745-6215-16-S2-P236 [DOI] [Google Scholar]
- 14. Goetz CG, Tilley BC, Shaftman SR, et al. Movement disorder Society-sponsored revision of the unified Parkinson's disease rating scale (MDS-UPDRS): scale presentation and Clinimetric testing results. Mov Disord. 2008;23:2129–70. 10.1002/mds.22340 [DOI] [PubMed] [Google Scholar]
- 15. Chan A-W, Tetzlaff JM, Altman DG, et al. Spirit 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med 2013;158:200–7. 10.7326/0003-4819-158-3-201302050-00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stacy M, Hauser R, Oertel W, et al. End-of-dose wearing off in Parkinson disease: a 9-question survey assessment. Clin Neuropharmacol 2006;29:312–21. [DOI] [PubMed] [Google Scholar]
- 17. Excellence NIfHaC Cardiovascular disease: risk assessment and reduction, including lipid modification (NICE guideline CG181), 2014. Available: http://guidance.nice.org/CG181 [PubMed]
- 18. Peninsula clinical trials unit. Available: https://www.plymouth.ac.uk/your-university/about-us/university-structure/faculties/medicine-dentistry/pupsmd-research/penctu
- 19. Aiman U, Najmi A, Khan RA. Statin induced diabetes and its clinical implications. J Pharmacol Pharmacother 2014;5:181–5. 10.4103/0976-500X.136097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crandall JP, Mather K, Rajpathak SN, et al. Statin use and risk of developing diabetes: results from the diabetes prevention program. BMJ Open Diab Res Care 2017;5 10.1136/bmjdrc-2017-000438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cederberg H, Stančáková A, Yaluri N, et al. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia 2015;58:1109–17. 10.1007/s00125-015-3528-5 [DOI] [PubMed] [Google Scholar]
- 22. WHO , 2011. Available: http://www.who.int/diabetes/publications/report-hba1c_2011.pdf
- 23. Shulman LM, Gruber-Baldini AL, Anderson KE, et al. The clinically important difference on the unified Parkinson's disease rating scale. Arch Neurol 2010;67:64–70. 10.1001/archneurol.2009.295 [DOI] [PubMed] [Google Scholar]
- 24. Tilley BC, Palesch YY, Kieburtz K, et al. Optimizing the ongoing search for new treatments for Parkinson disease: using futility designs. Neurology 2006;66:628–33. 10.1212/01.wnl.0000201251.33253.fb [DOI] [PubMed] [Google Scholar]
- 25. Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Invest. 2013;123:2730–6. 10.1172/JCI68295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. RCaC T. Biostatistics in clinical trials. Wiley, 2001. [Google Scholar]
- 27. Schulz KF, Altman DG, Moher D. Consort 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med 2010;152:726 10.7326/0003-4819-152-11-201006010-00232 [DOI] [PubMed] [Google Scholar]
- 28. Piaggio G, Elbourne DR, Altman DG, et al. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA 2012;308:2594–604. [DOI] [PubMed] [Google Scholar]
- 29. Calvert M, Blazeby J, Altman DG, et al. Reporting of patient-reported outcomes in randomized trials: the CONSORT pro extension. JAMA 2013;309:814–22. [DOI] [PubMed] [Google Scholar]
- 30. Eldridge SM, Chan CL, Campbell MJ, et al. Consort 2010 statement: extension to randomised pilot and feasibility trials. BMJ 2016;355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ioannidis JPA, Evans SJW, Gøtzsche PC, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 2004;141:781–8. 10.7326/0003-4819-141-10-200411160-00009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2019-029740supp001.pdf (43.2KB, pdf)