

Summary
Primary Sjögren’s syndrome (pSS) is a chronic inflammatory, autoimmune and systemic disorder commonly associated with dry eyes and a dry mouth. Recently, the hypothetical link between epithelial–mesenchymal transition (EMT)‐dependent salivary gland (SG) fibrosis and chronic inflammatory conditions has been suggested. In this study, we present data demonstrating a negative correlation of the epithelial marker E‐cadherin expression and a positive correlation of mesenchymal vimentin and collagen type I expression with increasing degrees of tissue inflammation in pSS SG specimens. In addition, as it is not clear whether dysregulated cytokines in pSS, interleukin (IL)‐17 and IL‐22 may also contribute to the EMT‐dependent fibrosis process, the effect of IL‐17 and IL‐22 treatment on EMT‐dependent SG fibrosis was evaluated in primary human salivary gland epithelial cells (SGEC) isolated from healthy subjects. Here we present data demonstrating that IL‐17 and IL‐22 can induce SGEC to undergo a morphological and phenotypical transition to a mesenchymal phenotype. In support of this, vimentin and collagen type I were up‐regulated while a decreased expression of E‐cadherin occurs after interleukin treatment, and co‐operation between IL‐17 and Il‐22 was required to induce the EMT.
Keywords: EMT, fibrosis, interleukin, salivary glands, Sjögren’s syndrome
Introduction
An inflammatory process and inflammatory factors are inseparably associated with the epithelial–mesenchymal transition (EMT) by intricate pathways in the chronic inflammatory condition characterizing several diseases. Nevertheless, the cellular mechanisms by which inflammatory factors promote the EMT are largely unknown. Primary Sjögren’s syndrome (pSS) is an autoimmune disease characterized by a severe chronic inflammatory process with the pre‐eminent hallmarks of intense dryness of the eyes and mouth and lymphocytic foci infiltrates in lacrimal and salivary gland (SG) tissue 1, 2. The pathogenic mechanisms are poorly understood, but several lines of evidence show that the disease is the consequence of an altered behaviour of the salivary gland epithelial cells (SGEC), in which the cell phenotype switches from the epithelial to mesenchymal fibroblast‐like cell shape 3, 4. Much evidence indicates that epithelial cells are also a crucial source of myofibroblasts in fibrosis 5, 6 and this transdifferentiation is considered as a specialized process of the EMT programme that may be a pivotal event in the SG fibrosis pathogenesis. Importantly, the chronic inflammatory microenvironment common to fibrotic and inflammatory cells has emerged as a decisive key element in triggering the pathological EMT programme 7, 8.
In recent years great attention has been paid to the chronic inflammatory aspects of pSS, and although inflammation is known to be linked with fibrosis, the mechanisms that lead to this disease and the role of the EMT in the connecting inflammation and salivary fibrosis remain unclear. Among the aberrant cytokines involved in the pathogenesis of several chronic autoimmune inflammatory diseases, interleukin‐ (IL)‐17 and IL‐22 result as important proinflammatory cytokines in systemic lupus erythematosus 9, 10, psoriasis 11, inflammatory bowel diseases 12, 13, rheumatoid arthritis 14, 15, 16, multiple sclerosis, rheumatic arthritis, Crohn’s disease and psoriasis 9, 17, 18. Currently, new evidence indicates that IL‐17 and IL‐22 are implicated in the pathogenesis of EMT‐dependent fibrosis, reporting that IL‐17 triggers the EMT in airway epithelial cells dependent on transforming growth factor (TGF)‐β1 19, 20, 21, and IL‐22 determines a dramatic up‐regulation of TGF‐β, α‐SMA gene expression, laminin, hyaluronic acid and collagen type IV in human hepatic stellate cells, promoting pathological liver fibrosis 22. Interestingly, elevated IL‐17 and IL‐22 levels are detected in SG tissue and serum of patients with pSS, clearly correlated with serological markers of pSS disease or the focus score (FS) 23, 24, 25, 26, 27, 28, suggesting their key participation in this disease pathogenesis 26, 29. As the EMT is induced by a dramatic up‐regulation of cytokines and has an important role in promoting the progression of fibrosis, it would be interesting to determine whether IL‐17‐/IL‐22‐dependent EMT occurs in pSS SG tissue and to correlate the EMT with the severity of the pSS inflammatory grade. Results obtained established that EMT‐related marker expression is correlated with an increased level of inflammation and pSS disease severity; furthermore, IL‐17 and IL‐22 treatment can promote the EMT in healthy SGEC in vitro, as evidenced by a fibroblast‐like morphological appearance through the loss of the epithelial marker E‐cadherin and augmented mesenchymal markers vimentin and collagen type I. The current research offers the exciting prospect of discovering IL‐17 and IL‐22 as new promoters of the EMT, and a plausible link between inflammation and fibrosis.
Material and methods
Reagents and antibodies
Recombinant reagents and antibodies used were as follows: human IL‐17 and recombinant human IL‐22 (both from Sigma‐Aldrich, St Louis, MO, USA), mouse anti‐human β‐actin monoclonal antibody (mAb) clone AC‐ 15 (1 : 100; Sigma‐Aldrich), mouse anti‐human E‐cadherin mAb (1 : 100; Dako, Santa Clara, CA, USA), mouse anti‐human vimentin mAb (1 : 100; ThermoFisher Scientific, Middletown, VA, USA) and rabbit anti‐human collagen type I polyclonal antibody (pAb) (1 : 500; ThermoFisher).
pSS biopsy selection
The Department of Pathology, University of Bari Medical School, selected 36 pSS minor salivary glands (MSG) biopsies (patients aged 63·4 ± 3·7 years) according to the 2016 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for pSS 30, based on evaluation of the following items: anti‐SSA/Ro antibody positivity and focal lymphocytic sialadenitis with a focus score of ≥1 foci/4 mm2, each scoring 3, an abnormal ocular staining score of ≥5 (or van Bijsterveld score of ≥4), a Schirmer’s test result of <5 mm/5 min and an unstimulated salivary flow rate of <0·1 ml/min, each scoring 1. For the diagnosis of pSS, all sections were randomly analyzed by two experts blinded to the clinical and molecular data. Each sample was independently evaluated, and any discrepancies were resolved by consensus. The pSS MSG biopsy specimens selected were assigned to three groups with different degrees of inflammation: SS‐I, mild MSG lesions (n = 8, 1+ biopsy score), SS‐II, intermediate MSG lesions (n = 8, 2+ biopsy score) and SS‐III, severe MSG lesions (n = 8, biopsies of 3+/4+ score). In all SS patients, the biopsy focus score (lymphocytic foci/4‐mm2 of tissue) was ≥1. Healthy subjects (n = 6) analysed for an abnormal salivary function and suspected pSS, but whose biopsy and other diagnostic tests were normal, were enrolled as controls. Participants gave informed consent to take part in the study, which followed the tenets of the Declaration of Helsinki and was approved by the local Ethics Committee. Healthy labial MSG were harvested according to the explant outgrowth technique 31, and both biopsy specimens and SGEC cultures were prepared as described below.
Histochemistry
Serial 3‐μm sections of healthy and pSS formalin‐fixed, paraffin‐embedded MSGs tissues were used for immunohistochemical staining. For this experimental procedure, paraffin‐embedded MSGs tissues from non‐specific sialadenitis were used as positive control. While rehydrating the deparaffinized sections in graded alcohol, the slides were immersed for 1 h in 70% ethanol supplemented with 0·25% NH3, and rehydration was resumed by immersion in 50% ethanol for 10 min. The slides were then washed in phosphate‐buffered saline (PBS) (pH 7.6 3 × 10 min) and immersed in ethylenediamine tetraacetic acid (EDTA) buffer (0·01 M, pH 8.0) for 20 min in a waterbath at 98°C to unmask antigens. The sections were immunolabelled according to the following procedure: blockade of endogenous peroxidase by treatment with 3% hydrogen peroxide solution in water for 10 min at room temperature (RT), rinsing for 3 × 10 min in PBS, pH 7.6, preincubation in non‐immune donkey serum (Dako LSAB kit; Dako, Carpenteria, CA, USA) for 1 h at RT and incubation overnight at 4°C with primary antibodies against human E‐cadherin and human vimentin at the dilutions indicated above. The sections were washed for 3 × 10 min in PBS and then incubated with the relative secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) diluted 1 : 200 in PBS for 1 h at RT, rinsing for 3 × 10 min in PBS; incubation with the streptavidin–peroxidase complex (Vector Laboratories, Burlingame, CA, USA) for 1 h at RT, incubation with the chromogen 3,3‐diaminobenzidine tetrahydrochloride (DAB) (Vector Laboratories) for 10 min at RT, then counterstaining with haematoxylin (Merck Eurolab, Dietikon, Switzerland). Negative controls of the immunoreactions were performed by replacing the primary antibody with donkey serum diluted 1 : 10 in PBS. After the addition of the secondary antibody, no specific immunostaining was observed in the negative controls (data not shown).
Histological procedures for collagen expression analysis
To detect collagen type I expression, serial 3‐μm sections of healthy, pSS and non‐specific sialadenitis formalin‐fixed, paraffin‐embedded MSGs tissues were stained with Masson’s trichrome stain.
Aperio digital analysis
Digital images of immunohistochemically (IHC)‐stained healthy and pSS slides were obtained using a whole slide scanner Aperio Scanscope CS2 (Leica Biosystems, Nussloch, Germany), and an archive of the digital high‐resolution images was created. Digital slides were analyzed with Aperio ImageScope version 11 software (Leica Biosystems, Wetzlar, Germany) at ×10 magnification and 10 fields with an equal area were randomly selected for analysis at ×40 magnification 32. The expression of E‐cadherin and vimentin was assessed with the positive pixel count algorithm embedded in the Aperio ImageScope software and reported as positivity percentage, defined as the number of positively stained pixels on the total pixels in the image. This approach allows a reliable automatic estimation of the amount of staining in the tissue and reduced variability associated with human error 33.
Cell culture and cytokine stimulation
Healthy MSGs, obtained by the explant outgrowth technique from the lower lip 31, were dissociated by enzymatic and mechanical means into suspensions of single cells and plated onto a culture flask. Healthy dispersal SGEC were resuspended in McCoy’s 5a modified medium supplemented with 1% heat‐inactivated (56°C for 30 min) fetal calf serum (FCS) (to avoid excessive growth during treatment with interleukins), 1% antibiotic solution, 2 mM L‐glutamine, 50 ng ml–1 epidermal growth factor (EGF; Promega, Madison, WI, USA), 0·5 μg ml−1 insulin (Novo, Bagsvaerd, Denmark) and incubated at 37°C, 5% CO2 in air. The contaminating fibroblasts were selectively removed with 0·02% EDTA treatment. Immunocytochemical confirmation of the epithelial origin of cultured cells was routinely performed 34. Subconfluent healthy SGEC were stimulated with IL‐17 (50 ng/ml) and IL‐22 (50 ng/ml) alone and in combination in the growth medium for 24–72 h and then harvested for future analyses. After 24 h, reverse transcriptase–polymerase chain reaction (RT–PCR) and quantitative real‐time PCR (qRT–PCR) for E‐cadherin, vimentin and collagen gene expression were performed; after 48 h of stimulation, E‐cadherin, vimentin and collagen protein expression was detected by Western blot and flow cytometry. After 72 h from treatment, SGEC morphological changes were evaluated. All experiments were performed in triplicate and repeated three times.
Assessment of morphological EMT‐related changes in SGEC
To confirm the activation of IL‐17‐ and IL‐22‐dependent EMT‐dependent fibrosis in pSS, in‐vitro analysis of the morphological changes indicative of the EMT was performed in 72‐h‐treated SGEC with IL‐17 (50 ng/ml), IL‐22 (50 ng/ml) or the combination of IL‐17 + IL‐22. Changes in cell morphology were assessed under phase‐contrast light microscopy.
Gene transcripts amplification by RT–PCR and qRT–PCR
Total RNA was extracted from untreated and variously treated healthy SGEC following the manufacturer’s protocol and reverse‐transcribed as previously reported 35, 36. Total RNA from untreated and variously treated healthy SGEC was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First‐strand cDNA was synthesized by M‐MLV reverse transcriptase (Promega, Madison, WI, USA) with 1 μg each of DNA‐free total RNA sample and oligo‐(dT)15 (Life Technologies, Grand Island, NY, USA). Equal amounts of cDNA were subsequently amplified by PCR in a 20‐μl reaction mixture containing 2 μM of each sense and anti‐sense primer, PCR buffer, 2·4 mM MgCl2, 0·2 mM each dNTP, 10 μl of transcribed cDNA and 0·04 U/μl Taq DNA polymerase. The primers used to amplify cDNA fragments were as follows: E‐cadherin, forward 5′‐ TTCCCTGCGTATACCCTGGT‐3′ and reverse 5′‐GCGAAGATACCGGGGGACACTCATGAG‐3′; vimentin, forward 5′‐AGGAAATGGCTCGTCACCTTCGTGAATA‐3′ and reverse 5′‐ GGAGTGTCGGTTGTTAAGAACTAGAGCT‐3′; and collagen type I, forward 5′‐ GAGCGGAGAGTACTGGATCG‐3′, reverse 5′‐TACTCGAACGGGAATCCATC‐3′.
Amplification products were electrophoresed on 1·5% agarose gel containing ethidium bromide and visualized under ultraviolet transillumination. For qRT–PCR, forward and reverse primers for all the genes tested and the internal control gene β2 microglobulin (part no. 4326319E; β2M) were purchased from Applied Biosystems (Assays‐On‐Demand; Applied Biosystems, Foster City, CA, USA). Each qPCR reaction was run in triplicate on ABI PRISM 7700 sequence detector (Applied Biosystems). Relative gene mRNA expression ratios were calculated using the ΔΔCt formula where Ct is the threshold cycle time value. The different expression of mRNA was deducted from 2−ΔΔ Ct.
Data evaluation and sequence analysis
mRNA quantification data were based on the average of a set of three independent experiments, and densitometric analysis was performed by gel image software (Bio‐Profil Bio‐1D; ltf Labortechnik GmbH, Wasserburg, Germany). The intensity values for each band relative to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (internal control for lane loading) were determined and expressed as arbitrary units. The identity of each PCR product was confirmed by the size, and the purified products were directly sequenced during the gene‐specific forward or reverse primers.
Western blot analysis
Lysates obtained from untreated and variously treated healthy SGEC were subjected to sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE), and proteins (30 μg/lane) with prestained standards (BioRad Laboratories, Hercules, CA, USA) were loaded on SDS‐polyacrylamide precast gels. After electrophoresis, the resolved proteins were transferred from gel to nitrocellulose membranes. A blot buffer [20 mM Tris/150 mM glycine, pH 8, 20% (v/v) methanol] was used for gel and membrane saturation and blotting. The blot conditions were the following: 200 mA (constant amperage), 200 V for 110 min. Blots were then blocked by PBS pH 7.2 with 0·1% (v/v) Tween 20, 5% w/v non‐fat dried milk for 1 h and washed three times with 0·1% (v/v) Tween 20–PBS 1 × (T‐PBS). Membranes were incubated for 90 min with mouse anti‐human E‐cadherin mAb, mouse anti‐human vimentin mAb and rabbit anti‐human collagen type I pAb, and subsequently for 30 min with the relative secondary conjugated antibody–horseradish peroxidase (HRP) (Santa Cruz Biotechnology). Proteins recognized by the antibodies were revealed using chemiluminescence luminal reagent (Santa Cruz Biotechnology), according to the manufacturer’s protocol. Immunoblots were then incubated with stripping buffer (Thermo Scientific, Middletown, VA, USA) and probed with mouse anti‐human β‐actin mAb clone AC‐15 as control.
Flow cytometry
Healthy and variously treated pSS SGEC were analysed for E‐cadherin, vimentin and collagen I expression in flow cytometry. Cells were incubated with the anti‐human E‐cadherin, vimentin and collagen I as primary antibodies and with secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen). Protein expression was analyzed by a Becton‐Dickinson (BD, Becton‐Dickinson, Kelberg, Germany) FACS Canto™ II flow cytometer and BD FACS Diva software. Indirect staining using only the secondary antibody was performed as negative standard, indicated in the figures as ‘negative’. The events were acquired, and data were analyzed as mean fluorescence intensity (MFI) using the FACS Diva software package (BD Biosciences). MFI values were compared with those observed in healthy untreated SGEC. This procedure was repeated for at least three passages.
Statistical analysis
Normalized data of mRNA, protein and enzymatic activity were processed to calculate mean values ± standard error (s.e.). Differences between parameters were evaluated using Student’s t‐test. Spearman’s correlation coefficients were calculated to evaluate associations between EMT molecular marker expression and pSS inflammatory grade. Differences were considered statistically significant at P < 0·05.
Results
Immunohistochemical findings
EMT activation in the pSS minor SG bioptic specimen groups with different degrees of inflammation, SS‐I, SS‐II and SS‐III was statistically evaluated by the immunohistochemical determination of E‐cadherin, vimentin and collagen type I expression, in comparison with healthy SGs tissues. Labelled vimentin and E‐cadherin proteins in the healthy and pSS biopsies quantification performed by the Aperio ScanScope was submitted to a computerized morphometric analysis software to measure pixel intensities (Fig. 1a,b); in this analysis the expression of vimentin was significantly increased (P < 0·01), while E‐cadherin expression was decreased in pSS. Collagen expression was assessed using Masson’s trichrome staining to evaluate it as a possible additional marker of fibrosis in pSS (Fig. 2). Quantifying the intensity of blue, fibrotic areas had a rich fibrillar collagen level, and the total collagen density was lower in healthy normal SG tissue (Fig. 2a) than in pSS specimens (Fig. 2b–d). Spearman’s rank correlation analysis of the histological fibrosis markers results and the inflammation grade demonstrated that there were significant differences in the distribution of fibrosis markers and inflammation grade between healthy subjects and the SS‐I, SS‐II and SS‐III groups. In fact, a significant inverse association between membranous staining for E‐cadherin and the histological inflammatory grade of pSS was reported (r = –0·234 for grade I, r = –0·387 for grade II and r = –0·574 for grade III; P < 0·001). Conversely, high cytoplasmic vimentin expression was closely associated with a high inflammatory degree (for vimentin, r = 0·20 for grade I, r = 0·54 for grade II and r = 0·674 for grade III; P < 0·01). Similarly, investigating the association of collagen content with the increased FC with Spearman’s correlation analysis, the collagen type I expression and the pSS inflammatory grade was significantly positively correlated (r = 0·29 for grade I, r = 0·47 for grade II and r = 0·774 for grade III; P < 0·001). In all these experimental procedures paraffin‐embedded MSGs tissues from non‐specific sialadenitis were used as positive control (Fig. 1e,j, Fig. 2e; in Fig. 1b, related to the Aperio ScanScope, quantification of this control was indicated as non‐SS).
Figure 1.
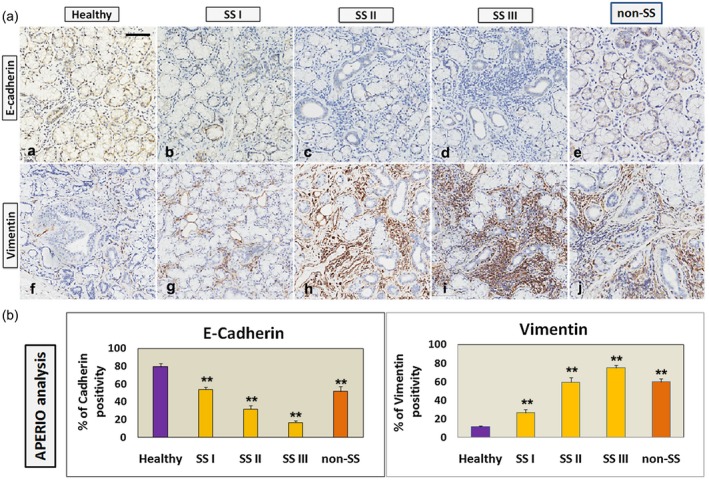
(a) Immunohistochemical localization of E‐cadherin (a–d) and vimentin (e–h) in healthy (a,e) and primary Sjögren’s syndrome (pSS) (b–d,f–h) salivary gland (SG) tissues classified according to different grades of inflammation (I, II, III). Paraffin‐embedded minor salivary glands (MSGs) tissues from non‐specific sialadenitis (non‐SS) were used as positive control (Fig. 1e,j). A significantly (P < 0·01) increased expression of vimentin and a reduction of E‐cadherin in pSS was evident, correlated with the increased inflammatory grade and FC. Brown staining shows positive immunoreactions; blue staining shows nuclei (a–h); bar = 20 μm. (b) All images were scanned and analyzed with the Aperio ImageScope instrument. The expression of labelled vimentin and E‐cadherin proteins in the healthy and pSS (grades I, II, III) biopsies obtained by the Aperio ScanScope was also quantified using computerized morphometric analysis software and expressed in terms of pixel/intensities. Histograms represent immunohistochemistry signal quantification of E‐cadherin and vimentin positivity performed by the Aperio ImageScope software on healthy and pSS (grades I, II, III) SGs sections. The positive control related to Aperio ScanScope quantification of non‐specific sialadenitis specimens was indicated as non‐SS. Absorbance measurements performed by Aperio confirmed the microscope observations, showing a significant (**P < 0·01) inverse association between membranous staining for E‐cadherin and the histological inflammatory grade of pSS, and high cytoplasmic vimentin expression, closely associated with a high inflammatory pSS degree, respectively (**P < 0·01).
Figure 2.
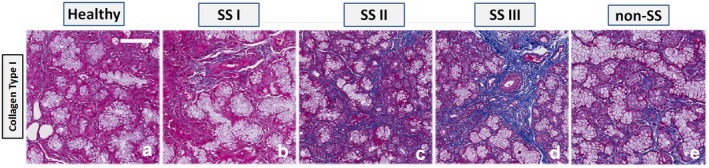
Histochemical analysis of collagen fibre deposits in salivary gland (SG) tissues from healthy controls (a) and primary Sjögren’s syndrome (pSS) (b) patient tissues classified according to different grades of inflammation (I, II, III). Paraffin‐embedded minor salivary glands (MSGs) tissues from non‐specific sialadenitis (non‐SS) were used as positive control (Fig. 2e). The slides were subjected to Masson’s trichrome staining to analyse collagen deposits (blue). At ×20 magnification, a remarkable deposition of collagen fibres were seen in the interstitial area around acinar and ductal cells in pSS SGs tissues (b–d) compared to controls (a). The figure shows an increased collagen deposition from low (b) to high (d) grades of inflammation. Scale bar = 20 μm.
Effects of IL‐17 and IL‐22 on the epithelial cell phenotype of healthy SGEC
As reported in the literature, IL‐22 and associated cytokine IL‐17 are highly present in the inflamed salivary glands of SS patients, both correlated with the degree of tissue inflammation 24. We therefore assessed the effects of IL‐17 and IL‐22 treatment in EMT‐induced fibrosis in human SGEC, in‐vitro treating SGEC with IL‐17, IL‐22 or IL‐17 + IL‐22 and conducting an analysis of epithelial cell phenotype modifications. As shown in Fig. 3, under the phase‐contrast microscope, IL‐17 or IL‐22 alone trigger a programme that involves the conversion of epithelial cells to a mesenchymal phenotype having a discernible effect on the morphology of cultured primary healthy SGEC (Fig. 3a–d). However, we noted that 3 days of IL‐17 + IL‐22 stimulation preferentially promoted the most complete change to a mesenchymal phenotype in cultures of healthy SGEC (Fig. 3d) that convert to spindle‐shaped cells.
Figure 3.
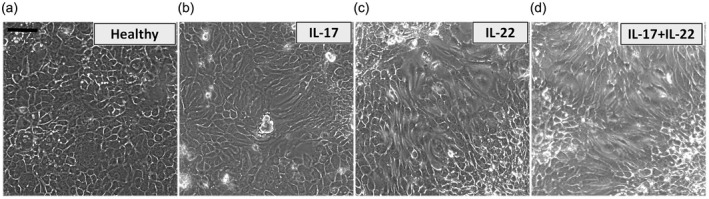
Morphological changes in in‐vitro‐cultured salivary gland epithelial cells (SGEC) treated with interleukin (IL)‐17, IL‐22 or IL‐17 + IL‐22. SGEC derived from salivary gland (SG) biopsies of healthy donors were incubated with 50 ng/ml interleukins for 72 h. (a) Untreated SGEC show a clear pebble‐like shape and cell–cell adhesion. IL‐17‐ and IL‐22‐treated cells (b,c) show a decrease in cell–cell contacts and a more elongated morphological shape. The most evident morphological changes toward a mesenchymal phenotype were observed in SGEC subjected to double stimulation with IL‐17 + IL‐22 (d) that showed a high proportion of spindle‐shaped cells. Changes in cell morphology were assessed under phase contrast light microscopy. Bar = 20 μm.
IL‐17 and IL‐22 induce mesenchymal gene expression in primary healthy SGEC
Cytokine‐mediated signalling mechanisms regulating EMT‐dependent fibrosis in SGs have been little addressed in the literature to date 37. To determine whether the increased expression of IL‐17 and IL‐22 recently linked to the pathogenesis of pSS tissue inflammation was involved in the regulation of epithelial and mesenchymal markers, the incubation of cultured healthy SGEC with IL‐17 and IL‐22 alone or in combination for 2 days was performed. Epithelial E‐cadherin and mesenchymal vimentin and collagen type I gene expression was determined by RT–PCR following 48 h of stimulation with IL‐17, IL‐22 or IL‐17 + IL‐22 (Fig. 4). All genes examined showed dramatic changes; in fact, stimulation promoted a significantly (P < 0·01) marked increase in the expression of vimentin and collagen type I mRNAs, while stimulation with IL‐17 and IL‐22 alone reduced the E‐cadherin gene transcription rate (Fig. 4a,b). Double stimulation with IL‐17 + IL‐22 had an additional effect on the expression of E‐cadherin mRNA (Fig. 4a,b) (P < 0·01). The levels of expressed genes were quantified by RT–PCR (Fig. 4c) and confirmed these results, demonstrating that the majority of healthy SGEC were negative for vimentin and collagen type I mRNAs but, under the IL‐17 and IL‐22 treatment condition, collagen type I and vimentin mRNA expression was induced. The effect was more evident in response to IL‐17 and IL‐22 double stimulation (Fig. 4a–c).
Figure 4.
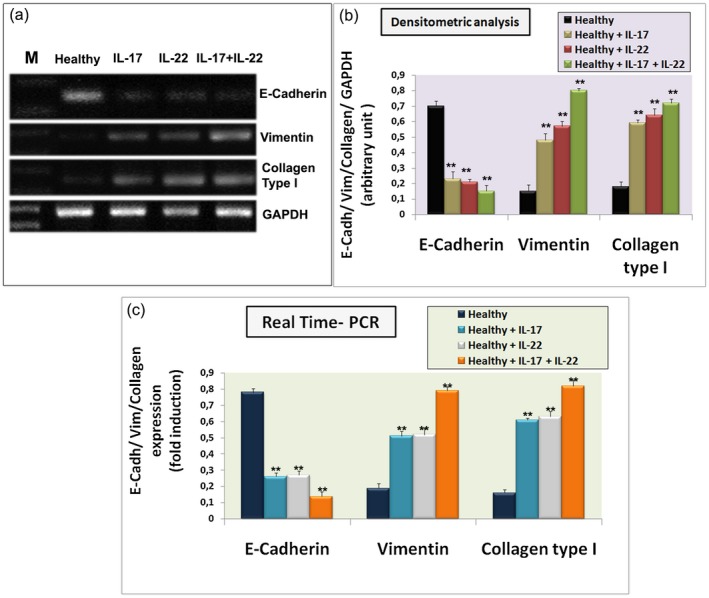
E‐cadherin, vimentin and collagen type I gene expression was quantified by semiquantitative reverse transcription–polymerase chain reaction (RT–PCR) (a) and real‐time PCR (c) in in‐vitro‐cultured healthy salivary gland epithelial cells (SGEC) treated for 24 h with interleukin (IL)‐17, IL‐22 or IL‐17 + IL‐22, and in untreated control healthy SGEC. The images show that interleukin treatment determined an increased gene expression of mesenchymal marker vimentin and collagen type I and decreased gene expression of epithelial marker E‐cadherin. IL‐17 and IL‐22 co‐operate to induce these effects. Bands intensities were analyzed by densitometric analysis performed by gel image software, normalized against that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and expressed in arbitrary units (b). Gene expression changes observed in real‐time PCR analysis (c) are in agreement with the results monitored by semiquantitative RT–PCR [data represent mean ± standard error (s.e.); n = 3]. Asterisks indicate statistical significance P < 0·01. M = marker.
IL‐17 and IL‐22 regulate the epithelial and mesenchymal protein marker expression in SGEC
We sought to investigate the differences in protein levels of E‐cadherin, vimentin and collagen type I between untreated and IL‐treated healthy SGEC. Cell lysates was collected from cultured cells after 3 days of treatment with IL‐17, IL‐22 or IL‐17 + IL‐22 and E‐cadherin, vimentin and collagen type I protein expression (Fig. 5a,b) was detected by Western blot. As shown, IL‐17 and IL‐22 stimulation on healthy SGEC efficiently reduced E‐cadherin expression at the protein level (Fig. 5a), with a trend towards a further decrease in E‐cadherin expression with IL‐17 + IL‐22 treatment (Fig. 5a; P < 0·01). In fact, semiquantitative analysis of the results (Fig. 5b) confirmed significant inhibitory effects of IL‐17 and IL‐22 on the expression of the epithelial marker (P < 0·01). The down‐regulated E‐cadherin expression was accompanied by a simultaneously increased expression of mesenchymal markers such as vimentin and collagen type I (Fig. 5). In the light of these data, we further determined by flow cytometry whether IL‐17 and IL‐ 22 treatment might affect vimentin, collagen type I and E‐cadherin expression in cultured SGEC. Figure 5c, in which the MFI values are reported, showed that the maintenance of E‐cadherin expression by SGEC is counteracted by the treatment of IL‐17 and IL‐22 alone and, in addition, combined treatment with IL‐17 + IL‐22 mainly enhances the vimentin and collagen type I expression in healthy SGEC at the expense of E‐cadherin (P < 0·01). These data strongly support the direct involvement of IL‐17 and IL‐22 in the process of SGs EMT mimicking what happens in the pSS disease, characterized by chronic over‐expression of these interleukins.
Figure 5.
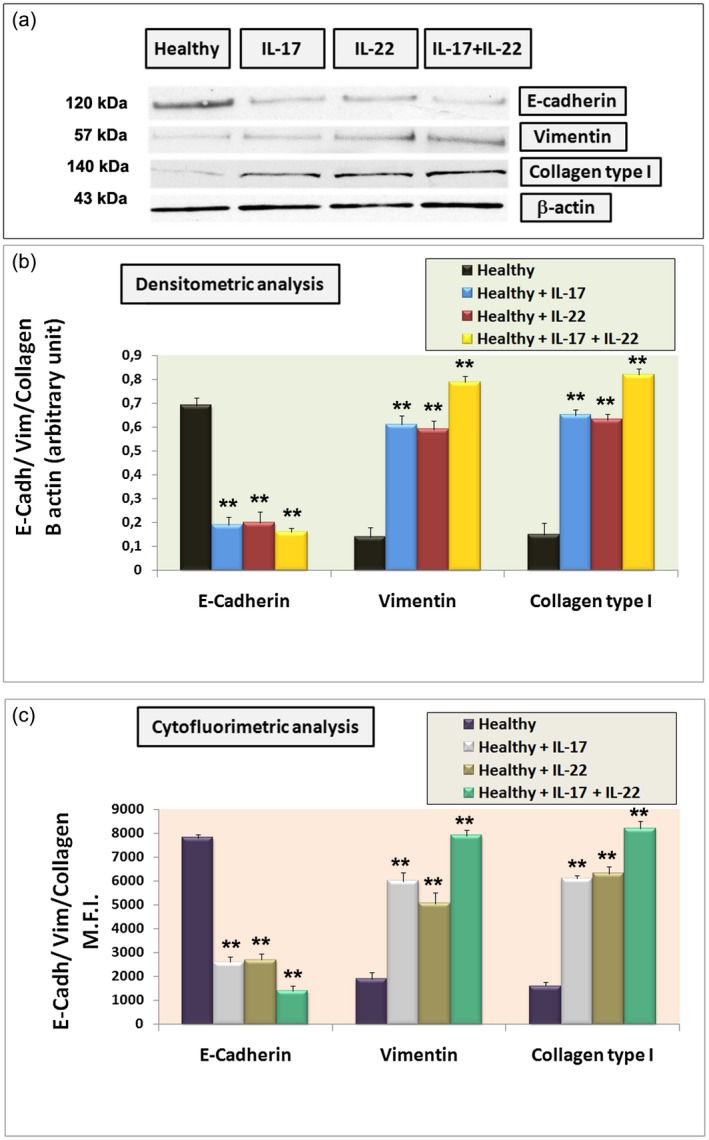
(a) Western blot analysis of the epithelial marker E‐cadherin and mesenchymal markers vimentin and collagen type I in cultured untreated salivary gland epithelial cells (SGEC) and in healthy SGEC treated with interleukin (IL)‐17 (50 ng/ml), IL‐22 (50 ng/ml) or IL‐17 + IL‐22 for 48 h. Lane 1: healthy control SGEC, lane 2: IL‐17‐treated healthy SGEC, lane 3: IL‐22‐treated healthy SGEC, lane 4: IL‐ 17 + IL‐22‐treated healthy SGEC. Protein expressions were quantified using ImageJ software (b). The data are expressed as relative intensity of proteins normalized to β‐actin expression. Values are considered statistically significant at *P < 0·05 or **P < 0·01. Interleukin treatment determined a significant increase in mesenchymal vimentin and collagen type I band intensity, while epithelial marker E‐cadherin expression was reduced compared with untreated control SGEC. All Western blots were repeated a minimum of three times. (c) Summary of flow cytometry analysis for E‐cadherin, vimentin and collagen type I SGEC expression in the presence of IL‐17, IL‐22 or IL‐17 + IL‐22. The mean fluorescence intensity (MFI) is shown. Treatment with IL‐17 + IL‐22 mainly interferes with E‐cadherin basal expression in healthy SGEC, while vimentin and collagen type I expression was increased following interleukin treatment in comparison with untreated control cells (**P < 0·01). Data summarize three independent experiments [mean ± standard error (s.e.)].
Discussion
Much research on EMT‐dependent fibrosis has been carried out relating to cancer; it is not clear whether EMT plays a role in autoimmune conditions and whether epithelial cells can undergo the EMT during autoimmune diseases. Furthermore, understanding the role of the EMT process in pathological fibrosis is of great significance, and thus it could lead to identifying approaches which could hamper the formation of fibrotic tissue in autoimmune disease. Our work reveals for the first time, to our knowledge, that in the markedly inflammatory microenviroment of pSS SGs, characterized by an intense increase of proinflammatory cytokines, the loss of E‐cadherin and acquisition of vimentin and collagen type I occur in SGEC in a correlated manner with the growing inflammatory grade. Additionally, to corroborate this observation, we demonstrated that IL‐17 and IL‐22 contribute to trigger the EMT‐dependent fibrotic process in healthy SGEC and, furthermore, combined treatment with IL‐17 and IL‐22 induces and accentuates EMT progression. Our findings provide important insight into the induction of the EMT in SGEC, and it seems clear that inflammatory mediators play a role in both the initiation and the progression of pathological SG fibrosis in pSS. Chronic inflammation leads to the release of a wide range of inflammatory mediators which can drive and regulate multiple aspects of inflammatory disease and contribute to the pathological fibrosis that, in turn, is the final consequence of many diseases with chronic inflammatory features 38, 39, 40. It should be noted that salivary fibrosis is a pathological characteristic of pSS related to inflammation in SG foci 41. TGF‐β1 is the main mediator that promotes pathological fibrosis in the majority of inflammatory disorders, involving organs such as the kidney 42, 43, 44, lung 45 and liver 46. Interestingly, TGF‐β1 over‐production or the effects of profibrotic boosting induce evident fibrotic changes and lead to an increase of pathological fibrotic tissue, affecting normal organ function 47, 48, 49, 50. Recently, it was discovered that the TGF‐β1 levels was augmented in SG tissues derived from biopsies of pSS patients, characterized by a sustained inflammatory disorder that determines an excess of fibrotic tissue, altering the functional morphology of SGs 37. In fact, the earliest changes in the SG structure appear to be due to an active EMT, which is associated with a cascade of changes in the expression of TGF‐β1‐dependent regulators, including Smads, Snail and profibrotic markers 37. The ability of other cytokines to directly drive the EMT and the mechanisms initiating or maintaining the EMT‐dependent fibrotic process involved in the pathogenesis of pSS remain poorly explored. Elevated levels of several cytokines and chemokines are detected in the peripheral blood, and in the affected SGs and pSS their alteration triggers both systemic and exocrine manifestations 51, 52. In addition, our previous reports have shown the presence of marked inflammatory cell components in functionally and structurally damaged SGs of pSS patients that contribute to an increased proinflammatory cytokine accumulation and thus to the amplification of the epithelial‐derived cytokine cascade 28, 35, 53. In addition, high levels of IL‐22, strictly associated with hyposalivation, have recently been shown in pSS patients’ sera 24 and a marked up‐regulation of IL‐17 and IL‐22 has been reported in biopsies isolated from patients affected by pSS, suggesting a potential direct involvement of IL‐17 and IL‐22 in the initiation and progression of pSS 24, 54. Moreover, a recent report has reported that IL‐22 and linked cytokine IL‐17 are strongly present in the inflamed SGs of pSS patients, both correlated with the degree of tissue inflammation 24, 55. In addition, contemporary expression of IL‐17 and IL‐22 in the inflamed pSS SG seems to suggest a predominant proinflammatory role of these cytokines. Increasing evidence has suggested that IL‐17 is clearly linked with the EMT in lung inflammatory diseases 56 and pulmonary fibrosis 56, 57. Recently, it was demonstrated that in obliterative bronchiolitis, IL‐17‐mediated type V collagen expression mediated by IL‐17 and the EMT may have an affect via TGF‐β1‐dependent signalling 57. Therefore, recent work 56 has provided evidence that an antagonist of IL‐17 is able to inhibit the chronic inflammatory response and pulmonary fibrosis in a TGF‐β1‐dependent way. Additionally, several findings have demonstrated that strong expression of IL‐17 and IL‐22 has a prospective role in the development of fibrosis in several organs, such as the heart 58, kidney 59 and liver 60. However, although up‐regulation of IL‐17 and IL‐22 has been reported in patients with pSS, little is known regarding their role in the regulation of the EMT and the underlying mechanisms as possible inducers of the EMT programme in SGs. To address this question, we examined, as the first step, the expression of epithelial and mesenchymal markers by immunohistochemical analysis in biopsies derived from pSS patients with different grades of inflammatory lesions and FC in comparison with controls. As expected, we observed the progressive loss of epithelial marker E‐cadherin and a growing increase in expression of the mesenchymal markers vimentin and collagen type I in the highest degrees of tissue inflammation and pSS FC. These findings contribute to supporting our emerging idea that SGs biopsies undergo EMT‐dependent fibrosis correlated with the degree of tissue inflammation.
To investigate whether IL‐17 and IL‐22 can induce the EMT programme in SGs, SGEC were cultured in vitro in the presence of IL‐17, IL‐22 and with a mix of IL‐17 + IL‐22. As expected, after cytokine treatment morphological changes from a rounded shape epithelial phenotype to a spindle‐like mesenchymal phenotype in SGEC were observed, which are indicative of the beginning of an EMT process. Furthermore, the effect was more evident in response to IL‐17 and IL‐22 double exposure. The morphological changes from an epithelial to a mesenchymal phenotype were determined by analyzing changes in the expression of E‐cadherin, vimentin and collagen type I by qualitative and quantitative RT–PCR, Western blotting and flow cytometry analysis. The results revealed that IL‐17 and IL‐22 treatment changes the phenotypical markers in SGEC, inducing an EMT process based on the down‐regulation of E‐cadherin expression and up‐regulation of vimentin and collagen type I expression compared with controls.
In conclusion, the present study demonstrated that IL‐17 and IL‐22 have a potential role in the EMT program of pSS SGEC, as revealed by the feature changes in the altered gene and protein expression of the EMT markers E‐cadherin, vimentin and collagen type I, as well as by characteristic morphological changes directing SGEC transdifferentiation towards a mesenchymal cell type. Therefore, these findings support the hypothesis that when the normal biological function of SGEC is impaired the progression of SGs fibrosis may be initiated, and suggest that IL‐17 and IL‐22 suppression may open new therapeutic avenues for pSS patients and could contribute to our overall understanding of the pSS pathogenesis.
Disclosure
All the authors have no conflicts of interest to disclose.
Author contributions
M. S. and S. L. performed the experiments and wrote the paper; L. L., R. T. and G. I. performed the experiments; D. R. designed the study and revised the paper.
Acknowledgement
We are grateful to M. V. C. Pragnell BA, a professional scientific text editor, for critical reading of the manuscript.
References
- 1. Humphreys‐Beher MG, Peck AB. New concepts for the development of autoimmune exocrinopathy derived from studies with the NOD mouse model. Arch Oral Biol 1999; 44:S21–S25. [DOI] [PubMed] [Google Scholar]
- 2. Moutsopoulos HM, Chused TM, Mann DL et al Sjogren’s syndrome (Sicca syndrome): current issues. Ann Intern Med 1980; 92:212–26. [DOI] [PubMed] [Google Scholar]
- 3. Aiello NM, Maddipati R, Norgard RJ et al EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell 2018; 45:681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell 2016; 166:21–45. [DOI] [PubMed] [Google Scholar]
- 5. Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 2006; 3:364–72. [DOI] [PubMed] [Google Scholar]
- 6. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 2007; 101:830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. López‐Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 2009; 1:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta 2013; 1832:1049–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan HF, Zhao XF, Yuan H et al Decreased serum IL‐22 levels in patients with systemic lupus erythematosus. Clin Chim Acta 2009; 401:179–18. [DOI] [PubMed] [Google Scholar]
- 10. Cheng F, Guo Z, Xu H, Yan D, Li Q. Decreased plasma IL22 levels, but not increased IL17 and IL23 levels, correlate with disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis 2009; 68:604–6. [DOI] [PubMed] [Google Scholar]
- 11. Wolk K, Haugen HS, Xu W et al IL‐22 and IL‐20 are key mediators of the epidermal alterations in psoriasis while IL‐17 and IFN‐ gamma are not. J Mol Med (Berl) 2009; 87:523–36. [DOI] [PubMed] [Google Scholar]
- 12. Schmechel S, Konrad A, Diegelmann J et al Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL‐22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis 2008; 14:204–12. [DOI] [PubMed] [Google Scholar]
- 13. Li LJ, Gong C, Zhao MH, Feng BS. Role of interleukin‐22 in inflammatory bowel disease. World J Gastroenterol 2014; 20:18177–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ikeuchi H, Kuroiwa T, Hiramatsu N et al Expression of interleukin‐22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum 2005; 52:1037–46. [DOI] [PubMed] [Google Scholar]
- 15. Geboes L, Dumoutier L, Kelchtermans H et al Proinflammatory role of the Th17 cytokine interleukin‐22 in collagen‐induced arthritis in C57BL/6 mice. Arthritis Rheum 2009; 60:390–5. [DOI] [PubMed] [Google Scholar]
- 16. da Rocha LF, Jr Duarte AL, Dantas AT et al Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol 2012; 39:1320–5. [DOI] [PubMed] [Google Scholar]
- 17. Holtta V, Klemetti P, Sipponen T et al IL‐23/IL‐17 immunity as a hallmark of Crohn’s disease. Inflamm Bowel Dis 2008; 14:1175–84. [DOI] [PubMed] [Google Scholar]
- 18. Blake SJ, Teng MW. Role of IL‐17 and IL‐22 in autoimmunity and cancer. Acta Dermosifiliogr 2014; 105:41–50. [DOI] [PubMed] [Google Scholar]
- 19. Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest 2009; 119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson JR, Nishioka M, Chakir J et al IL‐22 contributes to TGF‐β1‐mediated epithelial‐mesenchymal transition in asthmatic bronchial epithelial cells. Respir Res 2013; 14:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T‐cell‐mediated tolerance and contribute to airway remodeling. Mucosal Immunol 2012; 6:335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gao Y, Ren H, Meng F et al Pathological roles of interleukin‐22 in the development of recurrent hepatitis c after liver transplantation. PLOS ONE 2016; 11:e0154419. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Zhang LW, Zhou PR, Wei P, Cong X, Wu LL, Hua H. Expression of interleukin‐17 in primary Sjögren’s syndrome and the correlation with disease severity: a systematic review and meta‐analysis. Scand J Immunol 2018; 87:e12649. [DOI] [PubMed] [Google Scholar]
- 24. Ciccia F, Guggino G, Rizzo A et al Potential involvement of IL22 and IL‐22‐producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann Rheum Dis 2012; 71:295–301. [DOI] [PubMed] [Google Scholar]
- 25. Ciccia F, Guggino G, Rizzo A et al Interleukin (IL)‐22 receptor 1 is over‐expressed in primary Sjogren’s syndrome and Sjögren‐associated non‐Hodgkin lymphomas and is regulated by IL‐18. Clin Exp Immunol 2015; 181:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lavoie TN, Stewart CM, Berg KM, Li Y, Nguyen CQ. Expression of interleukin‐22 in Sjogren’s syndrome: significant correlation with disease parameters. Scand J Immunol 2011; 74:377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mieliauskaite D, Dumalakiene I, Rugiene R, Mackiewicz Z. Expression of IL‐17, IL‐23 and their receptors in minor salivary glands of patients with primary Sjögren’s syndrome. Clin Dev Immunol 2012; 2012:187258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lisi S, Sisto M, Lofrumento DD, D’Amore M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF‐κB. Lab Invest 2012; 92:615–24. [DOI] [PubMed] [Google Scholar]
- 29. Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin‐17 and linked cytokines associated with Sjögren’s syndrome immunopathogenesis. Am J Pathol 2009; 175:1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vitali C, Bombardieri S, Jonsson R et al Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American–European Consensus Group. European Study Group on Classification Criteria for Sjögren’s Syndrome. Ann Rheum Dis 2002; 61:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sens DA, Hintz DS, Rudisill MT, Sens MA. Spicer SS Explant culture of human submandibular gland epithelial cells: evidence for ductal origin. Lab Invest 1985; 52:559–67. [PubMed] [Google Scholar]
- 32. Jannink I, Bennen JN, Blaauw J, van Diest PJ, Baak JP. At convenience and systematic random sampling: effects on the prognostic value of nuclear area assessments in breast cancer patients. Breast Cancer Res Treat 1995; 36:55–60. [DOI] [PubMed] [Google Scholar]
- 33. Rizzardi AE, Johnson AT, Vogel RI et al Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn Pathol 2012; 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kapsogeorgou EK, Dimitriou ID. Abu‐Helu RF, Moutsopoulos HM, Manoussakis MN. Activation of epithelial and myoepithelial cells in the salivary glands of patients with Sjögren’s syndrome: high expression of intercellular adhesion molecule‐1 (ICAM.1) in biopsy specimens and cultured cells. Clin Exp Immunol 2001; 124:126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sisto M, Lisi S, Lofrumento DD, Ingravallo G, Mitolo V, D’Amore M. Expression of pro‐inflammatory TACE‐TNF‐α‐amphiregulin axis in Sjögren’s syndrome salivary glands. Histochem Cell Biol 2010; 134:345–53. [DOI] [PubMed] [Google Scholar]
- 36. Lisi S, Sisto M, Lofrumento DD et al Pro‐inflammatory role of Anti‐Ro/SSA autoantibodies through the activation of Furin–TACE‐amphiregulin axis. J Autoimmun 2010; 35:160–70. [DOI] [PubMed] [Google Scholar]
- 37. Sisto M, Lorusso L, Ingravallo G, Tamma R, Ribatti D, Lisi S. The TGF‐β1 signaling pathway as an attractive target in the fibrosis pathogenesis of Sjögren’s syndrome. Mediat Inflamm 2018; 2:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen L, Deng H, Cui H et al Inflammatory responses and inflammation‐associated diseases in organs. Oncotarget 2018; 9:7204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012; 18:1028–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leehan KM, Pezant NP, Rasmussen A et al Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin Exp Rheumatol 2018; 112:80–8. [PMC free article] [PubMed] [Google Scholar]
- 42. Meng XM, Tang PM, Li J, Lan HY. TGF‐β/Smad signaling in renal fibrosis. Front Physiol 2015; 6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meng XM, Nikolic‐Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol 2014; 10:493–503. [DOI] [PubMed] [Google Scholar]
- 44. Meng XM, Nikolic‐Paterson DJ, Lan HY. TGF‐β: the master regulator of fibrosis. Nat Rev Nephrol 2016; 12:325–38. [DOI] [PubMed] [Google Scholar]
- 45. Willis BC, Borok Z. TGF‐β‐induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 2007; 293:L525–34. [DOI] [PubMed] [Google Scholar]
- 46. Fabregat I, Moreno‐Càceres J, Sánchez A et al TGF‐β signalling and liver disease. FEBS J 2016; 283:2219–32. [DOI] [PubMed] [Google Scholar]
- 47. Kanzler S, Lohse AW, Keil A et al TGF‐β1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol 1999; 276:G1059–G1068. [DOI] [PubMed] [Google Scholar]
- 48. Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A. Transforming growth factor‐beta1 induces an epithelial‐to‐mesenchymal transition state in mouse hepatocytes in vitro . J Biol Chem 2007; 282:22089–101. [DOI] [PubMed] [Google Scholar]
- 49. Liu N, He S, Ma L et al Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF‐beta and EGFR signaling. PLOS ONE 2013; 8:e54001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)‐beta1 stimulates pulmonary fibrosis and inflammation via a Bax‐dependent, bid‐activated pathway that involves matrix metalloproteinase‐12. J Biol Chem 2007; 282:7723–32. [DOI] [PubMed] [Google Scholar]
- 51. Roescher N, Tak PP, Illei GG. Cytokines in Sjogren’s syndrome: potential therapeutic targets. Ann Rheum Dis 2010; 69:945–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reksten TR, Jonsson MV, Szyszko EA, Brun JG, Jonsson R, Brokstad KA. Cytokine and autoantibody profi ling related to histopathological features in primary Sjogren’s syndrome. Rheumatology (Oxf) 2009; 48:1102–6. [DOI] [PubMed] [Google Scholar]
- 53. Lisi S, Sisto M, Lofrumento DD, D’Amore M. Sjögren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF‐κB. Lab Invest 2012; 92:615–24. [DOI] [PubMed] [Google Scholar]
- 54. Ciccia F, Guggino G, Rizzo A et al Rituximab modulates IL‐17 expression in the salivary glands of patients with primary Sjögren’s syndrome. Rheumatology (Oxf) 2014; 53:1313–20. [DOI] [PubMed] [Google Scholar]
- 55. Ciccia F, Giardina A, Rizzo A et al Rituximab modulates the expression of IL‐22 in the salivary glands of patients with primary Sjogren’s syndrome. Ann Rheum Dis 2013; 72:782–3. [DOI] [PubMed] [Google Scholar]
- 56. Mi S, Li Z, Yang HZ et al Blocking IL‐17A promotes the resolution of pulmonary inflammation and fibrosis via TGF‐beta1‐dependent and ‐independent mechanisms. J Immunol 2011; 187:3003–14. [DOI] [PubMed] [Google Scholar]
- 57. Vittal R, Fan L, Greenspan DS et al IL‐ 17 induces type V collagen overexpression and EMT via TGF‐beta‐dependent pathways in obliterative bronchiolitis. Am J Physiol Lung Cell Mol Physiol 2013; 304:L401–L414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valente AJ, Yoshida T, Gardner JD, Somanna N, Delafontaine P, Chandrasekar B. Interleukin‐17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual‐specificity phosphatase MKP‐1/DUSP‐1. J Cell Signal 2012; 24:560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu L, Li FG, Yang M et al Effect of pro‐inflammatory interleukin‐17A on epithelial cell phenotype inversion in HK‐2 cells in vitro. Eur Cytokine Netw 2016; 27:27–33. [DOI] [PubMed] [Google Scholar]
- 60. Meng F, Wang K, Aoyama T et al Interleukin‐17 signaling in inflammatory. Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012; 143:765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]