

Supplemental Digital Content is available in the text
Keywords: exosome, label-free MS/MS, Parkinson disease, proteomics
Abstract
Exosomes are nanometer-sized vesicles with intercellular communication functions, and their encapsulated proteins may participate in the pathological process of neurodegenerative disorders. The aim of this study was to identify the protein changes of serum exosomes in Parkinson disease (PD) patients with different disease progress types, and to identify potential biomarkers. The exosomes of PD patients with different severity and healthy control group were isolated from serum. The exosome proteins were analyzed by mass spectrometry with label-free quantitative proteomics. A total of 429 proteins were identified, of which 14 were significantly different in mild and severe PD patients. The expression levels of 7 proteins, including pigmented epithelium-derived factor, afamin, apolipoprotein D and J, were significantly increased in PD patients. The expression levels of 7 proteins, including complement C1q and protein Immunoglobulin Lambda Variable 1-33 (IGLV1-33)Cluster -33, were decreased in PD patients. These differentially expressed proteins were analyzed by gene ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analysis, which confirmed that the interaction between prion diseases and ECM receptors was the most significant pathways of enrichment. The changes of proteins and pathways may be related to the pathophysiological mechanism of PD. Therefore, some of these proteins could be considered as potential biomarkers for early PD diagnosis.
1. Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease in the United States, with an estimated 640,000 elderly people aged 65 years and over (1.6% of elders) suffering from this disease.[1] Neuropathologically manifestations of PD patients are motor disorders, including changes in muscle rigidity, tremors, and gait.[2] In addition to the motor symptoms, nonmotor features of the disease include autonomic failure, urinary incontinence, hallucinations, and dementia.[3] Studies have shown that in the first clinical manifestations of PD, patients lost about 60% of their striatum dopaminergic nerve terminals, and about 30% of their substantia nigra (SN) neurons.[4] Since the disease has entered the advanced stage of neuropathology at the time of diagnosis, it is of great significance to find potential biomarkers that can identify the disease early in the experiment of disease modification therapy.
Exosomes are cell-derived vesicles between 30 and 100 nm in diameter, protected by lipid-rich binding membranes. These vesicles were originally considered to be vesicles produced by mature blood reticulocytes and secreted into extracellular milieu with topically expressed transferrin receptors.[5,6] They are secreted by hematopoietic and nonhematopoietic cells, and are present in serum, urine, cerebrospinal fluid (CSF), bronchi alveolar lavage, synovial fluid, and other body fluids due to their specific cell-derived components on their surfaces. Their components include proteins, lipids, and different amounts of DNA, RNA, and small cytoplasmic RNA. These components are known to may be involved in many physiological processes and the progression of diseases, such as neurodegenerative diseases. In neurodegenerative diseases, the progressive accumulation of protein aggregates and lack of sensitive biomarkers hamper the development of disease-specific therapies. In addition, although protein aggregates can be detected in CSF and blood, their extremely low levels limit their use as potent biomarkers. In view of these limitations, the focus of research on biomarkers in neurodegenerative diseases has shifted from proteins to the vesicles in biological fluids. The presence of disease-specific molecular signatures in exosomes makes them powerful diagnostic candidates.[7] As exosomes protect their cargo from degradation, screening exosomes in CSF and serum provides a way to identify the origin of their cells, thus providing insights into the cellular and pathogenic processes of the central nervous system. The exosomes produced by tumor cells in the brain display immunosuppressive and carcinogenic factors, which is a good example of disease-specific molecular characteristics in exosomes.[8]
In the present study, we quantitatively analyzed the serum exogenous proteomes of PD patients using mass spectrometry-based label-free technique and compared them with healthy controls. This study was designed to identify difference of exosome protein content between PD patients and control. A total of 429 proteins were identified. Some of them, including apolipoprotein J, pigment epithelium-derived factor (PEDF), and gelsolin, may be potential target for early PD pathogenesis.
2. Materials and methods
2.1. Participants and sample collection
All subjects received written informed consent for participation from their guardians directly or directly, and were approved by the institutional Ethics Committee of the Second People's Hospital of Lishui, Zhejiang province. All participants were assessed including medical history, physical and neurological examinations, and neuropsychological assessments. PD patients were diagnosed according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINDS-ADRDA) criteria, and their diagnoses were confirmed by the senior neurologist according to the United Kingdom Parkinson's Disease Society Brain Bank Clinical Diagnostic Criteria.[9] The sporadic PD group included 20 subjects (average age 78 years; 11 males; 9 females). According to the Hoehn and Yahr classification,[10] these PD patients were divided into 2 clinical subgroups: mild (score less than 3; n = 10), or severe (score greater than 3; n = 10). Healthy control subjects (average age 79 years; 6 males, 4 females) were community volunteers with good health. In the Cambridge Cognitive Examination (CAMCOG) scale, they have no signs or symptoms indicating cognitive impairment.[11] Whole blood samples were centrifuged to separate out serum. All serum samples were stored at −80°C until analysis.
2.2. Exosome isolation from serum
The initial volume of serum for the experiments was 5 mL per group. The samples were diluted with an equal volume of Phosphate Buffered Saline (PBS) to decrease the viscosity. The diluted serum samples were centrifuged at 2000 g for 10 minutes, 10,000 g for 30 minutes at 4°C to remove dead cells and cell debris. The supernatant was transferred into Ultra-Clear tubes (Beckman Coulter, Indianapolis, IN), and centrifuged at 100,000 g using a Beckman Optima XL-70 ultracentrifuge for 70 minutes at 4°C. The supernatant was removed by pipette, and the supernatant was remained 2 mm above the pellet. Five cycles of ultracentrifugation were needed to purify exosomes and remove serum proteins.[12] After centrifugation, the exosomes appeared as beige in the bottom of the vessel. The exosomes dissolved in 200 μL SDT buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl pH 8.0), and boiled for 5 minutes. Undissolved debris was removed by centrifugation at 14,000 rpm for 15 minutes. The supernatant were collected and quantified with a BCA Protein Assay Kit (Bio-Rad, CA, USA).
2.3. Exosomal protein digestions
Digested protein (50 μg for each sample) was performed according to the filter-aided sample preparation (FASP) procedure described previously.[13] Briefly, the detergent, DTT and other low molecular weight components were separated by centrifugation using 200 μL UA buffer (8 M urea, 150 mM Tris-HCl pH 8.0) by repeated ultrafiltration (pall, 10 kD). Then 100 μL of 0.05 M iodoacetamide in the UA buffer was added to the sample block to reduce cysteine residues and the samples were incubated for 20 minutes in darkness. The filter was washed with 100 μL UA buffer 3 times and then 100 μL 25 mM NH4HCO3 twice. Finally, the suspended protein was digested with 3 μg trypsin (Promega, Shanghai, China) in 40 μL 25 mM NH4HCO3 overnight at 37°C, and the resulting peptides were collected as a filtrate. Based on the frequency of tryptophan and tyrosine in vertebrate proteins, the content of the peptide was estimated by ultraviolet spectral density at 280 nm using a solution with an extinction coefficient of 0.1% (g/L).
2.4. Label-free quantitative proteomics
The peptide of each sample was desalted on C18 Cartridges (Empore SPE Cartridges C18 [standard density], bed I.D. 7 mm, volume 3 mL, Sigma-Aldrich, CA), and then concentrated by vacuum centrifugation and reconstituted in 40 μL of 0.1% (v/v) trifluoroacetic acid. MS experiments were performed on a Q Exactive mass spectrometer that was coupled to Easy nLC (Proxeon Biosystems, now Thermo Fisher Scientific, San Josse, CA). 5 μg peptide was loaded onto a C18-reversed phase column (Thermo Scientific Easy Column, 10 cm long, 75 μm inner diameter, 3 μm resin) in buffer A (2% acetonitrile and 0.1% formic acid), and separated with a linear gradient of buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 250 nL/minutes controlled by IntelliFlow technology over 120 minutes. MS data was acquired using a data-dependent top 10 method to dynamically select the most abundant precursor ions from the survey scan (300–1800 m/z) for High Energy Collisional Dissociation (HCD) fragmentation. Determination of the target value is based on predictive automatic gain control. Dynamic exclusion duration was 25 seconds. The resolution of survey scan was 70,000 at m/z 200, and the resolution for HCD spectrum was set to 17,500 at m/z 200. Normalized collision energy was 30 eV and the underfill ratio was defined as 0.1%, which specifies the minimum percentage of the target value that may be reached within maximum fill time.[14] The peptide recognition mode was enabled when the instrument was running. MS experiments were performed triply for each sample.
2.5. Sequence database searching and data analysis
The MS data were analyzed using MaxQuant (version 1.3.0.5) software. MS data were searched in the UniProt human database (148,327 total entries, downloaded 20,160,207). An initial search setting was 6 ppm in the precursor mass window. The search followed the enzymatic cleavage rule of trypsin/P, allowing for maximum deletion of 2 missed cleavage sites, and a mass tolerance of fragment ions was 20 ppm. Cysteine aminomethylation was defined as a fixed modification, and protein N-terminal acetylation and methionine oxidation were defined as variable modifications for database searches. The cutoff value of the global false discovery rate for peptide and protein identification was set to 0.01. Label-free quantification was carried out in MaxQuant as previously described.[15] Protein abundance was calculated on the basis of the normalized spectral protein intensity (LFQ intensity).
2.6. Bioinformatics
Differentially expressed proteins (DEPs) were annotated using the gene ontology (GO) program Blast2GO (https://www.blast2go.com/) to create a histogram of GO annotations, including cellular components, biological processes, and molecular functions. For pathway analysis, the differential proteins were mapped to the terms in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the KAAS program (http://www.genome.jp/kaas-bin/kaas_main).[16]
2.7. Statistical analysis
The Perseus software (version 1.3.0.5) was used to analyze the library files obtained by MaxQuant and compare the peak intensities to obtain quantitative data for all the peptides in the sample. The LFQ intensities of peptides from MaxQuant analysis were imported and converted into logarithmic scale with base 2. Student t test was used to calculate the quantification of protein and statistical significance. P < .05 was considered as a significant difference.
3. Results
3.1. Quantitative protein identification of serum-derived exosomes
Serum-derived exosome proteins were isolated and digested with trypsin. The peptides from the in-gel digestion were analyzed by LC-MS/MS, and the resulting data was submitted to database search using the Mascot (Matrix Science, London, UK) program. In total, 429 proteins were detected in all samples with high confidence. The quantization information on the identification of peptides was listed in the supporting file (Table S1). Their molecular weight is mainly distributed in the ranges 10 ∼ 70 kDa (Fig. S1), indicating that a group of middle molecular substances and the basic proteins are enriched. Among these 429 proteins, 355 proteins were found in exosome samples of all groups (Fig. 1A). Nine unique proteins, including protein S100, tyrosine protein kinase receptor, lactoferrin, dermcidin, platelet activating factor acetylhydrolase, and isocitrate dehydrogenase, exist only in serum exosomes of patients with severe PD. On the other hand, some common proteins in serum samples, including keratin 1, ryanodine receptor 2, alternative protein fibroblast growth factor receptor 2 (FGFR2), and Immunoglobulin G (IgG) chains, exist only in serum exosomes of healthy controls. Detailed information on unique proteins in serum exosomes of each group, including protein score, sequence coverage, and molecular weight, is provided in the supporting file (Table S2).
Figure 1.
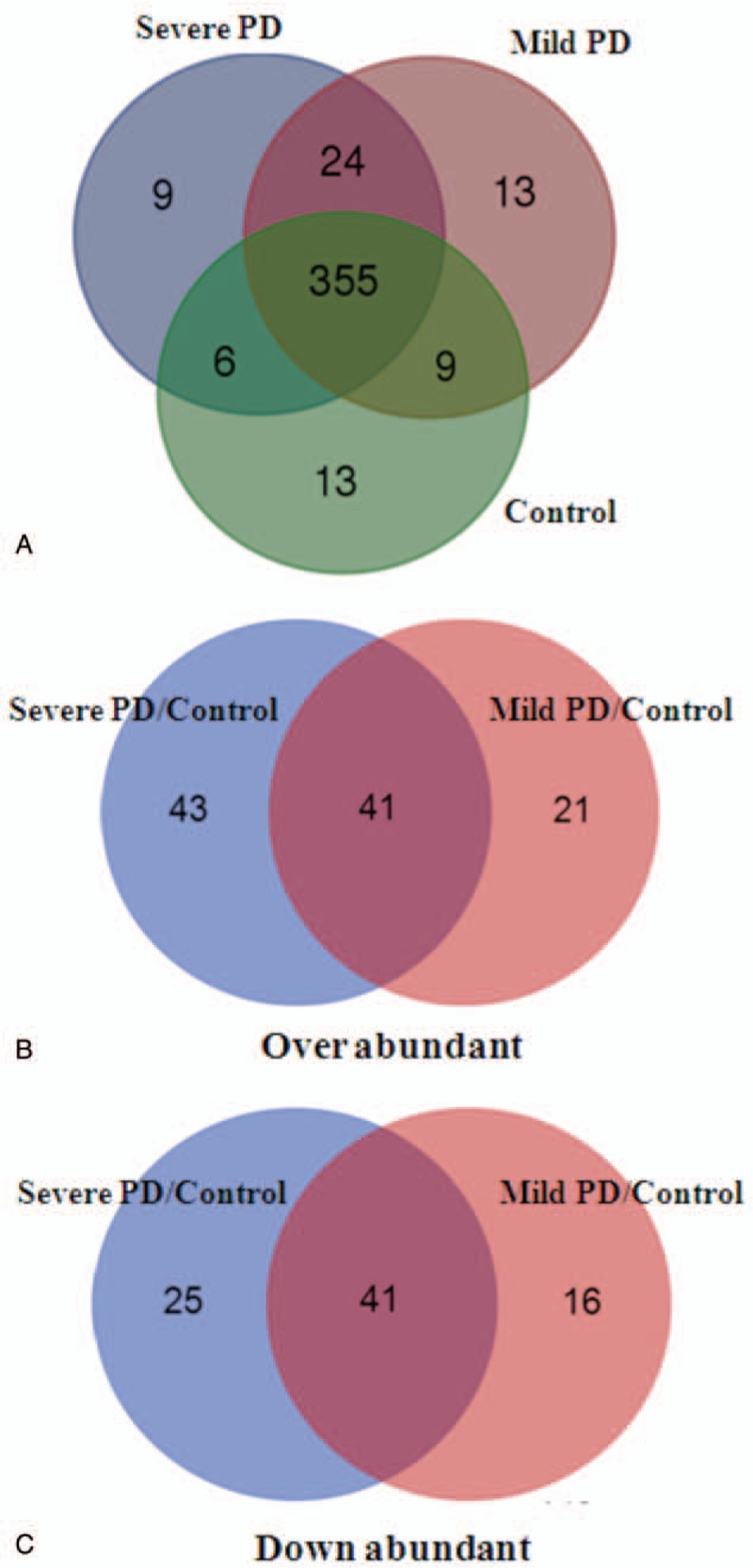
Summary of significant proteins in serum-derived exosomes of PD patients or controls. (A) The number of identified proteins in each group is represented by Venn diagram. The number of upregulated (B) and downregulated (C) proteins in mild and severe PD compared with healthy control. Up or downregulated of the 2 groups of proteins is at the intersection. PD = Parkinson disease.
3.2. Quantitative comparison of protein expression at different stages
To accurately evaluate the proteomic changes, more than twice the statistical value between 2 groups were defined as significant expressed proteins. In total, compared with healthy control, 62 and 84 proteins were upregulated in mild and severe PD groups, respectively, while 57 and 66 were downregulated. Among these significantly expressed proteins, 41 proteins were upregulated and 41 were downregulated in PD patients both with mild and severe stages (Fig. 1B and C). Details of these significantly expressed proteins are shown in Table S3. More importantly, clusterin, complement C1r subcomponent, afamin, angiotensinogen variant, apolipoprotein D (ApoD), gelsolin, and PEDF were progressively upregulated from mild PD to severe PD (Fig. 2A). In contrast, human neuroblastoma full-length cDNA clone CS0DD006YL02, precursor (AA −19 to 113), complement C1q subcomponent, myosin-reactive immunoglobulin kappa chain, Ig kappa chain V-III region, immunoglobulin mu chain, and immunoglobulin kappa variable 1 to 33 are gradually downregulated in the serum exosomes from mild PD to severe PD (Fig. 2B).
Figure 2.

Significant proteins in serum-derived exosomes of PD patients. Upregulation (A) or downregulation (B) of the protein is progressively from mild PD to severe PD. PD = Parkinson disease.
3.3. GO enrichment of DEPs
To obtain a global image of the proteomic changes during PD development, we performed the DEPs were annotated with GO terms based on the biological processes, molecular functions, and cellular component categories. For the classification of biological processes, the top 5 enriched annotation terms were response to stimulus, response to stress, regulation of stimulus response, defense against response, and immune system processes (Fig. 3A). The molecular functions of these proteins included receptor binding, calcium ion binding, cell adhesion molecule binding, growth factor activity, protein binding, and low-density lipoprotein particle binding (Fig. 3B). The cellular component classes of these changed proteins were the extracellular region, extracellular space, blood microparticle, fibrinogen complex, cell cortex, cytoplasmic region, and sarcoplasm (Fig. 3C).
Figure 3.

Gene ontology functional classification of serum-derived exosome proteomes. (A) Biological process, (B) molecular function, and cellular component (C). The numbers after comma indicate the number of hits for each subcategory.
3.4. Pathway analysis
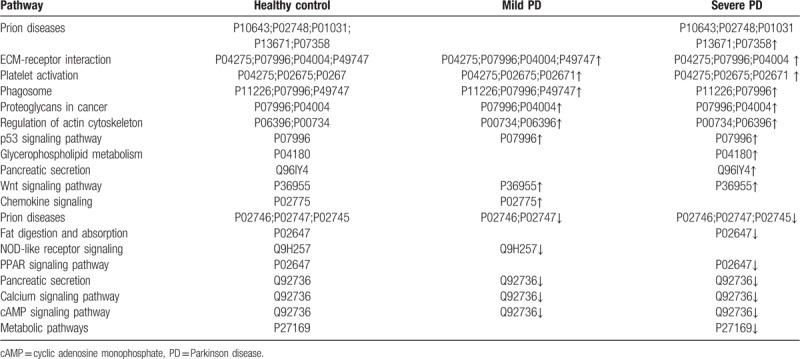
The label-free MS results were also evaluated using UniPathway. DEPs were divided into up and downregulated pathways. In the upregulated pathways of PD patients, the top 6 enrichment terms of biological process class were prion diseases, Extracellular Matrix (ECM)–receptor interaction, platelet activation, phagosome, proteoglycans in cancer, and regulation of actin cytoskeleton (Table 1). Compared with the healthy control, the expression of complement component C5–9 (P10643, P02748, P01031, P13671, and P07358) in prion diseases pathway, cartilage oligomeric matrix protein (P49747) in ECM–receptor interaction pathway, phosphatidylcholine–sterol acyltransferase (P04180) in glycerophospholipid metabolism, and carboxypeptidase B2 (Q96IY4) in pancreatic secretion were significantly increased in PD patients. Conversely, the 5 pathways were downregulated in PD patients, including prion diseases, fat digestion and absorption, Nucleotide-binding Oligomerization Domain (NOD)-like receptor signaling, and Peroxisome Proliferators-Activated Receptors (PPAR) signaling pathway (Table 1). Compared with the healthy control, complement C1q subcomponent subunit A–C (P02745, P027746, and P02747) in prion diseases pathway, apolipoprotein AI (P02647) in fat digestion and absorption pathway, caspase recruitment domain-containing protein 9 (Q9H257) in NOD-like receptor signaling pathway, and ryanodine receptor 2 (Q92736) in pancreatic secretion were significantly decreased in PD patients.
Table 1.
Differentially upregulated or downregulated protein pathway analysis in patients with mild and severe Parkinson disease compared with healthy control.
4. Discussion
Although exosomes play an important role in intercellular communication and influence both physiological and pathological processes, their function in PD remains still in its infancy. The working hypothesis was that serum-derived exosomes exhibited distinct biochemical compositions during the development of PD. This hypothesis is important, since the discovery of distinct protein changes in the exosomes might provide novel insights into the disease-specific systemic responses induced by the pathological processes of PD. In this research, we conducted a case–control study to explore the proteomic profiling of serum-derived exosomes in healthy subjects and patients with PD at different stages. The result revealed the development of distinct protein specific to the PD.
4.1. Proteomic characteristics of serum exosomes in patients with PD
The current studies have identified 383 proteins in human serum-derived exosomes from healthy samples. Previous studies evaluated the serum exosomes in healthy control and reported a total of 66 identified proteins.[17] Differences in the number of proteins detected by healthy subjects in different studies could be explained by the number of samples, where in the previous study only 2 healthy subjects was analyzed.[18] It has been demonstrated that when samples were aggregated from multiple subjects, the variability of samples reduced.[19] Perhaps, by increasing the number of samples, the range of detected proteins may increase. On the other hand, other studies have reported that circulating exosomes from healthy or diseased subjects contained more detected proteins.[20,21] In the study of Tomlinson et al,[20] 1033 human proteins were identified by nano-Ultra Performance Liquid Chromatography (UPLC)-MS/MS. Another study identified 1369 human proteins in healthy subjects by antibody-coupled monolithic silica.[21] Differences in the amount of identified proteins can also be explained by the MS technique and the number of peptides used to identify proteins. Studies using only 1 peptide to identify proteins may increase sensibility[22,23]; however, it may reduce specificity. In this study, we pooled serum-derived exosomes from PD patients at various developmental stages to reduce individual impact and used a peptide to identify proteins. Our results showed that the label-free proteomic approach successfully detected and quantified the overall protein profile alternations in the mild and severe stages of PD development.
4.2. Identifying significant protein alterations in serum exosomes of PD patients
The emergence and development of proteomic technology has brought us new hope for further investigation of its pathogenesis and treatment methods. There are some special advantages due to the unique accessibility of serum samples. This study used a relative quantitative proteomics approach to compare serum samples of healthy controls with those of patients with 2 different progressive types of PD. In total, 14 DEPs are progressively altered in exosomes derived from serum of patients with PD at different stages. According to previous studies, most of these proteins have already been associated with PD or other neurodegenerative disorders, which can be mentioned and analyzed below.
Studies have shown that immunologic systems are found in central neural system (CNS) which involved with PD, in addition, the serum inflammatory factors and blood T cells are involved in the immune dysregulation of PD and inspected as the potential clinic biomarkers for PD prediction.[24] The PD animal models showed that abundant CD3+, CD4+, CD8+ T lymph cells migrated from blood vessels into SN to attack dopamine neurons.[25,26] Therefore, it is reasonable to suppose that there may be some potential connections between PD stage and immune dysregulation in periphery blood. The complement system is believed to amplify the effectiveness of both the specific and nonspecific immunological defense system, which is related to the pathology of various neurodegenerative disorders including the pathogenesis of PD.[27] Complement C1q (C1q) is the first subcomponent of complement activation classical pathway C1 complex, which is essential for the elimination and connection of synapses. In our study, the level of C1q was significantly lower in PD patients. Recent evidence has shown that C1q can mediate the formation of central synapses and contribute to the neuroprotection of the central nervous system, which may be a beneficial therapeutic target in neurodegenerative disorders.[28] Interestingly, constitutive expression of C1q was restricted to microglia throughout the brain, and microglial C1q expression was early and transiently upregulated after treatment in the substantia nigra (SN) and striatum, but nigrostriatal dopaminergic injury may be not affected by C1q in PD model.[29] Therefore, combined with our observation and previous studies, C1q has complicated functions in the pathogenesis of PD.
Some exosomes with significant changes are also metabolic related and primarily expressed in other organs beside of the nervous system. ApoD is a lipocalin transporter of small hydrophobic molecules, which is expressed in and secreted from a variety of peripheral and brain tissues and plays an important role in several neurodegenerative diseases.[30] In healthy neural system, ApoD is expressed by glia cells and other nonneuronal cells (such as pericytes), which has a profound impact on the function and survival of neurons.[31] In addition to its role in extracellular lipid transport, ApoD regulates AA metabolism in an antioxidant and anti-inflammatory manner.[32] Apolipoprotein plays an important role in PD and exhibits vital function in the pathogenesis of PD. apolipoprotein J (or clusterin) is widely expressed in human tissues and has multiple functions in health and disease. Previous proteomic studies showed that the level of clustering protein in PD serum was higher than that in the control group.[33] Consistent with these results, our proteomic analysis further revealed that the level of cluster proteins in serum-derived exosomes gradually increased from mild to severe patients (Fig. 3). In addition, the dose-dependent manner of inducing oligomerization in patients with α-synuclein PD in CFS liquid has recently been observed.[34] In view of the close correlation between exosome and synuclein, we speculated that the higher level of exosomal clusterin in serum might modify the formation of synuclein-positive Lewy bodies and thus play a role in PD. In addition to clusterin and ApoD, a small hydrophobic lipocalin transporter was upregulated in PD glial cells, but not in dopaminergic neurons, suggesting its role in neuroprotection and the importance of glia in the amount of this protein in the central nervous system.[35] Our study further demonstrated the elevation of serum-derived exosome ApoD in PD patients at different stages, indicating its potential as a biomarker of PD.
PEDF is a member of the serpin superfamily. It is a nerve growth factor, which has been detected in the CNS and has interesting properties in terms of neuronal survival, as it can protect against oxidative stress and glutamate excitotoxicity. PEDF seems to be a neuroprotective factor for spinal motor neurons, hippocampal neurons, and cerebellar granule cells. Furthermore, PEDF has been shown to improve neuronal survival in a 6-hydroxydopamine PD mouse model. In this study, our results showed that the level of PEDF in serum of PD increased. One explanation for the increase in PEDF might be that PD patients have a compensatory mechanism against neuronal cell death. Similarly, another independent study also has revealed elevated levels of serum PEDF.[36] Taking together, the data indicated that PEDF seems to be an applicable biomarker.
Gelsolin is an 82 kDa actin-modulating protein regulated by Ca2+ and phosphoinositides. Gelsolin exists in 2 isoforms; a secreted protein found in plasma and CSF, which is considered to be part of the actin scavenging system, and the cytoplasmic form that regulated actin cytoskeleton.[37] Here, our proteomic analysis revealed that the levels of serum-derived gelsolin in PD patients were significantly higher than those of control, suggesting that gelsolin might play a potential role in PD. In line with our results, previous immunohistochemical studies showed that gelsolin co-occurred with α-synuclein Lewy bodies in PD patients and the presence of α-synuclein calcium chloride, which is closely associated with the aggregation rate.[38] Similarly, plasma gelsolin also showed binding to amyloid beta-peptide (Aβ) and inhibition of fibril formation. In addition, peripherally administrated plasma gelsolin Aβ levels reduced brain transgenic Alzheimer disease mouse model.[39] These observations, together with our studies, indicate that increased serum gelsolin could be involved in the aggregation process of α-synuclein and thus might affect the formation of Lewy bodies.
Exosomes are secreted membrane nanovesicles that not only transfer membrane components, but also deliver nucleic acids and proteins among different cells. Although this study identified 14 particular proteins in patients with PD, larger prospective cohort studies are required to validate the present results. Moreover, further studies are needed to investigate whether the change occurs originally or may just be the secondary effect of PD or even irrelevant diseases.
4.3. Assessing limitations
There is significant heterogeneity in gender and age risks for PD. Although there is good evidence that men are, in general, about 1.5 times more likely to suffer from PD than women, the results of different studies are not the same.[40] The ratio of male and female (M:F) in the incidence of PD is more significant in different regional populations, ranging from 1.01 to 2.1.[41] Similarly, the average age of onset fluctuated from 68 to 79.[42] According to these observations, the M:F was 1.2 (11/9), and the average age was 79 in our study. These patients were carefully selected within several years in line with strict clinical features and exclusion criteria. The clinical manifestations include motor impairments involving resting tremor, bradykinesia, and rigidity, along with nonmotor symptoms such as autonomic nervous, cognitive, and psychiatric problems. And the exclusion criteria were as follows: secondary Parkinson disease or Parkinsonism syndrome; (2) history of stroke, brain injury, or surgery, Alzheimer disease, motor neuron disease, or other central nervous system diseases; and coexistence of severe systemic disease (such as tumors, hepatitis, nephritis, chronic heart failure, or chronic obstructive pulmonary disease) or infectious diseases at the time of enrollment.[43] According to Hoehn and Yahr scores, patients were classified as moderate and severe, and those less than 3 were classified as mild, or more than 3 as severe. When the basic symptoms mentioned above and the clinical history of PD or any other disease did not exist, and the results of the detection of neurological diseases were negative, they were considered case control.
There are some limitations of this study that should be discussed. Firstly, small sample size was a limitation for our study, and further research with large sample size are needed to confirm these preliminary results. Secondly, the potential proteins identified in the study need be verified by other molecular methods including Western blot. Thus, further investigations are required to identify whether these proteins are promising biomarkers for PD. Although our sample size is not large, it was enough to show significant differences among the clinical groups through MS analysis. On the other hand, previous proteomic studies were also performed in relatively small sample size. For instance, Bostanci et al[44] studied 5 healthy subjects and 5 patients with aggressive periodontitis; and Kido et al[45] studied 8 patients with CP and 1 health periodontal subject. Additionally, some studies analyzed only 10 healthy subjects.[46]
5. Conclusions
In summary, our proteomics analysis demonstrates that serum-based proteomic can identify disordered pathways and diagnostic biomarkers. The limitation of this study is that the sample size is relatively small, so future work is required to validate potential candidate targets and determine whether the identified serum biomarkers can increase the performance value of PD at different stages and change clinical management. In all, the identification of serum exosome proteins in PD by proteomic should be the basis of future research. According to other proteomic analysis of these proteins reported in the literature, our findings suggest that these proteins can be used as potential biomarkers of PD.
Acknowledgments
We are grateful to all the patients who participated in the study, and their participation made this analysis possible.
Author contributions
Conceptualization: Ronghu Ke, Shuiyan Meng, Xiumei Yan.
Data curation: Ruilai Jiang, Ronghu Ke.
Formal analysis: Shuiyan Meng, Xiumei Yan.
Funding acquisition: Shaochang Wu.
Investigation: Xiumei Yan, Honglin Ke.
Project administration: Chunjiao Rong, Ronghu Ke.
Software: Chunjiao Rong, Shuiyan Meng, Honglin Ke, Shaochang Wu.
Validation: Xiumei Yan.
Visualization: Shuiyan Meng.
Writing – original draft: Ruilai Jiang, Chunjiao Rong, Honglin Ke.
Writing – review & editing: Ruilai Jiang, Chunjiao Rong, Honglin Ke, Shaochang Wu.
Supplementary Material
Footnotes
Abbreviations: Aβ = amyloid beta-peptide, ApoD = apolipoprotein D, CNS = central neural system, CSF = cerebrospinal fluid, DEPs = differentially expressed proteins, GO = gene ontology, M:F = ratio of male and female, PD = Parkinson disease, PEDF = pigment epithelium-derived factor, SN = substantia nigra.
How to cite this article: Jiang R, Rong C, Ke R, Meng S, Yan X, Ke H, Wu S. Differential proteomic analysis of serum exosomes reveals alterations in progression of Parkinson disease. Medicine. 2019;98:41(e17478).
RJ and CR contributed equally to this work.
The raw data used to support the finding of this study are available from the corresponding author upon reasonable request.
The study was funded by the National Natural Science Foundation of China (No.81401616) and the Lishui Science and Technology Bureau Foundation Committee (No.2017ZDYF03).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Allison WW, Evanoff BA, Min L, et al. Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology 2010;34:143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med 1998;339:1044–53. [DOI] [PubMed] [Google Scholar]
- [3].Hely MA, Reid WGJ, Adena MA, et al. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord 2008;23:837–44. [DOI] [PubMed] [Google Scholar]
- [4].Hsiao-Chun C, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Harding C, Stahl P. Transferrin recycling in reticulocytes: pH and iron are important determinants of ligand binding and processing. Biochem Biophys Res Commun 1983;113:650–8. [DOI] [PubMed] [Google Scholar]
- [6].Johnstone RM, Adam M, Hammond JR, et al. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 1987;262:9412–20. [PubMed] [Google Scholar]
- [7].Jan AT, Malik MA, Rahman S, et al. Perspective insights of exosomes in neurodegenerative diseases: a critical appraisal. Front Aging Neurosci 2017;9:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vos KEVD, Balaj L, Skog J, et al. Brain tumor microvesicles: insights into intercellular communication in the nervous system. Cell Mol Neurobiol 2011;31:949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. 1967. Neurology 2001;5710 Suppl 3:S11–26. [PubMed] [Google Scholar]
- [11].Roth M, Tym E, Mountjoy CQ, et al. A standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry 1986;149:698–709. [DOI] [PubMed] [Google Scholar]
- [12].Kim J, Tan Z, Lubman DM. Exosome enrichment of human serum using multiple cycles of centrifugation. Electrophoresis 2015;36:2017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wisniewski J, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6:359–62. [DOI] [PubMed] [Google Scholar]
- [14].Liming M, Peizeng Y, Shengping H, et al. Label-free proteomics reveals decreased expression of CD18 and AKNA in peripheral CD4+ T cells from patients with Vogt-Koyanagi-Harada syndrome. PLoS One 2011;6:e14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li L, Hu X, Xia Y, et al. Linkage of oxidative stress and mitochondrial dysfunctions to spontaneous culture degeneration in Aspergillus nidulans. Mol Cell Proteomics 2014;13:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Looze C, Yui D, Leung L, et al. Proteomic profiling of human plasma exosomes identifies PPARgamma as an exosome-associated protein. Biochem Biophys Res Commun 2009;378:433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Turay D, Khan S, Osterman CJD, et al. Proteomic profiling of serum-derived exosomes from ethnically diverse prostate cancer patients. Cancer Invest 2015;34:1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ngo LH, Veith PD, Chen YY, et al. Mass spectrometric analyses of peptides and proteins in human gingival crevicular fluid. J Proteome Res 2010;9:1683–93. [DOI] [PubMed] [Google Scholar]
- [20].Tomlinson PR, Zheng Y, Fischer R, et al. Identification of distinct circulating exosomes in Parkinson's disease. Ann Clin Transl Neurol 2015;2:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ueda K, Ishikawa N, Tatsuguchi A, et al. Antibody-coupled monolithic silica microtips for highthroughput molecular profiling of circulating exosomes. Sci Rep 2014;4:6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Baliban RC, Sakellari D, Li Z, et al. Novel protein identification methods for biomarker discovery via a proteomic analysis of periodontally healthy and diseased gingival crevicular fluid samples. J Clin Periodontol 2012;39:203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sachio T, Mamoru S, Hiroshi U, et al. Proteomic analysis of gingival crevicular fluid for discovery of novel periodontal disease markers. Proteomics 2012;12:2190–202. [DOI] [PubMed] [Google Scholar]
- [24].Chen L, Mo M, Li G, et al. The biomarkers of immune dysregulation and inflammation response in Parkinson disease. Transl Neurodegener 2016;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Odoardi F, Sie C, Streyl K, et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012;488:675–9. [DOI] [PubMed] [Google Scholar]
- [26].Wheeler CJ, Seksenyan A, Koronyo Yv. T-lymphocyte deficiency exacerbates behavioral deficits in the 6-OHDA unilateral lesion rat model for Parkinson's disease. J Neurol Neurophysiol 2014;5:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].More SV, Kumar H, Kim ISv. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson's disease. Mediators Inflamm 2013;2013:952375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benoit ME, Hernandez MX, Dinh ML, et al. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J Biol Chem 2013;288:654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Depboylu C, Schorlemmer K, Klietz M, et al. Upregulation of microglial C1q expression has no effects on nigrostriatal dopaminergic injury in the MPTP mouse model of Parkinson disease. J Neuroimmunol 2011;236:39–46. [DOI] [PubMed] [Google Scholar]
- [30].Waldner A, Dassati S, Redl B, et al. Apolipoprotein D concentration in human plasma during aging and in Parkinson's disease: a cross-sectional study. Parkinsons Dis 2018;2018:3751516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rassart E, Bedirian A, Do Carmo Sv. Apolipoprotein D. Biochim Biophys Acta 2000;1482:185–98. [DOI] [PubMed] [Google Scholar]
- [32].Provost PR, Villeneuve L, Weech PK, et al. Localization of the major sites of rabbit apolipoprotein D gene transcription by in situ hybridization. J Lipid Res 1991;32:1959–70. [PubMed] [Google Scholar]
- [33].Maarouf CL, Beach TG, Adler CH, et al. Cerebrospinal fluid biomarkers of neuropathologically diagnosed Parkinson's disease subjects. Neurol Res 2012;34:669–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stuendl A, Kunadt M, Kruse N, et al. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson's disease and dementia with Lewy bodies. Brain 2016;139:481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].OrdoEz C, Navarro A, Perez C, et al. Apolipoprotein D expression in substantia nigra of Parkinson disease. Histol Histopathol 2006;21:361–6. [DOI] [PubMed] [Google Scholar]
- [36].Halbgebauer S, Ãc P, Wirth K, et al. Protein biomarkers in Parkinson's disease: focus on cerebrospinal fluid markers and synaptic proteins. Mov Disord 2016;31:848–60. [DOI] [PubMed] [Google Scholar]
- [37].Yin HL, Kwiatkowski DJ, Mole JE, Cole FS. Structure and biosynthesis of cytoplasmic and secreted variants of gelsolin. J Biol Chem 1984;259:5271–6. [PubMed] [Google Scholar]
- [38].Welander H, Bontha SV, Näsström T, et al. Gelsolin co-occurs with Lewy bodies in vivo and accelerates α-synuclein aggregation in vitro. Biochem Biophys Res Commun 2011;412:32–8. [DOI] [PubMed] [Google Scholar]
- [39].Chauhan VP, Ray I, Chauhan A, Wisniewski HM. Binding of gelsolin, a secretory protein, to amyloid beta-protein. Biochem Biophys Res Commun 1999;258:241–6. [DOI] [PubMed] [Google Scholar]
- [40].Taylor KSM, Cook JA, Counsell CE. Heterogeneity in male to female risk for Parkinson's disease. J Neurol Neurosurg Psychiatry 2007;78:905–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dominique T, Perkins KSM, Carl C. Systematic review of incidence studies of Parkinson's disease. Mov Disord 2003;18:19–31. [DOI] [PubMed] [Google Scholar]
- [42].Benito-León J, Bermejo-Pareja F, Morales-González JM, et al. Neurological Disorders in Central Spain (NEDICES) Study Group. Incidence of Parkinson disease and Parkinsonism in three elderly populations of central Spain. Neurology 2004;62:734–41. [DOI] [PubMed] [Google Scholar]
- [43].Xu XM, Dong MX, Feng X, et al. Decreased serum proNGF concentration in patients with Parkinson's disease. Neurol Sci 2017;39:91–6. [DOI] [PubMed] [Google Scholar]
- [44].Bostanci N, Heywood W, Mills K, et al. Application of label-free absolute quantitative proteomics in human gingival crevicular fluid by LC/MS E (gingival exudatome). J Proteome Res 2010;9:2191–9. [DOI] [PubMed] [Google Scholar]
- [45].Kido J, Bando M, Hiroshima Y, et al. Analysis of proteins in human gingival crevicular fluid by mass spectrometry. J Periodontal Res 2012;47:488–99. [DOI] [PubMed] [Google Scholar]
- [46].Carneiro LG, Venuleo C, Oppenheim FG, et al. Proteome data set of human gingival crevicular fluid from healthy periodontium sites by multidimensional protein separation and mass spectrometry. J Periodontal Res 2012;47:248–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.