

Abstract
Fungi are known for their diverse biologically active secondary metabolites, compounds that have provided the basis for many landmark therapeutics in the last century. Due to ease of collection and culturing, the existing fungal chemical literature is vast, and fungal natural product isolation can often be hindered by the numerous nuisance and pan-toxic compounds that many strains produce. Dereplication efforts, aimed at identifying such compounds early in the purification, are imperative to reduce time and expense of rediscovery of known metabolites. The common practice of dereplication then deprioritizes samples containing nuisance compounds and often excludes them from the drug discovery workflow. We have implemented a two-step dereplication protocol that uses tandem mass spectrometry to identify nuisance compounds, followed by mass-directed chromatographic editing to remove them while leaving the remaining 'edited extract' in the drug discovery workflow. This two-step strategy facilitates rapid and more accurate evaluation of the chemical potential of high-throughput extract screening campaigns by consideration of bioactivity beyond that triggered by known metabolites. We demonstrate the isolation of a new natural product antibiotic from an otherwise toxic extract using the technique.
Keywords: Dereplication, tandem-MS, natural products, subtraction chromatography
1.1. Introduction
Fungi have played a significant role in the history of natural products chemistry: they have provided humankind the basis for some of our most important antibiotics, like the penicillins and cephalosporins [1], industrial chemicals, and crop and feed additives [2]. Fungi are also the producers of some of nature’s most potent toxins, like the aflatoxins, ergot alkaloids, and fusarium toxins [3]. In addition to mycotoxins, dozens of common nuisance metabolites bearing pan-active bioactivity profiles are known from fungi. Of the fungal genera most commonly isolated in the laboratory (Aspergillus, Penicillium, and Fusarium), the number of reported compounds can be well into the hundreds or even thousands (~1700, ~1700, and ~600 compounds, respectively, listed in AntiBase) [2]. In the face of rising drug resistance globally, the need for faster evaluation of the great diversity of fungal chemistry is imperative. While new methods are regularly developed to exploit fungal chemistry, nuisance compounds such as the mycotoxins remain a major obstacle in the discovery of new natural product drugs. As this dilemma has become more prevalent, the ability to analyze, identify, and predict the presence of known chemistry (dereplication) has become a critical stage of microbial natural product drug discovery [4-8].
Tandem MS has emerged as an essential technique for structural analysis of biological compounds, including peptides, proteins, and small molecule metabolites, and has subsequently formed the basis of most dereplication strategies employed in natural product research [9]. Tandem MS provides several pieces of data that are highly useful for characterization of secondary metabolites. The precursor ion and fragmentation pattern of each compound is unique: a single compound can be correctly identified independently from retention time, purity, and similarity to isomeric compounds [10]. While the tandem MS profile for each compound is unique, compounds with similar structures may have similar fragmentation patterns. Members of a compound class can be readily identified from similarities in fragmentation patterns, and structure determination for unreported derivatives can be aided by tandem MS data [2,10]. Tandem MS data has often been combined with other data, including chemotaxonomy [11], biological activity, and chemical novelty [6] to create versatile public and ad hoc databases for identification of known compounds and new metabolites, and for prediction of biological activity of components in crude extracts.
Our fungal extract library currently contains extracts from approximately 3000 fungal isolates, each of which we have grown in multiple culture conditions, yielding nearly 10,000 distinct extracts. Many of these extracts display biological activity against bacterial pathogens like the ESKAPE strains (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), Mycobacterium tuberculosis, and Clostridium difficile, as well as neglected infectious agents such as Leishmania sp., Acanthamoeba sp. and Naegleria fowleri. However, efforts to identify new bioactive compounds have been hindered by the large number of mycotoxins present in these fungal extracts. In order to more efficiently dereplicate these extracts and assess whether other bioactive components may be present, a two-step protocol was developed. The first step is a standard tandem MS spectral profile for each bioactive crude extract, using public and in-house fungal metabolite and toxin databases to identify known chemistry. Following dereplication, we sought to develop a method to efficiently remove nuisance compounds that might be masking the presence of new bioactive metabolites. To this end, any extracts with known or toxic compounds were then subjected to stage two, which involved a mass-guided separation of the known or toxic compounds from the crude extract. The remainder of the toxin-subtracted crude extract could then be rescreened for biological activity. This two-stage protocol enables early identification and removal of known and toxic compounds from a crude extract, allowing the rest of the extract components to be evaluated for biological activity and potential new chemistry (Scheme 1). This methodology can importantly be implemented with a high degree of automation, while simultaneously reducing the quantity of time-consuming biological and chemical evaluation techniques needed, ideal for high-throughput evaluation of large extracts libraries.
Scheme 1.

Mass spectrometry-based two-stage dereplication strategy workflow.
1.2. Methods and Materials
1.2.1. General Experimental Procedures.
Optical rotation was measured using an AutoPol IV polarimeter at 589 nm; UV absorption was measured by Agilent Cary 60 UV-Vis spectrophotometer. The IR spectrum was recorded with a PerkinElmer Spectrum Two equipped with a UATR (single reflection diamond) sample introduction system; NMR spectra were recorded at 298K on Varian Inova 400 or Varian Direct Drive 500 spectrometers. Chemical shifts are reported with the use of the residual MeOH-d4 signal (δH 3.31 ppm and δC 49.2 ppm) as internal standards. The 1H and 13C NMR assignments were supported by gCOSY, gHSQC/gHMQC, and gHMBC spectra. MPLC was performed using a Combiflash Rf 200i MPLC, using ELSD and UV detection with a RediSepRf 80 g silica column. Semi-preparative and analytical high performance liquid chromatography (HPLC) was performed on a Shimadzu LC-20 AT system equipped with an evaporative light scattering detector (ELSD) and an ultraviolet detector. Normal phase was completed with a gradient of hexanes to EtOAc on a semi-preparative Phenomenex silica column (10 μm, 100 Å, 250 × 10 mm) using ELSD and UV detection. Reversed phase HPLC was completed with a gradient of H2O to either ACN or MeOH on a semi-preparative Phenomenex C18 column (10 μm, 100 Å, 250× 10 mm), or on an analytical Phenomenex C18 column (5 μm, 100 Å, 250 × 4.6 mm).
1.2.2. Collection of Biological Material.
Fungal isolates were obtained from various mangrove trees found throughout Florida. Isolate KML14-75MG-C8 was isolated from the leaf of exotic Australian mangrove relative, Pandanus spiralis, found in the Fairchild Botanical Gardens in Miami, FL, on a growth media of Sabouraud dextrose agar, salt, and chloramphenicol, made according to manufacturer's specifications. Isolate HF14-37B-1B was isolated from the leaf of a red mangrove (Rhizophora mangle) in Tampa, FL, on a growth media of potato dextrose agar, salt, chloramphenicol, and cycloheximide, made according to manufacturer's specifications. The isolates were each regrown on 300 g of rice supplemented with broth media and treated with a 100 μM solution of 5-azacytidine, an epigenetic modulator [13]. Cultures were incubated at 28 °C for 21 days, then extracted with EtOAc for 24 hrs.
1.2.3. HRESIMS and Tandem MS.
High-resolution electrospray ionization mass spectra (HRESIMS) were obtained on an Agilent 6230A LC/TOF, and tandem mass spectra were obtained on an Agilent 6540 UHD Accurate-Mass QTOF LC/MS, each in positive ionization mode, with samples dissolved in MeOH and eluted with a gradient of H2O to ACN on a Phenomenex Kinetex C-18 column (2.6 μm, 100 Å, 150 × 3 mm). LC-MS/MS data was processed using Agilent MassHunter Qualitative Analysis B.05.00. Compounds were identified and spectra collected into a profile with software automated Compound Finder (Auto MS/MS) and MS/MS Spectral Extraction. All compounds within an extract were analyzed with public databases, METLIN-AMRT-PCDL and Mycotoxins-AMRT-PCDL, and our own in-house database, using MassHunter Qualitative Analysis as specified by manufacturer's instructions.
HRESIMS and liquid chromatography/tandem MS (LC-MS/MS) spectral profiles were generated for each crude extract. Extracts were subjected to LC-MS with a standardized elution gradient to generate a metabolite chromatographic and HRESIMS profile. Crude extracts were then subjected to an automated LC-MS/MS experiment which utilized the same standardized elution gradient as the LC-MS experiment, with collision-induced dissociation (CID) energies of 0 mV, 10 mV, and 40 mV selected to match data provided in most public databases [2, 9, 14].
1.2.4. Chromatographic Editing.
Extracts found to contain nuisance or pan-toxic metabolites were subjected to preparative mass-guided chromatography and edited to selectively remove such compounds. A standard, reverse phase elution gradient was used for each crude extract. A portion of the LC output was diverted to a fraction collector, and separated based on retention time and UV absorbance (220 nm). A portion of the eluent was simultaneously subjected to HRESIMS mass analysis to identify compounds in each fraction based on the molecular ion (M+H). Fractions containing previously identified nuisance compounds were maintained in separate collection vessels, while the remaining fractions and elution waste were recombined to form a 'nuisance compound subtracted' edited extract.
Preparative HPLC was performed on a Shimadzu LC-20 AT system equipped with an ultraviolet detector. Reverse Phase HPLC samples were dissolved in MeOH and eluted with a gradient of H2O to either ACN or MeOH on a semi-preparative Phenomenex C18 column (10 μm, 100 Å, 250× 10 mm). HRESIMS was obtained on an Agilent 6230A LC/TOF with positive mode ionization.
1.2.5. Biological Assay.
Extracts were assessed for their ability to inhibit growth of the multi-drug resistant ESKAPE (E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and E. cloacae) pathogens in a tiered approach beginning with 200 μg/mL. ESKAPE pathogens were grown in tryptic soy broth (TSB) overnight at 37 °C with shaking. Minimum inhibitory concentration (MIC) assays were performed in 96-well plate by diluting overnight cultures 1:1000 in TSB (Gram-positive organisms) or Mueller-Hinton broth (MHB) (Gram-negative organisms) and ensuring a total reaction volume of 100 μL was attained. Serial dilutions of extracts were screened, beginning at 200 μg/mL, until there was no inhibitory effect detected. Inhibition was determined via visual inspection and was apparent by lack of turbidity in the wells [12, 13]. Each round of assays were incubated in 96-well plates overnight at 37 °C with shaking.
1.2.6. Isolation of mycobenzoxazepine (1).
The crude HF14-37B-1B fungal extract (4.5 g) was first partitioned between hexane and 1:3 MeOH:H2O. The aqueous partition was subjected to tandem MS dereplication, and, following identification of toxic compounds, was subjected to edited chromatography. The toxin subtracted extract was recombined, mounted on silica gel, and subjected to normal phase MPLC with an elution gradient of hexane to EtOAc, following by a wash of 1:3 MeOH:EtOAc. The MPLC yielded eight fractions. Fraction H, which eluted with 1:3 MeOH:EtOAc, maintained biological activity, and, based on a secondary round of tandem MS dereplication, contained no known metabolites. Fraction H was purified using reversed phase analytical HPLC, with an elution gradient of H2O to MeOH over 30 min. The purified compound (1.0 mg) eluted with 1:9 H2O:MeOH.
Mycobenzoxazepine (3-methyl-4,1-benzoxazepine-2,5(1H,3H)-dione, (1): amorphous solid, (c 0.1, MeOH); UV (MeOH) λmax (log ε) 230 (1.78); IR (thin film) 3250, 3000, 2940, 1730, 1700, 1590, 1550, 1250 cm−1; 1H and 13C NMR Data, Table 1; HRESIMS m/z 174.0568 [M+H-H2O]+ (C10H8NO2, calculated 174.0555), m/z 192.0666 [M+H]+ (C10H10NO3, calculated 192.0660);
Table 1.
NMR Data (ppm) for mycobenzoxazepine (1) in MeOH-d4.
| Position | 13Ca, type | 1Hb | HMBC |
|---|---|---|---|
| 2 | 175.7, C | ||
| 3 | 68.8, CH | 4.20, q (6.8) | 2 |
| 5 | 172.2, C | ||
| 5a | 122, C | ||
| 6 | 128.4, CH | 7.70, dd (7.7, 1.4) | 5, 8, 9a |
| 7 | 123.5, CH | 7.14, m | 5a |
| 8 | 132.7, CH | 7.47, m | 6, 9a |
| 9 | 121.5, CH | 8.50, dd (8.0, 1.0) | 7, 9a |
| 9a | 138.7, C | ||
| 10 | 20.1, CH3 | 1.40, d (6.8) | 2 |
13C shifts taken from HMBC spectrum, multiplicity determined by HSQC.
400 MHz (multiplicity, (J in Hz)).
1.3. Results and Discussion
1.3.1. Development of the two stage Dereplication and Extract Editing Protocols.
Extracts containing known nuisance compounds previously isolated in our lab, and that displayed biological activity against one or more of the ESKAPE pathogens, were chosen to model the protocol. Two extracts, derived from fungal strains KML14-75MG-C8 ('KML14') and HF14-37B-1B ('HF14'), were identified with Staphylococcus aureus and Enterococcus faecium. LC-MS and LC-MS/MS dereplication (e.g., Figure 1) found both harbored the tetramic acids equisetin and ent-epi-equisetin, known pan-toxic fungal metabolites likely to contribute to the observed antibiotic activity. These two extracts therefore advanced to the chromatographic editing protocol.
Figure 1.
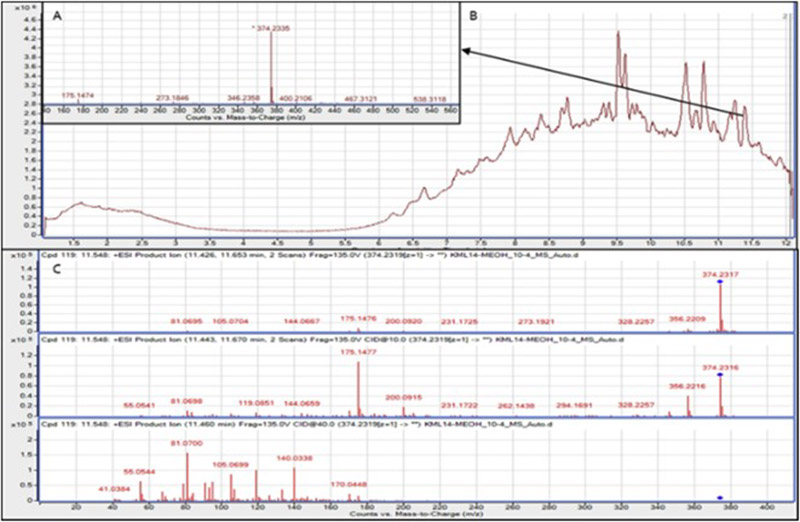
HRESIMS (A) and LC-MS/MS (C) spectral profiles for equisetin, derived from the crude extract of strain KML14 (B).
Fractions from a preparative HPLC separation of KML14 and HF14 were analyzed by HRESIMS mass analysis. Fractions bearing the identified tetramic acids (Figure 2) were separated, while the remaining fractions and elution waste were recombined to form a 'nuisance compound subtracted' edited extract. The scale and automation of instrumentation used allowed us to remove the nuisance compounds in a faster and more reliable method than in traditional separation methodologies driven by UV-vis or evaporative light scattering detection (ELSD) methods.
Figure 2.
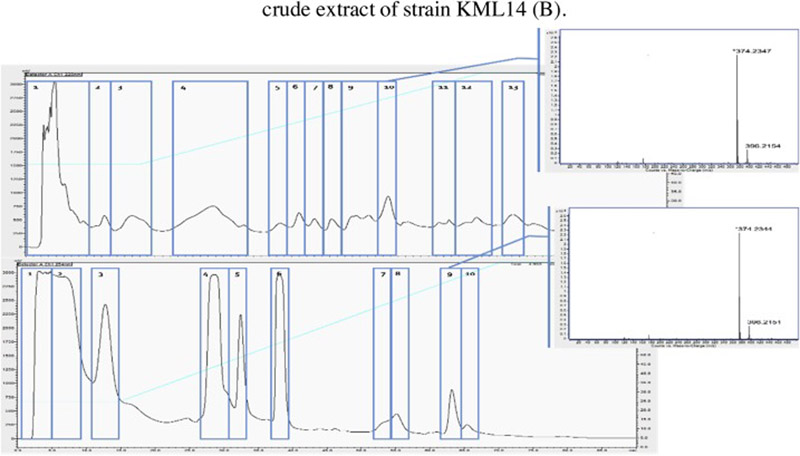
Chromatographic editing: extracts from strains (A) KML14 and (B) HF14 were subjected to an aqueous elution gradient of ACN (blue) and monitored based on 220 nm UV absorbance (black). HRESIMS spectra of eluting metabolites were used to identify nuisance compounds (fractions 10 and 9, respectively), which were separated from the remaining extract.
The edited extracts were then resubmitted for biological screening, in order to assess the potential of the remaining compounds in the extract. Unlike a traditional fractionation protocol, where a single crude extract can produce a myriad of fractions to be screened, our methodology ensures a minimal number of extracts needing to be screened, significantly reducing the time and effort required to screen and prioritize extracts. As presented in Figure 3, KML14 lost biological activity that was present in the crude extract. This recommended deprioritization of the extract, as activity could be solely attributed to the subtracted compounds. However, HF14 maintained activity after removal of the toxins and therefore was resubmitted for tandem MS dereplication, which confirmed the lack of other known compounds. HF14 then advanced in the antibiotic discovery workflow.
Figure 3.
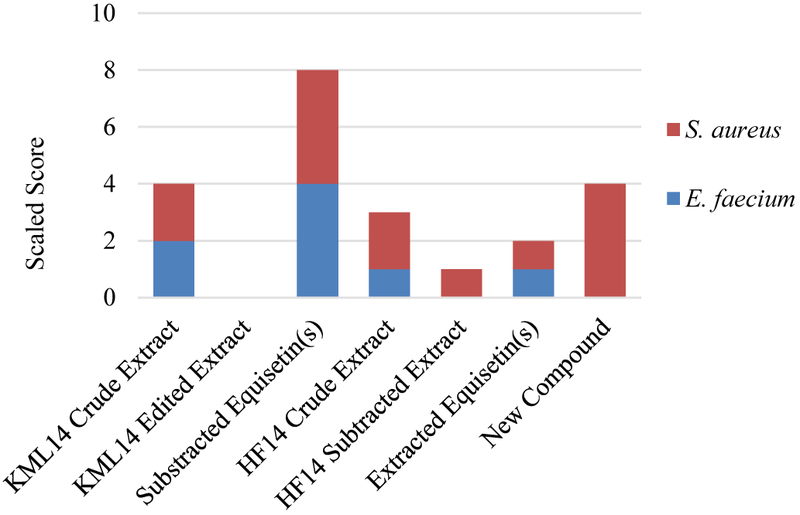
Comparison of biological activity of crude extracts, edited extracts, and subtracted nuisance compounds. Scaled score [15] relates MIC (μg/mL) against multiple ESKAPE pathogens as a single score.
1.3.2. Proof of Concept: Isolation of a New Natural Product Antibiotic from a Nuisance-Compound-Containing Extract
Bioassay-guided fractionation of HF14 resulted in the isolation of a metabolite retaining activity against Staphylococcus aureus. The metabolite was an amorphous solid determined to have a molecular formula of C10H9NO3 based on HRESIMS (m/z 192.0666 [M+H]+). Evaluation of the 1H and 13C NMR spectra indicated the presence of a 1,2-disubstituted benzene ring, two carbonyls, and a deshielded methyl and methine (Table 1). 2D NMR spectra (gCOSY, gHMBC) were used in the analysis of the structure. gCOSY correlations verified the 1,2-disubstituted aromatic ring among position 6-9, and the spin system comprising positions 3 and 10 (Figure 4). The carbon shift of positions 5 (δC 172.2) and 5a (δC 122.0) and gHMBC correlations between positions 5 and 6 indicated that position 5 was a benzylic ester carbonyl. Similarly, the shift of position 9a (δC 138.7) indicated a benzylic amine or amide. The chemical shifts of positions 3 (δC 68.8) and 10 (δC 20.1), and gHMBC correlations between positions 10 and 2 (δC 175.5) supported linking the benzylic ester and benzylic amide, providing the completed structure of the metabolite, which we named mycobenzoxazepine (1) (Figure 4). Position 3 represents the only stereocenter in mycobenzoxazepine. The specific rotation of 1 () was matched to a synthetic product [16] in which the stereocenter was derived from (R)-(+)-lactic acid, via SN2 inversion of the corresponding tosylate, suggesting mycobenzoxazepine bears the S-configuration at C-3. Mycobenzoxazepine was found to be active against MRSA with an MIC of 50 μg/mL.
Figure 4.
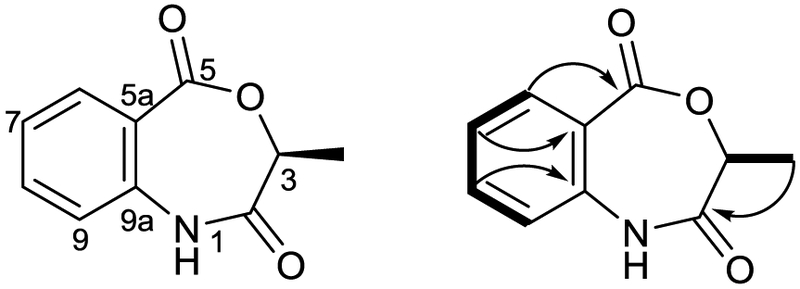
Structure of mycobenzoxazepine (1) Key COSY (—) and HMBC (→) correlations are shown.
1.4. Conclusions
The isolation and identification of mycobenzoxazepine (1) is significant for several reasons. Interestingly, though it is a new natural product, mycobenzoxazepine and related derivatives have been reported as an intermediates in the synthesis of various 4-quinazolinones [17]. The original synthesis was undertaken to produce natural product mimics, as 4-quinazolinones were determined to be important structural components to plant alkaloids [16]. Mycobenzoxazepine may represent a heretofore unreported intermediate in the 4-quinazolinone biosynthesis present in some fungal strains. Importantly, 1 represents a new compound that was isolated following our two-stage dereplication strategy, in a fraction that may otherwise have been deprioritized due to the abundance of potent cytotoxins present in the extract. In comparison to a traditional approach to new natural product isolation, which may require several rounds of fractionation, biological screening, and chemical analysis, 1 was isolated much more rapidly. Following a single round of nuisance compound extraction and biological screening, 1 was identified and targeted, and was isolated and purified with only one additional round of chromatography. It serves as a verification that our two-stage dereplication strategy can be used to evaluate the metabolite profile of extracts quickly, remove nuisance compounds, and mine the remaining extract for new bioactive chemistry. When used in conjunction with a large extract library, this two-stage dereplication strategy is a powerful tool to accelerate new natural product drug discovery efforts.
Supplementary Material
Highlights:
A two-stage, mass spectrometry-guided chromatographic editing strategy was developed
Chromatographic editing selectively subtracts nuisance compounds in crude extracts
Mycobenzoxozepine was efficiently recovered from a toxin-rich fungal extract
Chromatographic editing accelerates drug discovery from large extract libraries
ACKNOWLEDGMENTS
We would like to thank the staff at the University of South Florida’s Chemical Purification Analysis and Screening (USF CPAS) core facility, and at the Center for Drug Discovery and Innovation (CDDI) for making our chromatographic and spectroscopic chemical analysis possible. In particular, we would like to thank Andrew Shilling (CDDI) and Timothy Odom (USF CPAS) for their support and maintenance of the instruments that were critical to this work.
Funding Sources
This research was supported by research grant AI103715 (to LNS and BJB) and AI124458 (to LNS) from the National Institutes of Health. Facilities in the Center for Drug Discovery and Innovation were supported by a State of Florida Center of Excellence Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
The following files are available free of charge.
1D and 2D NMR spectra for mycobenzoxazepine (1) (PDF)
REFERENCES
- [1].Butler MS, The role of natural product chemistry in drug discovery. J. Nat. Prod. 67 (2004)2141–2153. [DOI] [PubMed] [Google Scholar]
- [2].Klitgaard A, Iversen A, Andersen MR, Larsen TO, Frisvad JC, Nielsen KF, Aggressive dereplication using UHPLC–DAD–QTOF: screening extracts for up to 3000 fungal secondary metabolites. Anal. Bioanal. Chem. 406 (2014) 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Miller JD, Mycotoxins in small grains and maize: old problems, new challenges. Food Addit. Contam. 25 (2008) 219–230. [DOI] [PubMed] [Google Scholar]
- [4].Jeffryes JG, Colastani RL, Elbadawi-Sidhu M, Kind T, Niehaus TD, Broadbelt LJ, Hanson AD, Fiehn O, Tyo KE, Henry CS, MINEs: open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. Cheminform. 7 (2015) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Allard P-M, Genta-Jouve G, Wolfender JL, Deep metabolome annotation in natural products research: towards a virtuous cycle in metabolite identification. Curr. Opin. Chem. Biol. 36 (2017) 40–49. [DOI] [PubMed] [Google Scholar]
- [6].Olivon F, Allard P-M, Koval A, Righi D, Genta-Jouve G, Neyts J, Apel C, Pannecouque C, Nothias LF, Cachet X, Marcourt L, Roussi F, Katanaev VL, Touboul D, Wolfender J, Litaudon M. Bioactive natural products prioritization using massive multi- informational molecular networks. ACS Chem. Biol. 12 (2017) 2644–2651. [DOI] [PubMed] [Google Scholar]
- [7].Bobzin S, Yang S, Kasten T, LC-NMR: a new tool to expedite the dereplication and identification of natural products. J. Ind. 25 (2000) 342–345. [DOI] [PubMed] [Google Scholar]
- [8].Nielsen KF, Må M, Rank C, Frisvad JC, Larsen TO, Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 74 (2011) 2338–2348. [DOI] [PubMed] [Google Scholar]
- [9].Sleno L, Volmer DA, Ion activation methods for tandem mass spectrometry. J. Mass. Spectrom. 39 (2004) 1091–1112. [DOI] [PubMed] [Google Scholar]
- [10].El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ, Oberlies NH, High-resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J. Nat. Prod. 76 (2013) 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Frisvad JC, Andersen B, Thrane U, Stadler M, The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycol. Res. 112 (2008) 231–240. [DOI] [PubMed] [Google Scholar]
- [12].Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina-Franco JL, Giulianotti MA, A Houghten R, Shaw LN. Combinatorial libraries as a tool for the discovery of novel, ,broad-spectrum antibacterial agents targeting the ESKAPE pathogens. J. Med. Chem. 58 (2015) 3340–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Demers DH, Knestrick MA, Fleeman R, Tawfik R, Azhari A, Souza A, Vesely B, Netherton M, Gupta R, Colon BL, Rice CA, Rodriguez-Perez MA, Rohde KH, Kyle DE, Shaw LN, Baker BJ, Exploitation of mangrove endophytic fungi for infectious disease drug discovery. Mar. Drugs. 16 (2018), 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wells JM, Mcluckey SA, Collision- induced dissociation (CID) of peptides and proteins. Methods Enzym. 402 (2005) 148–185. [DOI] [PubMed] [Google Scholar]
- [15].Scaled score calculated from the highest tested concentration divided by the MIC (μg/mL) for each pathogen, then summing the results from each pathogen (e.g.: a sample active against two ESKAPE pathogens, one with an MIC of 200 μg/mL, the second with an MIC of 100 μg/mL would receive a scaled score of 3: (200/200) + (200/100) = 3). See: Fleeman R; Lavoi TM; Santos RG; Morales A; Nefzi A; Welmaker GS; Medina-Franco JL; Giulianotti MA; Houghten RA; Shaw LN J. Med. Chem. 58 (2015) 3340–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bean CM, Kenyon J, Phillips H, J. Chem. Soc. 1936, 303–311. [Google Scholar]
- [17].Uskokovic M, Iacobelli J, Toome V, Wenner W, The conversion of 4,1-benzoxazepine-2,5(1H,3H)-diones into 2-(a-hydroxyalkyl)-4-quinazolinones. J. Org. Chem. 29 (1963) 582–584. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.