

Summary
Transient hypogammaglobulinaemia of infancy (THI) is a relatively rare disorder where there is an exaggeration of the physiological nadir of immunoglobulin (Ig)G between loss of transplacentally acquired maternal IgG and production by the infant. Patients may be vulnerable to infections during the period of hypogammaglobulinaemia. The precise time to recovery in all infants is currently unknown. We sought to determine the clinical features and time–course of recovery for patients with THI. We reviewed our experience with THI over the last three decades in order to describe clinical and laboratory features, as well as the time–course of recovery. Forty‐seven patients were identified with THI. Only thirty‐seven per cent remitted by 4 years of age, while some patients did not recover until the third or fourth decade. In keeping with previous studies, the majority (25 of 47) presented with recurrent infections, nine had a family history of immunodeficiency and 13 had adverse reactions to food as their dominant clinical manifestation. Chronic tonsillitis developed in 10 patients and symptoms improved following surgery. The group with food allergies recovered sooner than those presenting with infections or with a family history immunodeficiency. Eight patients failed to respond to at least one routine childhood vaccine. Two have IgA deficiency and four individuals recovering in adolescence and adulthood continue to have borderline/low IgG levels. None have progressed to common variable immunodeficiency disorders (CVID). THI is a misnomer, as the majority do not recover in infancy. Recovery from THI can extend into adulthood. THI must be considered in the differential diagnosis of adolescents or young adults presenting with primary hypogammaglobulinemia.
Keywords: common variable immunodeficiency disorders, hypogammaglobulinaemia, hypogammaglobulinaemia of uncertain significance, immunodeficiency, transient hypogammabulinemia of infancy
Introduction
Transient hypogammaglobulinaemia of infancy (THI) is a primary immunodeficiency disorder (PID) 1, 2. THI was first described in 1956, when two infants with low immunoglobulin levels recovered spontaneously without intervention 3. Infants with THI must be distinguished from those with permanent immune defects such as common variable immunodeficiency disorders (CVID), who usually require life‐long subcutaneous or intravenous immunoglobulin (SCIG/IVIG) replacement.
There are currently differing definitions of THI. The European Society for Immunodeficiency (ESID) requires THI patients to have reduced IgG levels, which recover around their fourth birthday 4. The World Health Organization/International Union of Immunological Societies (WHO/IUIS) definition, in contrast, mandates a reduction of both IgG and IgA to qualify 1. Given the difficulties in determining age‐appropriate IgA levels in infancy, most authors accept reduced IgG or globulins alone, which later recover, as sufficient evidence for a diagnosis of THI. All authors agree that children with THI should have no other explanation, including other PIDs, for their reversible hypogammaglobulinaemia.
The precise cause of THI is unknown, which may represent an exaggeration of the physiological nadir between loss of maternal IgG and the infant synthesizing his or her own IgG. Although familial cases have been described, no genetic marker has been identified for THI at this time. Given that most IgG is transferred through the placenta in the third trimester, preterm infants are susceptible to hypogammaglobulinaemia.
Because of the requirement for spontaneous recovery, THI is a retrospective diagnosis. It is also apparent that recovery can occur much later than a child’s fourth birthday. In several case–series, many children had not recovered by the end of the study period and were deemed to have unclassified hypogammaglobulinaemia (UCH) 4, 5, 6. This suggests that the follow‐up period, particularly in prospective studies, may be inadequate to accurately determine the time–course of recovery in these patients 7.
Here we describe a group of patients with confirmed THI who presented over three decades to the corresponding author. The key finding is that, while most patients recovered in childhood, some did not remit until the third or fourth decade of life. One patient, aged 32 years, who was on long‐term IVIG for presumed CVID, was able to successfully discontinue treatment after THI was diagnosed. THI will need to be considered in the differential diagnosis of children, adolescents and young adults presenting with hypogammaglobulinaemia.
Patients and methods
Patients recovering from THI were identified from a retrospective clinical patient safety audit following written correspondence with the Health and Disability Ethics Committee (HDEC) of the NZ Ministry of Health. The primary aim was to ensure that patients with hypogammaglobulinaemia were not lost to follow‐up to confirm that they were not at risk of serious disorders such as CVID. Unlike patients with THI who recover, patients with undiagnosed CVID are at risk of meningitis, sepsis, pneumonia and bronchiectasis. Similarly, patients with undiagnosed immunodeficiency are at increased risk of mortality from the current global measles pandemic.
Infants and children with primary hypogammaglobulinaemia were identified from records of the private and public practice of the lead author (R. A.). In order to fulfil the criteria for THI, patients were required to have at least one reduced IgG (n = 46) or globulin level (n = 1) under the age of 4 years. Patients must have made a spontaneous recovery, and lastly there was no other explanation for the hypogammaglobulinaemia. Unlike the WHO/IUIS definition, a reduction in IgA was not required for the diagnosis of THI in this study 1.
Clinical assessment included a careful history, examination and investigations directed by clinical features. Patients had detailed immunological studies including immunophenotype, vaccine responses and isohaemagglutinins. Other secondary causes of hypogammaglobulinaemia, such as medications, were also actively excluded at the time of consultation.
Because most patients were evaluated during infancy and early childhood while undergoing their primary immunization series, vaccine antibody results were available for the majority. All citizens and permanent residents in New Zealand are assigned a National Health Index (NHI) number. The NHI is linked to multiple health indices, including laboratory results, medications and immunizations.
All children were confirmed as having received their primary vaccination series according to the NZ immunization schedule through computerized records. All vaccine responses were evaluated during the period of hypogammaglobulinaemia prior to resolution of THI. Older children, adolescents and adults (n = 33) underwent additional vaccine challenges with the diphtheria–tetanus toxoid, Haemophilus influenzae type B conjugated vaccine (HIB) and the pneumococcal polysaccharide vaccine (PPV, Pneumovax®), depending on the chronology of the introduction of these vaccines 8.
We also assessed the quality of vaccine responses. Both protective levels as well as higher thresholds we have advocated in our CVID diagnostic criteria were evaluated 9. The vaccine responses in our CVID criteria are based on studies of normal adult individuals in the community and are higher than protective levels 9.
Patients were deemed to have recovered as soon as they had one normal IgG level, within the age‐specific reference interval. Infants and young children were followed closely until recovery, while older children and adults were reviewed approximately 6‐monthly. If patients were lost to follow‐up before spontaneous recovery of their IgG, they and/or their physicians were contacted and advised to undertake further testing to confirm recovery.
Results
This case–series includes infants as well as older children who had hypogammaglobulinaemia at the time of referral, which subsequently recovered 10. In the case of older children, who later remitted, the referring paediatricians from throughout New Zealand had already made the diagnosis of a probable PID based on IgG or globulin levels below the age‐specific reference interval.
Fifty‐two patients with possible THI were identified from 1988 to 2018. Two patients had a final IgG of 6·9 g/l and were deemed to have recovered. Ten were lost to follow‐up before their IgG levels normalized. After being contacted, five had repeat tests confirming remission and are included in Tables 1 and 2, but not in the Kaplan–Meier plots (Figs 1 and 2), as we could not establish the precise time of recovery. The five patients who did not undertake follow‐up immunoglobulin levels were excluded from the analysis.
Table 1.
Clinical features and treatment classified according to the subphenotype of THI
| THI subphenotype | Infections (25) | Food allergy (13) | Family history (9) | All (47) |
|---|---|---|---|---|
| Male/female | 20/5 | 9/4 | 6/3 | 35/12 |
| Pneumonia | 2/25 | 0/13 | 3/9 | 5/47 |
| Bronchiectasis | 1/25 | 0/13 | 0/9 | 1/47 |
| Meningitis/sepsis | 1/25 | 0/13 | 0/9 | 1/47 |
| Viral infections | 2/25 | 0/13 | 0/9 | 2/47 |
| Failure to thrive | 1/25 | 1/13 | 0/9 | 2/47 |
| ‘Food allergy’ | 3/25 | 13/13* | 2/9 | 18/47 |
| Asthma | 7/25 | 7/13 | 1/9 | 15/47 |
| Prophylactic abx | 7/25 | 2/13 | 1/9 | 10/47 |
| IVIG | 3/25 | 0/13 | 0/9 | 3/47 |
| Age at resolution mean: range (months) | 123·28 (10–264) | 62·4 (12–289) | 147·1 (15–371) | |
| Tonsillectomy | 7/25 | 2/13 | 1/9 | 10/47 |
| Adenoidectomy | 2/25 | 0/13 | 0/9 | 2/47 |
| Allergen‐specific IT | 2/25 | 1/13 | 0/9 | 3/47 |
| Tympanostomy tubes | 4/25 | 2/13 | 2/9 | 8/47 |
IVIG = intravenous immunoglobulin; IT = immunotherapy; abx = antibiotics; THI = transient hypogammabulinaemia of infancy.
One patient had food intolerances.
Table 2.
Laboratory results
| THI subphenotype | Infections (25) | Food allergy (13) | Family history (9) | All (47) |
|---|---|---|---|---|
| IgA deficiency (< 0·07 g/l)* | 1/25 | 1/13 | 0/9 | 2/47 |
| Reduced switched memory B cells (CD19+, CD27+) | 3/9 | 1/1 | 3 /4 | 7/14 |
| Dip < 0·1 IU/ml | 5/23 | 0/12 | 0/10 | 5/45 |
| Dip > 1 IU /ml | 11/23 | 9/12 | 8/10 | 28/45 |
| Tet < 0·1 IU/ml | 2/23 | 0/11 | 1/9 | 3/43 |
| Tet > 1 IU/ml | 18/23 | 9/11 | 6/9 | 33/43 |
| HIB < 0·15 µg/ml | 1/16 | 0/9 | 1/6 | 2/31 |
| HIB > 1 µg/ml | 10/16 | 9/9 | 5/6 | 24/31 |
| Pneumo < 16 µg/ml | 0/20 | 2/3 | 0 /8 | 2/31 |
| Isohaemagglutinins > 1 : 2 | 16/16 | 2/2 | 3/3 | 21/21 |
| IgG2 deficiency | 1 | 1** | 0 | 2 |
| IgG3 deficiency | 2 | 0 | 0 | 2 |
The denominator represents the number of patients tested. Vaccine responses are shown as both protective (above) and the higher thresholds (below) we have advocated for our CVID diagnostic criteria. Pneumococcal responses were assayed by the pooled Binding Site® assay (Birmingham, UK), which pools multiple serotypes and states the normal range as > 16 μg/ml. All vaccine antibody tests were performed during the period of hypogmammaglobulinaemia. The denominator represents the number of patients tested.
Dip = diphtheria; HIB = H. influenzae type B; pneumo = pneumococcal antibodies; Tet = tetanus; THI = transient hypogammabulinaemia of infancy.
Persistent immunoglobulin (Ig)A deficiency following recovery of IgG levels;
recovery could not be confirmed, as the patient emigrated.
Figure 1.
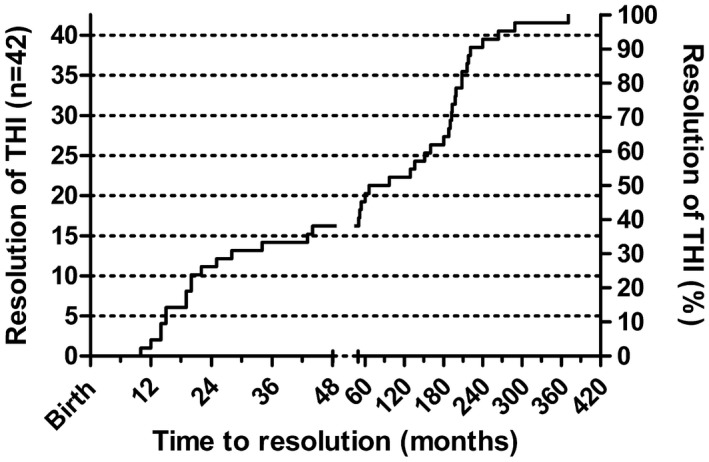
Kaplan–Meier plot of 42 transient hypogammabulinaemia of infancy (THI) patients, showing the time–course of immunoglobulin (Ig)G recovery from birth. The five patients lost to follow‐up but subsequently shown to have recovered are not included. The graph is presented in two unequal sections, one to age 4 years and the other extending into the fourth decade. Patient numbers are on the left axis and percentages recovering are on the right axis.
Figure 2.
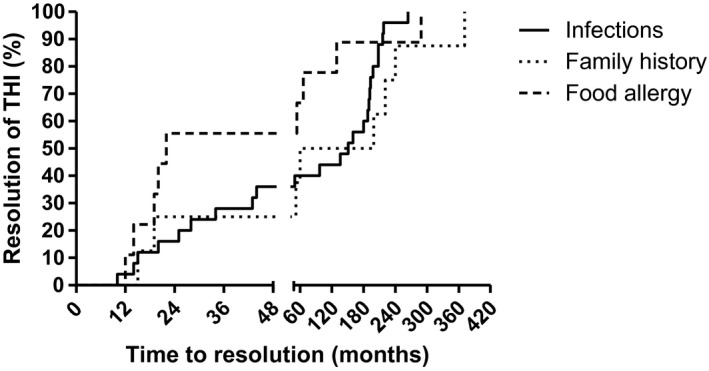
Kaplan–Meier plot showing time–course of immunoglobulin (Ig)G recovery in each of the three subphenotypes of transient hypogammabulinaemia of infancy (THI). The graph is presented in two unequal sections, one to age 4 years and the other extending into the fourth decade. Figures 1 and 2 are presented separately for clarity. Because of unequal numbers in each subphenotype, percentages are used on the y‐axis.
Clinical features are shown in Table 1. As in most other THI series, the majority were male. One patient was born at 34 weeks. Figure 3 shows the three overlapping phenotypic presentations of THI: predominant infections, predominant food allergy and those with a family history of an immune defect. Given this overlap, classification of the subphenotypes was based on the following hierarchy: family history, predominant infections and lastly the broad category of adverse reactions to foods, including allergies and intolerances (Fig. 3).
Figure 3.
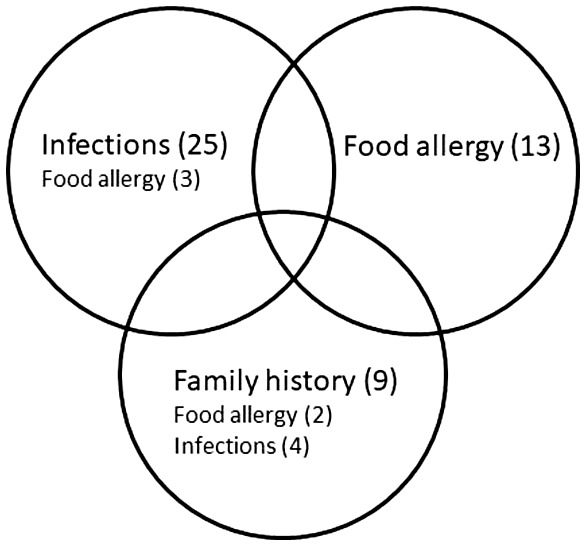
The three overlapping subphenotypes of transient hypogammabulinaemia of infancy (THI). The majority presented with recurrent infections, while others were identified during investigation for food allergy or because of a family history of immunodeficiency. Some patients with a family history were asymptomatic. See text and Table 1 for detailed explanation of numbers for those with overlapping subphenotypes.
Twenty‐five infants and children who later recovered were referred for recurrent or severe infections. Patients in the infections subphenotype experienced predominantly upper respiratory tract infections. Nine had chronic ear infections and four required tympanostomy tubes. One patient suffered meningitis and five had pneumonias. Four had chronic diarrhoea, which resolved. We were able to confirm normal albumin levels in three patients, indicating that loss of IgG through the gut was not the explanation of the hypogammaglobulinaemia. Two had severe viral infections (Table 1) and one patient had chronic candidiasis, but all three had normal T cell function and subsequently improved. Two patients had recurrent staphylococcal furunculosis. The parents of four children stated that infections responded poorly to antibiotics. Only one was hospitalized for infections. One patient whose IgG subsequently normalized developed bronchiectasis, most likely from severe gastroesophageal reflux. Three with predominant infections were subsequently found to have food allergies.
Thirteen were identified during evaluation of adverse reactions to foods. Most patients with a ‘food allergy’ presentation also had reduced globulins or recurrent infections, leading to immunodeficiency testing. These patients had relevant symptoms of type 1 hypersensitivity to foods (n = 11), which was confirmed by specific IgE testing. One had biopsy‐proven eosinophilic oesophagitis and one patient had severe reflux with probable food intolerances but negative allergy tests. These patients were classified as having the ‘food allergy’ subphenotype, as infections were not the dominant feature.
Nine had a family history of immunodeficiency in a first‐degree relative (n = 8) or grandparent (n = 1). Three patients with a family history were asymptomatic, having been identified because of an affected family member. There were three sets of siblings in this group. Two patients with a family history of immunodeficiency had food allergies.
Laboratory data, including vaccine responses and memory B cell data, are shown in Table 2. Isohaemagglutinins were measured in the majority and were considered normal if they had anti‐A or anti‐B antibodies in titres > 1 : 2, provided that they were not AB blood type. Four patients with deficiency of IgG2 and IgG3 subclasses were identified. While three patients recovered, one patient who emigrated had recovered total IgG but had IgG2 deficiency at the time of departure.
The time–course to recovery of total IgG levels is shown in Fig. 1. The longest duration of THI in this series was 32 years. The mean age at recovery in patients presenting with food allergy versus those with infections or a family history is shown in Table 1, and are graphically represented in the Kaplan–Meier plot (Fig. 2).
Ten patients were treated with prophylactic antibiotics pending recovery. Most patients belonged to the infections subphenotype. Of the predominant infection subgroup, three were treated with intravenous immunoglobulin. One was treated for a year, while a second patient was treated till 16 years of age. The oldest patient in this case–series (aged 32 years) was treated for 15 years before stopping treatment.
Several patients continued to have infective symptoms after resolution of THI. Of these, 10 underwent tonsillectomy for chronic tonsillitis, resulting in improvement of infective symptoms in nine. Five patients, four of whom are adults, continue to have mild hypogammaglobulinaemia (range 6–6·9 g/l) after recovery of their IgG into the normal range (7–14 g/l). Two are IgA‐deficient. Given that NHI numbers and SCIG/IVIG are linked in New Zealand, no patient has developed another disorder such as CVID during the three‐decade observation period.
Discussion
This study has examined the natural history of confirmed THI in a large number of patients referred from throughout New Zealand over three decades. Its strength lies in the uniform approach to diagnosis by a single practitioner trained in paediatric and adult immunology, which allowed long‐term follow‐up of patients transitioning from childhood to adulthood. As part of this patient safety audit, the NHI and linked computer records have allowed us to contact all but one patient lost to follow‐up before recovery of their IgG. Five patients did not undertake follow‐up immunoglobulin levels to confirm recovery and were excluded from the analysis.
Some data are incomplete in this retrospective study. Because the observation period spans three decades, some assays, such as immunophenotyping, memory B cells and vaccine responses, were not available in early cases. HIB became part of the NZ primary immunization schedule in 1994 11 and Prevenar® 7 was introduced in 2008. Patients seen prior to this time did not receive these vaccines or antibody assays. Some patients with impaired vaccine responses or reduced switched memory B cells chose not to arrange follow‐up once their IgG levels normalized. The denominator in Table 2 reflects these incomplete data.
In prospective studies of THI, many patients have not normalized their IgG by the end of the study period, which may underestimate the time to recovery 5, 6, 7, 10, 12, 13. In this case–series, approximately 37% of children recovered during the first 4 years of life (Fig. 1). This is a much lower proportion than what has been stated in the literature. This may be a result of the very long observation period of this study. It is apparent that some patients do not recover until the third or even fourth decade (Fig. 1). Given this time–course, prospective studies are unlikely to be practical in determining the natural history of THI.
Earlier in life, a range of potentially severe primary immunodeficiency disorders need to be considered in the differential diagnosis of possible THI. None of the patients were severely lymphopenic and all who were immunophenotyped had normal numbers of B and T cells (results not shown). This would help to identify patients with severe combined immunodeficiency (SCID). Protective vaccine antibody responses and normal in‐vitro lymphocyte proliferation to lectins would also help to exclude most SCID variants. With the advent of newborn screening, most patients with SCID would now be identified with low numbers of T cell receptor excision circles (TRECS) before they present with life‐threatening infections.
A definitive diagnosis of THI is only possible once the IgG levels normalize. THI, as well as other B cell defects presenting in early childhood, were an important consideration in raising the diagnostic age from 2 to 4 years for CVID. From the present study, however, it is apparent that the majority of THI patients do not remit during the first 4 years of life (Fig. 1). Thus, in older children with unresolved hypogammaglobulinaemia, the main differential diagnosis is CVID (Table 3). In 2013 we described new diagnostic criteria for CVID 9. Application of our criteria to patients with symptomatic hypogammaglobulinaemia may help to identify those with CVID with greater precision 14, 15, 16, 17. In accordance with our diagnostic criteria for CVID, we suggest that patients with hypogammaglobulinaemia who do not have a definitive diagnosis are classified as having hypogammaglobulinaemia of uncertain significance (HGUS) 16. Depending on whether or not they have symptoms, they can be designated sHGUS or aHGUS.
Table 3.
THI patients failing to respond to vaccines
| Vaccine failure | Absent vaccine response | THI: age at remission (months) | Vaccines received during THI period |
|---|---|---|---|
| P1 | Dip and Tet | 14 | Dip ×3 Tet ×3 |
| P2 | Dip | 15 | Dip ×3 |
| P3 | Dip | 193 | Dip ×3 |
| P4 | Dip and Tet | 34 | Dip ×3 Tet ×3 |
| P5 | Dip | 198 | Dip ×3 Tet ×3 |
| P6 | HIB | 19 | HIB ×3 |
| P7 | Tet | 60 | Tet ×3 |
| P8 | Pne | 19 | PCV ×3 |
| P9 | Pne | 12 | PCV ×0 |
| P10 | Pne | 28 | PCV ×0 |
Vaccine antibody responses were assayed during the period of hypogammaglobulinaemia. The number of vaccines administered prior to the antibody assay are as stated.
Dip = diphtheria; HIB = H. influenzae type B; Tet = tetanus; Pne = pneumococcal antibodies; PCV = pneumococcal conjugated vaccine; THI = transient hypogammabulinaemia of infancy. P9 and P10 did not respond to the pneumococcal vaccine, but they did not receive Prevenar®.
Application of our CVID diagnostic criteria 9 helped in identifying THI in a patient who had been placed on long‐term IVIG (Table 3) 18. He had symptomatic hypogammaglobulinaemia throughout childhood and had an IgG of 3·6 g/l (7–14 g/l) at age 17 years. He was placed on IVIG until age 32. Following discontinuation of IVIG, he experienced several months of hypogammaglobulinaemia before recovering his IgG into the normal range, but has continued to have mild intermittent hypogammaglobulinaemia. In his case, the normal IgA levels and normal switched memory B cells while on IVIG prompted review of the diagnosis. This observation has implications for other patients being treated with SCIG/IVIG. The original diagnosis should be carefully reviewed, particularly if the patient commenced SCIG/IVIG in early childhood.
Increasingly, Next Generation Sequencing (NGS) is being deployed in patients with CVID, particularly when there is a family history of similar disorders 19. The presence of a causative mutation such as NFKB1 or TCF3 defects would confirm that the patient was suffering from a CVID‐like disorder, which would make spontaneous recovery unlikely 19, 20. Such a mutation would exclude THI. None of the patients in this case–series underwent genetic testing 21. NGS could be considered for those with persisting severe hypogammaglobulinaemia, particularly if symptomatic 19.
We have documented vaccine antibody responses prior to the recovery of the IgG, where available (Table 2). While the majority of THI patients achieved protective levels, a proportion reached the higher thresholds we have advocated in our CVID diagnostic criteria (Table 2). Reaching protective levels prior to recovery of IgG levels will help to exclude SCID variants, while those reaching higher thresholds seen in the general population will make CVID less likely 7.
Although there are difficulties in interpreting vaccine antibody results 14, 22, 23, one of the cardinal features of THI prior to recovery of the IgG has been stated to be normal vaccine responses 24. The current case–series shows that eight patients who subsequently recovered did not achieve protective antibody responses during their period of hypogammaglobulinaemia (Table 2), despite receiving routine immunizations.
Thus, poor vaccine responses early in hypogammaglobulinaemia do not preclude THI. In contrast, all patients who were tested were able to generate isohaemagglutinins. This may be a more reliable marker than protective antibody levels in predicting THI in children with hypogammaglobulinaemia. The absence of isohaemagglutinins is one of our CVID diagnostic criteria, and their presence may suggest THI rather than CVID.
There may be other clues to CVID in children presenting with hypogammaglobulinaemia, such as the presence of persistently low memory B cells 6. Seven of 14 patients who were tested in this series had reduced switched memory B cells (Table 2). All seven subsequently recovered their IgG levels, confirming THI. They did not return for follow‐up to confirm normalization of switched memory B cells. A temporary reduction of memory B cells has been previously described in THI 6. There is, however, variability in the reported numbers of memory B cells in THI 6, 13, 25. Unlike patients with CVID, the memory B cells would be expected to recover in THI patients. Memory B cell assays should be repeated, as we have shown intra‐individual variability in CVID patients on repeat testing 26.
Our study thus confirms that a reduction in switched memory B cells or poor vaccine responses does not preclude a subsequent diagnosis of THI. Patients with persistently reduced switched memory B cells and not recovering their IgG will need to be monitored, as the differential diagnosis includes CVID, various hyper‐IgM (HIM) syndromes and a range of other PIDs.
Most patients with CVID have reduced or absent IgA and/or IgM. Reduced IgA and IgM levels are difficult to interpret in very young children, but only two children had persistent IgA deficiency after recovery of their IgG. Once the IgG recovers, THI is confirmed and CVID is excluded. Prior to recovery of IgG in THI patients, the presence of normal levels of IgA and IgM makes CVID less likely (Table 4). At this time, neither of the two IgA deficient patients have developed CVID.
Table 4.
Features differentiating CVID from THI before recovery
| Clinical features | THI before recovery | CVID |
|---|---|---|
| Infections | ++ | ++++ |
| Allergy | ++ | – |
| Autoimmunity | – | ++ |
| Bronchiectasis | +/– | ++ |
| Chronic sinus disease | + | +++ |
| Inflammatory disorders | – | ++ |
| Sarcoid‐like disorders | – | ++ |
| Family history | + | ++ |
| Laboratory features | ||
| IgG | Recovers | Persistently decreased |
| IgA and/or IgM | Recovers | Persistently decreased |
| IgG subclasses | If decreased, recovers | Persistently decreased |
| Vaccine responses | Impaired in some patients | May be impaired |
| Memory B cells | If impaired, recovers | Impaired |
| Isohaemagglutinins | Present | May be absent |
| Tests for autoimmunity | – | ++ |
| Histology | ? | Abnormal* |
| Genetic abnormalities | – | ++ |
Abnormal histology includes absent plasma cells, granulomatous disorder, nodular regenerative hyperplasia of the liver and nodular lymphoid hyperplasia of the gut.
CVID = common variable immunodeficiency disorder; THI = transient hypogammabulinaemia of infancy; Ig = immunoglobulin.
Even after recovery, four adult patients who had THI continue to have borderline IgG levels, although their symptoms were much improved. Our study spans three decades, but we cannot completely exclude the possibility that some patients might develop CVID later in life. It is known that CVID can present in the seventh or eighth decades of life. Children and adolescents with persistent hypogammaglobulinaemia should be transitioned to adult immunology services for long‐term follow‐up.
As in most case–series, the majority of THI patients (25 of 47) presented with recurrent infections and were then identified as having hypogammaglobulinaemia, and later recovered. Three patients with predominant infections were subsequently found to have food allergies, which may be a reflection of the increasing prevalence of food allergy in infants 27, 28.
The second group, comprising 13 patients presenting with adverse reactions to foods, were co‐incidentally found to have reduced globulin levels, or when investigated for recurrent infections 27. There was a striking difference in the mean time to recovery between those with predominant food allergy versus infections or family history (Table 1). Loss of IgG from the gut consequent to food allergy‐induced enteropathy has been suggested as a possible mechanism 27. However, albumin levels were confirmed as normal in 12 of these patients. Food allergies have been rarely described in CVID patients and IgE levels are generally low, and this may an important clue to THI 29. THI patients with food allergies had the highest rate of asthma, which is a known association (Table 1).
The last group, consisting of nine patients, was tested because of a family history of a PID or THI. Three patients were asymptomatic and were tested because of the family history of immunodeficiency. In general, children with THI identified from their family history have an excellent prognosis, although one of these patients did not recover until 32 years of age (Fig. 2).
It seems likely these subphenotypic patterns will vary, depending on the availability and expertise of clinical services 30. A food allergy clinic is less likely to be referred patients with predominant infections, while an immunodeficiency clinic may be less likely to consult on patients with mostly food allergies, who are subsequently found to have reduced immunoglobulin levels.
As seen here, the male predominance has been noted in multiple case–series, but the exact reason for this is not clear (Table 1). At this time, no mutation has been identified on the X chromosome, which could explain this observation.
Compared to SCID or CVID, THI is a relatively benign disorder. Only one patient had meningitis and five had pneumonia. Two patients had severe viral infections, one with adenovirus and the other with rubella and varicella, which initially suggested a significant T cell defect. Laboratory tests were, however, reassuring and all three recovered their IgG to the age‐specific reference interval. One patient developed bronchiectasis, but severe gastroesophageal reflux was the probable cause, rather than the immune defect. The reflux improved after Nissen funduplication. He had normal vaccine responses and his IgG also recovered. None of the patients had granulomatous lymphocytic interstitial lung disease (GLILD) or autoimmunity, which are hallmarks of CVID (Table 4).
Some authors have advocated either regular or intermittent SCIG/IVIG infusions during the symptomatic phase until recovery of IgG levels 30, 31. This study shows that most THI patients with recurrent infections can be successfully managed with antibiotic prophylaxis until they recover 32. Given the small number (n = 3), we could not determine if IVIG altered the prognostic trajectory of THI. It would seem reasonable to consider temporary SCIG/IVIG treatment in the event of breakthrough infections despite prophylactic antibiotics.
Patients who have made a biochemical recovery from THI may have other explanations for persistent infections. Three patients underwent allergen‐specific immunotherapy later in childhood, which reduced both upper respiratory tract allergies and infections. Two had an adenoidectomy and chronic tonsillitis was identified in 10 patients. Nine improved following tonsillectomy. One of these patients had removal of both adenoids and tonsils. Six of these patients with chronic tonsillitis were older than 4 years. It is possible that the hypogammaglobulinaemia predisposed to recurrent infections leading to chronic tonsillitis, which persisted after spontaneous recovery of IgG. Consistent with this hypothesis is the observation that the majority of patients needing tonsillectomy were from the infection subphenotype of THI. Chronic tonsillitis should be actively excluded in patients with symptomatic hypogammaglobulinaemia as well as those who have recovered from THI.
This case–series is representative of THI patients, as the three subphenotypes of THI described in the literature were identified (Fig. 2) 12, 24. This validates our novel observations, including delayed recovery of IgG into adulthood, the high incidence of chronic tonsillitis and poor vaccine responses in some THI children who subsequently made a full recovery.
This study confirms that THI is misnomer, given that a substantial number of patients recover in adolescence or adulthood 33. THI must be considered in the differential diagnosis of infants, children, adolescents and young adults presenting with hypogammaglobulinaemia.
Disclosures
The authors declare they have no conflicts of interest.
Acknowledgments
This clinical patient safety audit was conducted with the prior written guidance of the Health and Disability Ethics Committee (HDEC) of the NZ Ministry of Health. It complies with HDEC ethics requirements and individual consent was not required for this retrospective patient safety audit. The request to the HDEC also included permission to review clinical data and laboratory results and to publish the findings. The main purpose of this audit was to ensure that patients with hypogammaglobulinaemia had follow‐up laboratory tests to confirm that they had recovered. This was particularly important given the global measles pandemic. As per the terms of the audit and HDEC correspondence, physicians and/or patients caring for patients who were lost to follow‐up before recovery of IgG were contacted to advise follow‐up immunoglobulin testing.
References
- 1. Picard C, Al‐Herz W, Bousfiha A et al Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol 2015;35:696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stiehm RE. The four most common pediatric immunodeficiencies. Adv Exp Med Biol 2007;601:15–26. [DOI] [PubMed] [Google Scholar]
- 3. Gitlin D, Janeway CA. Agammaglobulinemia, congenital, acquired and transient forms. Prog Hematol 1956;1:318–29. [PubMed] [Google Scholar]
- 4. Seidel MG, Kindle G, Gathmann B et al The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract 2019. [DOI] [PubMed] [Google Scholar]
- 5. Keles S, Artac H, Kara R, Gokturk B, Ozen A, Reisli I. Transient hypogammaglobulinemia and unclassified hypogammaglobulinemia: ‘similarities and differences’. Pediatr Allergy Immunol 2010;21:843–51. [DOI] [PubMed] [Google Scholar]
- 6. Moschese V, Graziani S, Avanzini MA et al A prospective study on children with initial diagnosis of transient hypogammaglobulinemia of infancy: results from the Italian Primary Immunodeficiency Network. Int J Immunopathol Pharmacol 2008;21:343–52. [DOI] [PubMed] [Google Scholar]
- 7. Dalal I, Reid B, Nisbet‐Brown E, Roifman CM. The outcome of patients with hypogammaglobulinemia in infancy and early childhood. J Pediatr 1998;133:144–6. [DOI] [PubMed] [Google Scholar]
- 8. Empson M, Sinclair J, O’Donnell J, Ameratunga R, Fitzharris P, Steele R. The assessment and management of primary antibody deficiency. NZ Med J 2004;117:U914. [PubMed] [Google Scholar]
- 9. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol 2013;174:203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kutukculer N, Gulez N. The outcome of patients with unclassified hypogammaglobulinemia in early childhood. Pediatr Allergy Immunol 2009;20:693–8. [DOI] [PubMed] [Google Scholar]
- 11. Ameratunga SN, Lennon DR, Entwistle B, Robinson E, Ameratunga RV. The immunogenicity of Haemophilus influenzae: meningococcal protein conjugate vaccine in Polynesian and non‐Polynesian New Zealand infants. J Paediatr Child Health 1997;33:138–41. [DOI] [PubMed] [Google Scholar]
- 12. Kidon MI, Handzel ZT, Schwartz R, Altboum I, Stein M, Zan‐Bar I. Symptomatic hypogammaglobulinemia in infancy and childhood – clinical outcome and in vitro immune responses. BMC Fam Pract 2004;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cipe FE, Dogu F, Guloglu D et al B‐cell subsets in patients with transient hypogammaglobulinemia of infancy, partial IgA deficiency, and selective IgM deficiency. J Invest Allergol Clin Immunol 2013;23:94–100. [PubMed] [Google Scholar]
- 14. Ameratunga R, Brewerton M, Slade C et al Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ameratunga R, Gillis D, Steele R. Diagnostic criteria for common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2016;4:1017–8. [DOI] [PubMed] [Google Scholar]
- 16. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for CVID. Expert Rev Clin Immunol 2014;10:183–6. [DOI] [PubMed] [Google Scholar]
- 17. Ameratunga R, Becroft DM, Hunter W. The simultaneous presentation of sarcoidosis and common variable immune deficiency. Pathology 2000;32:280–2. [PubMed] [Google Scholar]
- 18. Ameratunga R, Storey P, Barker R, Jordan A, Koopmans W, Woon ST. Application of diagnostic and treatment criteria for common variable immunodeficiency disorder. Expert Rev Clin Immunol 2015;12:257–66. [DOI] [PubMed] [Google Scholar]
- 19. Ameratunga R, Lehnert K, Woon ST et al Review: Diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol 2018;54:261–8. [DOI] [PubMed] [Google Scholar]
- 20. Fliegauf M, L. Bryant V, Frede N, et al Haploinsufficiency of the NF‐κB1 Subunit p50 in common variable immunodeficiency. Am J Hum Genet 2015;97:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ameratunga R, Woon ST, Neas K, Love DR. The clinical utility of molecular diagnostic testing for primary immune deficiency disorders: a case based review. Allergy Asthma Clin Immunol 2010;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beck SC. Making sense of serotype‐specific pneumococcal antibody measurements. Ann Clin Biochem 2013;50:517–9. [DOI] [PubMed] [Google Scholar]
- 23. Ameratunga R, Ahn Y, Steele R, Woon S‐T. The natural history of untreated primary hypogammaglobulinemia in adults: implications for the diagnosis and treatment of common variable immunodeficiency disorders (CVID). Front Immunol 2019. doi: 10.3389/fimmu.2019.01541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whelan MA, Hwan WH, Beausoleil J, Hauck WW, McGeady SJ. Infants presenting with recurrent infections and low immunoglobulins: characteristics and analysis of normalization. J Clin Immunol 2006;26:7–11. [DOI] [PubMed] [Google Scholar]
- 25. Karaca NE, Aksu G, Gulez N, Yildiz B, Azarsiz E, Kutukculer N. New laboratory findings in Turkish patients with transient hypogammaglobulinemia of infancy. Iran J Allergy Asthma Immunol 2010;9:237–43. [PubMed] [Google Scholar]
- 26. Koopmans W, Woon ST, Zeng IS et al Variability of memory B cell markers in a cohort of common variable immune deficiency patients over six months. Scand J Immunol 2013;77:470–5. [DOI] [PubMed] [Google Scholar]
- 27. Walker AM, Kemp AS, Hill DJ, Shelton MJ. Features of transient hypogammaglobulinaemia in infants screened for immunological abnormalities. Arch Dis Child 1994;70:183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crooks C, Ameratunga R, Simmons G et al The changing epidemiology of food allergy – implications for New Zealand. NZ Med J 2008;121:74–82. [PubMed] [Google Scholar]
- 29. Ameratunga R. Assessing disease severity in common variable immunodeficiency disorders (CVID) and CVID‐like disorders. Front Immunol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Breslin ME, Lin JH, Roberts R, Lim KJ, Stiehm ER. Transient hypogammaglobulinemia and severe atopic dermatitis: open‐label treatment with immunoglobulin in a case series. Allergy Rhinol (Providence) 2016;7:69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duse M, Iacobini M, Leonardi L, Smacchia P, Antonetti L, Giancane G. Transient hypogammaglobulinemia of infancy: intravenous immunoglobulin as first line therapy. Int J Immunopathol Pharmacol 2010;23:349–53. [DOI] [PubMed] [Google Scholar]
- 32. Dogu F, Ikinciogullari A, Babacan E. Transient hypogammaglobulinemia of infancy and early childhood: outcome of 30 cases. Turk J Pediatr 2004;46:120–4. [PubMed] [Google Scholar]
- 33. McGeady SJ. Transient hypogammaglobulinemia of infancy: need to reconsider name and definition. J Pediatr 1987;110:47–50. [DOI] [PubMed] [Google Scholar]