

Summary
Various reports of disease‐related lung pathologies in common variable immunodeficiency disorder (CVID) patients have been published, with differing histological and high‐resolution computed tomography (HRCT) findings. Data were extracted from the validated Oxford Primary Immune Deficiencies Database (PID) database (1986–2016) on adult, sporadic CVID patients with suspected interstitial lung disease (ILD). Histology of lung biopsies was studied in relation to length of follow‐up, clinical outcomes, HRCT findings and chest symptoms, to look for evidence for different pathological processes. Twenty‐nine CVID patients with lung histology and/or radiological evidence of ILD were followed. After exclusions, lung biopsies from 16 patients were reanalysed for ILD. There were no well‐formed granulomata, even though 10 patients had systemic, biopsy‐proven granulomata in other organs. Lymphocytic infiltration without recognizable histological pattern was the most common finding, usually with another feature. On immunochemistry (n = 5), lymphocytic infiltration was due to T cells (CD4 or CD8). Only one patient showed B cell follicles with germinal centres. Interstitial inflammation was common; only four of 11 such biopsies also showed interstitial fibrosis. Outcomes were variable and not related to histology, suggesting possible different pathologies. The frequent nodules on HRCT were not correlated with histology, as there were no well‐formed granulomata. Five patients were asymptomatic, so it is essential for all patients to undergo HRCT, and to biopsy if abnormal HRCT findings are seen. Internationally standardized pathology and immunochemical data are needed for longitudinal studies to determine the precise pathologies and prognoses in this severe complication of CVIDs, so that appropriate therapies may be found.
Keywords: clinical outcomes, common variable immunodeficiency disorder, interstitial lung diseases, several pathologies, standardization of data
Introduction
Common variable immunodeficiency disorders (CVIDs) are the most frequent symptomatic primary immunodeficiencies and comprise a heterogeneous group of syndromes, characterized by antibody production failure resulting in bacterial infections. However, some CVID patients also develop non‐infectious complications, including enteropathy, polyclonal lymphocytic infiltration, lymphoid malignancy and/or autoimmune disorders, all associated with increased lethality 1, 2, 3, 4, 5, 6.
The chest complications of CVID patients include recurrent chest infections, bronchiectasis and non‐infectious interstitial lung disease (ILD), as extensively reviewed by Baumann et al. 7. All types of chronic lung disease can lead to recurrent hospital admissions and significant morbidity, and are a leading cause of death in these patients, particularly ILD. The issue of disease‐related lung pathologies in CVID is a matter of great concern among clinicians.
Severe infections are largely preventable with regular and appropriate immunoglobulin replacement therapy 6, 8. Bronchiectasis is a common complication of previous pneumonia 5, 9 and can lead to chronic, severe or even fatal infections if not recognized by HRCT at diagnosis of CVID. Polyclonal lymphocytic infiltration and other histological features of ILD, unrelated to acute infection or bronchiectasis, are the focus of this report. Diagnosis of ILD relies on HRCT and biopsy; hence, biopsy is essential for diagnosis and for determining the relevant pathology for treatment selection.
In most large series, HRCT, routinely obtained at diagnosis or in the assessment of chest symptoms, shows lung involvement in 30–60% of patients 9, 10, 11.
Several studies have attempted to characterize patients with CVID and ILD using HRCT or biopsy or both, but the HRCT findings do not appear to correlate with the pathological changes, and furthermore not all patients have been biopsied. Reticular, nodular or ground glass opacities are features found on HRCT 12, 13. Well‐formed granulomata have been described in lung biopsies in some CVID patients 10, 11, as have follicular bronchiolitis, lymphoid hyperplasia and lymphoid interstitial pneumonia. In 2004, in the first series of 67 CVID patients, there was radiographic evidence of ILD in 18 (26%), five of whom had granulomata, four had lymphoid interstitial pneumonitis, two had lymphoid hyperplasia, one had follicular bronchiolitis and one had B cell lymphoma 10. These authors coined the phrase ‘granulomatous‐lymphocytic interstitial lung disease’ (GLILD) to encompass all these features of ILD in CVID patients.
Carrillo et al. have suggested that there is a spectrum of reactive pathologies in the lung, which include peribronchial follicular bronchiolitis with lymphoproliferation followed by lymphocytic interstitial pneumonia (LIP) or nodular lymphoid hyperplasia, which do not necessarily result ultimately in B cell malignancy 14. Maglione et al. agreed with this range of pathologies but pointed out that granuloma were present in only a small proportion, preferring to call this group of conditions ‘pulmonary lymphoid hyperplasia’.
However, the potential for different types of ILD in different CVID populations remains unknown. Imaging findings are variable, and there are usually mixed histological abnormalities within a single biopsy 9, 11, 15, 16, 17, 18. It is our hypothesis that these are likely to represent different pathologies requiring different therapies.
The greatest obstacles for such studies arise from the rarity of the condition as well as the heterogeneity of CVID. CVID diagnosis is one of exclusion of other known causes of antibody failure, and the literature on CVID‐ILD has included heterogeneous groups of patients with unexplained antibody failure and various types of lung complications that may have resulted from other immune defects. Furthermore, the distinction between sporadic, late‐onset CVID patients with ‘infections only’ due to antibody failure and those with further disease complications 1, 5, 19 is important, as some supposed complex ‘CVID’ patients have been shown later to have a combined immune deficiency (CID) 2. ILD is not only found in CVID patients but also in patients with CID such as cytotoxic T lymphocyte antigen 4 (CTLA‐4) haploinsufficiency, lipopolysaccharide (LPS)‐responsive and beige‐like anchor protein (LRBA) deficiency and signal transducer and activator of transcription 3 (STAT‐3) gain‐of‐function (GOF). Interestingly, it is not found in hyperimmunoglobulin (IgM) syndromes or in congenital agammaglobulinaemic patients 9. Possible aetiologies include infection 20, hypersensitivity pneumonitis, immune dysregulation, lymphoma 21 or even autoimmunity. In addition, histological assessments were performed in different centres, using different scoring methods. Although there is now a proposal for an international consensus for HRCT findings 22, as yet there are none for the unified classification of pathological findings. Furthermore, only one study has provided longitudinal data 10, leaving the question concerning pathological processes, prognostic value and management guidelines open 11, 18, 23, 24, 25, 26.
The data presented here come from a longitudinal database of immunodeficiency patients in Oxford (1986–2016). An audit of lung histology was performed on sporadic CVID patients who were seen in the clinic from 1986 to 2016. We looked at histological findings in relation to symptoms, HRCT results and clinical outcomes in order to find some clues as to how to proceed in multi‐national studies and to look for evidence as to whether or not CVID‐ILD is a single disease or has several different pathologies.
Methods
Patient selection
Clinical, laboratory, radiological and histopathological information was extracted from the validated Oxford Primary Immune Deficiencies Database (PID) database 1 for which patients had consented; the search was restricted to those patients meeting the 1999 diagnostic criteria for CVID 27 without affected family members or abnormal circulating T cells at diagnosis. There were 29 such sporadic CVID patients with results from lung biopsy, lobectomy or post‐mortem samples, which had been assessed in the Histopathology Department. Patients were biopsied on the basis of the HRCT findings of suspected ILD. In our experience, lung biopsy based on HRCT findings was straightforward and without complications in these patients. All patients were on therapeutic doses of replacement immunoglobulin 8.
Data extracted
Information for each patient on the length of follow‐up, clinical phenotype, all disease‐related complications, time from CVID diagnosis to suspected ILD, patient outcomes, the different histopathological patterns reported, HRCT findings, symptoms in relation to ILD and all therapeutic interventions were noted. Spirometry results were not included, as these are rarely abnormal in CVID‐ILD 23, 28; other lung function tests included gas transfer in a limited number of patients. Immunocytochemistry reports were also included if available, although this had been performed on only six biopsies for historical reasons.
For reporting of HRCT‐guided transbronchial biopsy results, pathologists were asked to search for findings of ILD in particular, as well as any evidence of pathogens: special stains for pneumocystis, mycobacteria, fungi and polymerase chain reaction (PCR) for viral infections due to Epstein–Barr virus (EBV), cytomegalovirus (CMV) and human herpesvirus 8 (HHV8). Biopsies were reviewed at multi‐disciplinary team meetings of the clinicians and pathologists. Histological findings were analysed and recorded according to the findings of Rao et al. 18 (Table 1a). Thoracoscopy was not used.
Table 1.
Classifications used for histological and HRCT findings
| (a) Histological findings, analysed and recorded according to the findings of Rao et al.18 |
|---|
|
|
|
|
|
|
| (b) HRCT findings, using the classification of Hansell 30 |
|
|
|
|
|
HRCT = high‐resolution computed tomography.
The HRCT findings were reviewed by one radiologist (N.M.) as part of the European HRCT Study Group 29 using Hansell's classification 30 (Table 1b).
The patients with ‘infections only’ CVID phenotype and those who had developed lymphoid malignancy [Mucosa‐Associated Lymphoid Tissue lymphoma (MALToma) or non‐Hodgkin lymphoma (NHL)] were excluded from the final biopsy analysis of correlation with outcomes, to reduce the heterogeneity of CVID conditions 1, and outcomes from malignancies would invalidate the results. The remaining specimens from patients with suspected ILD were all lung biopsies (Table 2). The analysis was performed both with and without patients receiving corticosteroids prior to the lung biopsy, as the histological patterns might have altered the histology 10. Correlations of HRCT findings or biopsy results with patient outcomes were performed using Fisher's exact test.
Table 2.
Patient data on patients with available lung histology, ordered by clinical phenotype, namely infections‐only (OI) 37, 38, malignancy (MAL) or lymphoproliferation
| Number | Age at CVID diagnosis | Time CVID and death | Time: CVID to ILD symptoms | Age at death | Type of lung sample | CVID phenotype | Granulomata in other sites |
|---|---|---|---|---|---|---|---|
| 1 | 6 | 0 | –2 | n.a. | Bx | IO | No |
| 2 | 8 | 0 | –2 | n.a. | Lobectomy | IO | No |
| 3 | 10 | 7 | 3 | 17 | PM | IO | No |
| 4 | 25 | 10 | 11 | 35 | Bx | IO | No |
| 5 | 32 | 11 | 0 | 43 | Bx | MAL (LI to NHL) | No |
| 6 | 50 | 0 | 3 | n.a. | Bx | MAL (MALToma) | No |
| 7 | 61 | 17 | –11 | 78 | Bx | MAL (MALToma to DLCL) | No |
| 8 | 54 | 0 | 1 | 60 | Bx | MAL (LI to NHL) | No |
| 9 | 48 | 12 | 8 | 60 | Bx | MAL (MALToma to NHL) | No |
| 10 | 36 | 9 | 4 | 45 | PM | MAL (LI to NHL) | No |
| 11 | 27 | 19 | 6 | 46 | Bx | CYT and LP | No |
| 12 | 22 | 24 | 17 | 46 | PM | LP | Brain, lymph node |
| 13 | 27 | 27 | 21 | 54 | PM | CYT and LP | Skin, gut |
| 14 | 62 | 6 | 0 | 68 | Bx | LP | No |
| 15 | 38 | 14 | –4 | 52 | Bx | LP | Lymph nodes |
| 16 | 36 | 0 | 0 | n.a. | Bx | LP | No |
| 17 | 26 | 0 | –8 | n.a. | Bx | LP | No |
| 18 | 28 | 8 | 6 | 36 | Bx | LP | Liver, bone marrow |
| 19 | 14 | 0 | –1 | n.a. | Bx | LP | Lymph nodes |
| 20 | 39 | 0 | 1 | n.a. | Bx | LP | Lymph nodes |
| 21 | 55 | 0 | –4 | n.a. | Bx | LP | No |
| 22 | 54 | 0 | 0 | n.a. | Bx | CYT and LP | Liver |
| 23 | 35 | 40 | 25 | 75 | Bx | LP | No |
| 24 | 45 | 21 | 20 | 66 | Bx | LP | Liver, spleen, lymph nodes |
| 25 | 20 | 0 | –1 | n.a. | Bx | LP | Lymph nodes |
| 26 | 34 | 0 | 5 | n.a. | Bx | LP | Lymph nodes |
| 27 | 60 | 0 | –1 | n.a. | Bx | LP | No |
| 28 | 64 | 0 | 0 | n.a. | Bx | LP | No |
| 29 | 30 | 0 | –14 | n.a. | Bx | LP | No |
IO = infections only common variable immunodeficiency disorder (CVID) phenotype; MAL – lymphoid malignancy; LP = lymphoproliferative CVID phenotype; CTY = cytopenia CVID phenotype; Bx = biopsy; PM = post‐mortem findings; LI = lymphocytic infiltration; NHL = non‐Hodgkin lymphoma; DLCL = diffuse large cell lymphoma; n.a. = not applicable. Pretreatment with corticosteroids; included in final histological patterns.
Results
Patients
The ages of the patients at CVID diagnosis are given in Table 2 (only three were aged under 11 years at diagnosis and all three were of the ‘infections only’ phenotype), as are patient follow‐up times from CVID diagnosis to ILD symptoms and or death, type of lung sample for histology and clinical phenotype. Four patients had the ‘infections only’ CVID phenotype and six others developed lymphoid malignancies. The lung specimens from the four ‘infections only’ CVID phenotype patients showed severe infection and there was no evidence of ILD and they were excluded from the final histological versus outcome analysis. Five of the six patients who developed lymphoid malignancies (three NHL and three MALToma) eventually died of high‐grade lymphoma, and one patient remains alive after more than 20 years without progression. Two transformed from MALToma 12 and 17 years later and the other three progressed from lymphocytic infiltration 5, 5 and 11 years later (Table 2). Because patients dying from NHL could not be considered in relation to outcome of ILD, all six of the malignancy patients were excluded from the HRCT and histological analyses. We, and others, have suggested that these patients may have different pathologies and perhaps aetiologies 31. The flow diagram (Fig. 1) shows the exclusion groups and those used in the final analyses of histology or HRCT findings versus outcome.
Figure 1.
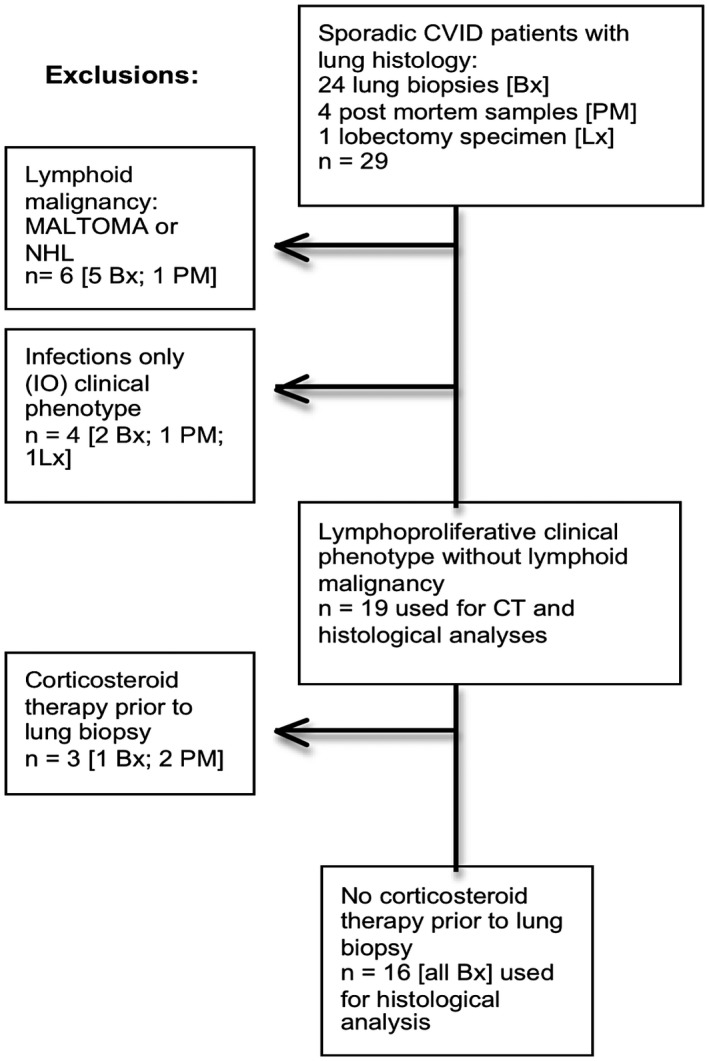
Flow diagram to show material used in CT and histological analyses. Lx = lobectomy; Bx = biopsy; PM = post‐mortem findings; NHL = non‐Hodgkin lymphoma; IO = infections only common variable immunodeficiency disorders (CVID) phenotype.
Absence of granuloma in lung specimens
The most striking finding was the absence of well‐formed granulomata in any of the 19 biopsy reports, despite the pathologists being asked to look for these. There was only one report of ‘poorly formed granuloma’ and one comment on ‘inflammatory aggregates’. Ten of these patients had well‐formed, biopsy‐proven granulomata elsewhere in varying anatomical sites, including: skin, brain, tongue, epiglottis, liver, bone marrow, gut, spleen or lymph nodes. The absence of granulomata in the lungs of these patients with systemic granuloma, in addition to the patients without suggestion of granuloma elsewhere, suggests that this feature may represent a different pathology.
Patterns of histological findings
We were interested to look for patterns of histological findings, in order to discern whether these were different stages of a single process or due to different pathological processes. We hypothesized that there were several pathologies of ILD in CVID, as most biopsies show more than one abnormality (see Table 3 and Fig. 2a,b). One patient had no abnormality suggestive of ILD (she had been investigated due to unexplained weight loss). Figure 2a shows the results based on the 16 biopsies from patients without previous corticosteroid therapy for a cytopenia, although the final analysis showed that exclusion of the three with previous corticosteroids (Fig. 1) had made no difference (Fig. 2a,b).
Table 3.
Histological findings in 19 patients with LP CVID phenotype (using outline from Rao et al. 19; see Table 1)
| Case | Age/sex | Unexplained inflammation | Lymphocytic infiltration | Granulomas | OP | Fibrosis | Outcome | Follow‐up (years) since CVID diagnosis | Prebiopsy treatment with corticosteroids | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bronchiolar | Interstitial | Interstitial fibrosis | Remodelling | No | |||||||
| 2 | 65/M | Y | N | Y | N | Y | Y | N | D, LIP and infection | 6 | No |
| 4 | 46/M | Y | Y | Y | N | N | Y | N | Well, off corticosteroids | 21 | No |
| 5 | 20/M | N | N | Y +E | N | N | Y (subpleural) | N | Well, off Ig | 26 | No |
| 8 | 41/F | Y | Y | Y‐ T | N | N | Y | N | D, lung disease | 14 | No |
| 11 | 36/F | Y | Y | Y | N loose aggregates only | N | Y | N | D, lung/liver fail | 8 | No |
| 13 | 14/M | Y | N | Y | N | Y | N | N | Well, off corticosteroids | 26 | No |
| 14 | 43/M | Y | N | Y | N | Y | N | N | A, on MMF | 16 | No |
| 16 | 55/M | N | N | N | N | N | N | N | A, LIP on corticosteroids | 8 | No |
| 21 | 54/F | N | Y pleural | Y | N | Y | N | N | A, liver transplant after biopsy | 8 | No |
| 22 | 70/M | N | Y | Y‐T | N | N | N | N | D, respiratory failure | 40 | No |
| 23 | 40/F | N | Y | Y‐T | N | N | N | N | D, gut perforation after corticosteroids | 21 | No |
| 25 | 22/M | N | Y | Y+E | Poorly formed | Y | N | N | A, needs MMF | 4 | No |
| 26 | 40/F | N | Y | Y‐B follicles +T | N | N | N | N | A, on MMF | 9 | No |
| 27 | 59/F | N | Y | Y | N | N | N | N | Well | 7 | No |
| 28 | 65/M | N | Y | N | N | Y | Y | Y‐pneumocytes | A, on hydrocortisone for MG | 6 | No |
| 29 | 31/M | Y | N | N | N | N | N | N | Well | 7 | No |
| 9 | 46/F | N | N | Y | N | N | Y | N | D, infective pericarditis | Not included in statistics | YES |
| 19 | 55/M | N | N | N | N | N | N | Y | Fibrosis | Not included in statistics | YES |
| 1 | 44/F | N | Y | Y‐T | N | N | Y | Y honeycombing | D, lung fibrosis | Not included in statistics | YES |
OP = organizing pneumonia; CVID = common variable immunodeficiency disorder; LP CVID = lymphoproliferative CVID phenotype; N = no; Y = yes; A = alive; D = died; E = eosinophils; Y‐T = T cell infiltration; Y‐B = B cell infiltration; LIP = lymphoid interstitial pneumonitis; MMF = mycofenalate mofetil; MG = myasthenia gravis; Ig = immunoglobulin therapy.
Figure 2.
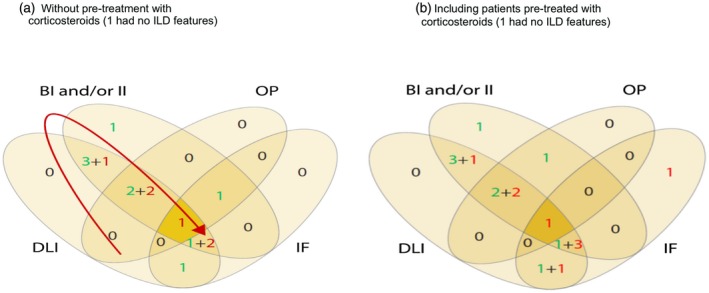
Venn diagrams of overlapping histological findings (a) based on 16 patients without pre‐biopsy corticosteroid therapy, (b) including all 19 patients despite previous corticosteroid therapy in three. The numbers in green represent patients who are alive and those in red are deceased. The red arrow shows the possible progressive changes over time. Note that there is no overlap between organizing pneumonia (OP) and interstitial fibrosis (IF), suggesting possible different pathological processes.
Diffuse lymphocytic infiltration was the most common finding (Table 3 and Fig. 2). Sheets of lymphocytes were reported in 12 of the 16 biopsies, usually with another finding; only four of these 12 had only lymphocytic infiltration. Of the five biopsies on which immunochemistry was performed, diffuse lymphocytic infiltrations without a recognizable histological pattern were due to T cells (Fig. 3a,b). Only one biopsy showed neofollicles with germinal centres; immunochemistry showed that this lymphocytic infiltrate to be CD20‐positive B cells (as expected) with a diffuse T cell infiltrate, indicating a possible different aetiology. Two other biopsies showed B cell follicles without germinal centres but with additional T cell infiltration: CD8+ T cells in one and CD4+ T cells in the other, again suggesting possible different pathologies.
Figure 3.
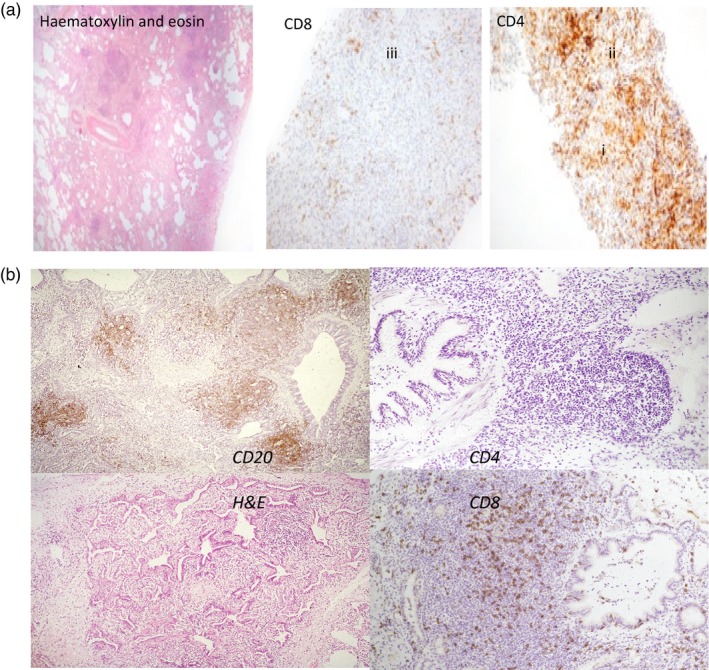
Representative histological specimens. (a) Lung biopsy stained with haematoxylin and eosin to show architecture, particularly loss of alveolar space. Staining with monoclonal antibodies to show CD4 T cell infiltration (i,ii), and very few CD8 T cells (iii). This patient had shortness of breath but had not received previous corticosteroids. (b) Lung biopsy to show CD20 B cells in follicles but few CD4 T cells despite lymphocytic infiltration around the bronchiole. Excess CD8 T cells (using monoclonal antibody) shown outside the follicle and in the interstitium.
Regarding bronchial and or interstitial inflammation, 10 biopsies showed interstitial inflammation (Fig. 2 shows both bronchial and interstitial inflammation together). Of these, nine also showed lymphocytic infiltration and one showed organizing pneumonia; four also showed interstitial fibrosis. Interstitial fibrosis without inflammation was reported in biopsies from six patients, three of whom were dead within 2 years. Bronchiolar inflammation was present in seven biopsies; only one of these did not show lymphocytic infiltration as well.
The six with bronchiolar inflammation and diffuse lymphocytic infiltration showed other features: three biopsies showed organizing pneumonia and four showed interstitial fibrosis; none showed organizing pneumonia alone. This might suggest that either interstitial fibrosis or organizing pneumonia represent late stages in one pathological process in ILD (as suggested by the arrow in Fig. 2) or that the presence of organizing pneumonia indicates a separate condition with a different aetiology.
We wanted to determine if these ILD findings related to outcome, which was possible given the lengthy follow‐up times for these patients.
Patient outcomes
Outcomes in terms of survival after ILD were hugely variable. Overall, 15 of 29 patients died during the follow‐up period (see Table 3). There were two deaths in the ‘infections only’ clinical phenotype: one due to terminal pneumonia and the other unrelated to immunodeficiency. As stated, those patients who developed malignancies were excluded from this analysis. Of the 19 remaining patients used for the analysis, the time between diagnosis of ILD (symptoms or suggestive HRCT) in relation to the initial CVID diagnosis was very variable, ranging from 14 years before CVID diagnosis to more than 20 years after CVID diagnosis (see Table 2). However, four symptomatic patients with the longest time‐periods (17–25 years) were those in whom there was a late biopsy due to lack of availability of /refusal to undergo a biopsy at the time; they were eventually biopsied. Without these four, the range of times from CVID diagnosis to ILD diagnosis was still lengthy, from –14 years to +11 years.
Histopathological findings in relation to patient outcomes
Table 4 shows that, of the six patients whose biopsy showed organizing pneumonia, five remain well (mean follow‐up = 14·7 years; range = 4–26 years); two of these are currently receiving mycofenalate mofetil (MMF), two are on corticosteroid therapy and one receives immunosuppressive therapy following a liver transplant (after the chest biopsy). Of the six patients showing interstitial fibrosis, three died of respiratory failure. Of the three surviving patients, one is well with a normal HRCT despite no longer receiving prednisolone, suggesting recovery of ILD associated with a different aetiology, perhaps in relation to an excess of eosinophils seen in the biopsy. One patient received hydrocortisone for late‐developing myasthenia gravis.
Table 4.
Histological findings versus clinical outcomes by individual pathological finding in 16 patients
| Yes | No | Total | |
|---|---|---|---|
| (a) Organizing pneumonia versus survival (P = not significant) | |||
| Well/A | 5 | 5 | 10 |
| Dead | 1 | 5 (4 died of respiratory failure) | 6 |
| Total | 6 | 10 | 16 |
| Yes | No | Total | |
| (b) Interstitial fibrosis versus survival (P = not significant) | |||
| Well/A | 3 | 8 | 11 |
| Dead | 3 | 2 | 5 |
| Total | 6 | 10 | 16 |
The seven patients whose biopsies showed bronchiolar inflammation had mixed fortunes: of the six patients with additional lymphocytic infiltration (follow‐up mean = 16·7 years; range = 6–26 years), three died of respiratory failure (6, 8 and 14 years after CVID diagnosis), two are off prednisolone (21 and 26 years later) and one remains on MMF. One biopsy was reported with bronchiolar inflammation without excess lymphocytes, and the patient is well without corticosteroids.
Two patients had an excess of eosinophils: the one who recovered (above) and the other who also had lymphocytic infiltration and organizing pneumonia and remains on MMF; he presented with ILD of unknown aetiology (as described previously by the DEFI study group) 32 14 years prior to investigation for CVID.
Chest HRCT findings
One patient had an early HRCT that was not high resolution and another had no abnormal features to indicate ILD on HRCT, but had a biopsy in view of excessive weight loss to rule out lymphoid malignancy (an excess of mesothelial cells was found in this patient, but no features of malignancy or ILD, and she remains under careful follow‐up). These two patients were excluded from the HRCT analysis. Therefore, the analysis of HRCT findings in relation to histology was undertaken on 17 patients, those with the ‘infections only’ phenotype or lymphoid malignancy having already been excluded. Those with prior corticosteroid/immunosuppressive therapy were included, as these therapies were more than 10 years before the HRCT. There were no abnormal chest HRCT findings in the patients with the ‘infections only’ clinical CVID phenotype other than bronchiectasis and/or severe infection.
There was a mixture of ILD findings in 15 of 17 patients, usually more than two findings in any one patient. Six patients had two abnormal HRCT findings, four patients had three abnormal features and five patients had four features. No particular patterns were associated, suggesting possible different specific aetiologies.
The most common feature was lymphadenopathy in the chest (n = 14 of 17), but this is very non‐specific and might indicate infection, malignancy or ILD. Five of these 11 patients also showed marked interlobular septa and six of 11 had ground glass; only one had all three features. Ground glass areas were present on HRCT in nine of 17 patients, of whom only two of nine also had marked interlobular septa and six also showed ‘nodules’ on the images. There was no significant association between the presence of ground glass on HRCT and the finding of organizing pneumonia on histology, although the numbers are small. There was no correlation between frequent nodules on HRCT and granuloma, despite attempts to ensure that the biopsy included the abnormal tissues.
In the 17 patients, the only HRCT abnormality that correlated with poor survival was diffuse infiltrates (P = 0·04) (Table 5). When the other findings were considered, either together or separately, there was no excess of deaths in the positive or negative groups.
Table 5.
Correlations of CT findings of diffuse infiltrates with patient outcome (P = 0·0408) by Fisher's exact test
| Diffuse infiltrates | Yes | No | |
|---|---|---|---|
| Alive | 1 | 10 | 11 |
| Dead | 5 | 3 | 8 |
| 6 | 13 | 19 |
Despite the lack of symptoms, the five asymptomatic patients had variable suggestive findings of ILD on HRCT: lymphadenopathy and ground glass in two patients’ scans; lymphadenopathy, ground glass and nodules in one patient; lymphadenopathy, ground glass, nodules and marked interlobular septa in one other patient.
Symptoms and findings in relation to outcome
We looked to see if there was any correlation between symptoms of ILD and histological findings. Symptoms noted in the 19 patients with suspicion of ILD (without malignancy or infections‐only) included fatigue, mild/moderate shortness of breath (SOB) or weight loss. We did not consider cough, as this is more typical of bronchiectasis, a fairly common complication of pneumonia in CVID patients 1; bronchiectasis was present on biopsy in all four of the infections‐only patients and in one from a long‐standing lymphoproliferative patient.
Five patients had no symptoms; they were investigated on the basis of abnormal findings in the chest HRCT performed at diagnosis of CVID. Of the symptomatic patients, six patients had only one symptom; namely, mild/moderate SOB in one, fatigue in three and weight loss in two patients. Of the three patients with two symptoms, mild/moderate SOB with fatigue presented in two patients and weight loss with fatigue in one. Five patients had all three symptoms. Overall, neither diffuse infiltrates nor ground glass correlated with SOB. Furthermore, there was no correlation between symptoms and survival in the 29 patients with lung samples available (see Table 6).
Table 6.
Lack of correlation between symptoms and survival in all patients with lung samples available
| SOB | Yes | No | |
|---|---|---|---|
| (a) No correlation between mild/moderate shortness of breath (SOB) and survival | |||
| Alive | 3 | 8 + 1* + 2 † | 14 |
| Dead | 4 + 1* | 4 + 4* + 2* | 15 |
| 8 | 21 | 29 | |
| (b) No correlation between lack of symptoms and survival | |||
| Symptoms | Yes | No | |
| Alive | 7 | 4 + 1* + 2* | 14 |
| Dead | 7 + 3* | 1 + 2* + 2* | 15 |
| 17 | 12 | 29 | |
Malignancy;
infections only (IO).
Discussion
We identified ILD in 19 of the 29 sporadic CVID patients undergoing lung histology and/or HRCT investigation for ILD, in the database running for more than 30 years (1986–2016). We examined the clinical, histopathological and radiological features and attempted to correlate these with clinical outcomes.
The most striking histological finding was that there were no well‐formed granulomata in any of the biopsies, in contrast to other centres 15. This absence of granulomata was despite 10 patients having well‐formed, biopsy‐proven, non‐caseating granulomata in other anatomical sites, including skin, brain, tongue, epiglottis, liver, bone marrow, gut, spleen or lymph nodes. This suggests that, in our patients, the aetiology of systemic granulomata, regardless of anatomical site, is separate from that of ILD and that we should avoid the term ‘granulomatous lymphocytic interstitial lung disease’ (GLILD), as it can be confusing if granulomata are not necessarily present in the lung 9, 11.
The other surprising finding was that only three biopsies showed B cells in follicles with diffuse T cell lymphocytic infiltrates. Furthermore, in the limited immunochemistry findings, the T cells in a given biopsy were either CD4‐positive or CD8‐positive. This suggests that there may be several different possible aetiologies. In a recent paper from Mount Sinai, Maglione et al. have suggested at least two different types of CVID‐ILD, one stable and one progressive, with the latter due to B cell activating factor (BAFF)‐driven chronic B cell hyperplasia. They have demonstrated increased levels of BAFF in the blood and lungs of patients with progressive B cell ILD (defined as B cell follicles present in the biopsy as well as increasing serum IgM levels) 33.
Mooney et al. have recently reviewed microbial and other non‐B cell immune aetiologies 34, and suggested different possible causes of ILD in CVID patients. In our study histological findings were variable, and there was no consistent pattern. Previous studies have suggested that these features are part of a spectrum, culminating in lymphoid malignancy 9, 21, and this may be true for B cell ILD, but we provide some evidence for different pathologies, associated with varying outcomes and not always with malignancy, as a possible alternative. For example, the outcomes of those patients with organizing pneumonia were strikingly different from those with interstitial fibrosis or interstitial inflammation. Bronchial inflammation was also present in a third of these biopsies and while this may represent disease progression, as three died within a relatively short time, two others have been without corticosteroid therapy for more than 6 years; these widely different outcomes may reflect different pathologies. In addition, of the two patients with an excess of eosinophils on biopsy, one has recovered entirely. Given the variable outcomes, we suggest several different pathological processes with varying aetiologies and therefore prognoses 9. It is important for future longitudinal studies for patients to undergo lung biopsy at an early stage if HRCT indicates ILD abnormalities, and to undergo repeat biopsy if ILD remains resistant to treatment, especially if lymphoid malignancy is suspected.
The length of time for non‐infectious complications to arise in CVID patients was noted previously in our large multi‐centre study, in which cytopenias or polyclonal lymphoproliferation (other than splenomegaly) were detected both before or after CVID diagnosis, in three‐quarters of patients up to more than 20 years later 5. This was in contrast to enteropathy and hepatomegaly, which were diagnosed subsequent to the CVID diagnosis in almost all, possibly reflecting susceptibility to infection. In the current study there was a similar range for ILD presenting after CVID diagnosis (Fig. 4).
Figure 4.
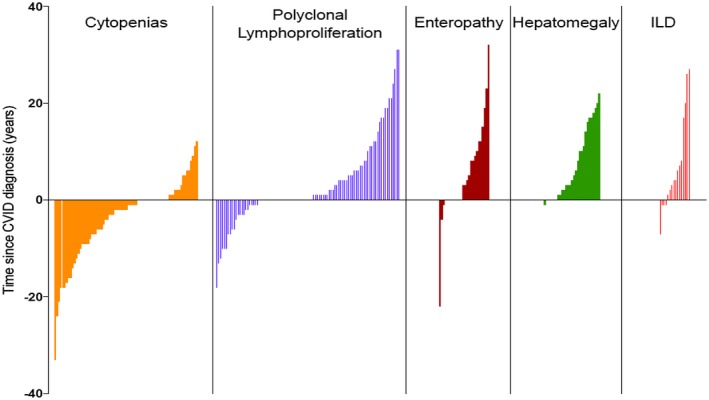
Data from reference 5 to show time from common variable immunodeficiency disorder (CVID) diagnosis to onset of other disease‐related complications in CVID patients in Oxford compared with interstitial lung disease (ILD) patients in this study. Each line represents a single patient.
Almost a quarter of the patients had no symptoms of ILD, highlighting the need for baseline HRCT at diagnosis rather than reliance on clinical findings. Furthermore, there was no consistency in the multiplicity of symptoms indicating that symptoms are unreliable, as reported in a recent series from Italy 22. As well as HRCT at diagnosis of CVID, interim HRCT should be performed as indicated either by new chest symptoms, rising serum beta 2‐microglobulin 1, 13, IgM 33 or reduced gas transfer 5, 19. In our experience and that of others 10, 20, neither spirometry nor chest X‐ray are useful as independent surrogates of significant lung deterioration. HRCT is the most sensitive indication, but should be used sparingly in view of the risks of HRCT radiation dose. Other indicators include unexplained changes in serum immunoglobulin levels, particularly serum IgM and BAFF levels, 33 IgA 20 and mannose‐binding lectin (MBL) 20.
With regard to outcomes, next to lymphoid malignancy the most common cause of death in CVID patients with ILD was lung fibrosis or respiratory failure; interstitial fibrosis on biopsy was a poor prognostic feature. The lengths of time from CVID diagnosis to lymphoid malignancy were extremely variable in our patients (see Table 7); there were patients with long periods since CVID diagnosis in which malignancy has not developed. Whether or not progression to malignancy is inevitable if patients survive other complications long enough is debatable 10, and will need well‐standardized, long‐term data to determine. This small audit of a validated longitudinal database over 30 years’ duration considers outcomes other than malignancy in relation to histology or imaging. As there is no consistency or standardization of histological classifications in the literature, we used the same classification used by Rao et al. 18 for comparison. There needs to be international standardization of pathological findings 35 for all uncommon complications in CVIDs. Fortunately, there has recently been an attempt to standardize the HRCT findings in CVID‐ILD 22. Only sporadic CVID patients with lung samples were included here (abnormal T cells excludes a diagnosis of CVID in our cohort), as most children with antibody failure in our cohort have been found recently to have a CID on next‐generation sequencing studies 36, 37, 38.
Table 7.
Cause of death in 15 CVID patients with interstitial lung disease
| Cause of death | CVID phenotype |
|---|---|
| Marked myocardial hypertrophy and sudden death | IO |
| Terminal bronchopneumonia | IO |
| Chest infection and NHL after benign nodular lymphoid hyperplasia 9 years previously | MALIG |
| DLCL after MALToma 11 years previously | MALIG |
| NHL ILD diagnosis 15 years previously | MALIG |
| NHL ILD diagnosis 12 years previously | MALIG |
| NHL ILD diagnosis 5 years previously | MALIG |
| End stage fibrotic lung | LP and CYTO |
| Streptococcus pneumoniae pericarditis and lung fibrosis | LP |
| Septicaemia and lung fibrosis | LP and CYTO |
| Widespread lymphocytic infiltration; CT showed LIP still | LP |
| Cirrhosis and pulmonary fibrosis | LP |
| Liver failure and ILD | LP |
| Respiratory failure | LP |
| Gut perforation after steroids given subsequent to lung bx | LP |
IO = infections only CVID phenotype; MALIG = lymphoid malignancy; LP = lymphoproliferative CVID phenotype; CTY = cytopenia CVID phenotype. Pretreatment with corticosteroids; included in final histological patterns
We support the conclusions of the UK consensus document, in that not only is biopsy essential but that scores to measure symptoms and pathological findings are needed 17 in all longitudinal long‐term studies. It is not possible to understand the various possible aetiologies, and therefore appropriate treatments, where there are few data on outcomes, no clear consensus on investigation or no agreed guidelines for the interpretation of histological findings 17. Many different immunosuppressive drugs have been used but corticosteroids remain the most efficacious, as found by the French study 32. It is important to find new biomarkers of the disease that can be useful to stratify the patients and new molecules that can be targeted with specific drugs.
As suggested by others 9, we request that the overarching term ‘granulomatous lymphocytic interstitial lung disease’ (GLILD) not be used, as CVID‐ILD may involve several different pathological processes (perhaps in these vulnerable patients from different populations worldwide), and it is important to determine the aetiology of each so that appropriate therapies are found. For this, biopsy of the abnormal tissue, based on HRCT findings, is essential for further investigations 17.
Conclusions
There were no well‐formed granulomata on biopsy in our study.
Most biopsies showed excessive unorganized lymphocyte infiltration of T cells of variable immunophenotype.
Interstitial fibrosis was a poor prognostic finding in most, but not all, patients.
Organizing pneumonia may be a different condition, associated with longer survival.
There appears to be more than one pathological process in terms of histological findings related to outcomes, suggesting different possible aetiologies, e.g. infection, immune dysregulation or incipient malignancy.
HRCT findings do not indicate the precise nature of the ILD, so biopsy, based on the HRCT findings, should be performed so that the abnormal tissue can be examined histologically.
Disclosures
The authors declare no conflicts of interest.
Author contributions
Database curators: M. L., S. P. and H. C.; HRCT analysis: H. C. and N. M.; design of data analysis: H. C., S. P. and C. A.; all authors contributed to the paper.
Acknowledgements
No specific funding was obtained for this study so there are no conflicts of interest. We are grateful to the clinicians of the Department of Clinical Immunology in Oxford, in particular to Drs Siraj Misbah, Rashmi Jain and Jenny Lortan, the trainees at the time, Drs Malini Bhole and Dilani Arnold; also to the nursing and office staff for their care of the patients, to the staff of the Histopathology and Radiology departments for their reports and discussions, but most of all to the patients who agreed for their data to be entered into the Oxford Immunodeficiency Databases.
This article is published with the permission of the Controller of HMSO and the Queen's Printer for Scotland.
References
- 1. Chapel H, Lucas M, Lee M et al Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008; 112:277–86. [DOI] [PubMed] [Google Scholar]
- 2. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract 2013; 1:545–56. [DOI] [PubMed] [Google Scholar]
- 3. Cunningham‐Rundles C. The many faces of common variable immunodeficiency. Hematology Am Soc Hematol Educ Program. 2012; 2012:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malphettes M, Gérard L, Carmagnat M et al, DEFI Study Group . Late‐onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis 2009; 49:1329–38. [DOI] [PubMed] [Google Scholar]
- 5. Chapel H, Lucas M, Patel S et al Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol 2012; 130:1197–8.e9. [DOI] [PubMed] [Google Scholar]
- 6. Resnick ES, Moshier EL, Godbold JH, Cunningham‐Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119:1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baumann U, Routes JM, Soler‐Palacín P, Jolles S. The lung in primary immunodeficiencies: new concepts in infection and inflammation. Front Immunol 2018; 9:1837–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lucas M, Lee M, Lortan J, Lopez‐Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol 2010; 125:1354–60. [DOI] [PubMed] [Google Scholar]
- 9. Schussler E, Beasley MB, Maglione PJ. Lung disease in primary antibody deficiencies. J Allergy Clin Immunol Pract 2016; 4:1039–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous‐lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114:415–21. [DOI] [PubMed] [Google Scholar]
- 11. Maglione PJ, Overbey JR, Cunningham‐Rundles C. Progression of CVID interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunol Pract 2015; 3:941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van de Ven A, de Jongm PA, Hoytema van Konijnenburg DP et al Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol 2011; 165:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maglione P, Overbey JR, Radigan L, Bagiella E, Cunningham‐Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy Asthma Immunol 2014; 113:452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carrillo J, Restrepo CS, Rosado de Christenson M, Ojeda Leon P, Lucia Rivera A, Koss MN. Lymphoproliferative lung disorders:a radiologic‐pathologic overview. Part I: reactive disorders. Semin Ultrasound CT MR 2013; 34:525–34. [DOI] [PubMed] [Google Scholar]
- 15. Chase N, Verbsky JW, Hintermeyer MK et al Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013; 33:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartono S, Motosue MS, Khan S et al Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy Asthma Immunol 2017; 118:614–20. [DOI] [PubMed] [Google Scholar]
- 17. Hurst J, Verma N, Lowe D et al Consensus statement on the definition, diagnosis and management of granulomatous‐lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency disorders (CVID). J Allergy Clin Immunol Pract 2017; 5:938–45. [DOI] [PubMed] [Google Scholar]
- 18. Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency—histologic and immunohistochemical analyses of 16 cases. Hum Pathol 2015; 46:1306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chapel H, Cunningham‐Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol 2009; 145:709–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gregersen S, Mogens Aaløkken T, Mynarek G et al Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol 2010; 104:503–10. [DOI] [PubMed] [Google Scholar]
- 21. Maglione P, Ko HM, Beasley MB, Strauchen JA, Cunningham‐Rundles C. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol 2014; 133:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cereser L, Girometti R, d’Angelo P, De Carli M, De Pellegrin A, ZuianI C. Humoral primary immunodeficiency diseases: clinical overview and chest high‐resolution computed tomography (HRCT) features in the adult population. Clin Radiol 2017; 72:534–42. [DOI] [PubMed] [Google Scholar]
- 23. Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high‐resolution CT scan findings. Q J Med 2002; 95:655–62. [DOI] [PubMed] [Google Scholar]
- 24. Cereser L, Girometti R, d’Angelo P, De Carli M, De Pellegrin A, Zuiani C. Humoral primary immunodeficiency diseases: clinical overview and chest high‐resolution computed tomography (HRCT) features in the adult population. Clin Radiol 2017; 72:534–42. [DOI] [PubMed] [Google Scholar]
- 25. Maarschalk‐Ellerbroek LJ, de Jong PA, van Montfrans JM et al CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol 2014; 34:642–54. [DOI] [PubMed] [Google Scholar]
- 26. Maarschalk‐Ellerbroek LJ. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol 2014; 34:642–54. [DOI] [PubMed] [Google Scholar]
- 27. Conley M, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Clin Immunol 1999; 93:190–7. [DOI] [PubMed] [Google Scholar]
- 28. Touw C, van de Ven AA, de Jong PA et al Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy Immunol 2009; 21:793–805. [DOI] [PubMed] [Google Scholar]
- 29. Schütz K, Alecsandru D, Babar J et al Imaging of bronchial pathology in primary antibody deficiencies: data from the European Chest CT Group. J Clin Immunol 2019; 39:45–54. [DOI] [PubMed] [Google Scholar]
- 30. Hansell D, Bankier AA, MacMahon H, McLoud TC, Mu¨ller, NL , Remy, J . Glossary of terms for thoracic imaging. Radiology 2008; 246:697–722. [DOI] [PubMed] [Google Scholar]
- 31. da Silva SP, Resnick E, Lucas M et al Lymphoid proliferations of indeterminate malignant potential arising in adults with common variable immunodeficiency disorders: unusual case studies and immunohistological review in the light of possible causative events. J Clin Immunol 2011; 31:784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boursiquot J, Gérard L, Malphettes M et al, the DEFI Study Group . Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol 2013; 33:84–95. [DOI] [PubMed] [Google Scholar]
- 33. Maglione PJ, Gyimesi G, Montserrat C et al BAFF‐driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight 2019; 4:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mooney D, Edgar D, Einarsson G, Downey D, Elborn S, Tunney M. Chronic lung disease in common variable immune deficiency (CVID): a pathophysiological role for microbial and non‐B cell immune factors. Crit Rev Microbiol 2017; 43:508–19. [DOI] [PubMed] [Google Scholar]
- 35. Kokosi M, Nicholson AG, Hansell DM, Wells AU. Rare idiopathic interstitial pneumonias: LIP and PPFE and rare histologic patterns of interstitial pneumonias: AFOP and BPIP. Respirology 2016; 21:600–14. [DOI] [PubMed] [Google Scholar]
- 36. Kienzler A, van Schouwenburg PA, Taylor J et al Hypomorphic function and somatic reversion of DOCK8 cause combined immunodeficiency without hyper‐IgE. Clin Immunol 2016; 163:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lopez‐Granados E, Stacey M, Kienzler AK et al A mutation in X‐linked inhibitor of apoptosis (G466X) leads to memory inflation of Epstein–Barr virus‐specific T cells. Clin Exp Immunol 2014; 178:470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dhalla F, Murray S, Sadler R et al Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58‐year‐old man with XMEN disease. J Clin Immunol 2015; 35:112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]