

Summary
Systemic lupus erythematosus (SLE) is an autoimmune chronic inflammatory disease of unknown etiology, although genetic and environmental factors appear to contribute to its pathogenesis. Specifically, infectious processes are associated with SLE onset and exacerbation. However, we are far from a complete understanding of the interactions between infectious agents and the host, explaining the interest in gathering updated scientific information on this topic. According to the literature, the pathogens most frequently associated with SLE are viruses, notably human endogenous retroviruses, Epstein–Barr virus, parvovirus B19, cytomegalovirus and human immunodeficiency virus type 1, alongside certain bacterial components that can also trigger activation of the immune system. The mechanisms underlying autoreactivity remain unclear but various explanations have been proposed, including immunological changes responsible for infectious processes or molecular mimicry between host structures and those of infectious agents.
Keywords: bacteria, etiology, infection, systemic lupus erythematosus, virus
Abbreviations
- CMV
cytomegalovirus
- EBV
Epstein–Barr virus
- HERVs
human endogenous retroviruses
- HIV‐1
human immunodeficiency virus
- HTLV‐1
human T‐cell lymphotropic virus
- SLE
systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune and multisystemic chronic inflammatory disease that can affect various organs, including skin, joints, kidneys, lungs and the nervous system, and is characterized by periods of remission and exacerbation.1 It is one of the most common autoimmune diseases, especially among Afro‐American, Hispanic and Asian populations. It can begin at any age, although its onset is more frequent during the first 5 years of life and especially during the next 10 years, mainly among women.2, 3
The pharmacological treatment of SLE is based on four main types of drug: non‐steroidal anti‐inflammatory drugs, antimalarial drugs for skin and joint lesions, anti‐inflammatory corticosteroids, and cytotoxic or immunosuppressant drugs for more severe forms of the disease. Other biological therapies have been developed, targeting either B or T cells.4
The etiology of SLE is unknown, although both exogenous and endogenous factors have been found to influence its pathogenesis. Exogenous factors include estrogenic hormones, ultraviolet light, drug and tobacco consumption, pollution, vaccines, vitamin D deficiency, and various viruses and microorganisms. Endogenous influences include genetic factors, which can in turn be determined by retroviruses that were acquired during evolution or became part of the human genome through the molecular mimicry of some viruses.3 Hence, the scientific evidence suggests that SLE develops through interaction between exogenous and endogenous factors, with infectious diseases playing an important role in genetically predisposed individuals.5, 6
The objective of this study was to analyze the relationship of infectious processes with the onset and development of SLE.
Infection and autoimmunity
The capacity of the immune system to differentiate between host and foreign cells is impaired in autoimmune diseases, leading to the generation of autoantibodies that attack host structures, such as the cell nucleus and especially the DNA, and circulate in the blood. As in other autoimmune diseases, SLE is triggered by the combined action of genetic, immunological and environmental factors. With respect to the latter, there have been studies on the possible role of certain microbial agents (viral, bacterial, parasitic or fungal) (Fig. 1) in the breakdown of immunological tolerance towards host antigens and the subsequent development of immune dysfunction (Table 1), mainly attributable to multiple polyclonal autoantibodies.7 Patients with SLE are known to have a dysfunction in humoral and cellular components of the innate and adaptive immune response. In addition, autoantibodies characteristic of other rheumatic diseases are detected in blood alongside SLE‐specific autoantibodies and have been described as highly useful biomarkers for SLE diagnosis.4 Autoreactivity in patients with SLE is attributable to the activation of T cells, with γδ T cells playing a major pathogenic role, secreting cytokines (especially proinflammatory factors) that act on B cells and thereby promote antibody production. A higher percentage of γδ T cells has been found in skin lesions of patients with SLE comapared with healthy individuals.8, 9
Figure 1.
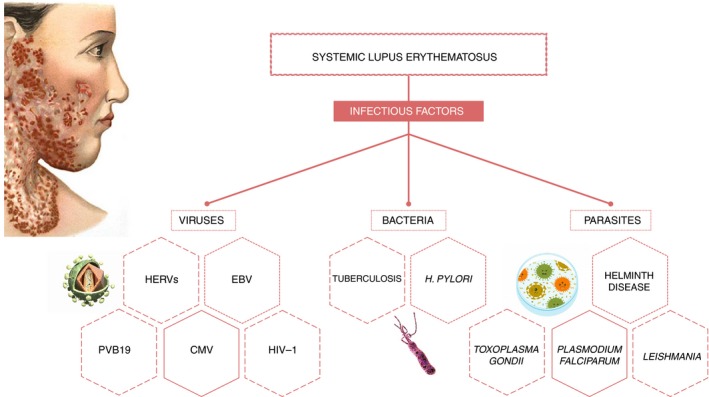
Association of systemic lupus erythematosus and the infectious agents.
Table 1.
References table
| References | Infectious agent | Objective | Methodology | Main results |
|---|---|---|---|---|
| Esposito et al.2 | Virus (Epstein–Barr virus, parvovirus B19, cytomegalovirus and retroviruses) | To define the role of infectious agents in the pathogenesis of SLE. | Review of the literature | Viral infections are highly associated with SLE. Specifically, Epstein–Barr virus, parvovirus B19, retrovirus, and cytomegalovirus infections might play a crucial pathogenic role. |
| Nelson et al.3 | Virus (Epstein–Barr virus and HERVs) | To clarify the role of viruses as a potential pathogenic agent in SLE. | Review of the literature | Viruses have been implicated in the pathogenesis of SLE and other autoimmune diseases. In particular, HERVs are a potential exogenous and endogenous cause of autoimmunity disease. |
| Doaty et al.4 |
|
To update the knowledge of infectious risk in patients with SLE. | Review of the literature | The major treatments used in the management of SLE increase the risk of infection and infectious agents have been shown to play a role in the pathogenesis of SLE. |
| Rigante and Esposito, 30 Frenzel and Hermine7, 30 | Virus (Epstein–Barr virus, Parvovirus B19, cytomegalovirus and retroviruses) | To understand a causative link between infectious agents in the pathogenesis of SLE. | Review of the literature | Viruses such as Epstein–Barr virus, parvovirus B19, cytomegalovirus and retroviruses are implicated in SLE pathogenesis. However, protozoan infections might even protect from autoimmune processes and rescind an ongoing B‐cell activation. |
| Wu et al.8 | – | To define the role of γδ T cells in the pathogenesis of SLE. | Review of the literature | γδ T cells play an important role in SLE, presenting the capacity of secretion of proinflammatory cytokines, immunomodulatory properties and ability to promote antibody production. |
| Romero et al.9 | – | To describe the relationship between cytokines and SLE. | Review of the literature | T cells stimulate secretion of cytokines (IL‐1, IL‐6 and TNF‐α) which, at the same time, trigger B‐cell activation. |
| Manderson et al.10 | – | To explain the connection between complement deficiency the development of SLE. | Review of the literature | Deficient or decreased complement levels can be associated with the development of SLE because of the relation with the physiological waste‐disposal mechanisms of dying cells and immune complexes, or because of the activation of B and T lymphocytes. |
| Lingh et al.11 | Virus | To specify the pathway associated with C1q complement protein and viral infection in SLE. | In vitro study | This paper shows that C1q, but not C3, restrains the response to self‐antigens by modulating the mitochondrial metabolism of CD8+ T cells. C1q deficiency also triggers an exuberant effector CD8+ T‐cell response to chronic viral infection. |
| Valentine et al.12 | – | To explore the CD8 T‐cell function in the germinal center. | In vitro study | CD8 T cells take a CD4 T follicular helper profile in the deficiency of functional regulatory T cells in both the IL‐2‐deficient and scurfy mouse models. Depletion of CD8 T cells mitigates autoimmune pathogenesis in IL‐2‐deficient mice. |
| Burbano et al.13 | – | To determine the role of microparticles and immune complexes as potential immunomodulators in the context of autoimmune responses and diseases. | Review of the literature | Microparticles can configure immune complexes, amplifying the proinflammatory response and tissue damage, being implicated in the pathogenesis of SLE. |
| Shirdel et al.14 | Virus: HTLV‐1 | To assess the correlation between HTLV‐1 infection and SLE. | Cross‐sectional study | There is no association between HTLV‐1 and SLE (P = 0·049). |
| Quaresma et al.15 | Virus: HTLV‐1 | To discuss the immunological changes in HTLV‐1 infection and its association with autoimmune diseases. | Review of the literature | HTLV‐1 modifies the behavior of CD4+ T cells on infection and alters their cytokine production. |
| Draborg et al.16 | Virus: Epstein–Barr virus | To clarify the cytokine response to Epstein–Barr virus in patients with SLE. | Cross‐sectional study | The study shows decreased levels of IL‐12, IFN‐γ, IL‐17 and IL‐6 in Epstein–Barr virus nuclear antigen 1 in patients with SLE. |
| Cui et al.17 | Virus: Epstein–Barr virus | To determine the link between Epstein–Barr virus infection and SLE in China. | Cross‐sectional study | This study shows an association between Epstein–Barr virus infection elevated serum liver enzymes, nasopharyngeal carcinoma and SLE. |
| Pisetky 18 | Virus: Epstein–Barr virus | To assess the connection between Epstein–Barr virus and SLE. | Review of the literature | An increase of anti‐Epstein–Barr virus antibodies has been shown in patients with SLE. Epstein–Barr virus is also associated with other diseases such as rheumatoid arthritis. |
| Yamazaki et al.19 | Virus: cytomegalovirus | To report a case of childhood SLE with concomitant cytomegalovirus infection. | Case report | Ganciclovir treatment is not completely safe, and there are no clear clinical guidelines regarding its use in patients with SLE triggered by cytomegalovirus infection. |
| Guo et al.20 | Virus: cytomegalovirus | To elucidate the human cytomegalovirus gene expression profiles in the peripheral blood mononuclear cells of SLE. | In vitro study | Cytomegalovirus infection involves significant changes in gene expression in different cell types at different stages of infection. |
| Mody et al.21 | Virus: HIV | To determine the effect of SLE and HIV. | Review of the literature | Although HIV infection is associated with a reduction in SLE disease activity, it did not prevent these patients from presenting active SLE. In this study lupus flares recorded in our patients were unrelated to the use of antiretroviral treatment, as a consequence of nuclear factor‐κB2 activation and US31 interaction. |
| Carugati et al.22 | Virus: HIV | To analyze the association between HIV and SLE. | Case report and literature review | The study shows that an autoimmune disease such as SLE might occur despite the loss of immunocompetence caused by HIV infection. Moreover, SLE and HIV infection influence each other possibly through immunological mechanisms determining awkward manifestations. |
| Georges et al.23 | Human parvovirus B19 | To describe the relationship between erythematosus and HIV infection | Case report | The patients showed an elevation of serum IgM antibodies for parvovirus B19 and detection of parvovirus B19 DNA on renal biopsy. |
| Page et al.24 | Human parvovirus B19 | To determine the pathophysiological role of human parvovirus B19 and autoimmune diseases. | Review of the literature | Parvovirus B19 is highly suspected to be involved in rheumatoid arthritis and SLE, caused by the production of numerous autoantibodies and cytokines. |
| Rigante et al.25 | Virus (Epstein–Barr virus, parvovirus B19, cytomegalovirus and retroviruses) | To describe the role of infectious agents in the pathogenesis of SLE, in the last 15 years. | Review of the literature | The increased viral load of Epstein–Barr virus, parvovirus B19, cytomegalovirus, retroviruses and transfusion‐transmitted viruses might be triggers for SLE. Nevertheless, some other infectious agents might exert a protective effect from autoimmunity. |
| Balbi et al.26 | Tuberculosis | To reveal relevant aspects of TB infection in patients with SLE | Review of the literature | Molecular and epidemiological data suggest that TB may be involved in the pathogenesis of SLE. |
| Fischer et al.27 | Toxoplasma gondii | To investigate the seroprevalence and clinical correlation of anti‐T. gondii antibodies in patients with rheumatoid arthritis and SLE. | Cross‐sectional study | The study shows a higher seroprevalence of anti‐T. gondii IgG antibodies in rheumatoid arthritis patients, compared with patients with SLE (P = 0·01). |
| Hirako et al.28 | Plasmodium falciparum | To determine the importance of DNA‐containing immunocomplexes in activation of innate immune cells and pathogenesis of malaria. | In vitro study | A novel report is giving in this study where evidence for the role of the proinflammatory activity of immunocomplexes by demonstrating their ability to induce inflammasome assembly and caspase‐1 activation in human monocytes. |
| Santana et al.29 | Leishmania | To evidence the relationship between SLE and visceral leishmaniasis infection. | Case report | In this study hepatosplenomegaly or isolated splenomegaly was identified in the majority of the reported cases where Visceral leishmaniasis occurred, leading to inappropriate suspicions of SLE or mimicking an SLE flare. |
Abbreviations: HERV, human endogenous retroviruses; HIV, human immunodeficiency virus type 1; HTLV‐1, human T‐cell lymphotropic virus; IFN‐γ, interferon‐γ; IL‐1, interleukin‐1; SLE, systemic lupus erythematosus; TB, tuberculosis; TNF‐α, tumor necrosis factor‐α.
Further research is needed to elucidate the mechanisms underlying the high autoreactivity in patients with SLE. However, it has been hypothesized that autoreactive T‐cell and B‐cell activation is mediated by the molecular mimicry of various infectious agents, leading T cells to attack host tissues because of similarities between host and microbial proteins. ‘Bystander’ or non‐specific activation has also been proposed, in which infection‐activated antigen‐presenting cells interact with and activate autoreactive T cells. This implies that an expansion of epitopes is at the root of autoimmune diseases, with the development of antibodies against a protein generating increasing numbers of antibodies against similar molecules in neighboring organelles or tissues; this can lead to clonal anergy, with lymphocytes becoming incapable of responding against specific antigens.2, 7
C1q, initiator of the classical complement pathway, is known to be associated with SLE development, while C3 deficiency is not a predisposing factor.10 In a mouse model of SLE, Ling et al.11 demonstrated that C1q but not C3 restricts the response to autoantigens by modulating the mitochondrial metabolism of CD8+ T cells, which can in turn propagate autoimmunity. Hence, a C1q deficit may trigger an increased response by effector CD8+ T cells to chronic viral infection, leading to immune disease. This study explores the mechanism by which C1q protects against SLE and examines its role in the capacity of viral infections to perpetuate autoimmunity.
It was recently observed that CD8+ T cells function as T follicular helper cells in the germinal center during infection;12 the CD8+ T cells in the follicle of B cells express B‐cell co‐stimulating proteins and promote B‐cell differentiation and antibody isotope changes. According to these data, involvement of autoreactive CD8+ T cells in autoimmune diseases is in part mediated by the differentiation and functionality of follicular CD4 cells.
A further mechanism is related to immune complex deposits in tissues, which generate an increased inflammatory response that can cause chronic damage in various organs. These immune complexes mainly derive from autoantigens formed by the interaction of microparticles generated during the activation and death of certain cells (e.g. lymphocytes or platelets) with their specific antibodies.13
Viruses as causal agents of SLE
It is necessary to differentiate between endogenous and exogenous viruses related to SLE.
Human endogenous retroviruses involved in SLE
Human endogenous retroviruses (HERVs) are endogenous viruses that have been present in the genome of vertebrates for 30–40 million years.2 They are transmitted from generation to generation and recognized as autoantigens by the immune system. They may be responsible for the loss of immunological tolerance and the triggering of autoimmune diseases such as rheumatoid arthritis or SLE. Human T‐cell lymphotropic virus (HTLV‐1) has mainly been associated with leukemia/HTLV‐1‐related T‐cell lymphoma and with HTLV‐1‐associated myelopathy or tropical spastic paraparesis in adults. However, it can deregulate the immune system and has been associated by some authors with other autoimmune diseases, including SLE, although these associations remain controversial.14 This retrovirus acts by infecting CD4+ T lymphocytes, which may explain the immunological changes in acquired immunity, triggering inflammatory reactions that break immunological tolerance and cause autoimmunity. The underlying mechanisms remain unclear, although some studies have implicated molecular mimicry in autoimmunity.15 The HERVs can be activated by hormones (e.g. estrogens), solar light exposure (UVB), or DNA hypomethylation.3, 4
Epstein–Barr virus as potential causal agent of SLE
Epstein–Barr virus (EBV) is frequently found in humans and is associated with the pathogenesis of Hodgkin lymphoma. It mainly causes infectious mononucleosis, which courses asymptomatically or with mild symptoms at early ages. A large percentage of the population carries antibodies against this virus, which is more frequent in patients with SLE than in healthy individuals and is considered the main causal agent of SLE.7, 16, 17 In addition, this virus remains latent within the cells of individuals who have had the disease and can reactivate at any time, deregulating components of innate and acquired immunity. Epstein–Barr virus is a potent activator of autoreactive B cells, which proliferate and generate antibodies with low affinity for host antigens that attack cells and tissues. With respect to T cells, EBV acts as a superantigen that stimulates numerous T cells and interacts with the major histocompatibility complex class II molecules of antigen‐presenting cells using a pathway other than the normal activation of T cells, through peptide presentation. The numerous activated CD4+ T lymphocytes segregate large amounts of cytokines, generating the chronic inflammatory response characteristic of SLE.3 In this sense recently, Pisetsky18 analyzed the mechanisms by which EBV can trigger this disease.
Cytomegalovirus as possible trigger for SLE
Like EBV, cytomegalovirus (CMV) belongs to the herpes virus family and is considered to trigger SLE, with the observation of CMV DNA and high serum IgM anti‐CMV levels in patients with initial symptoms2, 3, 7 or exacerbation of the disease.4 However, it has not yet been clearly established whether CMV infection triggers SLE or occurs simultaneously with or after SLE onset. In a report on a 12‐year‐old female diagnosed with SLE, Yamazaki et al.19 reported high anti‐CMV IgG levels, signaling previous infection, alongside a high concentration of CMV protein 65, indicating active infection; however, they were unable to establish whether CMV infection was the potential trigger of SLE. Guo et al.20 recently described a role for CMV protein US31 in the induction of inflammation by monocytes–macrophages through the nuclear factor‐κB activation pathway, which contributes to SLE pathogenesis and development.
Human immunodeficiency virus type 1 and its relationship with SLE
The retrovirus human immunodeficiency virus type 1 (HIV‐1) can favor SLE by apoptotic dysregulation and disequilibrium of T helper lymphocytes. A similar mechanism was previously reported for HERVs, with autoimmunity being generated by molecular mimicry.2
Mody et al.21 attributed the simultaneous onset of SLE and HIV‐1 in some patients to a possible reactivation of SLE by antiretroviral therapy through its restoration of the immune system. However, other researchers have reported that autoimmune diseases such as SLE can progress despite HIV‐induced immunosuppression infection and that the infrequent association of SLE and HIV may be due to the targeting by HIV of (lymphoid tissue) lymphocytes T CD4+ cells, which play an important role in SLE pathogenesis.22
Parvovirus B19 and its involvement in SLE pathogenesis
Clinical and serological manifestations are similar between parvovirus B19 and SLE, but parvovirus B19 infection does not complete the four diagnostic criteria for SLE of the American College of Rheumatology,4, 7, 23 permitting its differential diagnosis. Parvovirus B19 infection triggers the production of cytokines and autoantibodies that are also present in the serum of patients with SLE, e.g. rheumatoid factor and antinuclear and antiphospholipid antibodies.24 According to some authors, the production of these antibodies may induce transient autoimmunity. However, the question as to whether parvovirus B19 infection triggers SLE or imitates/exacerbates its clinical characteristics as an autoimmunity adjuvant remains under study.4, 24
Bacterial infections in SLE development
Bacteria have also been implicated in SLE pathogenesis through activation of the immune system by their products (e.g. lipopolysaccharides or nucleic acids). In this complex process, bacteria and bacterial products interact with Toll‐like receptors, stimulating B, T and antigen‐presenting cells, and finally inducing the secretion of proinflammatory cytokines and the production of antibodies.25
Researchers have proposed an autoimmunity hypothesis based on bacterial amyloids, which are aggregates of nanostructured proteins produced within cells and are able to bind to elements of the host cell. They may therefore interact with Toll‐like receptors and generate large amounts of proinflammatory cytokines. However, these phenomena have been observed in mice, and no evidence has been published in humans.4
Molecular and epidemiological data suggest that tuberculosis may be involved in SLE pathogenesis, being more prevalent in patients with SLE and therefore considered a risk factor for its development.26 Helicobacter pylori has also been studied in patients with SLE, who were found to have low rates of seropositivity for H. pylori‐specific antibodies, suggesting that this bacterial infection may reduce the risk of SLE or delay its onset.2, 4, 7
SLE and parasitic infections
Less consistent data have been published on the association between parasitic infections and the development of autoimmune diseases. On one hand, some authors reported a protective role for helminth infections in certain autoimmune diseases, including SLE, attributable to the activation of lymphocytes T helper 2 and secretion of interleukin‐10.25 On the other hand, Toxoplasma gondii, related to rheumatoid arthritis, has been associated with certain autoimmune diseases,27 although it appears to have a protective effect against the progression of SLE.4
The effect on SLE pathogenesis of Plasmodium falciparum, the causal agent of malaria, has also been controversial. Some epidemiological studies have reported a higher SLE prevalence among descendants of people from Africa, where there is a high percentage of malaria‐positive individuals;2 however, a protective effect has been described by other authors, based on the high serum levels in malaria patients of tumor necrosis factor‐α, which activates macrophages, controlling parasitic replication and reducing the likelihood of SLE onset.28
Leishmania infection triggers an immune response in the host that imitates the clinical symptoms of SLE outbreaks, with a large amount of mainly antinuclear antibodies and hypergammaglobulinemia resulting from the polyclonal activation of B cells and molecular mimicry between Leishmania antigens and human ribonucleoproteins.29
Conclusions
In summary, the studies reviewed here report a clear association of infectious processes, especially viral infections, with the onset of SLE. Self‐reactivation can be triggered by molecular mimicry with host structures, attributable to the induction by infection of proinflammatory cytokines alongside the presence of viral or bacterial immunocomplexes or fractions. However, further research is warranted to elucidate the etiology of SLE and the molecular mechanisms involved in order to develop preventive approaches.
Disclosures
The authors declare that there is no conflict of interest. This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
Acknowledgements
We thank the research group BIO277 (Junta de Andalucía) and the Department of Nursing (University of Granada) for the support to carry out this research.
References
- 1. Espinosa G, Cervera R. Lupus eritematoso sistémico In: Cervera R, Espinosa G, Ramos M, Hernández-Rodríguez J, Cid MC, eds. Enfermedades Autoinmunes Sistémicas. Diagnóstico y Tratamiento. Madrid, Spain: Editorial Médica Panamericana S.A., 2014:1–27. [Google Scholar]
- 2. Esposito S, Bosis S, Semino M, Rigante D. Infections and systemic lupus erythematosus. Eur J Clin Microbiol Infect Dis 2014; 33:1467–75. [DOI] [PubMed] [Google Scholar]
- 3. Nelson P, Rylance P, Roden D, Trela M, Tugnet N. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus 2014; 23:596–605. [DOI] [PubMed] [Google Scholar]
- 4. Doaty S, Agrawal H, Bauer E, Furst DE. Infection and lupus: which causes which? Curr Rheumatol Rep 2016; 18:13. [DOI] [PubMed] [Google Scholar]
- 5. Abbas AK, Lichtman AH, Pillai S. Immunologic tolerance and autoimmunity In: Abbas AK, Lichtman AH, Pillai S, eds. Cellular and Molecular Immunology. Philadelphia, PA: Elsevier, 2017:319–43. [Google Scholar]
- 6. Dale RC, Nosadini M. Infection‐triggered autoimmunity: the case of herpes simplex virus type 1 and anti‐NMDAR antibodies. Neurol Neuroimmunol Neuroinflammation 2018; 5:e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rigante D, Esposito S. Infections and systemic lupus erythematosus: binding or sparring partners? Int J Mol Sci 2015; 16:17331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu M, Yang J, Li X, Chen J. The role of γδ T cells in systemic lupus erythematosus. J Immunol Res 2016; 2016:2932531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romero TB, Garcia E, Leal J. Citocinas y lupus eritematoso sistémico. Gac Médica Caracas 2009; 117:196–211. [Google Scholar]
- 10. Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol 2004; 22:431–56. [DOI] [PubMed] [Google Scholar]
- 11. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens N et al C1q restrains autoimmunity and viral infection by regulating CD8+ T cell metabolism. Science 2018; 360:558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valentine KM, Davini D, Lawrence TJ, Mullins GN, Manansala M, Al-Kuhlani M et al CD8 follicular T cells promote B cell antibody class switch in autoimmune disease. J Immunol Baltim Md 1950 2018; 201:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burbano C, Rojas M, Vásquez G, Castaño D. Microparticles that form immune complexes as modulatory structures in autoimmune responses. Mediators Inflamm 2015; 2015:267590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shirdel A, Hashemzadeh K, Sahebari M, Rafatpanah H, Hatef M, Rezaieyazdi Z et al Is there any association between human lymphotropic virus type I (HTLV‐I) infection and systemic lupus erythematosus? An original research and literature review. Iran J Basic Med Sci 2013; 16:252–7. [PMC free article] [PubMed] [Google Scholar]
- 15. Quaresma JAS, Yoshikawa GT, Koyama RVL, Dias GAS, Fujihara S, Fuzii HT. HTLV‐1, immune response and autoimmunity. Viruses 2015; 8:5 10.3390/v8010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Draborg AH, Sandhu N, Larsen N, Lisander Larsen J, Jacobsen S, Houen G. Impaired cytokine responses to Epstein–Barr virus antigens in systemic lupus erythematosus patients. J Immunol Res 2016; 2016:6473204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cui J, Yan W, Xu S, Wang Q, Zhang W, Liu W et al Anti‐Epstein–Barr virus antibodies in Beijing during 2013–2017: What we have found in the different patients. PLoS ONE 2018; 13:e0193171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pisetsky DS. Role of Epstein–Barr virus infection in SLE: gene–environment interactions at the molecular level. Ann Rheum Dis 2018; 77:1249–50. [DOI] [PubMed] [Google Scholar]
- 19. Yamazaki S, Endo A, Iso T, Abe S, Aoyagi Y, Suzuki M et al Cytomegalovirus as a potential trigger for systemic lupus erythematosus: a case report. BMC Res Notes 2015; 8:487 10.1186/s13104-015-1520-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo G, Ye S, Xie S, Ye L, Lin C, Yang M et al The cytomegalovirus protein US31 induces inflammation through mono‐macrophages in systemic lupus erythematosus by promoting NF‐κB2 activation. Cell Death Dis 2018; 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mody GM, Patel N, Budhoo A, Dubula T. Concomitant systemic lupus erythematosus and HIV: case series and literature review. Semin Arthritis Rheum 2014; 44:186–94. [DOI] [PubMed] [Google Scholar]
- 22. Carugati M, Franzetti M, Torre A, Giorgi R, Genderini A, Strambio de Castilla F et al Systemic lupus erythematosus and HIV infection: a whimsical relationship. Reports of two cases and review of the literature. Clin Rheumatol 2013; 32:1399–405. [DOI] [PubMed] [Google Scholar]
- 23. Georges E, Rihova Z, Cmejla R, Decleire PY, Langen C. Parvovirus B19 induced lupus‐like syndrome with nephritis. Acta Clin Belg 2016; 71:423–5. [DOI] [PubMed] [Google Scholar]
- 24. Page C, François C, Goëb V, Duverlie G. Human parvovirus B19 and autoimmune diseases. Review of the literature and pathophysiological hypotheses. J Clin Virol Off Publ Pan Am Soc Clin Virol 2015; 72:69–74. [DOI] [PubMed] [Google Scholar]
- 25. Rigante D, Mazzoni MB, Esposito S. The cryptic interplay between systemic lupus erythematosus and infections. Autoimmun Rev 2014; 13:96–102. [DOI] [PubMed] [Google Scholar]
- 26. Balbi GGM, Machado‐Ribeiro F, Marques CDL, Signorelli F, Levy RA. The interplay between tuberculosis and systemic lupus erythematosus. Curr Opin Rheumatol 2018; 30:395–402. [DOI] [PubMed] [Google Scholar]
- 27. Fischer S, Agmon‐Levin N, Shapira Y, Porat Katz BS, Graell E, Cervera R et al Toxoplasma gondii: bystander or cofactor in rheumatoid arthritis. Immunol Res 2013; 56:287–92. [DOI] [PubMed] [Google Scholar]
- 28. Hirako IC, Gallego‐Marin C, Ataide MA, Andrade WA, Gravina H, Rocha BC et al DNA‐containing immunocomplexes promote inflammasome assembly and release of pyrogenic cytokines by CD14+ CD16+ CD64high CD32low inflammatory monocytes from malaria patients. mBio 2015; 6:e01605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santana IU, Dias B, Nunes EA, Rocha FA, Silva FS Jr, Santiago MB. Visceral leishmaniasis mimicking systemic lupus erythematosus: case series and a systematic literature review. Semin Arthritis Rheum 2015; 44:658–65. [DOI] [PubMed] [Google Scholar]
- 30. Frenzel L, Hermine O. Mast cells and inflammation. Jt Bone Spine Rev Rhum 2013; 80:141–5. [DOI] [PubMed] [Google Scholar]