

Epstein-Barr virus is a pathogen that causes persistent infection with potential consistent viral DNA shedding. The enhancement of production of proinflammatory cytokines by viral DNA itself may contribute to autoimmune disease development or exacerbation. In this project, we identified that endosomal Toll-like receptors are involved in triggering proinflammatory mediators in response to viral DNA. Pathways and receptors involved may serve as future therapeutic targets for autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus.
KEYWORDS: Epstein-Barr virus, IL-17A, TLR3, TLR7, TLR9, human herpesviruses, interleukins, Toll-like receptors
ABSTRACT
We previously demonstrated that Epstein-Barr virus (EBV) DNA increases the production of the proinflammatory cytokine interleukin-17A (IL-17A) in mice. This property may contribute to the established association between EBV and autoimmune diseases. The objective of the present study was to elucidate mechanisms through which EBV DNA modulates IL-17A levels in mice. To determine whether endosomal Toll-like receptors (TLRs) played a role in this pathway, the expression of TLR3, -7, or -9 was assessed by real-time reverse transcription-PCR in mouse spleens after injection of EBV DNA. Moreover, specific inhibitors were used for these TLRs in mouse peripheral blood mononuclear cells (PBMCs) cultured with EBV DNA and in mice injected with this viral DNA; IL-17A levels were then assessed using an enzyme-linked immunosorbent assay. The expression of the endosomal receptors TLR3, -7, and -9 was increased in mice injected with EBV DNA. When mouse immune cells were cultured with EBV DNA and a TLR3, -7, or -9 inhibitor or when mice were injected with the viral DNA along with either of these inhibitors, a significant decrease in IL-17A levels was detected. Therefore, endosomal TLRs are involved in the EBV DNA-mediated triggering of IL-17A production in mice. Targeting these receptors in EBV-positive subjects with autoimmunity may be useful pending investigations assessing whether they play a similar role in humans.
IMPORTANCE Epstein-Barr virus is a pathogen that causes persistent infection with potential consistent viral DNA shedding. The enhancement of production of proinflammatory cytokines by viral DNA itself may contribute to autoimmune disease development or exacerbation. In this project, we identified that endosomal Toll-like receptors are involved in triggering proinflammatory mediators in response to viral DNA. Pathways and receptors involved may serve as future therapeutic targets for autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus.
INTRODUCTION
Epstein-Barr virus (EBV), also referred to as human herpesvirus 4 (HHV-4), is a human gammaherpesvirus (double-stranded DNA [dsDNA] virus) known as the cause of infectious mononucleosis (IM), a lymphoproliferative disorder observed in young adults. It is considered one of the most common viruses in humans, infecting up to 90% of the population and persisting for the lifetime of an individual (1). The EBV genome is a linear double-stranded DNA that is approximately 172 kb long and carries more than 85 genes (2).
EBV is mostly transmitted via saliva, but it could also be transmitted sexually and through blood transfusions. Primary EBV infection is characterized by a self-limiting acute febrile illness where high rates of activated EBV-specific CD8+ T cell responses eliminate virus-infected B cells. It has also been associated with several types of B cell malignancies, including posttransplant lymphoma, Burkitt’s lymphoma, and Hodgkin lymphoma, in addition to epithelial cell malignancies, which include nasopharyngeal carcinoma and some gastric carcinomas (1, 3). Moreover, infection with EBV is associated with a higher risk of certain autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and multiple sclerosis (MS) (4). RA patients have a considerable increase in the EBV DNA load in peripheral blood mononuclear cells (PBMCs) compared with that in controls (5, 6). Moreover, RA patients have high numbers of circulating EBV-infected B cells (7) and EBV DNA loads in saliva (8). Antibody titers against EBV antigens are also elevated in SLE and MS patients compared to those in healthy controls (9, 10).
One of the most studied theories explaining how infections can be the cause of autoimmunity is molecular mimicry. This was suggested by Fujinami and Oldstone, who proposed that similarities between microbial and self-antigens on the molecular or structural level are the reason behind the cross-reactivity of T cells, B cells, and antibodies (11, 62). Concerning EBV association with autoimmune diseases, several studies were conducted to explore the link between EBV infection and autoimmunity. For example, EBV nuclear antigen 1 (EBNA-1), a viral protein of EBV, seems to share epitope similarities with multiple human nuclear antigens, and antibodies against EBNA-1 were shown to cross-react with dsDNA (12). EBNA-1 DNA complexes make a good source of antigens that may induce cross-reactive antibodies (12). Moreover, other studies showed a cross-reaction between autoantibodies against epitopes on SmB′ and SmD1 and different domains of EBNA-1 in SLE (13, 14). This may indicate that molecular mimicry underlies the association between EBV infection and SLE. Other studies indicated possible epitope similarities between EBV antigens and myelin basic protein (MBP), the autoantigen in MS (15, 16). The strongest genetic risk factor for MS is the HLA class II allele DRB1*1501. This association has been reported in Caucasian populations (17). However, in other populations, other HLA associations were detected instead of the DRB1*1501 allele (18–20). There is a strong linkage disequilibrium between DRB1*1501 and DRB5*0101, where these two alleles may be involved in molecular mimicry between EBV and MBP. It was reported that the Hy.2E11 T cell receptor (TCR) from an MS patient cross-recognized a DRB1*1501-restricted MBP peptide and a DRB5*0101-restricted EBV DNA polymerase peptide (15). Moreover, Lunemann et al. demonstrated that MS patients have EBNA-1-specific T cells in blood. These T cells recognize myelin antigens more than other tested autoantigens and produced both gamma interferon (IFN-γ) and interleukin-2 (IL-2) (16). Another candidate autoantigen in MS is the small heat shock protein αB-crystallin. It is expressed in MS lesions but not in normal white matter. Normally, it is not expressed by human B cells, but upon EBV infection, it was shown to be upregulated in EBV-infected B lymphocytes (21). Such expression of αB-crystallin in virus-infected human B cells results in the presentation of the protein via HLA-DR to T cells (21). This contributed to a hypothesis designated “mistaken self” (22), whereby virus-generated T cells specific for αB-crystallin cause cross-reactivity with central nervous system (CNS) myelin. Thus, peripheral EBV infection of B cells activates the human T cell repertoire to microbial antigens as well as to de novo-expressed αB-crystallin in infected lymphoid cells. Most RA patients have anti-citrullinated protein antibodies (ACPAs) in their sera. Citrullinated proteins are products of peptidyl arginine deiminase (PAD), which catalyzes the conversion of arginine residues into citrulline (23). PAD can be activated in the inflamed synovium due to high calcium concentrations. Thus, it may act on EBV proteins leading to posttranslational citrullination and thereby convert them to possible targets for ACPA. Antibodies specific for a citrullinated EBNA-1 peptide (amino acids [aa] 35 to 58) were detected in about 50% of RA sera, in comparison to <5% of normal and disease control sera (24).
In a previous study, we reported that EBV DNA enhances the production of the proinflammatory cytokine IL-17A as well as IL-23, tumor necrosis factor (TNF-α), and IFN-γ when injected into mice (25, 65). IL-17A plays a role in host defenses against bacterial and fungal infections (26). Despite this rather beneficial function, IL-17A has a pathogenic role in several autoimmune diseases such as psoriasis, inflammatory bowel disease, RA, MS, and SLE, whereby high levels of IL-17A were found in sera and tissues of patients with these diseases (27). In another study, we found that the average serum IL-17A levels were higher in RA patients than in non-RA subjects. In addition, we detected a linear correlation between EBV DNA levels and serum IL-17A levels in RA patients but not in controls (6).
The genomes of some viruses, such as herpes simplex virus 1 (HSV 1) and EBV, are rich with CpG motifs, which can activate the immune system, promoting Th1-like responses (28–30). The EBV genome has the third highest percentage of cytidine and guanidine (C+G) nucleotides (60%) among other human herpesviruses. It is second to HSV-1 and HSV-2 in GC percentages (69% and 68%, respectively) (31). Therefore, the EBV genome possesses a high ability to activate innate immune receptors. We previously indicated that Toll-like receptor 9 (TLR9), an endosomal receptor known for its recognition of CpG-rich DNA, may play a role in the modulation of IL-17A levels upon EBV DNA challenge of immune cells ex vivo (6). Here we examine the involvement of TLR9 along with TLR3 and -7, other endosomal receptors known to recognize microbial nucleic acid molecules, in this response both ex vivo and in vivo in mice.
RESULTS
EBV DNA-triggered IL-17A increase from mouse PBMCs is dependent on the endosome.
To assess the mechanism by which EBV DNA triggers IL-17A expression in mice, we examined the involvement of the endosome in this pathway. The endosome encompasses receptors capable of recognizing nucleic acids; therefore, we examined the response of mouse PBMCs to EBV DNA in the presence or absence of chloroquine, a lysosomotropic agent that prevents endosomal acidification (32). Incubation of mouse PBMCs with EBV DNA resulted in an 8.21-fold increase (P = 0.0006) in the level of IL-17A. On the other hand, culturing these cells with EBV DNA after preincubation with chloroquine led to a significant decrease in IL-17A production, with the highest fold decrease, 131-fold, observed with 40 μM chloroquine (P = 0.0004) (Fig. 1). Cells were also cultured with EBV DNA in the presence of DNase to ensure that EBV DNA was the sole factor in the preparation resulting in increased IL-17A levels. Culturing PBMCs with the EBV DNA preparation treated with DNase I resulted in a 7.21-fold decrease (P = 0.0031) in IL-17A levels compared to those in cells incubated with EBV DNA alone. These findings indicate that the endosome plays a role in enhancing the production of IL-17A from mouse immune cells upon treatment with EBV DNA.
FIG 1.
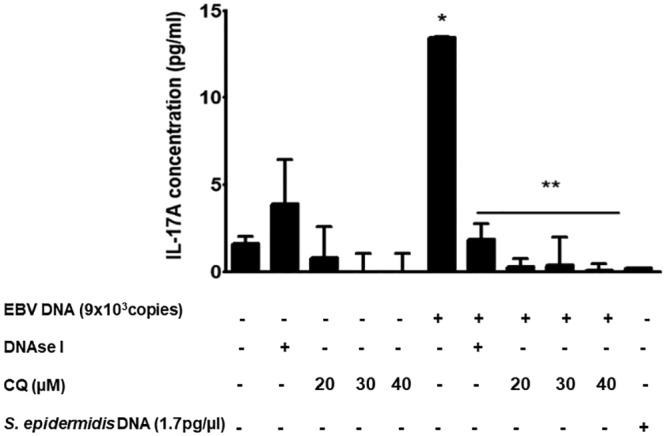
IL-17A levels in BALB/c mouse PBMCs treated with EBV DNA, DNase I, and chloroquine. PBMCs were cultured with EBV DNA (9 × 103 copies), EBV DNA plus DNase I, DNase I alone, EBV DNA plus different concentrations of the endosomal maturation inhibitor chloroquine (CQ), different concentrations of chloroquine alone, or Staphylococcus epidermidis DNA. Untreated cells were examined as controls. After a 24-h culture period, IL-17A levels were assessed in the culture medium by an ELISA. *, P < 0.05 compared to nontreated cells; **, P < 0.05 compared to EBV DNA-treated cells.
EBV DNA enhances the expression of endosomal TLRs in mouse splenic tissue.
To examine whether TLRs were the endosomal component mediating the enhanced IL-17A response to EBV DNA, we started by assessing whether this viral DNA affected the gene expression of TLR3, -7, and -9. These were examined in splenic tissues of mice injected with EBV DNA and in mock-treated mice. TLR3 gene expression was significantly increased by 19.5% (P = 0.0173) on day 6 and by 2.63-fold (P = 0.0001) on day 9 postinjection (Fig. 2A) but not on day 3 postinjection. On the other hand, TLR7 expression was increased by 9.43-fold (P = 0.0018), 5.15-fold (P = 0.0012), and 3.48-fold (P = 0.0079) on days 3, 6, and 9 postinjection, respectively (Fig. 2B). As for TLR9, its expression was increased by 3.02-fold (P = 0.0206), 2.72-fold (P = 0.008), and 2.93-fold (P = 0.0015) on days 3, 6, and 9 postinjection, respectively (Fig. 2C).
FIG 2.
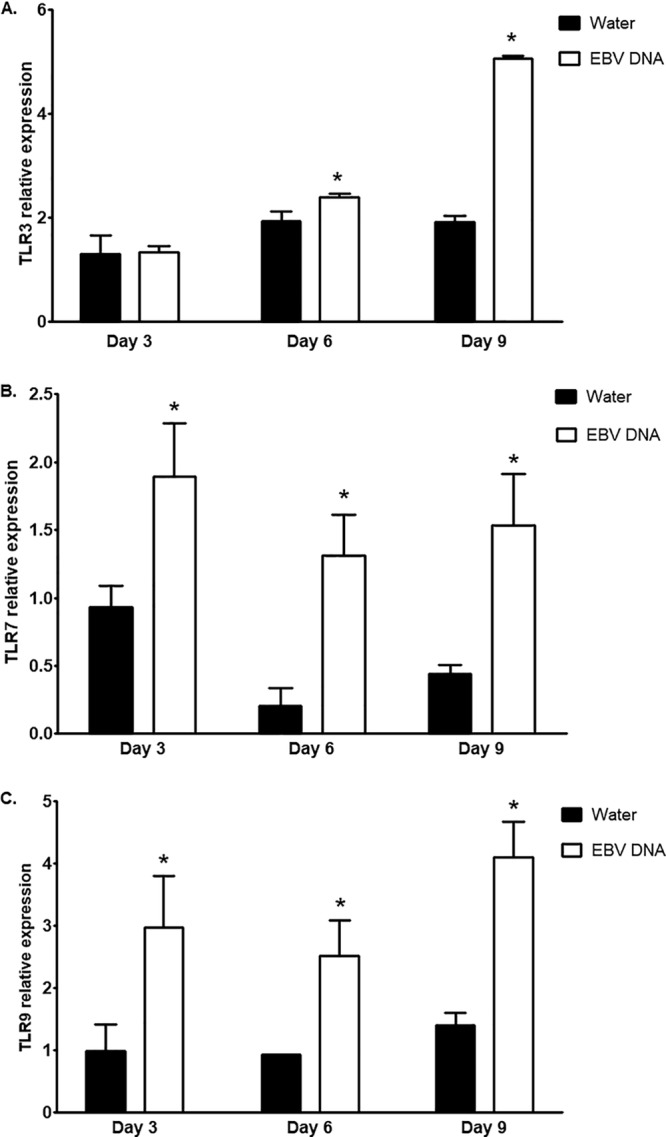
Relative expression levels of TLR3, -7, and -9 in splenic tissue from BALB/c mice treated with EBV DNA. BALB/c mice were intraperitoneally injected with sterile distilled water or EBV DNA (144 × 103 copies). Spleens were collected on days 3, 6, and 9 postinjection, and relative gene expression levels of TLR3 (A), TLR7 (B), and TLR9 (C) were assessed by reverse transcriptase real-time PCR. Levels were normalized to the level of β-actin. *, P < 0.05 compared to the water-injected group on the respective day of injection.
Endosomal TLR3, -7, and -9 play a role in EBV DNA-triggered IL-17A production ex vivo and in vivo.
To assess the roles of TLR3, -7, and -9 in mediating the response to EBV DNA, we employed specific oligonucleotides that act as inhibitors of these TLRs. A 2.31-fold increase (P = 0.0316) in the level of IL-17A was detected in the culture supernatant upon incubation of mouse PBMCs with EBV DNA. However, when introducing a specific TLR3 inhibitor in addition to EBV DNA, we detected a decrease in IL-17A levels by 4.2-fold (P = 0.0048) (Fig. 3). Upon incubation of mouse PBMCs with EBV DNA along with a specific TLR7 inhibitor, the IL-17A level was decreased by 23% (P = 0.0493) (Fig. 4). Consistent with our previous observations (6), incubation of mouse PBMCs with a specific TLR9 inhibitor and EBV DNA led to a significant decrease in IL-17A levels by 6.18-fold (P = 0.0039) compared to treatment with EBV DNA alone (Fig. 5). Culturing mouse PBMCs with a nonviral DNA from Staphylococcus epidermidis showed no significant modulation in IL-17A levels, thus highlighting the specific activity of EBV DNA in inducing an increase in IL-17A production. Interestingly, culturing cells with a TLR7 inhibitor alone caused a significant enhancement of IL-17A levels. However, TLR3 and TLR9 inhibitors alone did not result in significant changes.
FIG 3.
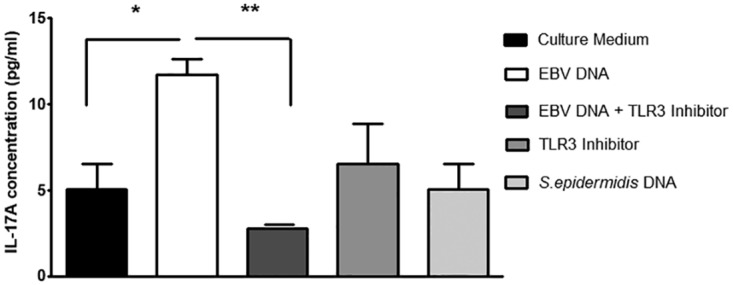
IL-17A levels in BALB/c mouse PBMCs cultured with EBV DNA and a TLR3 inhibitor. PBMCs were cultured with EBV DNA (9 × 103 copies), EBV DNA plus the TLR3 inhibitor (10 μM), the TLR3 inhibitor (10 μM) alone, or Staphylococcus epidermidis DNA (1.7 pg). After a 24-h culture period, IL-17A levels were assessed in the culture medium by an ELISA. *, P < 0.05 compared to nontreated cells; **, P < 0.05 compared to EBV DNA-treated cells.
FIG 4.
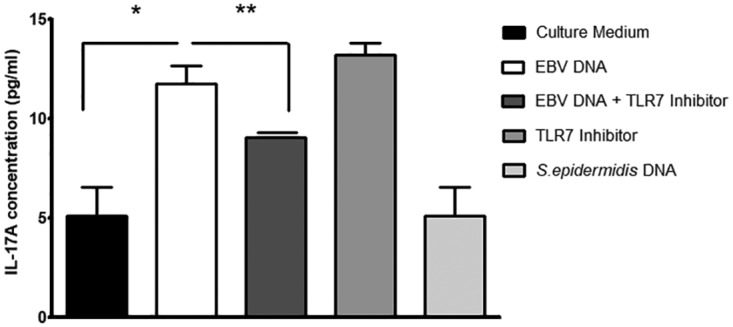
IL-17A levels in BALB/c mouse PBMCs cultured with EBV DNA and a TLR7 inhibitor. PBMCs were cultured with EBV DNA (9 × 103 copies), EBV DNA plus the TLR7 inhibitor (2.8 μM), the TLR7 inhibitor (2.8 μM) alone, or Staphylococcus epidermidis DNA (1.7 pg). After a 24-h culture period, IL-17A levels were assessed in the culture medium by an ELISA. *, P < 0.05 compared to nontreated cells; **, P < 0.05 compared to EBV DNA-treated cells.
FIG 5.
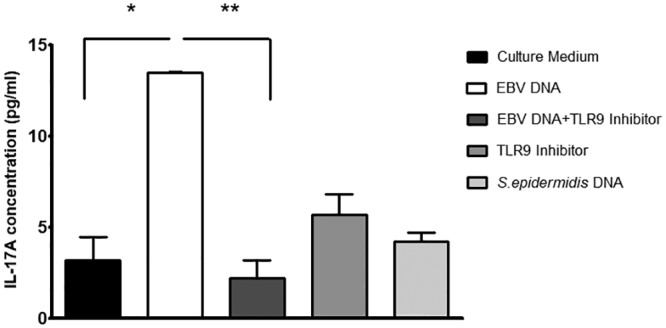
IL-17A levels in BALB/c mouse PBMCs cultured with EBV DNA and a TLR9 inhibitor. PBMCs were cultured with EBV DNA (9 × 103 copies), EBV DNA plus the TLR9 inhibitor (1.4 μM), the TLR9 inhibitor (1.4 μM) alone, or Staphylococcus epidermidis DNA (1.7 pg). After a 24-h culture period, IL-17A levels were assessed in the culture medium by an ELISA. *, P < 0.05 compared to nontreated cells; **, P < 0.05 compared to EBV DNA-treated cells.
To rule out that the detected decrease in IL-17A levels from mouse cells treated with the TLR inhibitors in the presence of EBV DNA was the result of apoptotic processes, treated cells were stained with annexin and 7-aminoactinomycin D (7-AAD). Early and late apoptotic cell population levels in the EBV- and TLR inhibitor-treated PBMCs were not higher than in those treated with EBV DNA alone. This indicates that the decrease in IL-17A levels seen in our ex vivo experiments is due to the inhibition of TLR3, -7, and -9 and not due to PBMCs undergoing apoptosis.
To determine whether the assessed TLRs play a similar role in vivo, mice were treated with EBV DNA in addition to the TLR3, -7, and -9 inhibitors and S. epidermidis DNA. IL-17A levels were significantly increased in mouse sera upon injection of EBV DNA; however, upon injection of the specific TLR3 inhibitor in addition to EBV DNA, we detected decreases in IL-17A levels by 24.3% (P = 0.0046), 31.5% (P = 0.0057), and 17.4% (P = 0.0421) on days 3, 6, and 9 postinjection, respectively (Fig. 6). A similar significant decrease in IL-17A levels was observed upon treating mice with the TLR7 inhibitor in addition to EBV DNA; the IL-17A levels were decreased by 19.3% (P = 0.0015), 26.8% (P = 0.0076), and 24.2% (P = 0.0064) on days 3, 6, and 9 postinjection, respectively, in comparison to the EBV DNA-injected groups (Fig. 7). IL-17A levels were also significantly decreased upon injection of EBV-treated mice with a specific TLR9 inhibitor; on days 3, 6, and 9 postinjection, IL-17A levels declined by 8.5% (P = 0.0068), 19.3% (P = 0.0139), and 6.6% (P = 0.0373), respectively, compared to the EBV DNA-injected groups (Fig. 8). No significant modulation of IL-17A levels was observed in the sera of mice injected with S. epidermidis DNA. TLR3 and TLR7 inhibitors alone did not result in significant changes. However, there was a remarkable increase in IL-17A levels in sera from groups injected with the TLR9 inhibitor alone (Fig. 8).
FIG 6.
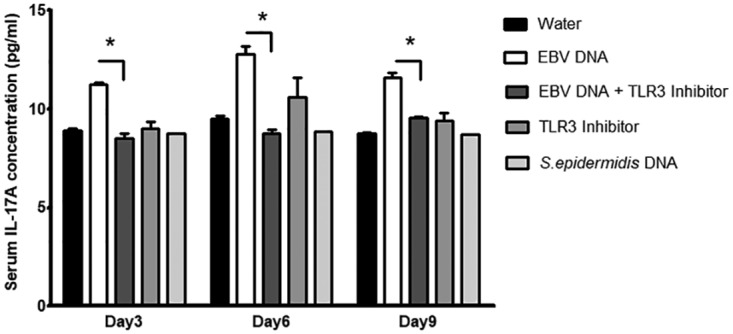
Serum IL-17A levels in BALB/c mice injected with EBV DNA and a TLR3 inhibitor. BALB/c mice were intraperitoneally injected with sterile distilled water, EBV DNA (144 × 103 copies), EBV DNA plus 250 μg of the TLR3 inhibitor (ODN2006), the TLR3 inhibitor alone, or Staphylococcus epidermidis DNA (27.2 pg). Sera were collected on days 3, 6, and 9 postinjection, and IL-17A levels were assessed by an ELISA. *, P < 0.05 compared to the EBV DNA-injected group on the respective day of injection.
FIG 7.
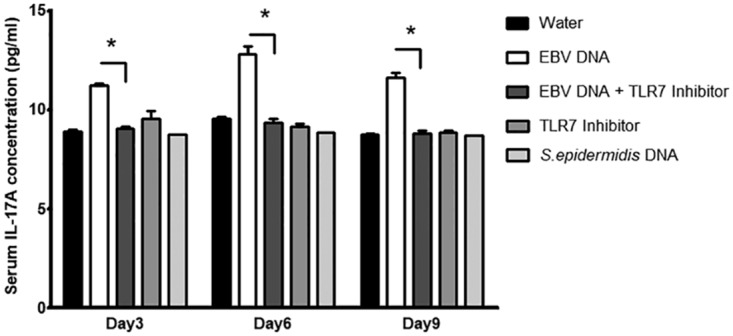
Serum IL-17A levels in BALB/c mice injected with EBV DNA and a TLR7 inhibitor. BALB/c mice were intraperitoneally injected with sterile distilled water, EBV DNA (144 × 103 copies), EBV DNA plus 57.5 μg of the TLR7 inhibitor (IRS661), the TLR7 inhibitor alone, or Staphylococcus epidermidis DNA (27.2 pg). Sera were collected on days 3, 6, and 9 postinjection, and IL-17A levels were assessed by an ELISA. *, P < 0.05 compared to the EBV DNA-injected group on the respective day of injection.
FIG 8.
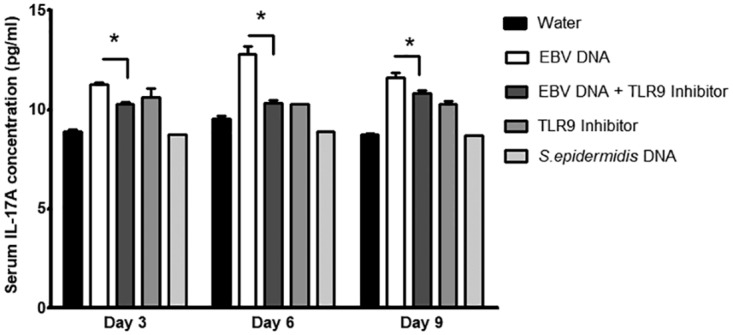
Serum IL-17A levels in BALB/c mice injected with EBV DNA and a TLR9 inhibitor. BALB/c mice were intraperitoneally injected with sterile distilled water, EBV DNA (144 × 103 copies), EBV DNA plus 56 μg of the TLR9 inhibitor (ODN2088), the TLR9 inhibitor alone, or Staphylococcus epidermidis DNA (27.2 pg). Sera were collected on days 3, 6, and 9 postinjection, and IL-17A levels were assessed by an ELISA. *, P < 0.05 compared to the EBV DNA-injected group on the respective day of injection.
DISCUSSION
EBV is a virus that affects the majority of the population, establishing latency in the host, with frequent recurrent infection and viral DNA shedding. EBV is thought to be involved in several autoimmune diseases since it appears to modulate human immune responses (33). We previously reported that EBV DNA was capable of enhancing the production of the proinflammatory cytokine IL-17A when injected into mice (25); moreover, we observed a linear correlation between EBV DNA and serum IL-17A levels in RA patients (6). Since IL-17A plays a critical role in the pathogenesis of a variety of inflammatory autoimmune diseases, EBV DNA may thus contribute to autoimmune disease development or exacerbation. Therefore, the objective of this study was to elucidate the mechanisms through which EBV DNA modulates the levels of IL-17A.
We previously reported that TLR9 inhibition in mouse PBMCs results in a decreased IL-17A response to EBV DNA (6). The activation of monocytes by TLR9 agonists was previously reported to result in the secretion of transforming growth factor β (TGF-β), IL-6, IL-1, IL-23, and IL-21 (30). These factors may then promote IL-17A production. Moreover, PBMCs from patients with acute IM were reported to have significantly elevated TLR9 expression compared to cells from controls or subjects in recovery from acute infection. Acute IM patients also had a higher Th17/CD4+ T cell ratio, a higher Th17/Treg ratio, as well as elevated levels of IL-17A and IL-22 (34).
In this study, we aimed at assessing whether other endosomal TLRs have a role in this pathway. Examination of endosomal TLR3, -7, and -9 using specific inhibitors, employed both ex vivo in PBMCs and in vivo, indicated that these mediators play an essential role in enhancing the production of IL-17A in response to EBV DNA.
Ligands traditionally reported to activate TLR3 and TLR7 are double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA), respectively; hence, our data may indicate indirect activation of these receptors or the possibility that EBV DNA itself may directly interact with TLR3 and -7. A suggested role for TLR3 in recognizing a Herpesviridae dsDNA was previously described. TLR3 and -9 were shown to be expressed in human corneal fibroblasts upon HSV DNA transfection; moreover, HSV DNA induction of IL-6 production in mouse corneal cells was dependent on TLR3 and -9 (35). Moreover, Martin et al. reported that both UV-inactivated and untreated EBV upregulated the expression of TLR7 in naive B cells (36). These findings indicate that both TLR3 and -7 can be activated by viral factors independent of viral replication and RNA transcripts; our data suggest that viral DNA itself may be this factor.
ODN2088, the TLR9 inhibitor employed in this study, was shown to be a potent inhibitor specific to TLR9. It could block activation by TLR9 ligands at very low concentrations (37, 38). Responses to TLR9 ligand induction were completely inhibited in mouse B cells, macrophages, and dendritic cells (DCs) (38, 39). Oligodeoxynucleotides (ODNs) comprising the mouse inhibitory motif for TLR9, like ODN2088 and others, were reported to be active in human B cells, plasmacytoid DCs (40, 41), as well as TLR9-transfected HEK cells (42). As for the TLR7 antagonist IRS661, it was developed by Barrat et al. as the prototypic inhibitor of TLR7-induced innate activation. This ODN specifically blocked IL-6 secretion by splenocytes induced by the TLR7/8 agonist R848 but was ineffective against TLR9 ligand-induced activation (43, 44). Moreover, the use of those inhibitory ODNs as well as others developed by different research groups showed no inhibitory activity against TLR2, -3, -4, and -5, even when using high concentrations (38, 44). On the other hand, ODN-2006, employed in our study, was reported to block TLR3-specific activation of the NF-κB reporter gene in cultured human bronchial epithelial cells and in human PBMCs that express TLR3 (45). ODN2006 has been demonstrated to colocalize with TLR3 in cells. Moreover, ODN2006 has been demonstrated to have no inhibitory effects on TLR4 and TLR9 (45). In our study, IRS661 interestingly triggered an increase in IL-17A levels upon incubation with mouse PBMCs; however, no significant changes were detected upon its injection into mice. On the other hand, ODN2088, the TLR9 antagonist that we used, caused an increase in IL-17A levels when administered by itself to mice. Conversely, when mouse PBMCs were incubated with ODN2088 alone, no significant change was observed in IL-17A levels. In a study conducted by Landrigan et al., ODN2088 was found to induce increases in IL-17A levels in human CD4+ T cells stimulated with anti-CD3 and anti-CD28 (46). This observation suggests that ODN2088, while inhibiting TLR9 and the MyD88 pathway, can stimulate cytokine release by activating other mediators. This observation may be due to pattern recognition receptor (PRR) cross talk, as proposed by Underhill; hence, innate immune receptors can collaborate either positively or negatively in regulating the immune response against a pathogen (47). Research studies conducted in the last few years demonstrated the existence of multiple receptors that detect DNA viruses in the cytosol. These receptors include DNA-dependent activator of interferon (IFN)-regulatory factors (DAI) (DLM-1/ZBP1), a 40-kDa cytosolic protein that binds dsDNA, resulting in an interferon response involving TANK-binding kinase 1 (TBK1) (48). Other reports demonstrated the involvement of a 39-kDa DNA-binding protein named absent in melanoma 2 (AIM2), which has a role in promoting cell death through activating IL-1β and caspase-1 via apoptosis-associated speck-like protein (ASC) (49–51). Other signaling proteins that respond to DNA have also been reported, like cyclic GMP-AMP synthase (cGAS) (52) and the IFN-inducible protein IFI16 (53). These receptors converge on a common signaling protein called STING (stimulator of interferon genes). STING is a transmembrane protein expressed on endoplasmic reticulum membranes and the outer mitochondrial membrane (54). When activated, STING recruits TBK1, an IκB kinase (IKK)-related kinase that phosphorylates the transcription factors IFN-regulatory factor 3 (IRF3) and IRF7 (54, 55), leading to proinflammatory responses from innate immune cells. These studies show that DNA-sensing mechanisms are overlapping and reveal the existence of various DNA sensors. Hence, the complexity of sensors, pathways that respond to nucleic acids, and types of cells that employ these pathways may underlie the discordant responses to the above-mentioned TLR inhibitors; however, the consistency in the decrease in the levels of IL-17A in response to EBV DNA both in vivo and ex vivo upon using these inhibitors confirms the roles played by these TLRs in this response.
Our data indicate the involvement of TLR-3, -7, and -9 in enhancing IL-17A levels in response to EBV DNA. These receptors may thus serve as future therapeutic targets for patients suffering from autoimmune diseases that may be exacerbated or triggered by an EBV infection. Oligonucleotide-based TLR antagonists referred to as immunoregulatory DNA sequences have already shown potential utility in therapeutic approaches to autoimmune diseases. One example is IRS954, which has a dual function, targeting both TLR7 and TLR9. This agent exerted an inhibitory effect on IFN-α production by human plasmacytoid dendritic cells in response to viral DNA and RNA as well as immune complexes from SLE patients (44). In addition, it showed a reduction of autoantibodies and improvement of disease symptoms in lupus-prone mice (43). IMO-3100, another oligonucleotide TLR7/9 dual antagonist, significantly reduced the expression of inflammatory genes such as IL-17A, β-defensin, and CXCL1 in a mouse model of IL-23-induced psoriasis (56). Moreover, this compound had an inhibitory effect on disease progression in a lupus-prone mouse model (57). Finally, IMO-8400, which is capable of inhibiting TLR7, -8, and -9, showed high efficacy in preventing inflammation and disease development in both lupus and psoriasis mouse models (56–58). IMO-3100 and IMO-8400 are currently in phase II clinical trials in patients with moderate to severe plaque psoriasis, and their efficacy is being evaluated (59).
In conclusion, our findings indicate the involvement of three endosomal receptors in EBV DNA recognition and the consequent enhancement of IL-17A levels from mouse immune cells as well as in vivo in mice. Understanding the roles of the plethora of cellular receptors that may potentially respond to viral DNA, including ones not assessed in the present study, may aid in designing prophylactic as well as therapeutic approaches to virally triggered inflammatory diseases, including autoimmune disorders.
MATERIALS AND METHODS
Mice.
Female BALB/c mice, 4 to 6 weeks of age, were obtained from the Animal Care Facility at the American University of Beirut (AUB) and were treated according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at AUB.
Evaluation of the role of the endosome in IL-17A level modulation by EBV DNA in mouse PBMCs.
To examine if the endosome is involved in EBV DNA-triggered IL-17A production, mouse PBMCs from female BALB/c mice, 4 to 6 weeks of age, were separated using the Ficoll-Isopaque method and then used in culture. PBMCs were cultured in 96 wells whereby each well contained 25 × 104 PBMCs in 250 μl of RPMI 1640 culture medium (Sigma-Aldrich Chemie GmbH, Munich, Germany) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich Chemie GmbH, Munich, Germany) and 1% penicillin-streptomycin (Lonza, Basel, Switzerland). Cells were cultured with EBV DNA (9 × 103 copies) (Advanced Biotechnologies, Columbia, MD), with different concentrations of chloroquine (Sigma-Aldrich, St. Louis, MO), with both EBV DNA and chloroquine, or with 1.7 pg of S. epidermidis DNA (equivalent to the weight of 9 × 103 copies of EBV DNA). The EBV DNA used in all experiments is the purified complete genome of EBV strain B95-8, which is a type 1 EBV strain. Isolated from nascent EBV particles, this genome is linear and rich in unmethylated CpG motifs. The 172,282 bp of the B95-8 genome were sequenced by Baer et al. (60). Chloroquine concentrations used were 20 μM, 30 μM, and 40 μM. For double-treatment conditions, cells were incubated with chloroquine 30 min before the addition of EBV DNA. As a control, the EBV DNA preparation was treated with a working concentration of 1 mg/ml of DNase I (Sigma-Aldrich, St. Louis, MO) and cultured with the mouse PBMCs as described above; the DNase by itself was also incubated with the PBMCs to exclude any effects of this treatment. In addition, untreated cells grown in culture medium were examined. Triplicates were performed for each treatment. Cells were incubated for 24 h at 37°C with 5% CO2. Supernatants were then collected and assessed by an enzyme-linked immunosorbent assay (ELISA) (Abcam, Cambridge, UK) for mouse IL-17A level assessment.
Assessment of gene expression of endosomal TLRs in splenic tissue from EBV DNA-treated mice.
To assess whether EBV DNA alters the expression of the endosomal TLRs, TLR3, -7, and -9, we conducted gene expression studies. For each TLR, 2 mouse groups were used: one injected with sterile distilled water as the negative control and the other injected with 144 × 103 copies of EBV DNA. Each group consisted of 9 mice, which were treated via intraperitoneal injection; 3 mice from each group were sacrificed on days 3, 6, and 9 after injection. Upon sacrifice, mouse spleens were collected and homogenized, and splenocytes were isolated for RNA extraction using Qiazol (Qiagen, Hilden, Germany). cDNA synthesis from the extracted RNA was performed using the QuantiTect reverse transcription kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Real-time PCR was then carried out on these samples to detect the relative gene expression levels of TLR3, -7, and -9 normalized to that of β-actin per sample. Primers were obtained from Macrogen (Seoul, South Korea). Primers, annealing temperatures, and product lengths are detailed in Table 1. Relative expression levels per sample normalized to that of the water-injected calibrator group on day 3 were then calculated using the ΔΔCT method (61, 63, 64). Assessments were performed in triplicates.
TABLE 1.
Primers used for relative gene expression assessment by real-time reverse transcription-PCR
| Gene | Primera | Annealing temp (°C) | Product length (bp) | Reference |
|---|---|---|---|---|
| TLR3 | F, 5′-GGTGTTTCCAGACAATTGGCAAG-3′ | 60.7 | 217 | 53 |
| R, 5′-TGGAGGTTGTTGTAGGAAAGATCG-3′ | ||||
| TLR7 | F, 5′-CCACAGGCTCACCCATACTTC-3′ | 62.4 | 129 | 54 |
| R, 5′-GGGATGTCCTAGGTGGTGACA-3′ | ||||
| TLR9 | F, 5′-ACTGAGCACCCCTGCTTCTA-3′ | 60.8 | 198 | 55 |
| R, 5′-AGATTAGTCAGCGGCAGGAA-3′ | ||||
| β-Actin | F, 5′-GGCATTGTTACCAACTGGGACGAC-3′ | 58.6 | 218 | 56 |
| R, 5′-CCAGAGGCATACAGGGACAGCACAG-3′ | ||||
F, forward; R, reverse.
Examination of the role of endosomal TLR3, -7, and -9 in EBV DNA-triggered IL-17A production ex vivo and in vivo.
We next assessed if endosomal TLRs, which are known to detect nucleic acids, play a role in EBV DNA detection leading to IL-17A production. Specific oligonucleotide inhibitors were used to assess the role of TLR3, -7, and -9 in this response to EBV DNA.
Mouse PBMCs from female BALB/c mice, 4 to 6 weeks of age, were incubated with 9 × 103 copies of EBV DNA, with each of the TLR inhibitors alone, with both EBV DNA and each of the inhibitors, or with S. epidermidis DNA (1.7 pg, equivalent to the weight of 9 × 103 copies of EBV DNA) as a nonviral DNA control. PBMCs (25 × 104) in 250 μl of RPMI 1640 culture medium supplemented with 10% FBS and 1% penicillin-streptomycin were used per treatment. The TLR3 inhibitor ODN2006 was used at a concentration of 10 μM, the TLR7 inhibitor IRS661 was used at a concentration of 2.8 μM, and the TLR9 inhibitor ODN2088 was used at a concentration of 1.4 μM. These concentrations were selected based on experiments previously conducted to assess the optimal concentrations to inhibit IL-17A release from PBMCs challenged with EBV DNA. The sequence of ODN2006 is 5′-TCGTCGTTTTGTCGTTTTGTCGTT-3′ (45), that of IRS661 is 5′-TGCTTGCAAGCTTGCAAGCA-3′ (44), and that of ODN2088 is 5′-TCCTGGCGGGGAAGT-3′ (40). Inhibitory oligonucleotides were designed to resist degradation by nucleases through the introduction of phosphorothioate linkages instead of the phosphodiester backbone. The TLR inhibitors were obtained from Integrated DNA Technologies (Coralville, IA). Untreated cells were included in the analysis as controls. Cells were then incubated for 24 h at 37°C with 5% CO2. After 24 h, cell supernatants were collected, and an ELISA (Abcam, Cambridge, UK) was performed to determine IL-17A levels. A duplicate analysis of the experiment was conducted.
To examine whether ex vivo observations could be replicated in vivo, 4- to 6-week-old female BALB/c mice were injected with EBV DNA in the presence or absence of treatment with the TLR inhibitors. Nine groups, each containing 9 mice, were used. Group 1 served as the negative control and was injected with 100 μl of sterile distilled water. Group 2 was injected with 144 × 103 copies of EBV DNA in 100 μl of solution. Groups 3 to 5 were injected with both EBV DNA and a TLR inhibitor. Groups 6 to 8 were injected with each of the TLR inhibitors alone. Group 9 was injected with 27.2 pg of genomic DNA from S. epidermidis (equivalent to the weight of 144 × 103 copies of EBV DNA) as a nonviral DNA control. TLR inhibitor doses were based on the concentrations of inhibitors used in ex vivo experiments. Doses were calculated depending on mouse weight and the number of cells in the BALB/c mouse strain. For each injection, 250 μg of ODN2006 in 200 μl of sterile water was given. As for IRS661 and ODN2088, the doses used were 57.5 μg and 56 μg in 100 μl of sterile water, respectively. All injections were performed intraperitoneally. Three mice per group were sacrificed after 3, 6, and 9 days of injection, their blood was collected, and the sera were grouped per time point from each group. An ELISA (Abcam, Cambridge, UK) was then used to assess serum levels of IL-17A. A duplicate analysis of the experiment was conducted.
Apoptosis analysis.
Mouse PBMCs were treated with EBV DNA in the presence or absence of the TLR3, -7, and -9 inhibitors as described above for the ex vivo analysis. Untreated PBMCs were included as well. After 24 h, cells were harvested, washed, and stained using the eBioscience annexin V apoptosis detection kit with fluorescein isothiocyanate (FITC) (Invitrogen) to detect early apoptotic cells and eBioscience 7-AAD viability staining solution (Invitrogen) to detect late apoptotic cells. After staining, live early and late apoptotic populations were assessed using the Guava Easycyte 8 flow cytometer (Merck Millipore), and analysis was performed using Guava InCyte software (Merck Millipore).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism. Data were analyzed using two-tailed paired Student’s t test (Wilcoxon) to compare means. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
This study was funded by a grant from the Medical Practice Plan (MPP) at the American University of Beirut. The funder did not have any contribution to study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Hislop AD. 2015. Early virological and immunological events in Epstein-Barr virus infection. Curr Opin Virol 15:75–79. doi: 10.1016/j.coviro.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Depper JM, Zvaifler NJ. 1981. Epstein-Barr virus. Its relationship to the pathogenesis of rheumatoid arthritis. Arthritis Rheum 24:755–761. doi: 10.1002/art.1780240601. [DOI] [PubMed] [Google Scholar]
- 3.Cohen JI. 2000. Epstein-Barr virus infection. N Engl J Med 343:481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 4.Toussirot E, Roudier J. 2008. Epstein-Barr virus in autoimmune diseases. Best Pract Res Clin Rheumatol 22:883–896. doi: 10.1016/j.berh.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Balandraud N, Meynard JB, Auger I, Sovran H, Mugnier B, Reviron D, Roudier J, Roudier C. 2003. Epstein-Barr virus load in the peripheral blood of patients with rheumatoid arthritis: accurate quantification using real-time polymerase chain reaction. Arthritis Rheum 48:1223–1228. doi: 10.1002/art.10933. [DOI] [PubMed] [Google Scholar]
- 6.Salloum N, Hussein HM, Jammaz R, Jiche S, Uthman IW, Abdelnoor AM, Rahal EA. 2018. Epstein-Barr virus DNA modulates regulatory T-cell programming in addition to enhancing interleukin-17A production via Toll-like receptor 9. PLoS One 13:e0200546. doi: 10.1371/journal.pone.0200546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tosato G, Steinberg AD, Yarchoan R, Heilman CA, Pike SE, De Seau V, Blaese RM. 1984. Abnormally elevated frequency of Epstein-Barr virus-infected B cells in the blood of patients with rheumatoid arthritis. J Clin Invest 73:1789–1795. doi: 10.1172/JCI111388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newkirk MM, Watanabe Duffy KN, Leclerc J, Lambert N, Shiroky JB. 1994. Detection of cytomegalovirus, Epstein-Barr virus and herpes virus-6 in patients with rheumatoid arthritis with or without Sjogren’s syndrome. Br J Rheumatol 33:317–322. doi: 10.1093/rheumatology/33.4.317. [DOI] [PubMed] [Google Scholar]
- 9.Berkun Y, Zandman-Goddard G, Barzilai O, Boaz M, Sherer Y, Larida B, Blank M, Anaya JM, Shoenfeld Y. 2009. Infectious antibodies in systemic lupus erythematosus patients. Lupus 18:1129–1135. doi: 10.1177/0961203309345729. [DOI] [PubMed] [Google Scholar]
- 10.Shirodaria PV, Haire M, Fleming E, Merrett JD, Hawkins SA, Roberts SD. 1987. Viral antibody titers. Comparison in patients with multiple sclerosis and rheumatoid arthritis. Arch Neurol 44:1237–1241. doi: 10.1001/archneur.1987.00520240019006. [DOI] [PubMed] [Google Scholar]
- 11.Fujinami RS, Oldstone MB, Wroblewska Z, Frankel ME, Koprowski H. 1983. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci U S A 80:2346–2350. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav P, Tran H, Ebegbe R, Gottlieb P, Wei H, Lewis RH, Mumbey-Wafula A, Kaplan A, Kholdarova E, Spatz L. 2011. Antibodies elicited in response to EBNA-1 may cross-react with dsDNA. PLoS One 6:e14488. doi: 10.1371/journal.pone.0014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James JA, Harley JB. 1992. Linear epitope mapping of an Sm B/B′ polypeptide. J Immunol 148:2074–2079. [PubMed] [Google Scholar]
- 14.Sabbatini A, Bombardieri S, Migliorini P. 1993. Autoantibodies from patients with systemic lupus erythematosus bind a shared sequence of SmD and Epstein-Barr virus-encoded nuclear antigen EBNA I. Eur J Immunol 23:1146–1152. doi: 10.1002/eji.1830230525. [DOI] [PubMed] [Google Scholar]
- 15.Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, Hjorth P, Sondergaard L, Svejgaard A, Wucherpfennig K, Stuart DI, Bell JI, Jones EY, Fugger L. 2002. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- 16.Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, Munz C. 2008. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med 205:1763–1773. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt H, Williamson D, Ashley-Koch A. 2007. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am J Epidemiol 165:1097–1109. doi: 10.1093/aje/kwk118. [DOI] [PubMed] [Google Scholar]
- 18.Caballero A, Alves-Leon S, Papais-Alvarenga R, Fernandez O, Navarro G, Alonso A. 1999. DQB1*0602 confers genetic susceptibility to multiple sclerosis in Afro-Brazilians. Tissue Antigens 54:524–526. doi: 10.1034/j.1399-0039.1999.540511.x. [DOI] [PubMed] [Google Scholar]
- 19.Marrosu MG, Murru MR, Costa G, Murru R, Muntoni F, Cucca F. 1998. DRB1-DQA1-DQB1 loci and multiple sclerosis predisposition in the Sardinian population. Hum Mol Genet 7:1235–1237. doi: 10.1093/hmg/7.8.1235. [DOI] [PubMed] [Google Scholar]
- 20.Samaha H, Rahal EA, Abou-Jaoude M, Younes M, Dacchache J, Hakime N. 2003. HLA class II allele frequencies in the Lebanese population. Mol Immunol 39:1079–1081. doi: 10.1016/S0161-5890(03)00073-7. [DOI] [PubMed] [Google Scholar]
- 21.van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM. 1999. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol 162:129–135. [PubMed] [Google Scholar]
- 22.van Noort JM, Bajramovic JJ, Plomp AC, van Stipdonk MJ. 2000. Mistaken self, a novel model that links microbial infections with myelin-directed autoimmunity in multiple sclerosis. J Neuroimmunol 105:46–57. doi: 10.1016/S0165-5728(00)00181-8. [DOI] [PubMed] [Google Scholar]
- 23.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. 2003. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25:1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 24.Pratesi F, Tommasi C, Anzilotti C, Chimenti D, Migliorini P. 2006. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum 54:733–741. doi: 10.1002/art.21629. [DOI] [PubMed] [Google Scholar]
- 25.Rahal EA, Hajjar H, Rajeh M, Yamout B, Abdelnoor AM. 2015. Epstein-Barr virus and human herpes virus 6 type A DNA enhance IL-17 production in mice. Viral Immunol 28:297–302. doi: 10.1089/vim.2014.0129. [DOI] [PubMed] [Google Scholar]
- 26.Iwakura Y, Nakae S, Saijo S, Ishigame H. 2008. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev 226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 27.Costa VS, Mattana TC, da Silva ME. 2010. Unregulated IL-23/IL-17 immune response in autoimmune diseases. Diabetes Res Clin Pract 88:222–226. doi: 10.1016/j.diabres.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Wagner H. 1999. Bacterial CpG DNA activates immune cells to signal infectious danger. Adv Immunol 73:329–368. doi: 10.1016/S0065-2776(08)60790-7. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg P, Welander P, Han X, Cantin E. 2003. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol 77:11158–11169. doi: 10.1128/JVI.77.20.11158-11169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiola S, Gosselin D, Takada K, Gosselin J. 2010. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J Immunol 185:3620–3631. doi: 10.4049/jimmunol.0903736. [DOI] [PubMed] [Google Scholar]
- 31.Kieff E, Dambaugh T, Heller M, King W, Cheung A, van Santen V, Hummel M, Beisel C, Fennewald S, Hennessy K, Heineman T. 1982. The biology and chemistry of Epstein-Barr virus. J Infect Dis 146:506–517. doi: 10.1093/infdis/146.4.506. [DOI] [PubMed] [Google Scholar]
- 32.Steinman RM, Mellman IS, Muller WA, Cohn ZA. 1983. Endocytosis and the recycling of plasma membrane. J Cell Biol 96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lunemann JD, Kamradt T, Martin R, Munz C. 2007. Epstein-Barr virus: environmental trigger of multiple sclerosis? J Virol 81:6777–6784. doi: 10.1128/JVI.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao A, Shao YW. 2017. Investigation for the roles of TLR2, TLR9 and Th17/Treg in the pathogenesis of infectious mononucleosis. Int J Clin Exp Med 10:14946–14953. [Google Scholar]
- 35.Hayashi K, Hooper LC, Chin MS, Nagineni CN, Detrick B, Hooks JJ. 2006. Herpes simplex virus 1 (HSV-1) DNA and immune complex (HSV-1-human IgG) elicit vigorous interleukin 6 release from infected corneal cells via Toll-like receptors. J Gen Virol 87:2161–2169. doi: 10.1099/vir.0.81772-0. [DOI] [PubMed] [Google Scholar]
- 36.Martin HJ, Lee JM, Walls D, Hayward SD. 2007. Manipulation of the Toll-like receptor 7 signaling pathway by Epstein-Barr virus. J Virol 81:9748–9758. doi: 10.1128/JVI.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenert P, Stunz L, Yi AK, Krieg AM, Ashman RF. 2001. CpG stimulation of primary mouse B cells is blocked by inhibitory oligodeoxyribonucleotides at a site proximal to NF-kappaB activation. Antisense Nucleic Acid Drug Dev 11:247–256. doi: 10.1089/108729001317022241. [DOI] [PubMed] [Google Scholar]
- 38.Stunz LL, Lenert P, Peckham D, Yi AK, Haxhinasto S, Chang M, Krieg AM, Ashman RF. 2002. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol 32:1212–1222. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Lenert P, Yi AK, Krieg AM, Stunz LL, Ashman RF. 2003. Inhibitory oligonucleotides block the induction of AP-1 transcription factor by stimulatory CpG oligonucleotides in B cells. Antisense Nucleic Acid Drug Dev 13:143–150. doi: 10.1089/108729003768247600. [DOI] [PubMed] [Google Scholar]
- 40.Duramad O, Fearon KL, Chang B, Chan JH, Gregorio J, Coffman RL, Barrat FJ. 2005. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J Immunol 174:5193–5200. doi: 10.4049/jimmunol.174.9.5193. [DOI] [PubMed] [Google Scholar]
- 41.Lenert P. 2005. Inhibitory oligodeoxynucleotides—therapeutic promise for systemic autoimmune diseases? Clin Exp Immunol 140:1–10. doi: 10.1111/j.1365-2249.2004.02728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashman RF, Lenert P. 2007. Structural requirements and applications of inhibitory oligodeoxyribonucleotides. Immunol Res 39:4–14. doi: 10.1007/s12026-007-0065-4. [DOI] [PubMed] [Google Scholar]
- 43.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. 2007. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol 37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 44.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. 2005. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranjith-Kumar CT, Duffy KE, Jordan JL, Eaton-Bassiri A, Vaughan R, Hoose SA, Lamb RJ, Sarisky RT, Kao CC. 2008. Single-stranded oligonucleotides can inhibit cytokine production induced by human Toll-like receptor 3. Mol Cell Biol 28:4507–4519. doi: 10.1128/MCB.00308-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landrigan A, Wong MT, Utz PJ. 2011. CpG and non-CpG oligodeoxynucleotides directly costimulate mouse and human CD4+ T cells through a TLR9- and MyD88-independent mechanism. J Immunol 187:3033–3043. doi: 10.4049/jimmunol.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Underhill DM. 2007. Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immunol Rev 219:75–87. doi: 10.1111/j.1600-065X.2007.00548.x. [DOI] [PubMed] [Google Scholar]
- 48.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 49.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. 2009. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 50.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroder K, Muruve DA, Tschopp J. 2009. Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome. Curr Biol 19:R262–R265. doi: 10.1016/j.cub.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 52.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hornung V, Latz E. 2010. Intracellular DNA recognition. Nat Rev Immunol 10:123–130. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 56.Suarez-Farinas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG. 2013. Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS One 8:e84634. doi: 10.1371/journal.pone.0084634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu FG, Jiang W, Bhagat L, Wang D, Yu D, Tang JX, Kandimalla ER, La Monica N, Agrawal S. 2013. A novel antagonist of Toll-like receptors 7, 8 and 9 suppresses lupus disease-associated parameters in NZBW/F1 mice. Autoimmunity 46:419–428. doi: 10.3109/08916934.2013.798651. [DOI] [PubMed] [Google Scholar]
- 58.Jiang W, Zhu FG, Bhagat L, Yu D, Tang JX, Kandimalla ER, La Monica N, Agrawal S. 2013. A Toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J Invest Dermatol 133:1777–1784. doi: 10.1038/jid.2013.57. [DOI] [PubMed] [Google Scholar]
- 59.Balak DM, van Doorn MB, Arbeit RD, Rijneveld R, Klaassen E, Sullivan T, Brevard J, Thio HB, Prens EP, Burggraaf J, Rissmann R. 2017. IMO-8400, a Toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin Immunol 174:63–72. doi: 10.1016/j.clim.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 60.Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 61.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 62.Ajib R, Janbazian L, Rahal E, Matar GM, Zaynoun S, Kibbi AG, Abdelnoor AM. 2005. HLA allele associations and V-beta T-lymphocyte expansions in patients with psoriasis, harboring toxin-producing Staphylococcus aureus. J Biomed Biotechnol 2005:310–315. doi: 10.1155/JBB.2005.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rahal EA, Kazzi N, Sabra A, Abdelnoor AM, Matar GM. 2011. Decrease in Shiga toxin expression using a minimal inhibitory concentration of rifampicin followed by bactericidal gentamicin treatment enhances survival of Escherichia coli O157:H7-infected BALB/c mice. Ann Clin Microbiol Antimicrob 10:34. doi: 10.1186/1476-0711-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matar GM, Rahal E. 2003. Inhibition of the transcription of the Escherichia coli O157:H7 genes coding for shiga-like toxins and intimin, and its potential use in the treatment of human infection with the bacterium. Ann Trop Med Parasitol 97:281–287. doi: 10.1179/000349803235002146. [DOI] [PubMed] [Google Scholar]
- 65.Sherri N, Salloum N, Mouawad C, Haidar-Ahmad N, Shirinian M, Rahal EA. 2018. Epstein-Barr virus DNA enhances diptericin expression and increases hemocyte numbers in Drosophila melanogaster via the immune deficiency pathway. Front Microbiol 9:1268. doi: 10.3389/fmicb.2018.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]