

Abstract
Purpose
To investigate the molecular pathways that drive thyroid stimulating hormone receptor (TSHR)–induced cellular proliferation in orbital fibroblasts (OFs) from thyroid eye disease (TED) patients.
Methods
Orbital fibroblasts from TED and non-TED patients were treated with TSH and changes in gene expression and proliferation were measured. To determine the role of TSHR, TSHR-specific siRNA was used to deplete TSHR levels. Proliferation was measured by bromodeoxyuridine (BrdU) incorporation. PI3K/Akt activation was analyzed by Western blot. The PI3K inhibitor LY294002 was used to investigate PI3K/Akt signaling in OF proliferation. Expression of TSHR, inflammatory cytokines, proliferation related genes and miR-146a and miR-155 were measured by qPCR.
Results
Orbital fibroblasts from TED patients proliferate significantly more than non-TED OFs in response to TSH. TSH-induced proliferation was dependent upon TSHR expression and required the PI3K/Akt signaling cascade. TSHR activation stimulated miR-146a and miR-155 expression. TED OFs produced significantly more miR-146a and miR-155 than non-TED OFs. MiR-146a and miR-155 targets, ZNRF3 and PTEN, which both limit cell proliferation, were decreased in TSH treated OFs.
Conclusions
These data reveal that TSHR signaling in TED OFs stimulates proliferation directly through PI3K/Akt signaling and indirectly through induction of miR-146a and miR-155. MiR-146a and miR-155 enhance TED OF proliferation by reducing expression of target genes that normally block cell proliferation. TSHR-dependent expression of miR-146a and miR-155 may explain part of the fibroproliferative pathology observed in TED.
Keywords: Thyroid eye disease, thyroid stimulating hormone receptor, TSHR, TSH, PI3K/Akt, Orbital fibroblast, microRNA, miR-146a, miR-155, proliferation
Thyroid eye disease (TED) is the most common orbital pathology seen and occurs in up to 50% of patients with Graves' disease, an autoimmune disorder characterized by hyperthyroidism.1 The hallmark features of TED are inflammation, excessive hyaluronan deposition, and increased fat and/or extraocular muscle/scar tissue. Orbital tissue remodeling and enlargement causes eye protrusion; neuropathy; double vision; and in severe cases, vision loss.2 While TED is a destructive inflammatory disease, it is unclear how and why TED develops and there are few effective treatments.
Fibroblasts are sentinel cells that display tissue specific gene expression and can differ in their response to external stimuli based on tissue and/or disease origin.3,4 Fibroblasts are also key effector cells in tissue repair, inflammation, disease progression and immune function.5 Resident orbital fibroblasts (OFs) and fibrocytes that accumulate in the orbit of TED patients are essential mediators of disease pathology.6,7 OFs from TED patients proliferate at a higher rate and differentiate into lipid accumulating fat cells more readily than OFs from non-TED patients.8 The molecular mechanisms whereby OFs from TED patients differ from other fibroblasts are unclear.
The thyroid stimulating hormone receptor (TSHR) is the primary autoantigen in Graves' disease and TED.9,10 In addition to being expressed on thyrocytes, TSHR is expressed by OFs from both normal and TED patients.11 Thus, many aspects of TED pathophysiology may be caused by TSHR autoantibody-OF interactions. TSHR is a G-protein coupled receptor (GPCR) that induces cAMP production and the PI3K/Akt signaling cascade.12 TSHR activation occurs through binding its natural ligand, thyroid stimulating hormone (TSH). In TED, TSHR signaling is activated by stimulatory autoantibodies that bind to the TSHR. While TSH and stimulatory antibodies activate similar TSHR signaling pathways, the pharmacodynamics of the activating antibodies cause sustained activation of the receptor.13 TSHR signaling promotes hyaluronan production and adipogenesis in OFs from TED patients.14,15 In thyrocytes and certain cancers, TSHR signaling induces cellular proliferation through cAMP- and PI3K/Akt-dependent pathways.12,16 Production of cAMP leads to protein kinase A and cAMP response element-binding protein (CREB) activation, which in turn contribute to inflammatory mediator production and increased cell proliferation.17,18 Likewise, the PI3K/Akt cascade drives inflammatory gene expression, cell survival and proliferation.19,20
In order to understand how TSHR stimulates adverse and unwanted proliferation in TED, we asked whether TSHR signaling activates expression of specific inflammatory and proliferation-inducing microRNAs (miRNAs). MiRNAs are endogenous, small RNAs that regulate physiology by suppressing target mRNA translation and/or increasing target mRNA degradation.21 Aberrant expression of miRNAs is linked with fibrosis, cancer, cardiovascular disease, and autoimmunity.22 Recently, altered miRNA expression has been detected in Graves' disease and TED.23–25 Interestingly, miR-146a and miR-155 are upregulated in TED orbital tissue compared to non-TED orbital tissue.25 Currently, why these miRNA are upregulated in TED tissue is unclear. Both miR-146a and miR-155 have key roles in the inflammatory response and miR-155 overexpression promotes autoimmunity in mice.26–28 Both miR-146a and miR-155 increase proliferation and cell survival by promoting PI3K/Akt signaling in follicular thyroid carcinoma and psoriasis, respectively.29,30 Therefore, we hypothesize that sustained TSHR signaling increases OF proliferation through the PI3K/Akt cascade directly, and also indirectly, by promoting expression of miR-146a and miR-155, which contributes to the proliferative pathophysiology of TED.
Methods
Cell Culture
Primary human OFs were isolated and cultured using established explant techniques.7 Samples were from either TED patients undergoing orbital decompression surgery (herein referred to as Graves' OF or GOFs) or non-TED patients undergoing unrelated orbital surgery (herein referred to as non-TED or NOFs) at the Flaum Eye Institute. The non-TED patients did not have any inflammatory orbital diseases. Relevant clinical information including history of smoking, steroid use, and radiation treatment are listed in the Table. Once established, fibroblasts strains were characterized as previously described.3,8,31 Sample collection followed the tenets of the Declaration of Helsinki and were approved by the Research Subjects Review Board at the University of Rochester Medical Center. Informed, written consent was obtained from all patients before surgeries. Explanted OFs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. All media and supplements were from Gibco (Carlsbad, CA, USA) and FBS was from Hyclone (Logan, UT, USA). LY294002, a specific PI3K/Akt inhibitor, was from Peprotech (Rocky Hill, NJ, USA).
Table.
Demographic Information for Clinical Subjects Used in This Study
| Status |
Age at Surgery |
Smoking Status |
Previous Steroid Use |
Radiation |
| Non-TED | 45 | Yes | N/A | N/A |
| Non-TED | 58 | No | N/A | N/A |
| Non-TED | 55 | No | N/A | N/A |
| Non-TED | N/A | N/A | N/A | N/A |
| TED | 47 | No | Oral Prednisone | None |
| TED | 65 | No | Oral Prednisone | None |
| TED | 27 | Previously* | None | None |
| TED | 60 | No | Oral Prednisone | None |
| TED | 54 | Previously† | Oral Prednisone | None |
| TED | 63 | Previously‡ | Oral Prednisone | None |
Subject stopped smoking 18 months before surgery.
Subject stopped smoking 5 months before surgery.
Subject stopped smoking 20 years before surgery.
Cell Proliferation Assay
NOF and GOF strains were seeded in a 96-well plate at a density of 2500 to 5000 cells/well as previously described.7 Cells were treated in triplicate with either 1X PBS (untreated), or bovine thyroid stimulating hormone (TSH, MilliporeSigma, Danvers, MA, USA) for 24 to 48 hours before addition of bromodeoxyuridine (BrdU) for 18 to 24 hours. The use of bovine TSH to stimulate the TSHR in orbital fibroblasts has been described previously.15,32,33 Proliferation was measured with the BrdU cell proliferation assay kit (MilliporeSigma). Briefly, samples were fixed and stained with an anti-BrdU antibody followed by incubation with a corresponding horseradish peroxidase (HRP) conjugated secondary antibody. After incubation with HRP substrate, BrdU incorporation was assessed using a microplate reader (Varioskan Flash; Thermo Fisher Scientific, Waltham, MA, USA).
Gene Expression Knockdown Using siRNA
TSHR siRNA 1 and 2 (s1145 and s1146) were from Applied Biosystems (Silencer Select predesigned siRNAs; Applied Biosystems, Foster City, CA, USA). These siRNA target distinct sequences of the TSHR mRNA. A nonspecific, control siRNA (negative control #1, Applied Biosystems) was used as a control. Cells were grown to 70% to 80% confluence in 96-well plates (for proliferation assay) or six-well plates (for RNA) and treated with the siRNAs mixed with RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA) in Opti-MEM I (Invitrogen) at a final concentration of 100 nM for 24 hours. Cells were incubated in DMEM containing 0.1% FBS for 24 hours prior to treatment as described.
Introduction of miRNA Mimics
MiRNA mimics (miR-146a, Cat. 4464066 ID “MC10722”; miR-155, Cat. 4464066 ID “MC28440”; and control Cat. 4464058) were obtained from Applied Biosystems. OFs were grown to 70% confluence in six-well plates and treated with miRNA mimics mixed with RNAiMAX in Opti-MEM I at 100 nM for 24 hours. Cells were incubated in DMEM containing 0.1% FBS for a further 24 hours before harvest.
Quantitative Real-Time PCR (qPCR) Detection of mRNA Levels
RNA was extracted using a commercial kit (miRNeasy; Qiagen, Valencia, CA, USA). Purified RNA concentrations were measured with a spectrophotometer (NanoDrop 1000; Thermo Scientific). We used 150 ng of total RNA to generate cDNA using a reverse transcription kit (iScript; Bio-Rad Laboratories, Hercules, CA, USA). Gene expression was quantified with gene specific primers and a universal PCR master mix (SsoFast Evergreen; Bio-Rad Laboratories) using a real-time PCR detection system (CFX Connect; Bio-Rad Laboratories). Forward and reverse gene specific sequences are as follows: IL-6: GTACATCCTCGACGGCATC and ACCTCAAACTCCAAAAGACCAG; IL-8: GAGAGTGATTGAGAGTGGACC and ACTGATTCTTGGATACCACAGAG; MCP1: TGTCCCAAAGAAGCTGTGATC and ATTCTTGGGTTGTGGAGTGAG; ZNRF3: GAATATGGCTGGGTAGGAGTG and CTGGGTTTTCAGACACATCAAAG; PTEN: GGATTATAGACCAGTGGCACT and TCGTGTGGGTCCTGAATTG; GAPDH: ATGGAAATCCCATCACCATCTT and CGGCCCACTTGATTTTGG; and 18S rRNA: TGAGAAACGGCTACCACATC and ACTACGAGCTTTTTAACTGC. TSHR mRNA expression was analyzed using a TaqMan Probe set (Cat no 4331182, Assay ID Hs01053846_m1) and Universal PCR master mix (both from Applied Biosystems). Both 18S rRNA and GAPDH mRNA levels were used to normalize mRNA levels of TSHR, IL6, IL8, MCP1, ZNRF3, and PTEN.
Analysis of miRNA Expression
MiRNA cDNA was generated from 50 ng of total RNA using a TaqMan microRNA reverse transcription kit (Applied Biosystems) and qPCR was performed using TaqMan Universal PCR master mix. TaqMan primers were from Applied Biosystems (miR-146a-5p: 4427975 “000468”, miR-155-5p: 4440886 “467534_mat”, miR-16-5p: 4427975 “000391”and U6 snRNA: 4427975 “001973”). Both miR-16 and U6 snRNA levels were used to normalize miR-146a and miR-155 levels.
Western Blot Analysis
Cells were homogenized with total cell lysis buffer (50 mM Tris-HCl pH 6.8, 2% sodium dodecyl sulfate) containing 1X protease inhibitor cocktail (Cell Signaling Technology, Inc., Danvers, MA, USA). Protein concentration was determined with the detergent compatible protein detection assay (Bio-Rad Laboratories). We separated 5 μg of protein via SDS-PAGE, transferred onto 0.45-μm PVDF membrane (Millipore, Danvers, MA), and blocked with 5% non-fat dry milk in 0.1% Tween 20 in 1X TBS. Antibodies targeting phospho-Akt (phospho-Ser473, rabbit anti-phospho-Akt cat. # 4060) and total Akt (rabbit anti-Akt cat. # 4691) were from Cell Signaling Technologies (Danvers, MA, USA). HRP-conjugated secondary antibodies were from Jackson Immunoresearch (West Grove, PA, USA). Chemiluminescent signals were captured using a VersaDoc imaging system (Chemidoc MP; Bio-Rad Laboratories). Densitometric analysis was performed with analysis software (ImageLab; Bio-Rad Laboratories).
Statistical Analysis
All data was analyzed and graphed using graphing software (GraphPad Prism, version 8; GraphPad Software, La Jolla, CA, USA). All values are presented as mean ± SEM unless otherwise noted. Experiments were conducted in triplicate unless stated otherwise. Student's t-test and 1-way and 2-way ANOVA were used for statistical analysis where appropriate. Statistical significance is stated with values of P < 0.05 (* or #); P < 0.01 (** or ##); and P < 0.001 (*** or ###).
Results
TSHR Signaling Induces Proliferation Significantly More in TED Fibroblasts Compared to non-TED OFs
TSHR signaling drives multiple downstream events including cell growth and proliferation.12,16 Orbital fibroblasts (OFs) from both TED (herein referred to as Graves' OFs or GOFs) and non-TED patients (non-TED OFs or NOFs) express TSHR.34,35 To investigate the role of TSHR signaling in OF proliferation the canonical TSHR ligand, TSH was used. Importantly, bovine TSH was used in these studies as it has a higher affinity for the TSHR than human TSH and thus results in stronger and more robust signaling.36 Both NOF and GOF strains were stimulated with bovine TSH (1–50 mU/mL) for 48 hours. The cells were cocultured with the thymidine analog BrdU to serve as a measure of DNA synthesis and cell proliferation. Both NOFs and GOFs show a dose-dependent increase in TSH-induced proliferation (Fig. 1A). GOFs show a significantly enhanced response to TSH compared to NOFs. At the lowest TSH dose used (1 mU/mL), NOFs failed to incorporate more BrdU than untreated samples while GOFs incorporated more than 2-fold the amount of BrdU. At 50 mU/mL TSH, GOFs incorporated BrdU over 7-fold baseline levels whereas NOFs only 3-fold baseline. Since GOFs respond more robustly to TSHR activation than NOFs, we investigated whether or not TSHR expression was different in the two groups (Fig. 1B). Total RNA was extracted from untreated GOFs and NOFs and TSHR mRNA levels were analyzed by qPCR. GOFs expressed significantly higher levels of TSHR than NOFs.
Figure 1.

TSH and TSHR signaling stimulate proliferation in orbital fibroblasts. (A) OFs explanted from patients with TED (GOFs, black bars) or without TED (NOFs, clear bars) were treated with 1, 10, or 50 mU/mL TSH (or without TSH, untreated) for 48 hours. The nucleotide analog BrdU was added for 24 hours to measure DNA synthesis. After culture, cells were fixed and the BrdU label was detected by ELISA as described in the Methods section. TSH treatment resulted in a dose-dependent increase in OF proliferation. GOFs accumulated significantly more BrdU than NOFs at all TSH doses. Results are presented as means ± SEM from triplicate wells repeated in n = 4 NOF strains and n = 5 GOF strains. #P < 0.05, ##P< 0.01, ###P < 0.001 versus untreated samples. **P< 0.01, ***P < 0.001 in GOF versus NOF samples of the same treatment. (B) Expression of TSHR mRNA was detected by qPCR in untreated NOFs (n = 4) and GOFs (n = 5). TSHR mRNA levels were normalized to GAPDH mRNA and 18S rRNA levels. GOFs had ∼1.25-fold more TSHR mRNA than NOFs, *P < 0.05. Relevant information on patient samples is given in the Table.
TSHR Expression and PI3K Signaling Are Required for TSH-Induced Proliferation
As TSH-induced proliferation is greater in GOFs than NOFs and because GOFs express higher levels of TSHR, we examined whether TSH-driven proliferation was dependent upon TSHR expression. To test this, GOFs were treated with nonspecific control siRNA or TSHR specific siRNAs for 48 hours to reduce TSHR expression. TSHR-specific siRNA dramatically reduced TSHR mRNA levels (∼85% knockdown) compared to control siRNA (Fig. 2A). GOFs were then incubated with or without TSH (50 mU/mL) for an additional 48 hours to analyze cellular proliferation levels (Fig. 2B). In control siRNA samples, TSH addition led to a significant increase in cellular proliferation. TSHR siRNA dramatically attenuated TSH-driven proliferation revealing that TSHR expression is required. Furthermore, baseline proliferation was reduced in TSHR-siRNA–treated samples suggesting that TSHR is important for basal proliferation in GOFs.
Figure 2.

TSH-induced proliferation requires TSHR expression. (A) GOFs were treated with control or TSHR-specific siRNA for 48 hours and then TSH (1, 10, or 50 mU/mL) for an additional 48 hours. Afterwards, cells were collected and TSHR mRNA levels were detected by qPCR. TSHR mRNA levels were normalized as above. TSHR siRNA reduced TSHR mRNA levels by ∼85% compared to control siRNA, **P < 0.01. Similar results were obtained with a second TSHR siRNA that targets a different nucleotide sequence of the TSHR mRNA (data not shown). (B) GOFs treated with control or TSHR specific siRNA were also treated with or without 50 mU/mL TSH for 48 hours. Proliferation was quantified with BrdU incorporation as described. GOFs treated with TSHR siRNA incorporated significantly less BrdU than control siRNA samples. Results are presented as means ± SEM from triplicate wells and represent experiments repeated in n = 3 GOF strains. ##P < 0.01 compared to vehicle sample. ***P < 0.001 compared to control siRNA-TSH–treated sample.
We next examined the molecular mechanism(s) driving TSHR-dependent proliferation in GOFs. TSHR signaling can be coupled to Gα and Gq proteins that activate production of the secondary messengers, cAMP and PIP3.15,37 Since PIP3 activates the PI3K/Akt pathway to drive proliferation in many cell types including highly proliferative malignant cells,38,39 we tested whether PI3K/Akt signaling is needed for TSHR-induced proliferation in GOFs. The pharmacologic PI3K inhibitor, LY294002 was used to examine this.40 GOFs were treated with vehicle or LY294002 in the presence or absence of TSH (10 or 50 mU/mL) for 48 hours to allow cellular proliferation (Fig. 3A). TSH induced proliferation in a dose-dependent manner. Inhibition of PI3K using LY294002 resulted in a block of cell proliferation. PI3K/Akt signaling was monitored by analyzing Akt phosphorylation by Western blot (Fig. 3B). TSH treatment resulted in a dose-dependent increase in phospho-Akt levels. LY294002 blocked Akt phosphorylation.
Figure 3.

TSHR-driven proliferation is PI3K/AKT dependent in GOFs. (A) GOFs were treated with TSH (10 or 50 mU/mL) for 48 hours in the presence of BrdU to measure cell proliferation. Some cells were also exposed to the specific PI3K/Akt inhibitor, LY294002 (10 μM) to block Akt signaling. LY294002 completely blocks TSH-induced OF proliferation. Results are presented as means ± SEM from triplicate wells and represent experiments repeated in n = 3 GOF strains. ###P < 0.001 versus vehicle. ***P < 0.001 versus TSH only. (B) Representative Western blot showing TSH induces a dose-dependent increase in the phosphorylation of AKT and this phosphorylation is blocked by LY294002. Total Akt levels serve as a loading control.
TSHR Signaling Induces Cytokine and Inflammation-Related miRNA Production
While TSHR signaling drives proliferation through PI3K/Akt signaling directly, we examined other pathways that may be important for promoting proliferation and/or proinflammatory signaling related to TED pathophysiology. Cytokines such as IL-6, IL-8, and MCP-1 are produced at high levels by OFs from TED patients.20,41,42 The cytokine IL-6 stimulates proliferation and TSHR activation has been shown to increase IL-6 expression in fibrocytes from TED patients.43 Thus, the role of TSHR in regulating these inflammatory mediators in GOFs was examined. TSHR activation results in a significant upregulation of IL6, IL8, and MCP1 gene expression in GOFs (Fig. 4). Since these canonical inflammatory genes were induced through TSHR signaling in GOFs, the expression of other inflammatory-related factors was analyzed. MiRNAs are important regulatory mediators that influence inflammation and cellular proliferation. Both miR-146a and miR-155 are important inflammatory mediators that also regulate proliferation.27,29,30 To determine if miR-146a and miR-155 are stimulated by TSH, GOFs were incubated with TSH (1–50 mU/mL) for 48 hours and then RNA extracted, and miRNA levels were analyzed by qPCR. TSH treatment led to a robust increase in miR-146a levels (Fig. 5A) and a moderate, yet significant increase in miR-155 levels (Fig. 5B).
Figure 4.

TSHR signaling in GOFs induces inflammatory cytokine expression. GOFs were treated with or without TSH (1, 10, 50 mU/mL) for 48 hours and then total RNA was extracted and subjected to analysis by qPCR for inflammatory cytokine mRNA expression. Expression was normalized as described above. (A) IL6, (B) IL8, and (C) MCP1 mRNAs were dose dependently induced by the TSHR ligand, TSH (*P < 0.05, **P < 0.01, ***P < 0.001 versus untreated control samples). Data are expressed as mean ± SEM of triplicate measurements and are representative of three independent experiments performed in three different GOF strains.
Figure 5.
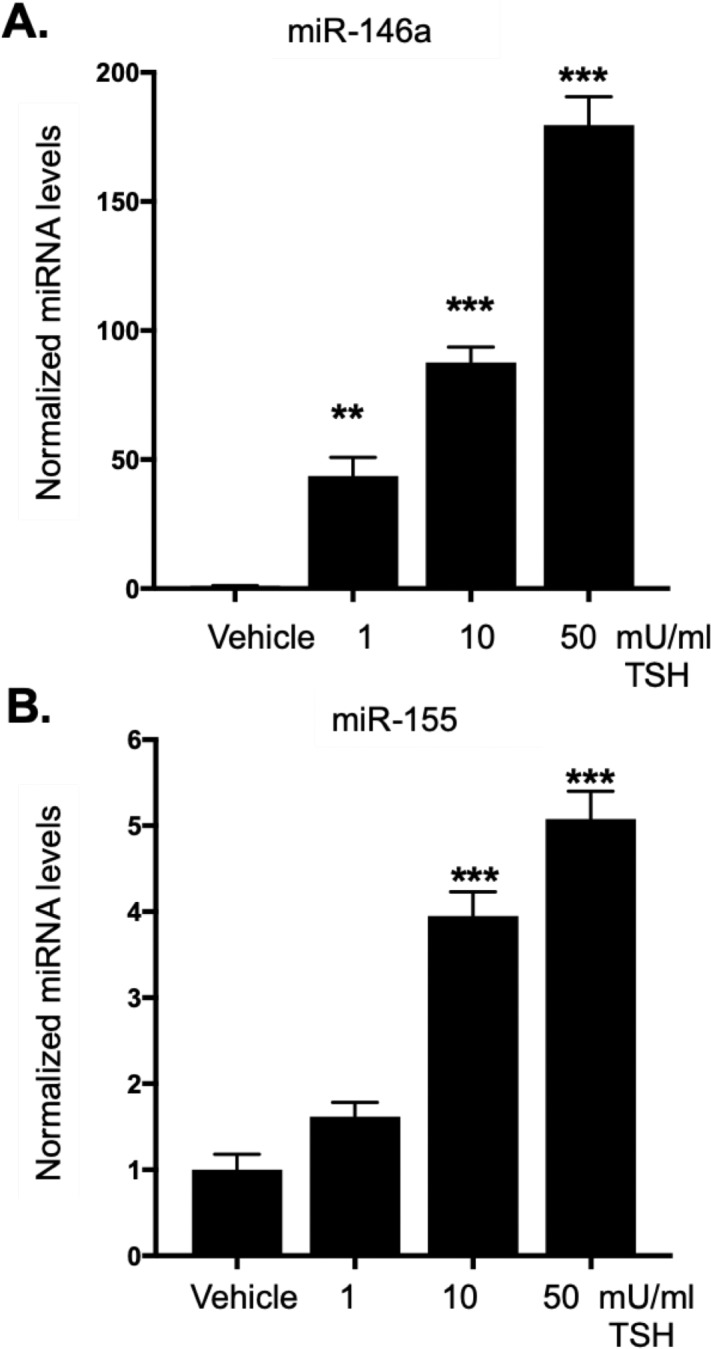
TSHR signaling in GOFs induces miR-146a and miR-155 expression. GOFs were treated with or without TSH (1, 10, or 50 mU/mL) for 48 hours and then total RNA was extracted and subjected to analysis by qPCR for miRNA expression. miR-146a and miR-155 expression levels were normalized to U6 snRNA and miR-16 levels. (A) miR-146a and (B) miR-155 were dose-dependently induced by TSH. (**P < 0.01, ***P < 0.001 versus untreated control samples). Data are expressed as mean ± SEM of triplicate measurements and are representative of three independent experiments performed in three different GOF strains.
TSH-Mediated Induction of MiR-146a and MiR-155 Requires TSHR Expression
Since TSH induced miR-146a and miR-155 levels in GOFs, we tested whether this was TSHR-dependent. GOFs were treated with either nonspecific control siRNA or TSHR specific siRNA followed by treatment with 1 to 100 mU/mL TSH for 48 hours. After treatment, RNA was isolated and miRNA expression analyzed by qPCR. MiR-146a expression was induced by TSH in control siRNA treated samples (Fig. 6A). Depletion of TSHR expression completely blocked miR-146a induction. MiR-155 levels were induced at 10, 50, and 100 mU/ml TSH in control siRNA-treated samples (Fig. 6B). TSH-induced miR-155 levels were attenuated in cells depleted of TSHR.
Figure 6.
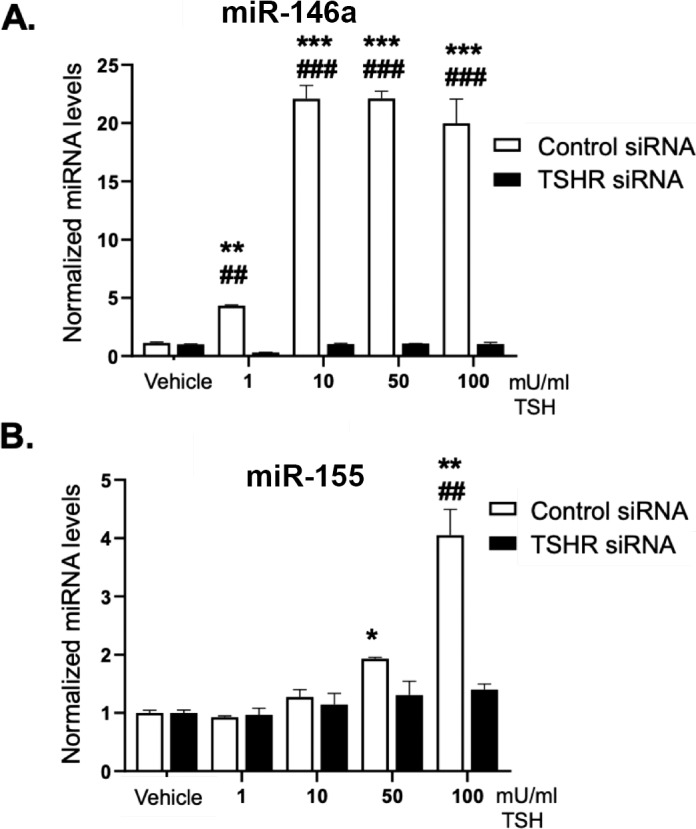
TSH-mediated induction of miR-146a and miR-155 is TSHR-dependent. GOFs treated with control or TSHR specific siRNA for 48 hours were then incubated with or without 1, 10, 50, or 100 mU/mL TSH for 48 hours. Total RNA was then extracted and analyzed by qPCR for miR-146a, miR-155 and U6 snRNA (control). While (A) miR-146a and (B) miR-155 were dose-dependently induced by TSH in the control siRNA samples, TSHR siRNA samples did not show TSH induced miRNA expression. Results are presented as means ± SEM from triplicate wells and represent experiments repeated in n = 3 GOF strains. *P < 0.05, **P < 0.01, ***P < 0.001 versus no TSH control. ##P < 0.01, ###P < 0.001 versus control siRNA-TSH–treated samples.
Since TSH induces proliferation in GOFs significantly more than in NOFs, and as TSHR signaling increases miR-146a and miR-155, we asked whether GOFs upregulate miR-146a and miR-155 expression to a greater extent than NOFs. To accomplish this, both GOFs and NOFs were incubated with TSH (1–50 mU/mL) for 48 hours. Afterwards, total RNA was isolated and miR-146a and miR-155 levels were measured. MiR-146a was not induced at 1 mU/mL TSH in NOFs but was increased ∼3-fold in GOFs (Fig. 7A). At both 10 and 50 mU/mL TSH, miR-146a was induced in NOFs; however, the induction in GOFs was significantly greater. While miR-155 expression was significantly induced in GOFs at 10 and 50 mU/mL TSH, miR-155 levels were not induced in NOFs (Fig. 7B). Therefore, miR-146a and miR-155 are significantly elevated by TSH in OFs from TED patients compared to non-TED patient OFs.
Figure 7.

TED OFs induce higher levels of miR-146a and 155 than non-TED OFs. OFs from patients with TED (GOFs, black bars) or without TED (NOFs, clear bars) were treated with 1, 10, or 50 mU/mL TSH (or without TSH, untreated) for 48 hours. Then, total RNA was extracted and analyzed by qPCR for miR-146a, miR-155 and U6 snRNA (control). miRNA levels were normalized to U6 snRNA levels. (A) MiR-146a levels were induced by TSH in NOF and GOF samples. (B) MiR-155 levels were induced in GOF samples but not in NOF samples. Furthermore, in TSH-treated GOF samples, both miR-146a and miR-155 were significantly higher than in TSH treated NOF samples. Results are presented as means ± SEM from triplicate wells and represent experiments repeated in n = 3 NOF and n = 3 GOF strains. *P < 0.05, **P < 0.01, ***P < 0.001 versus no TSH control. #P < 0.05, ###P < 0.001 versus NOF versus GOF samples of the same TSH treatment.
MiR-146a and MiR-155 Target ZNRF3 and PTEN to Promote Proliferation in TED OFs
Because miR-146a and miR-155 are significantly elevated in TSH-treated GOFs compared to NOFs and since these miRNAs promote cellular proliferation in other cells, key miR-146a and miR-155 target gene expression was analyzed. GOFs were treated with TSH as described for 48 hours and total RNA was isolated. As shown above, miR-146a and miR-155 levels were induced by TSH (Fig. 8A). ZNRF3, a miR-146a target gene that inhibits cell proliferation in osteosarcoma cells,44 was analyzed by qPCR in untreated and TSH treated GOFs (Fig. 8B, left panel). Interestingly, ZNRF3 was dose dependently decreased by TSH in GOFs. Next, PTEN, a miR-155 target gene and inhibitor of PI3K/Akt induced proliferation, was also analyzed by qPCR (Fig. 8B, right panel). Like ZNRF3, PTEN was reduced by TSH in a dose-dependent manner in GOFs. To test if ZNRF3 and PTEN are targeted by miR-146a and miR-155 in GOFs, control or miRNA mimics were introduced into GOFs and after 48 hours, RNA isolated and analyzed by qPCR (Fig. 8C). ZNRF3 mRNA levels were significantly reduced by miR-146a. Likewise, PTEN mRNA levels were attenuated by exogenous expression of miR-155.
Figure 8.

TSHR-induced miR-146a and miR-155 downregulate their target genes ZNRF3, and PTEN to increase proliferation. OFs were treated with 1, 10, or 50 mU/mL TSH for 48 hours. Afterwards, cells were harvested and total RNA isolated and analyzed by qPCR. (A) MiR-146a and miR-155 levels were analyzed as described above. Both miR-146a and miR-155 were dose-dependently induced by TSH. (B) MiR-146a target, ZNRF3, and miR-155 targets PTEN mRNA levels were analyzed by qPCR. Expression was normalized by 18S rRNA levels. The expression of ZNRF3 and PTEN were all dose-dependently reduced by TSH. (C) GOFs were treated with either control, miR-146a or miR-155 miRNA mimics for 48 hours. Total RNA was then extracted and analyzed by qPCR for ZNRF3 and PTEN. ZNRF3 mRNA levels were reduced by miR-146a over-expression and PTEN mRNA levels were reduced by miR-155 over-expression. Results are presented as means ± SEM from triplicate wells and represent experiments repeated in n = 3 GOF strains. *P < 0.05, **P < 0.01, ***P < 0.001 versus no TSH control.
Discussion
Taken together, the data presented herein demonstrate that TSHR signaling in GOFs stimulates PI3K/Akt-dependent proliferation directly and indirectly through induction of miR-146a and miR-155 (Fig. 9). These two miRNAs enhance proliferation by reducing target gene expression (ZNRF3 and PTEN) that would attenuate cellular proliferation. Additionally, miR-146a and miR-155 are induced to a greater extent in OFs from TED patients than those from non-TED patients. This may explain, in part, the proliferative pathophysiology observed in TED. Additionally, these studies present new genes that may be involved in TED pathophysiology. Namely, ZNRF3, a GSK3β/β-catenin pathway inhibitor,44 and PTEN, a tumor suppressor and inhibitor of PI3K signaling.29,45 Future studies defining the role of these genes (and the loss of their expression) in TED are warranted.
Figure 9.

TSHR mediated signaling induced OF proliferation through Akt signaling and induction of miR-146a and miR-155. The model shows that TSHR signaling promotes cell proliferation in TED through multiple pathways. The TSHR canonical ligand, TSH or stimulatory anti-TSHR antibodies (as detected in the majority of TED patients) signal through the receptor and activate downstream G protein signaling inducing both PIP3 and cAMP secondary messengers. PIP3 activates the canonical PI3K/Akt pathway resulting in phosphorylation of Akt and subsequent cellular proliferation pathways (Akt phosphorylation can be blocked by the PI3K inhibitor, LY294002). TSHR signaling also upregulates expression of inflammatory cytokines and the inflammation related miRNAs, miR-146a and miR-155. These miRNAs in turn regulate cell proliferation inhibitors including PTEN and ZNRF3. The resultant downregulation of these target genes leads to an enhanced proliferative response, which is a major pathophysiological response observed in TED.
While the canonical ligand of TSHR is TSH, activation of the receptor in TED occurs through interactions with stimulatory anti-TSHR autoantibodies that accumulate in the orbit of afflicted individuals.13,46 Stimulatory anti-TSHR antibodies, like TSH, activate cAMP and PI3K cascades. However, due to different pharmacodynamics and increased stability of the autoantibodies, they lead to a longer period of sustained activation of the receptor.13,15 In the current study, TSH was used to activate TSHR. As TSHR stimulatory antibodies drive similar pathways with persistent activation, miR-146a and miR-155 levels may be even further induced by antibody-receptor interactions. It may be that miR-146a and miR-155 are elevated in TED orbital tissue due to the presence of stimulatory antibodies in the orbit.25 Additionally, basal TSHR-induced signaling can occur, even in the absence of detectable ligands or stimulatory antibodies.17,18 Since GOFs express more TSHR than NOFs, they likely have increased basal signaling activity that can increase TSHR-mediated proliferation and signaling. This is consistent with previous reports showing that basal proliferation rates are increased in GOFs compared to NOFs.8 Furthermore, our data support the concept that TSHR expression is important for basal proliferation rates as TSHR knockdown reduced proliferation, in both untreated and TSH-treated cells.
Excessive proliferation of OFs is a central component of TED and drives further pathophysiology.10 As the number of fibroblasts in the orbit increases, their ability to drive orbital remodeling through continued activation, hyaluronan and inflammatory mediator production also increases.41,47 Furthermore, fibroblasts can differentiate into either adipocytes, which make up extraocular fat compartments, or scar forming myofibroblasts that form extraocular muscle components.7,8,48 Here, we show that TSHR driven miR-146a and miR-155 expression promote proliferation by targeting genes that normally inhibit cell growth. Increased expression of miR-155 promotes proliferation and apoptosis in psoriasis.29 Furthermore, overexpression of miR-155 in hematopoietic stem cells causes myeloproliferative disease in mice.49 Thus, in addition to driving proliferation in TED, miR-155 is an important factor in other inflammatory diseases.
MiR-155 may play additional roles in TED pathophysiology. In macrophages, miR-155 increases expression of inflammatory cytokines such as IL-6 and TNFα by targeting the suppressor of cytokine signaling 1 (SOCS1) gene.50 MiR-155 promotes collagen production in systemic sclerosis fibroblasts and increases myofibroblast formation.51 In contrast, miR-155 limits adipogenesis and fat cell formation in 3T3-L1 cells.52 Therefore, it is possible that in TED, miR-155 drives proliferation, inflammatory signaling and myofibroblast formation to promote extraocular muscle enlargement in TED as opposed to increasing fat cell formation.
While the impact of increased miR-155 points to TED pathophysiology, the effects of increased miR-146a expression in GOFs may be more heterogeneous. Here, we show that miR-146a levels promote increases in OF proliferation. Interestingly, miR-146a can block expression of the PI3K inhibitor, ST8SIA4 in follicular thyroid carcinoma cells, thereby increasing their proliferation rate.30 Recently, miR-146a overexpression was shown to limit TGFβ-induced myofibroblast formation in GOFs by targeting SMAD4.24 Additionally, miR-146a was shown to limit LPS induced inflammatory signaling and autoimmunity in mice.53 Thus, miR-146a expression may limit additional features of TED pathophysiology including scarring and inflammation. This suggests that there may be a critical balance between miR-146a and miR-155 levels in TED. When miR-155 signaling overcomes miR-146a, disease symptoms may develop and/or worsen. In contrast, when miR-146a signaling overcomes miR-155, inflammation and scarring may be attenuated. Therefore, in TED pathophysiology, miR-155 functions may outweigh anti-inflammatory miR-146a targets. Interestingly, this concept fits with our data showing that when TSHR signaling is activated, NOFs produce lower levels of miR-155 than similarly treated GOFs. A similar clash between miR-155 and miR-146a networks was found in T cell-mediated immunity.54
Although this study demonstrates that TSHR signaling in GOFs stimulates PI3K/Akt-dependent proliferation directly and indirectly through induction of two miRNAs, there are limitations. Even as OFs are key meditators of TED, there are several other cell types that play a role in disease. T cells, B cells, and macrophages also infiltrate the orbit during disease.55 T cells can activate OFs to proliferate and differentiate into adipocytes and/or scar forming cells.56,57 It may be interesting to test how T cells interact with OFs when TSHR signaling is activated by TSH or anti-TSHR antibodies. Additionally, activated OFs may further stimulate T cells through cytokine production. Further studies involving animal models or primary human orbital tissue will help determine the role of TSHR signaling and miRNA expression in vivo. Nonetheless, these studies are the first to show that TSHR signaling induces changes in miRNA expression in orbital fibroblasts and highlight the power of miRNA to regulate pathology of TED.
Acknowledgments
Supported by the National Institutes of Health Grants EY027308, HL13376 and an unrestricted grant from Research to Prevent Blindness.
Disclosure: C.F. Woeller, None; E. Roztocil, None; C. Hammond, None; S.E. Feldon, None
References
- 1.Smith TJ, Hegedüs L. Graves' disease. N Engl J Med. 2016;375:1552–1565. doi: 10.1056/NEJMra1510030. [DOI] [PubMed] [Google Scholar]
- 2.Bahn RS. Graves' ophthalmopathy. N Engl J Med. 2010;362:726–738. doi: 10.1056/NEJMra0905750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xi X, McMillan DH, Lehmann GM, et al. Ocular fibroblast diversity: implications for inflammation and ocular wound healing. Invest Ophthalmol Vis Sci. 2011;52:4859–4865. doi: 10.1167/iovs.10-7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baglole CJ, Ray DM, Bernstein SH, et al. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol Invest. 2006;35:297–325. doi: 10.1080/08820130600754960. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Atkins SJ, Fernando R, Wei RL, Smith TJ. Pentraxin-3 Is a TSH-inducible protein in human fibrocytes and orbital fibroblasts. Endocrinology. 2015;156:4336–4344. doi: 10.1210/en.2015-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woeller CF, Roztocil E, Hammond CL, Feldon SE, Phipps RP. The aryl hydrocarbon receptor and its ligands inhibit myofibroblast formation and activation: implications for thyroid eye disease. Am J Pathol. 2016;186:3189–3202. doi: 10.1016/j.ajpath.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuriyan AE, Woeller CF, O'Loughlin CW, Phipps RP, Feldon SE. Orbital fibroblasts from thyroid eye disease patients differ in proliferative and adipogenic responses depending on disease subtype. Invest Ophthalmol Vis Sci. 2013;54:7370–7377. doi: 10.1167/iovs.13-12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies TF, Latif R. Editorial: TSH receptor and autoimmunity. Front Endocrinol (Lausanne) 2019;10:19. doi: 10.3389/fendo.2019.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuriyan AE, Phipps RP, Feldon SE. The eye and thyroid disease. Curr Opin Ophthalmol. 2008;19:499–506. doi: 10.1097/ICU.0b013e3283131557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer S, Bahn R. Immunopathogenesis of Graves' ophthalmopathy: the role of the TSH receptor. Best Pract Res Clin Endocrinol Metab. 2012;26:281–289. doi: 10.1016/j.beem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.García-Jiménez C, Santisteban P. Thyroid-stimulating hormone/cAMP-mediated proliferation in thyrocytes. Expert Rev Endocrinol Metab. 2008;3:473–491. doi: 10.1586/17446651.3.4.473. [DOI] [PubMed] [Google Scholar]
- 13.Michalek K, Morshed SA, Latif R, Davies TF. TSH receptor autoantibodies. Autoimmun Rev. 2009;9:113–116. doi: 10.1016/j.autrev.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Zeijl CJ, Fliers E, van Koppen CJ, et al. Thyrotropin receptor-stimulating Graves' disease immunoglobulins induce hyaluronan synthesis by differentiated orbital fibroblasts from patients with Graves' ophthalmopathy not only via cyclic adenosine monophosphate signaling pathways. Thyroid. 2011;21:169–176. doi: 10.1089/thy.2010.0123. [DOI] [PubMed] [Google Scholar]
- 15.Kumar S, Nadeem S, Stan MN, Coenen M, Bahn RS. A stimulatory TSH receptor antibody enhances adipogenesis via phosphoinositide 3-kinase activation in orbital preadipocytes from patients with Graves' ophthalmopathy. J Mol Endocrinol. 2011;46:155–163. doi: 10.1530/JME-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67:8002–8006. doi: 10.1158/0008-5472.CAN-07-2471. [DOI] [PubMed] [Google Scholar]
- 17.Latif R, Lau Z, Cheung P, Felsenfeld DP, Davies TF. The “TSH Receptor Glo Assay” – a high-throughput detection system for thyroid stimulation. Front Endocrinol (Lausanne) 2016;7:3. doi: 10.3389/fendo.2016.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morshed SA, Latif R, Davies TF. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology. 2009;150:519–529. doi: 10.1210/en.2008-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev. 2001;22:631–656. doi: 10.1210/edrv.22.5.0444. [DOI] [PubMed] [Google Scholar]
- 20.Gillespie EF, Raychaudhuri N, Papageorgiou KI, et al. Interleukin-6 production in CD40-engaged fibrocytes in thyroid-associated ophthalmopathy: involvement of Akt and NF-kappaB. Invest Ophthalmol Vis Sci. 2012;53:7746–753. doi: 10.1167/iovs.12-9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 23.Bernecker C, Lenz L, Ostapczuk MS, et al. MicroRNAs miR-146a1, miR-155_2, and miR-200a1 are regulated in autoimmune thyroid diseases. Thyroid. 2012;22:1294–1295. doi: 10.1089/thy.2012.0277. [DOI] [PubMed] [Google Scholar]
- 24.Jang SY, Park SJ, Chae MK, Lee JH, Lee EJ, Yoon JS. Role of microRNA-146a in regulation of fibrosis in orbital fibroblasts from patients with Graves' orbitopathy. Br J Ophthalmol. 2018;102:407–414. doi: 10.1136/bjophthalmol-2017-310723. [DOI] [PubMed] [Google Scholar]
- 25.Jang SY, Chae MK, Lee JH, Lee EJ, Yoon JS. Role of miR-146a in the regulation of inflammation in an in vitro model of Graves' orbitopathy. Invest Ophthalmol Vis Sci. 2016;57:4027–4034. doi: 10.1167/iovs.16-19213. [DOI] [PubMed] [Google Scholar]
- 26.Mann M, Mehta A, Zhao JL, et al. An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat Commun. 2017;8:851. doi: 10.1038/s41467-017-00972-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doxaki C, Kampranis SC, Eliopoulos AG, Spilianakis C, Tsatsanis C. Coordinated regulation of miR-155 and miR-146a genes during induction of endotoxin tolerance in macrophages. J Immunol. 2015;195:5750–5761. doi: 10.4049/jimmunol.1500615. [DOI] [PubMed] [Google Scholar]
- 28.O'Connell RM, Kahn D, Gibson WS, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Leng H, Shi X, Ji J, Fu J, Leng H. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed Pharmacother. 2017;90:524–530. doi: 10.1016/j.biopha.2017.03.105. [DOI] [PubMed] [Google Scholar]
- 30.Ma W, Zhao X, Liang L, et al. miR-146a and miR-146b promote proliferation, migration and invasion of follicular thyroid carcinoma via inhibition of ST8SIA4. Oncotarget. 2017;8:28028–28041. doi: 10.18632/oncotarget.15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehmann GM, Woeller CF, Pollock SJ, et al. Novel anti-adipogenic activity produced by human fibroblasts. Am J Physiol Cell Physiol. 2010;299:C672–C678. doi: 10.1152/ajpcell.00451.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith TJ, Padovani-Claudio DA, Lu Y, et al. Fibroblasts expressing the thyrotropin receptor overarch thyroid and orbit in Graves' disease. J Clin Endocrinol Metab. 2011;96:3827–3837. doi: 10.1210/jc.2011-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turcu AF, Kumar S, Neumann S, et al. A small molecule antagonist inhibits thyrotropin receptor antibody-induced orbital fibroblast functions involved in the pathogenesis of Graves ophthalmopathy. J Clin Endocrinol Metab. 2013;98:2153–2159. doi: 10.1210/jc.2013-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bahn RS. TSH receptor expression in orbital tissue and its role in the pathogenesis of Graves' ophthalmopathy. J Endocrinol Invest. 2004;27:216–220. doi: 10.1007/BF03345269. [DOI] [PubMed] [Google Scholar]
- 35.Bahn RS, Dutton CM, Joba W, Heufelder AE. Thyrotropin receptor expression in cultured Graves' orbital preadipocyte fibroblasts is stimulated by thyrotropin. Thyroid. 1998;8:193–196. doi: 10.1089/thy.1998.8.193. [DOI] [PubMed] [Google Scholar]
- 36.Mueller S, Kleinau G, Szkudlinski MW, Jaeschke H, Krause G, Paschke R. The superagonistic activity of bovine thyroid-stimulating hormone (TSH) and the human TR1401 TSH analog is determined by specific amino acids in the hinge region of the human TSH receptor. J Biol Chem. 2009;284:16317–16324. doi: 10.1074/jbc.M109.005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agretti P, De Marco G, De Servi M, et al. Evidence for protein and mRNA TSHr expression in fibroblasts from patients with thyroid-associated ophthalmopathy (TAO) after adipocytic differentiation. Eur J Endocrinol. 2005;152:777–784. doi: 10.1530/eje.1.01900. [DOI] [PubMed] [Google Scholar]
- 38.Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- 39.Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, Iyer S, Bauer H, Coenen M, Bahn RS. A stimulatory thyrotropin receptor antibody enhances hyaluronic acid synthesis in Graves' orbital fibroblasts: inhibition by an IGF-I receptor blocking antibody. J Clin Endocrinol Metab. 2012;97:1681–1687. doi: 10.1210/jc.2011-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khong JJ, McNab AA, Ebeling PR, Craig JE, Selva D. Pathogenesis of thyroid eye disease: review and update on molecular mechanisms. Br J Ophthalmol. 2016;100:142–150. doi: 10.1136/bjophthalmol-2015-307399. [DOI] [PubMed] [Google Scholar]
- 42.Spinelli SL, Xi X, McMillan DH, et al. Mapracorat, a selective glucocorticoid receptor agonist, upregulates RelB, an anti-inflammatory nuclear factor-kappaB protein, in human ocular cells. Exp Eye Res. 2014;127:290–298. doi: 10.1016/j.exer.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Raychaudhuri N, Fernando R, Smith TJ. Thyrotropin regulates IL-6 expression in CD34+ fibrocytes: clear delineation of its cAMP-independent actions. PLoS One. 2013;8:e75100. doi: 10.1371/journal.pone.0075100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou C, Jiang CQ, Zong Z, Lin JC, Lao LF. MiR-146a promotes growth of osteosarcoma cells by targeting ZNRF3/GSK-3beta/beta-catenin signaling pathway. Oncotarget. 2017;8:74276–74286. doi: 10.18632/oncotarget.19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue X, Liu Y, Wang Y, et al. MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget. 2016;7:84508–84519. doi: 10.18632/oncotarget.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khoo TK, Bahn RS. Pathogenesis of Graves' ophthalmopathy: the role of autoantibodies. Thyroid. 2007;17:1013–1018. doi: 10.1089/thy.2007.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dik WA, Virakul S, van Steensel L. Current perspectives on the role of orbital fibroblasts in the pathogenesis of Graves' ophthalmopathy. Exp Eye Res. 2016;142:83–91. doi: 10.1016/j.exer.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Woeller CF, O'Loughlin CW, Pollock SJ, Thatcher TH, Feldon SE, Phipps RP. Thy1 (CD90) controls adipogenesis by regulating activity of the Src family kinase, Fyn. FASEB J. 2015;29:920–931. doi: 10.1096/fj.14-257121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Connell RM, Rao DS, Chaudhuri AA, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye J, Guo R, Shi Y, Qi F, Guo C, Yang L. MiR-155 regulated inflammation response by the SOCS1-STAT3-PDCD4 axis in atherogenesis. Mediators Inflamm. 2016;2016:8060182. doi: 10.1155/2016/8060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther. 2017;19:144. doi: 10.1186/s13075-017-1331-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu S, Yang Y, Wu J. TNFalpha-induced up-regulation of miR-155 inhibits adipogenesis by down-regulating early adipogenic transcription factors. Biochem Biophys Res Commun. 2011;414:618–624. doi: 10.1016/j.bbrc.2011.09.131. [DOI] [PubMed] [Google Scholar]
- 53.Boldin MP, Taganov KD, Rao DS, et al. MiR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huffaker TB, Hu R, Runtsch MC, et al. Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell Rep. 2012;2:1697–709. doi: 10.1016/j.celrep.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehmann GM, Feldon SE, Smith TJ, Phipps RP. Immune mechanisms in thyroid eye disease. Thyroid. 2008;18:959–965. doi: 10.1089/thy.2007.0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feldon SE, O'loughlin CW, Ray DM, et al. Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am J Pathol. 2006;169:1183–1193. doi: 10.2353/ajpath.2006.060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feldon SE, Park DJ, O'Loughlin CW, et al. Autologous T-lymphocytes stimulate proliferation of orbital fibroblasts derived from patients with Graves' ophthalmopathy. Invest Ophthalmol Vis Sci. 2005;46:3913–3921. doi: 10.1167/iovs.05-0605. [DOI] [PubMed] [Google Scholar]