

Abstract
In this era of high‐throughput biology, bioinformatics has become a major discipline for making sense out of large‐scale datasets. Bioinformatics is usually considered as a practical field developing databases and software tools for supporting other fields, rather than a fundamental scientific discipline for uncovering principles of biology. The KEGG resource that we have been developing is a reference knowledge base for biological interpretation of genome sequences and other high‐throughput data. It is now one of the most utilized biological databases because of its practical values. For me personally, KEGG is a step toward understanding the origin and evolution of cellular organisms.
Keywords: KEGG, KEGG MEDICUS, KEGG module, pathway mapping, reaction module
1. INTRODUCTION
The advent of DNA sequencing methods1, 2 in the mid‐1970s was followed by an accumulation of DNA and translated protein sequence data in published literature, leading to the establishment of sequence databases and new activities of data‐driven computational biology, later called bioinformatics. The trend of developing high‐throughput experimental technologies and generating large‐scale datasets was accelerated by the Human Genome Project in the 1990s and continues to the present. In the meantime, bioinformatics has become a major discipline for handling and analyzing large‐scale biological data with advanced informatics technologies. I had two opportunities to become part of these developments, first in 1979 for the startup of Los Alamos Sequence Library,3 which later became GenBank, and second in 1989 for the startup of the Japanese Human Genome Project, which eventually lead to the conception and development of the KEGG (Kyoto Encyclopedia of Genes and Genomes) database resource (www.kegg.jp).4 This review chronicles how KEGG has been expanded over the past 25 years enabling different types of bioinformatics research.
2. BLUEPRINTS OF LIFE
The KEGG database project was initiated in 1995 under the Japanese Human Genome Project, foreseeing the need for a reference resource that would enable computational reconstruction of the biological systems, including the cell, the organism, and the ecosystem, from the genome information. In the traditional view, the genome is a blueprint of life containing all necessary information that would make up an organism. In my view, however, the genome specifies only the molecular building blocks, while the cell, the basic unit of life, contains information about how they interact and react to form a system.5 What we inherit is not just the genome, but the entire cell, and there is a cellular continuity of the germline leading to the origin of life. From this perspective, knowledge of cellular functions and other high‐level features of organisms has been captured from experimental observations reported in published literature and organized in the form of KEGG pathway maps and other types of molecular networks. By integrating the molecular wiring diagrams encoded in the cell (chemical blueprint of life) with the molecular building blocks encoded in the genome (genetic blueprint of life) for all available cellular organisms, KEGG has become a reference resource for deciphering the genome.
3. PATHWAY MAPPING
The KEGG PATHWAY database is a collection of manually drawn KEGG pathway maps for metabolic pathways, signaling pathways, pathways involved in various cellular processes and organismal systems, and perturbed pathways associated with human diseases. The KEGG pathway map is a simply designed graphical diagram, in which rectangles represent gene products, mostly proteins, and circles represent all other molecules, mostly chemical compounds. Interactions and reactions among these molecules are represented by different types of arrows. Figure 1 shows an example map00220 for the metabolic pathway of arginine biosynthesis. The purpose of KEGG pathway maps was to establish links from genes in the genome to gene products in the pathway. This was the idea of KEGG pathway mapping, the main motivation of developing the original KEGG database.
Figure 1.
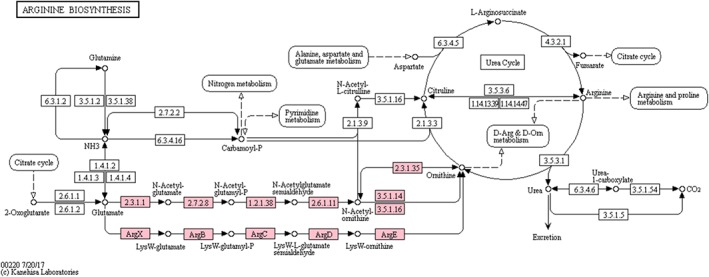
The KEGG metabolic pathway map for arginine biosynthesis (map00220), where rectangles and circles represent enzymes and chemical compounds (substrates and products), respectively. Enzymes are identified by functional orthologs called KOs, although EC numbers and gene names are displayed. The KEGG pathway module is a functional unit in the metabolic pathway defined by a set of KOs. Two modules M00028 (upper) and M00763 (lower) are shown by pink‐colored rectangles
Generally speaking, it is easier to collect everything when developing a database, than to extract or summarize essential contents. Two types of abstractions were used when designing KEGG pathway maps: generalized protein‐protein interactions and functional grouping later called KO (KEGG Orthology). The generalized protein–protein interactions represent how gene products are connected in three ways: enzyme–enzyme relations for successive reaction steps in metabolic pathways, gene expression relations for transcription factors and transcribed gene products, and direct protein–protein interactions. While the generalized protein–protein interaction was a simple rule to implement, the KO grouping required more effort. KO is a manually defined functional ortholog, and the KEGG pathway maps are drawn with KOs as nodes (rectangles) rather than individual genes or proteins, in order to extend experimental knowledge in specific organisms to other organisms. Once genes in a genome are given KO identifiers by the KEGG annotation procedure, they can be used to select nodes of matching KOs (marked by coloring) to generate organism‐specific KEGG pathway maps, enabling interpretation of cellular functions and other high‐level features of organisms. With the KO grouping, KEGG has become a generic resource that can be applied to any cellular organism.
4. INTEGRATION OF GENOMICS AND CHEMISTRY
Metabolism is one of the most basic attributes of cellular organisms. The KEGG resource contains knowledge of metabolic pathways represented by different types of KEGG pathway maps, called global map, overview map, and regular map, and also by KEGG pathway modules for conserved functional units of metabolic pathways. The global and overview maps are summaries of multiple regular maps, drawn with circles and lines representing chemical compounds (metabolites) and enzyme KOs, respectively. Figure 2 is the most utilized KEGG map, map01100 for the global map of metabolic pathways with highlighted parts corresponding to the regular map of arginine biosynthesis shown in Figure 1.
Figure 2.
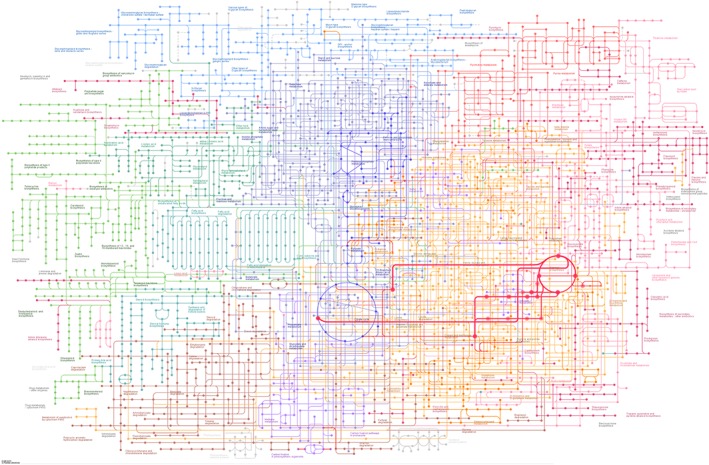
The global map of metabolic pathways (map01100), which contains 3,000 chemical compounds and 4,000 enzyme KOs. The coloring distinguishes categories of metabolic pathways as defined by KEGG. The arginine biosynthesis pathway of Figure 1 is highlighted with thick red lines
The regular metabolic pathway map is drawn with circles and rectangles for chemical compounds and enzyme KOs, respectively (see Figure 1), and may be viewed as a dual network. First, the map shows the chemical network of how chemical compounds are transformed, which is the common view of metabolism. Second, the map also shows a genomic network of how enzyme genes are connected, which is the view of enzyme‐enzyme relation networks. For the latter view, a set of genes encoded as a gene cluster (operon‐like structure) in the genome is known to sometimes correspond to a set of enzymes that catalyze successive reaction steps in the metabolic pathway.6 The pathway module is a functional unit of the metabolic pathway, including such an enzyme cluster. It is defined by a set of KOs, or more precisely by a logical expression of KOs for automated interpretation of its presence or absence. In Figure 1 two modules, M00028 and M00763, are defined for the enzyme sets that catalyze the same overall reaction from glutamate to ornithine, but the reaction steps are somewhat different, M00028 involving N‐acetylation and M00763 mediated by a carrier protein, LysW, and found in archaea.
To capture the similarity of reactions, local chemical structure transformation patterns between a substrate and a product are used to define the reaction class (RC).7 RC is a functional grouping of reactions, as compared to KO as a functional grouping of genes. The reaction module is then defined by a set of RCs as a conserved unit of similar reaction steps in the metabolic pathway viewed as a chemical structure transformation network. The two pathway modules M00028 and M00763 are found to belong to the same reaction module RM002. Furthermore, another pathway module, lysine biosynthesis module M00031 from 2‐aminoadipate to lysine mediated by LysW, also belongs to RM002. This and other related reaction modules are illustrated in Figure 3 and fully shown in the overview map of map01210 for 2‐oxocarboxylic acid metabolism. Thus, KEGG enables integrated analysis of genomics and chemistry, for example, to explore co‐evolution of chemical and genomic networks.8
Figure 3.
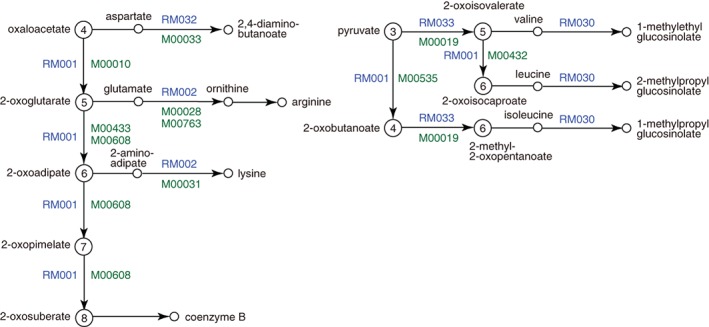
Correspondence of pathway modules (M numbers in green) defined by KOs and reaction modules (RM numbers in blue) defined by RCs. Reaction modules are more general functional units, each of which may correspond to multiple pathway modules, such as RM001 for 2‐oxocarboxylic chain extension and RM002 and RM033 for conversion to basic and branched‐chain amino acids, respectively
5. MEDICUS EXTENSION
Table 1 shows a history of how KEGG was expanded, starting from only four databases in 1995 and now consisting of 18 databases. The BRITE database was an important addition to KEGG, enabling mapping against hierarchical classification systems in a similar way to GO enrichment analysis.9 Although KEGG pathway mapping allows more detailed interpretation of molecular pathways involved, BRITE enrichment analysis has significantly expanded the repertoire of KOs for functional interpretation.
Table 1.
The KEGG database resource
| Category | Database | Content | Release |
|---|---|---|---|
| Systems information | PATHWAY | KEGG pathway maps | 1995 |
| BRITE | BRITE hierarchies and tables (classification systems) | 2005 | |
| MODULE | KEGG modules | 2006 | |
| Genomic information | KO | KO groups for functional orthologs | 2002 |
| GENOME | KEGG organisms (complete genomes) | 2000 | |
| GENES | Genes and proteins | 1995 | |
| SSDB | Sequence similarity among GENES entries | 2001 | |
| Chemical information | COMPOUND | Metabolites and other small molecules | 1995 |
| GLYCAN | Glycans | 2003 | |
| REACTION/RCLASS | Biochemical reactions/reaction classes | 1998/2010 | |
| ENZYME | Enzyme nomenclature | 1995 | |
| Health information | NETWORK/VARIANT | Disease‐related network variants/gene variants | 2017 |
| DISEASE | Human diseases | 2008 | |
| DRUG/DGROUP | Drugs/drug groups | 2005/2014 | |
| ENVIRON | Crude drugs and health‐related substances | 2010 |
The three categories in Table 1, systems, genomic and chemical information categories, constitute a generic resource used mostly for basic research. In contrast, the health information category is a human‐specific resource aimed at practical applications for use in society. This category is called KEGG MEDICUS, and also includes outside databases of drug labels: Japanese drug labels provided by Japan Pharmaceutical Information Center (JAPIC) and incorporated into KEGG and FDA drug labels linked to the DailyMed database (dailymed.nlm.nih.gov). KEGG MEDICUS is a resource for translational bioinformatics in two ways: one for helping research communities to develop practical applications and the other for helping society to better understand the scientific basis of diseases and drugs. KEGG MEDICUS is now one of the major drug information resources available on the web within Japan, heavily accessed through web search engines.
As a translational bioinformatics resource for scientific communities, the DISEASE and DRUG databases were developed from the viewpoints of molecular networks. Diseases are viewed as perturbed states of molecular networks caused by perturbants including gene variants, viruses and other pathogens, and various environmental factors. Drugs are different types of perturbants designed to change the states of molecular networks. These views are now explicitly implemented as network variants in the KEGG NETWORK database with a new type of KEGG map called network variation map.10 Efforts are being made to make KEGG more useful for understanding molecular mechanisms of diseases and for supporting drug discovery and development.
6. DATA, INFORMATION, KNOWLEDGE, AND PRINCIPLE
Big data analysis by businesses has been wildly successful, but how much success can we expect from big data analysis in biology? The data‐driven approach we have been promoting is to use reference knowledge that would never be extracted from big data alone. Genomics data are big data, but they contain only information about parts list. We need knowledge about wiring diagrams, which has been accumulated over the years by traditional model‐driven or hypothesis‐driven approaches. KEGG incorporates manually verified experimental results, but no computational predictions or simply processed data from high‐throughput experiments. In the hierarchy of data, information, and knowledge, I consider associations mined from big data as just information, but when they are manually verified with additional data I would call them knowledge.11 Eventually, a body of knowledge may lead to uncovering basic principles of biological systems in concordance with the principles of physics and chemistry. My goal is to develop KEGG into a knowledge base for analyzing biological systems toward this direction.
ACKNOWLEDGMENTS
I thank all the members, past and present, involved in the development of KEGG. I also thank Institute for Chemical Research, Kyoto University and Human Genome Center, Institute of Medical Science, University of Tokyo for the continued support of the KEGG project.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Science. 2019;28:1947–1951. 10.1002/pro.3715
Minoru Kanehisa is the winner of the 2019 Carl Branden Award.
REFERENCES
- 1. Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain‐terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanehisa MI. Los Alamos sequence analysis package for nucleic acids and proteins. Nucleic Acids Res. 1982;10:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kanehisa M (2000) Post‐genome Informatics, Oxford University Press.
- 6. Ogata H, Fujibuchi W, Goto S, Kanehisa M. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 2000;28:4021–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muto A, Kotera M, Tokimatsu T, Nakagawa Z, Goto S, Kanehisa M. Modular architecture of metabolic pathways revealed by conserved sequences of reactions. J Chem Inf Model. 2013;53:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kanehisa M. Chemical and genomic evolution of enzyme‐catalyzed reaction networks. FEBS Lett. 2013;587:2731–2737. [DOI] [PubMed] [Google Scholar]
- 9. Gene Ontology Consortium . Gene ontology consortium: Going forward. Nucleic Acids Res. 2015;43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47:D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]