

Abstract
The Hippo signaling pathway controls cell-cell contact, cell proliferation, as well as organ size by integrating changes in the cellular microenvironment. In recent years, the pivotal role of Hippo signaling in cancers has been well recognized. Inhibition of the pathway promotes the translocation of the major Hippo pathway effectors, the yes-associated protein (YAP) and its paralog TAZ, to the nucleus, where they interact with the transcription factor family transcriptional enhancer associate domain (TEAD), thus coactivating the expression of downstream genes, leading to cell transformation, tissue overgrowth, and tumor development. Therefore, the interruption of the YAP–TEAD transcriptional complex represents a novel opportunity for the treatment of cancer. Here, we established a fluorescence polarization (FP)-based assay for the identification and evaluation of YAP–TEAD protein–protein interface (PPI) inhibitors at the YAP Ω-loop binding region of TEAD, which is also called interface 3 at the YAP–TEAD binding surface. Furthermore, a patented small molecule (Patent-22) was evaluated by the FP assay, which confirmed that it was a YAP–TEAD PPI inhibitor at interface 3. Possessing great application value, this FP method is reliable, robust, and economical for inhibitor assessment and drug discovery.
Keywords: Hippo signaling pathway, YAP, TEAD, Cyclic peptide, Protein–protein interaction (PPI), Ω-loop, Fluorescence polarization (FP), Ki, Small molecule inhibitor, Drug discovery
1. Introduction
The Hippo signaling pathway was first discovered by mosaic genetic screens on Drosophila for mutants exhibiting tissue overgrowth in the 1990s [1]. In mammalian cells, the evolutionarily conserved pathway also plays pivotal roles in controlling cell growth and survival during development [2-4], whereas in adults, its tumor-suppression effects [5] have been recognized and studied since the last decade [3]. The core of the Hippo pathway is a kinase cascade consisting of STE20-like protein kinase 1 or 2 (MST1/2), which phosphorylate and activate the large tumor suppressors LATS1/2 with the help of adaptor protein Salvador homolog 1 (SAV1) and MOB kinase activators (MOB1A/MOB1B) [1,6]. The activated LATS1/2 phosphorylate and inactivate the yes-associated protein (YAP) and its paralog TAZ (the transcriptional coactivator with PDZ-binding motif) by promoting their cytoplasmic localization and triggering their degradation by recruiting 14-3-3 protein [2,5,7]. When the Hippo pathway is repressed, active or unphosphorylated YAP/TAZ will translocate and accumulate in the nucleus, where they interact with the growth-promoting transcription factor transcriptional enhancer associate domain (TEAD) protein family [8] by forming transcriptional complexes, consequently inducing the expression of target genes such as connective tissue growth factor (CGRF) [1,9,10].
The activation of YAP promotes cell proliferation and overcomes cell contact inhibition [11], which results in cell differentiation [2] and tumor development. It was shown that YAP expression levels and nuclear localization are strongly elevated in many human cancers [11-16]. In addition, hyperactivation of the Hippo pathway by upstream tumor suppressors or genetic and pharmacological disruption of the YAP–TEAD complex suppresses the oncogenic activity of YAP [11,17-19]. These findings suggest that inhibiting YAP–TEAD interaction is a pharmacologically feasible strategy against YAP-driven tumorigenesis [1,20].
Two crystal s th the YAP-binding domain (YBD) of human TEAD1 and TEAD4 were released in 2009 [21,22], while the apo structure of TEAD2 was resolved in 2010 [23]. The TEAD protein family contains four members, TEAD1–4, which are highly structurally conserved. All four TEADs contain an N-terminal TEA domain that binds to DNA and a C-terminal YBD that binds to the TEAD-binding domain (TBD) of YAP [24]. Structural studies have revealed that intrinsically disordered YAP–TBD becomes structured [25] by wrapping around TEAD–YBD and forms three highly conserved interfaces with extensive contacts [21] (Fig. 1). Interface 1 is an antiparallel β-sheet formed between YAP residues 52–58 and a β-sandwich from TEAD, mediated by seven intermolecular hydrogen bonds [21]. Interface 2 is formed by a conserved LXXLF α-helix motif (YAP residues 61–73) packing into a hydrophobic groove on TEAD [22,26]. The third interface is called the Ω-loop binding site, where YAP residues 86–100 form an Ω-shaped loop with side chains inserting into a comparatively deep pocket on the surface of TEAD [21,23]. Multiple molecular interactions are established at interface 3 to strengthen the binding interaction, such as intermolecular hydrogen bonds and van der Waals contacts between YAP and TEAD residues, as well as internal hydrophobic interactions within the YAP Ω-loop region which fix and stabilize the loop [26]. Experimental results showed that mutations at interface 3 significantly destabilize the YAP–TEAD complex [21-23,27]. Therefore, the interface at the Ω-loop binding site is the key for the YAP–TEAD interaction [25].
Fig. 1.
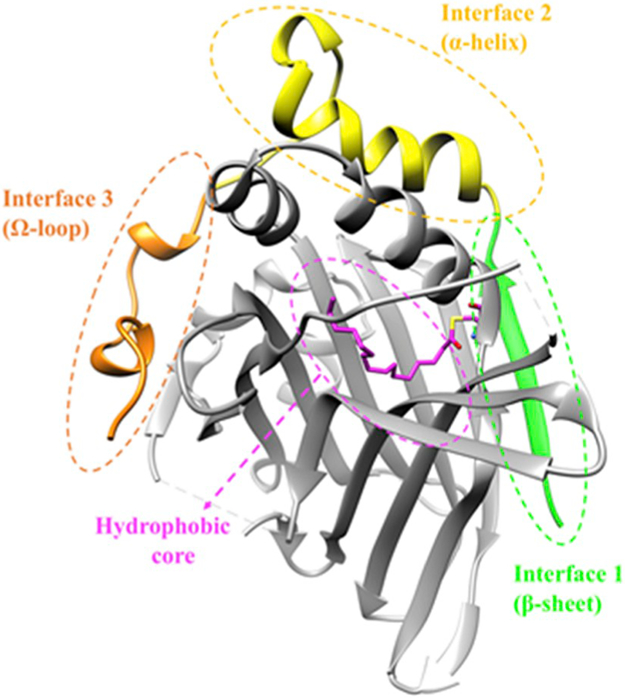
Crystal structure of YAP–TEAD complex. Interface 1–3 on YAP (PDB: 3KYS) are colored in green, yellow, and orange, respectively. TEAD1 (PDB: 3KYS) is shown in grey with a magenta palmitate molecule extracted and superimposed from palmitoylated TEAD2 (PDB: 5EMV). Structural figures were generated with Chimera. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
1.1. Current peptide and small molecule inhibitors targeting the YAP–TEAD complex
In general, there are two different strategies to disrupt the YAP–TEAD transcriptional activation complex, one of which is direct inhibition at either the Ω-loop or the α-helix binding interface [28] because of the importance of molecular interactions at these areas [23]. This protein–protein interface (PPI) inhibition might be accomplished either by inhibitory peptides or small molecules (Fig. 2). In 2014, Zhang et al. developed a potent cyclic peptide inhibitor YAP84–100−17 by applying conformational constraints within the Ω-loop region (YAP84–100), which was later confirmed by X-ray crystallography by Zhou et al. [29,30]. Combined with other beneficial mutations from an alanine scanning study, the replacement of R87 with homocysteine and F96 with cysteine and the introduction of a disulfide bond between the two new residues greatly stabilized the twisted-coil conformation, therefore significantly improving the binding affinity by nearly 1500-fold compared with the parent peptide YAP84–100 [20]. Another type of peptide inhibitor is designed to bind to interfaces 1 and 2 on TEAD protein by mimicking a transcriptional repressor called vestigial-like protein 4 (Vgll4), which is a tumor-suppressor protein [31,32] directly competing with YAP for its binding to TEADs in the nucleus [10].
Fig. 2.

Chemical structures of YAP84–100−17 and example small-molecule inhibitors targeting YAP–TEAD. α-Carbons of mutated residues in the cyclic peptide are marked by an asterisk.
Besides peptide inhibitors, small molecules are also believed to be functional in YAP–TEAD disruption and therapeutically promising. A series of aromatic heterocyclic molecules (e.g., Verteporfin (VP)) was first discovered in a luciferase reporter assay by Liu-Chittenden et al., in 2012 [19]. It was later determined that VP selectively binds to YAP [33-35], thus altering YAP conformation and abrogating its interaction with TEAD in vitro [36]. Alternatively, several small molecules are reported to disrupt YAP–TEAD binding by binding to the TEAD surface. Fragment-1 was screened out by thermal shift assay (TSA) screening and the crystal structure revealed its binding to the TEAD α-helix binding interface [37]. Fragment-2 is a fragment hit identified by NMR that binds to the N-terminal of the Ω-loop region on TEAD [38]. However, the binding affinity of Fragment-1 and -2 were quite low and no cellular assay results were disclosed. Recently, Gibault et al. identified Hit-2 as a micromolar-level inhibitor in a luciferase reporter assay through virtual screening conducted in a surface pocket at the Ω-loop region of TEAD [39]. Also, Smith et al. identified another YAP–TEAD interaction inhibitor CPD3.1 at interface 3 by in silico molecular docking, which showed antiproliferation and antimigratory effects in cellular assays [40]. Another moderate inhibitor family was identified and patented by Inventiva in 2017 for the treatment of malignant mesothelioma [10,41,42]. Among these patented molecules, Patent-22 showed potent inhibition activity in AlphaScreen assays with an IC50 of 83 nM [42], which is so far the most potent YAP–TEAD PPI small molecule inhibitor.
The second strategy to disrupt the binding between YAP and TEAD is not by targeting the interfaces but acting in an allosteric manner by inserting into the hydrophobic core of TEADs [10,24,28,38]. It was found that both intrinsic and purified TEADs were partially autopal-mitoylated in the central core of the β-sandwich through covalent linkage to a cysteine residue at the entrance of the core [43,44]. While palmitoylation facilitates YAP–TEAD activity by stabilizing TEAD [44,45], small molecule inhibitors such as flufenamic acid (FA) [24,38] and MGH-CP1 [28] suppress TEAD activity by competitively binding into the TEAD central pocket [46]. TED-347 [47] forms a covalent bond with a conserved cysteine within the palmitate binding pocket of TEADs and leads to allosteric inhibition of the YAP–TEAD protein–protein interaction. More small molecules have been designed this way recently.
1.2. Biochemical methods for YAP–TEAD inhibitor study
Many biochemical or biophysical methods have been applied in the study of YAP–TEAD binding inhibitors (Fig. 2). Isothermal titration calorimetry (ITC) is a useful technique to determine the thermodynamic parameters of binding interactions; however, there are also limitations, such as the requirement of high concentrations of both protein and ligand for weak bindings and the solubility issue of certain small molecules. Therefore, ITC is often used in the study of peptide inhibitor binding analysis for YAP–TEAD [23,24,37,38]. Another widely used method is based on protein unfolding, thermal shift assay (TSA), or differential scanning fluorimetry (DSF), in which the binding of the ligand stabilizes the protein receptor. It has been used in fragment screening for hit identification because of its high-throughput screening (HTS) ability [24,37,38,48]. However, binding of small molecules to the protein surface is usually so weak that no significant melting temperature (Tm) change can be obtained. Direct molecule binding can be analyzed by surface plasmon resonance (SPR) or biolayer interferometry (BLI). These are biosensor-based methods in which all the kinetic parameters, such as KD, kon, and koff, can be measured without labeling and in a high-throughput manner. Several groups have used this method for the binding study of truncated YAP peptides to TEAD [29,31,45,49], while a small-molecule inhibitor screening by SPR was carried out by Inventiva [42]. Gibault et al. used a microscale thermophoresis (MST) method to study the binding behavior of their virtual screening compounds. The binding of the YAP50–102 peptide to the protein receptor TEAD was validated by this method; nevertheless, no saturation was observed due to the solubility limit of their hit compounds [39] (e.g., Hit-2). Smith et al. established a Western-based method to characterize the inhibition activity of their in silico molecule CPD3.1 by using the isolated endogenous human YAP protein [40]. YAP50–114 peptides were utilized in an AlphaScreen-based HTS trial carried out by Inventiva, allowing the determination of IC50 values of inhibitors [28]. The inhibition activity of their patent molecules ranged from 83 nM to 105 μM; for example, Patent-22 had an IC50 of 83 nM [42].
Even though multiple methods have been used to characterize the binding behavior of YAP–TEAD inhibitors, there is no biochemical assay that is able to specify the specific binding area of these inhibitory molecules so far, especially for small-molecule inhibitors. NMR and X-ray crystallography could be applied to reveal structural information; however, they are limited by their low-throughput and time-consuming properties. Here, we have established and optimized a fluorescence polarization (FP)-based competition assay system for the discovery and assessment of YAP–TEAD PPI inhibitors specifically binding to the Ω-loop region on TEAD. Instead of using full-length YAP TBD (residues 50–114), we developed a fluorescent probe (YAP84–100−17) based on the cyclic peptide designed by Zhang et al., which specifically competed with inhibitors at interface 3. In addition, palmitoylated human TEAD2 (Pal-TEAD2) was used in our assay as the protein receptor to eliminate the interference of hydrophobic core inhibitors. Through assay condition optimization, the consumption of the peptide probe and TEAD protein was reduced to the nanomolar level. Therefore, our FP method is valuable and economical in identifying molecules that specifically target the Ω-loop binding interface.
2. Methods and materials
2.1. Protein expression and purification
The coding region of human TEAD2 YBD (217–147) (TEAD2_HUMAN, UniProtKB Q15562) was codon optimized, synthesized, and inserted into the pET-28a(+)-TEV vector between restriction sites NdeI and XhoI (GenScript). The final construct contained a tobacco etch virus (TEV) protease-cleavable His6-tag at the N-terminal of TEAD2217–447 and was verified by DNA sequencing (Genewiz). For protein expression, the pET-28a-TEV-TEAD2 plasmid was transformed into the Escherichia coli strain BL21 (DE3) CodonPlus competent cell (Invitrogen) and cultured (shaking) in LB media with antibiotics at 37 °C until OD 600 reached 0.5–0.6. Expression was induced by adding IPTG to a final concentration of 0.1mM and culturing (shaking) for 5 h at 16 °C. Harvested cells were resuspended and lysed by ultrasonication in lysis buffer (50mM Tris-HCl, 300mM NaCl, 10% glycerol, 1% Triton-X100, and 10mM imidazole; pH 7.4). The protein was first purified by metal ion affinity chromatography by loading to a Ni [2]-NTA IMAC cartridge (Bio-Rad) equilibrated in lysis buffer. The column was washed with buffer A (50mM Tris-HCl, 500mM NaCl, 10% glycerol, and 20mM imidazole; pH 7.4) and the protein was eluted in buffer B (50mM Tris-HCl, 500mM NaCl, 10% glycerol, and 250mM imidazole; pH 7.4). To completely occupy the hydrophobic core of TEAD2, the eluted protein was concentrated and palmitoylated by incubating with 2 eq. of alkyne palmitoyl-CoA (Cayman Chemical) for 1 h at room temperature (RT) in a buffer containing 50mM MES, pH 6.4 [43,44]. The palmitoylated or untreated TEAD2 were further purified by size exclusion chromatography on a Superdex75 (GE) gel filtration column in buffer C (50mM Tris-HCl, 100mM NaCl, 2mM MgCl2, 1mM TCEP, and 5% glycerol; pH 8.0). Protein purity was analyzed by SDS-PAGE. The purified protein was concentrated to 10–12 mg/ml, frozen in liquid nitrogen, and stored at −80 °C. The molecular weight of the palmitoylated or untreated TEAD2 was determined by intact protein LC-MS (Supplementary Fig. 1).
2.2. Synthesis of YAP peptides and fluorophore-labeled peptide probes
All the peptides used in this study were purchased from GenScript. The synthesized peptides and probes (Table 1) were analyzed by reversed-phase HPLC and MS.
Table 1.
Probe (fluorophore-labeled YAP peptide) design for FP assay.
 |
Abbreviations for some special amino acids: Hey, homocysteine; Nle, norleucine
2.3. Differential scanning fluorimetry (DSF)
Pal-TEAD2 and untreated TEAD2 proteins were buffer exchanged to three indicated buffers with different pH values. The final protein concentration for each sample was 20–200 μg/ml. Real-time fluorescence measurement was done by a Tycho NT.6 (NanoTemper). The capillary tubes were heated from 35 to 95 °C at a ramp rate of 0.2 °C/s and the ratio of fluorescence intensity at 350/330 nm was recorded in real time. First derivatives of the melting curve were used to determine the melting temperatures of proteins in different buffers. Experiments were performed in triplicate. Data are represented as mean ± SD, n = 3.
2.4. Fluorescence polarization (FP) measurement
All FP measurements were performed on a Synergy Neo2 HTS multimode microplate reader (BioTek) in black opaque 384-well microplates (Corning #3820). All experiments were performed in quadruplicate with 10 μL of the assay solution per well. Two FP filter cubes were used for the measurement of different fluorophore labels. The tetramethylrhodamine (TMR)-labeled probes were measured with FP 530/590 filter cube (530 nm excitation and 590 nm emission), and an FP 485/530 filter cube (485 nm excitation and 530 nm emission) was used for the measurement of the 5-carboxyfluoroscein (5-FAM)-labeled probe. The FP values were calculated according to the following equation [50,51]:
where V and H are the vertical and horizontal components of the intensity (I) of emitted light, respectively, when exited by vertically plane-polarized light [50]. Experiments were performed in quadruplicate. All FP measurement data are represented as mean ± SEM, n = 4.
2.5. FP binding assay
In the FP binding assays, different probes were titrated with palmitoylated TEAD2 in the optimized assay buffer: 20mM HEPES and 150mM NaCl (pH 7.4) with additives 0.1% BSA and 0.01% Tween-20. The reaction solution was incubated at RT for 1 h before FP signal measurement. During the optimization process, different buffers (20mM MES (pH 6.0) and 20mM Tris-HCl (pH 8.0) with or without additives) and different reaction conditions (temperature, incubation time, and DMSO tolerance) were tested. The binding affinity (dissociation constant, Kdapp) and the assay window (ΔmP) [52] were obtained by fitting the fluorescence polarization (mP) values and the corresponding protein concentrations into a nonlinear regression model in GraphPad Prism 8.
2.6. FP competition assay and Z′-factor determination
FP competition assays were carried out in the optimized buffer with 200 nM Pal-TEAD2 mixing with increasing concentrations of peptide inhibitors, while 5% DSMO was included for small molecules. The protein–inhibitor mixture was incubated at RT for 30 min, then 10 nM of probe 3 was added to the solution. The FP signal was recorded after 1 h incubation at RT. The FP values were plotted against the log of inhibitor concentrations, and the IC50 values were calculated from a four-parameter sigmoidal inhibition model in GraphPad Prism 8. The inhibition constant (Ki)) was calculated by a web-based tool [53-55]. For the Z′-factor determination assay, FP signals of 180 wells of negative control samples (no inhibitor was added to the competition assay) and 180 wells of positive control samples (100 μM YAP84–100-mut-3 peptide was added) were measured in the same 384-well plate. The high-throughput screening assay indicator Z′-factor was calculated based the following equation:
where σ and μ represent the standard deviations and means of the positive and negative controls, respectively [52,56].
3. Results
3.1. Probe design
Due to the focus on the characterization of interface-3-specific inhibitors, a truncated YAP peptide (residues 82–100, Ω-loop region) was first chosen as the fluorophore-labeled probe (Probe 1) for the binding of TEAD–YBD (residues 217–224). A minimal probe concentration of 50 nM was needed to maintain a stable and reliable FP signal. Purified, serial-diluted Pal-TEAD2 protein was mixed with the probe, and the binding of the probe to the protein resulted in an increased FP signal (Fig. 3). However, the binding curve could not reach a plateau even though 40 μM of Pal-TEAD2 was used. Since the required protein and probe concentrations in an FP assay depends on the binding affinity [57], we designed Probes 2 and 3 to carry more than one beneficial mutation based on the research of Zhang et al. Here, we found that the introduction of a disulfide bond greatly improved the probe binding affinity and also increased the assay sensitivity by reducing the amount of probe needed in the assay from 50 to 10 nM. The binding affinity Kdapp = 31.0 nM) of Probe 3 in our FP assay was consistent with the Kd of 15 nM previously determined by SPR [29]. The binding affinity was significantly improved; however, the ΔmP decreased from 141 to 100, which was still acceptable. To study the effect of different fluorophores, we tried 5-FAM instead of TMR and covalently linked it to the N-terminal of YAP peptides. Unfortunately, this modification did not improve the probe sensitivity but did result in a lower assay window (ΔmP = 50) and weaker binding affinity (Kdapp = 87 nM). Based on the above results, Probe 3 was chosen for use in the following assay optimization work as well as in the FP competition assays.
Fig. 3.

FP binding curves of designed probes. 50 nM of Probe 1 and 10 nM of Probes 2, 3, 4 were used, respectively, in the Pal-TEAD2 titration curves.
3.2. Optimization of buffer and pH
To improve the assay sensitivity (detection limit) and reduce the amount of biomolecular material used in this assay, we needed to reduce the probe concentration as much as possible while still maintaining adequate sensitivity of detection. Probe 3 was titrated in different buffer conditions with no protein to determine the lowest probe concentration required in the assay. Theoretically, the FP signal produced by the probe should not be affected by the probe concentration [50]. When there was no additive in the assay buffer, the FP signal of the probe fluctuated, especially when diluted below 100 nM (Fig. 4A). To solve this problem, two additives were added: 0.1% BSA (reducing nonspecific binding [52]) and 0.01% Tween-20 (reducing protein adsorption to the surfaces of the wells [57]). It was also suggested that the combination of more than one additive with distinct chemical properties could more effectively decrease compound aggregations in HTS [51,58]. Our results showed that the addition of BSA or Tween-20 alone did not help in this situation; instead, the FP signal considerably increased as the probe concentration decreased (Fig. 4B and C). However, the combination of two additives greatly stabilized the FP signal of the probe and lowered the lowest required concentration to 1 nM (Fig. 4D). Since an extremely low probe concentration resulted in a low signal intensity which could be masked by compound inhibitors, especially in a situation where much higher concentrations (> 1 μM) of small molecules are introduced [57], a probe concentration of 10 nM was chosen for the following FP binding and FP competition assays to minimize the background noise and compound interference.
Fig. 4.

Buffer optimization for FP assays. (A–D) The FP signals were plotted as a function of Probe 3 concentration in HEPES buffers (pH 7.4) in the absence or presence of additives 0.1% BSA or 0.01% Tween-20. (E) Melting temperatures of untreated or palmitoylated TEAD2 in different buffers were determined by DSF. The average Tm values were calculated and are shown. The error bars represent SD. (F) FP binding results of Pal-TEAD2 in different buffer pH values. All FP binding assays were performed by incubation at room temperature for 1 h after mixing the probe and protein. The assay window was plotted on the left y-axis in blue, while the apparent Kd was plotted on the right y-axis in red. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The effects of buffer pH were studied by DSF (Supplementary Fig. 2), and FP binding assays were conducted in different buffers with additives (0.1% BSA and 0.01% Tween-20). From the DSF results, the palmitoylation treatment increased the protein Tm by ~1 °C in all three tested buffers (Fig. 4E), which meant palmitoylation stabilized TEAD by occupying the central pocket. For the FP binding assay, serial-diluted Pal-TEAD2 proteins were mixed with the probe in different buffers, and the assay window and binding affinity values were evaluated (Fig. 4F). Since Pal-TEAD2 showed the best stability in HEPES pH 7.4 buffer with a Tm of 68.2 °C in the DSF experiment, as well as a decent ΔmP and Kdapp in the FP binding assay, we performed the following FP-based assays in the optimized assay buffer: 20mM HEPES and 150mM NaCl (pH 7.4) with additives 0.1% BSA and 0.01% Tween-20.
3.3. Optimization of temperature, incubation time and DMSO tolerance
To evaluate the stability of the FP binding assay, the reaction solutions were incubated at different temperatures (16, 25, 30, and 37 °C) in the optimized buffer (Fig. 5A). The binding affinity remained stable at temperatures below 30 °C, while higher temperatures might destabilize the protein, resulting in a weaker binding affinity. A slightly decreased assay window was also observed as the temperature increased. As shown in Fig. 5B, the FP signal for the probe-protein mixture was recorded every 1 h (at room temperature), then the assay window and binding affinity were plotted against incubation time. As the incubation time increased, the apparent Kd value increased in a time course probably because of the decreased protein stability. The assay window remained stable for the first 3 h but slightly increased at 4 h. Combining these observations, an incubation temperature of 25 °C (RT) and an incubation time of 1 h was used for future FP binding and FP competition assays.
Fig. 5.

Optimization of temperature, incubation time and DMSO tolerance. (A) Optimization of incubation temperature. The assay window and binding affinity derived from FP binding curves were plotted against temperature. (B) Optimization of incubation time. ΔmP and Kdapp were plotted against incubation time. (C) DMSO tolerance evaluation. The effect of up to 10% DMSO on ΔmP and Kdapp was negligible. (D) FP binding curve of 10 nM Probe 3 with Pal-TEAD2 titrated in the optimized assay condition (no DMSO).
To optimize this assay to be compatible with small molecules which are commonly dissolved in DMSO, the FP binding curve was evaluated in the presence of up to 10% DMSO in the optimized buffer (Fig. 5C). The results showed that the effect of DMSO was negligible even though 10% DMSO was added to the buffer, which made this assay a promising method for high-throughput screening of compounds with solubility issues. An example binding curve of 10nM Probe 3 with Pal-TEAD2 titrated in the optimized assay condition (no DMSO) is shown in Fig. 5D. Eighty percent of the probe was bound at a 200nM Pal-TEAD2 concentration; thus, 200nM Pal-TEAD2 was used in the following FP competitive assay.
3.4. FP competition assay for peptide inhibitors
Serial diluted inhibitory peptides were mixed with 200nM Pal-TEAD2 at RT. After 30 min of incubation, 10nM Probe 3 was added, mixed well, and incubated at RT for 1 h. According to the FP competition binding curves (Fig. 6), the FP signal decreased to the background level as the concentration of the inhibitory peptides increased. The inhibition constant Ki of YAP61–100 (interface 2 and 3 regions of YAP) was ~115-fold lower than YAP82–100 (interface 3 only), which meant YAP61–100 showed a much stronger binding affinity to Pal-TEAD2 than YAP82–100. Inhibition activities of the other designed YAP mutant peptides were also tested and their IC50 and Ki values are summarized in Table 2. Corresponding FP competition binding curves can be found in Supplementary Fig. 3.
Fig. 6.

FP competition binding curves of peptides (A) YAP82–100 (interface 3) and (B) YAP61–100 (interfaces 2 and 3). Different concentrations of peptides were titrated into competition assay solution containing 200 nM Pal-TEAD2 protein and incubated at RT for 30 min, then mixed with 10 nM Probe 3 and incubated at RT for 1 h before FP reading.
Table 2.
FP competitive inhibition results of designed peptides.
 |
Mutated residues are shown m bold.
3.5. FP competition assay for small-molecule inhibitors and Z′-factor determination
In 2017, a series of small molecules was patented as YAP–TEAD protein–protein interface inhibitors at interface 3 by Inventiva [42] (e.g., Patent-22). The inhibition behavior of this molecule was evaluated in our FP competition assay. Dose-dependent binding curves of Patent-22 were determined in assay buffer with or without Pal-TEAD2. Fig. 7A shows that the high concentrations of the tested molecule did not affect the FP reading of the probe (red), which meant the result was reliable. Also, a decreased FP signal was obtained in the buffer with Pal-TEAD2 (blue) as expected, which demonstrated that Patent-22 was indeed a YAP–TEAD PPI inhibitor at interface 3. However, the Ki value was 101 μM, which was far from the nanomolar level potency reported in the patent (discussed later).
Fig. 7.

FP competition assay for small-molecule inhibitor Patent-22 and Z′-factor determination. (A) FP competition binding curve of Patent-22 in the presence (blue) and absence (red) of Pal-TEAD2 in assay buffer with 5% DMSO. (B) Z′-factor determination from 180 negative control samples (no inhibitor added) and 180 positive control samples (YAP84–100-mut-3 peptide was added at a concentration of 100 μM) in one 384-well plate with 5% DMSO in assay buffer. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To evaluate the possibility and reliability of using this FP assay in high-throughput screening, we determined the statistical parameter Z′-factor (Fig. 7B). The assay solution of the negative control samples contained 200 nM Pal-TEAD2, 10 nM Probe 3, and 5% DMSO. An additional inhibitory peptide YAP84–100-mut-3 was added at a final concentration of 100 μM in the positive control samples. The resulting Z′-factor value was 0.523, which was better than the recommended value of 0.5 for high-throughput screening assays [52], suggesting that this is a reliable assay and could be used in HTS for future applications.
4. Discussion
Here, we established an FP-based competition assay for the evaluation of YAP–TEAD PPI inhibitors at the Ω-loop region. TMR-labeled cyclic peptide YAP84–100−17 (Probe 3) was selected as the best probe because of its high binding affinity (Kdapp=31.0 nM) to Pal-TEAD2. The final concentrations of the protein and probe used in the FP competition assay were: 200 nM of Pal-TEAD2 and 10 nM of Probe 3. The extremely low consumption of both components allows for large-scale peptide assessment or even HTS for small molecules. The utilization of the FP competitive assay was first validated by a series of truncated or mutated YAP peptides. The Ki value of YAP62–100 (interfaces 2 and 3) was 77.2 nM, which was at the same level as the Kd of 96 nM obtained by ITC in reported results [23]. The truncation of YAP62–100 to YAP82–100 (interface 3) greatly weakened its inhibition potency by more than 100-fold, which was also consistent with previous results [24,29]. The mutation of Met86 to Phe had slightly improved the inhibition activity from a Ki of 5.06 to 4.13 μM, while the substitution of special amino acid meta-Chloro substituted phenylalanine (3-Cl-Phe) at this position significantly decreased the Ki by 10-fold-634 nM. Unlabeled YAP84–100−17 cyclic peptide was also tested in our FP competition assays, where an IC50 of 23.0 nM was obtained. Based on the principle proposed by Huang, the range of resolvable inhibitor potency is limited by the affinity of the fluorescent probe [59], which means that the low end of inhibitor Ki values that can be resolved should be the Kd value of the fluorescent probe. In this case, the IC50 of YAP84–100−17 and its corresponding Ki were much lower than the Kdapp value (31.0 nM) of Probe 3; therefore, no reliable Ki could be obtained for YAP84–100−17 from this assay and the low end of inhibitor potency resolvable in this assay would be a Ki around 30 nM. All the other peptides or small molecules evaluated in this study resulted in Ki values above 30 nM, suggesting that the results were reliable.
Furthermore, we have validated the capability of the patent molecule Patent-22 to bind to the Ω-loop region on Pal-TEAD2 as a YAP–TEAD PPI inhibitor in vitro. This serves as direct evidence of its binding to interface 3 at the molecular level. Different from the nanomolar potency obtained in the AlphaScreen assay conducted by Inventiva, the Ki from our FP assay was much higher (~100 μM). The reason for this might be the usage of full-length YAP peptide and the lack of TEAD palmitoylation in the AlphaScreen assay. Based on a binding analysis by ITC, the binding affinity of full-length YAP2–268 (Kd = 33 nM) was only three times higher than that of YAP62–100 (Kd = 96 nM) [23], while in the FP assay, the affinity of the probe was at least 30 times stronger than YAP62–100. A weaker competitor may result in a more sensitive result in the AlphaScreen assay. However, it is a misconception that tight-binding fluorescent probes should be avoided to assess small-molecule inhibitors of low or intermediate potency. It had been demonstrated that the higher the affinity of the fluorescent probe, the wider the range of inhibitor potency that can be resolved [59]. Moreover, the usage of full-length YAP would complicate the analysis of the actual binding site in the AlphaScreen competition assay. Also, unpalmitoylated TEAD could interfere with the results, especially in a screening trial, since the small molecules might suppress YAP–TEAD interaction by binding to the central pocket rather than the interface, such as FA. Therefore, the FP assay is not only an economical but also a valuable tool for the identification and evaluation of small molecules specifically targeting YAP–TEAD interface 3 and can be used in HTS applications for screening large chemical libraries. The criteria or parameters for evaluating the “suitability” of an HTS assay for hit identification can be simplified as a coefficient called the “Z’-factor” [56]. While a Z′-factor value higher than 0.7 is considered to be an excellent assay, our FP assay with a Z′-factor of 0.523 is also decent and recommended for HTS assay. The only concerns would be the solubility of library compounds and the fluorescent interferences. The FP signal from a low concentration of a labeled probe (10 nM in our case) can be masked by some compounds that are introduced at much higher concentrations [57]. Further, high compound concentrations may result in aggregations which interfere with probe–protein binding. Therefore, dose-dependent curves and counter screenings are necessary for evaluating compounds of interest.
The patent molecule Patent-22 tended to aggregate at millimolar concentrations, so we were not able to take measurements at such high concentrations. However, the current data were still meaningful and useful for our understanding of its direct binding to TEAD interface 3.
Fig. 8 was a docking result of Patent-22 to the Ω-loop region on palmitoylated TEAD2 (PDB: 5EMV). The compound fell into a shallow pocket on the surface of TEAD2 at interface 3. While two hydrogen bonds were formed with the main chain residues, its benzothiazole dioxide group inserted into a hydrophobic subpocket. Also, the superimposition of YAP84–100 from PDB: 3KYS showed that the beneficial mutation on the Met86 residue side chain to meta-Cl-phenyl ring might increase the binding affinity by protruding into the same hydrophobic subpocket. These evidences suggest that this Ω-loop region is a targetable and druggable “spot” for YAP–TEAD PPI inhibitor discovery and development.
Fig. 8.

Predicted binding mode of Patent-22 on YAP–TEAD interface 3. (A) TEAD2 surface is shown in grey (PDB: 5EMV); YAP84–100 segment shown in orange is extracted from YAP–TEAD1 complex (PDB: 3KYS) and aligned to apo TEAD2, the key residue MET86 side chain is represented in stick; Patent-22 (in green stick) was docked to the TEAD2 Ω-loop binding interface by Autodock4. (B) Protein–ligand interaction predicted by docking. Two hydrogen bonds (dashed line) were formed between Patent-22 and TEAD main chain O or N. The benzothiazole dioxide group of Patent-22 fell into a hydrophobic pocket on the TEAD surface. Figures were generated with Chimera. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
5. Conclusion
The FP-based competition assay we established and optimized here is a valuable and economical tool for the identification and evaluation of peptide or small molecule inhibitors at YAP–TEAD protein–protein interface 3. We have verified the direct binding of a patented molecule Patent-22 with palmitoylated TEAD2 and confirmed that it disrupted YAP–TEAD protein-protein interaction specifically at interface 3. This assay is reliable, robust, and can be widely applied in the YAP–TEAD PPI drug discovery process, for example, high-throughput screenings of lead compounds from chemical libraries and lead optimizations.
Supplementary Material
Acknowledgements
We thank the Interdisciplinary Center for Biotechnology Research (ICBR) Proteomics & Mass Spectrometry Core of University of Florida for the mass spectrometry work. This work was partially supported by NIH R01NS088437 (Li) and the University of Florida Health Cancer Center Fund (Li).
Footnotes
The authors declare no competing financial interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ab.2019.113413.
References
- [1].Guo L, Teng L, YAP/TAZ for cancer therapy: opportunities and challenges (review), Int. J. Oncol 46 (4) (2015) 1444–1452. [DOI] [PubMed] [Google Scholar]
- [2].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S, Role of YAP/TAZ in mechanotransduction, Nature 474 (7350) (2011) 179–183. [DOI] [PubMed] [Google Scholar]
- [3].Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL, TEAD mediates YAP-dependent gene induction and growth control, Genes Dev. 22 (14) (2008) 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Astone M, Lai JKH, Dupont S, Stainier DYR, Argenton F, Vettori A, Zebrafish mutants and TEAD reporters reveal essential functions for Yap and Taz in posterior cardinal vein development, Sci. Rep. Uk 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD, Sun Y, Hudmon A, Wells CD, Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of Angiomotin by the LATS1/2 protein kinases, Proc. Natl. Acad. Sci. U. S. A 110 (43) (2013) 17368–17373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, Jeong KJ, Tsang YH, Moreno D, Bhavana VH, Peng X, Ge Z, Chen H, Li J, Chen Z, Zhang H, Han L, Du D, Creighton CJ, Mills GB, N. Cancer Genome Atlas Research, Camargo F, Liang H, Comprehensive molecular characterization of the Hippo signaling pathway in cancer, Cell Rep. 25 (5) (2018) 1304–1317 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen LM, Loh PG, Song HW, Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway, Protein Cell 1 (12) (2010) 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML, TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm, Genes Dev. 15 (10) (2001) 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Totaro A, Panciera T, Piccolo S, YAP/TAZ upstream signals and downstream responses, Nat. Cell Biol 20 (8) (2018) 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gibault F, Sturbaut M, Bailly F, Melnyk P, Cotelle P, Targeting transcriptional enhanced associate domains (TEADs), J. Med. Chem 61 (12) (2018) 5057–5072. [DOI] [PubMed] [Google Scholar]
- [11].Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Haider G, Lai ZC, Guan KL, Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control, Genes Dev. 21 (21) (2007) 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bossier P, Fernandes L, Rocha D, Rodrigues-Pousada C, Overexpression of YAP2, coding for a new yAP protein, and YAP1 in Saccharomyces cerevisiae alleviates growth inhibition caused by 1,10-phenanthroline, J. Biol. Chem 268 (31) (1993) 23640–23645. [PubMed] [Google Scholar]
- [13].Avruch J, Zhou D, Bardeesy N, YAP oncogene overexpression supercharges colon cancer proliferation, Cell Cycle 11 (6) (2012) 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu JY, Li YH, Lin HX, Liao YJ, Mai SJ, Liu ZW, Zhang ZL, Jiang LJ, Zhang JX, Kung HF, Zeng YX, Zhou FJ, Xie D, Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder, BMC Cane. 13 (2013) 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A, Wen W, Zhu Q, Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells, PLoS One 8 (6) (2013) e65539. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [16].Chen KH, He J, Wang DL, Cao JJ, Li MC, Zhao XM, Sheng X, Li WB, Liu WJ, Methylationassociated inactivation of LATS1 and its effect on demethylation or overexpression on YAP and cell biological function in human renal cell carcinoma, Int. J. Oncol 45 (6) (2014) 2511–2521. [DOI] [PubMed] [Google Scholar]
- [17].Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D, The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals, Dev. Cell 19 (1) (2010) 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL, Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein, Genes Dev. 25 (1) (2011) 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan DJ, Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP, Genes Dev. 26 (12) (2012) 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Santucci M, Vignudelli T, Ferrari S, Mor M, Scalvini L, Bolognesi ML, Uliassi E, Costi MP, The Hippo pathway and YAP/TAZ-TEAD protein-protein interaction as targets for regenerative medicine and cancer treatment, J. Med. Chem 58 (12) (2015) 4857–4873. [DOI] [PubMed] [Google Scholar]
- [21].Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, Xu Y, Structural insights into the YAP and TEAD complex, Genes Dev. 24 (3) (2010) 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen L, Chan SW, Zhang X, Walsh M, Lim CJ, Hong W, Song H, Structural basis of YAP recognition by TEAD4 in the Hippo pathway, Genes Dev. 24 (3) (2010) 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tian W, Yu J, Tomchick DR, Pan D, Luo X, Structural and functional analysis of the YAP-binding domain of human TEAD2, Proc. Natl. Acad. Sci. U. S. A 107 (16) (2010) 7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pobbati AV, Han X, Hung AW, Weiguang S, Huda N, Chen GY, Kang C, Chia CS, Luo X, Hong W, Poulsen A, Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy, Structure 23 (11) (2015) 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mesrouze Y, Bokhovchuk F, Izaac A, Meyerhofer M, Zimmermann C, Fontana P, Schmelzle T, Erdmann D, Furet P, Kallen J, Chene P, Adaptation of the bound intrinsically disordered protein YAP to mutations at the YAP:TEAD interface, Protein Sci. 27 (10) (2018) 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li Z, Zhao B, Wang P, Chen F, Dong ZH, Yang HR, Guan KL, Xu YH, Structural insights into the YAP and TEAD complex, Genes Dev. 24 (3) (2010) 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mesrouze Y, Bokhovchuk F, Meyerhofer M, Fontana P, Zimmermann C, Martin T, Delaunay C, Erdmann D, Schmelzle T, Chene P, Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Crawford JJ, Bronner SM, Zbieg JR, Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review, Expert Opin. Ther. Pat 28 (12) (2018) 867–873. [DOI] [PubMed] [Google Scholar]
- [29].Zhang Z, Lin Z, Zhou Z, Shen HC, Yan SF, Mayweg AV, Xu Z, Qin N, Wong JC, Zhang Z, Rong Y, Fry DC, Hu T, Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP-TEAD protein-protein interaction, ACS Med. Chem. Lett 5 (9) (2014) 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhou Z, Hu TS, Xu ZH, Lin ZH, Zhang ZS, Feng T, Zhu LC, Rong YP, Shen H, Luk J, Zhang XW, Qin N, Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides (vol 29, pg 724, 2015), FASEB J. 31 (4) (2017) 1767–1767. [DOI] [PubMed] [Google Scholar]
- [31].Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, Wang X, Guo T, Li P, Zhao Y, Ji H, Zhang L, Zhou Z, A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer, Cancer Cell 25 (2) (2014) 166–180. [DOI] [PubMed] [Google Scholar]
- [32].Zhang WJ, Gao YJ, Li PX, Shi ZB, Guo T, Li F, Han XK, Feng Y, Zheng C, Wang ZY, Li FM, Chen HQ, Zhou ZC, Zhang L, Ji HB, VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex, Cell Res. 24 (3) (2014) 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brodowska K, Al-Moujahed A, Marmalidou A, Horste MMZ, Cichy J, Miller JW, Gragoudas E, Vavvas DG, The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation, Exp. Eye Res 124 (2014) 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Feng JT, Gou JH, Jia J, Yi T, Cui T, Li ZY, Verteporfin, a suppressor of YAP-TEAD complex, presents promising antitumor properties on ovarian cancer, OncoTargets Ther. 9 (2016) 5371–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wei HL, Wang FH, Wang Y, Li T, Xiu P, Zhong JT, Sun XY, Li J, Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex, Cancer Sci. 108 (3) (2017) 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Santucci M, Vignudelli T, Ferrari S, Mor M, Scalvini L, Bolognesi ML, Uliassi E, Costi MP, The Hippo pathway and YAP/TAZ-TEAD protein-protein interaction as targets for regenerative medicine and cancer treatment, J. Med. Chem 58 (12) (2015) 4857–4873. [DOI] [PubMed] [Google Scholar]
- [37].Kaan HYK, Sim AYL, Tan SKJ, Verma C, Song H, Targeting YAP/TAZ-TEAD protein-protein interactions using fragment-based and computational modeling approaches, PLoS One 12 (6) (2017) eOl78381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li Y, Liu S, Ng EY, Li R, Poulsen A, Hill J, Pobbati AV, Hung AW, Hong W, Keller TH, Kang C, Structural and ligand-binding analysis of the YAP-binding domain of transcription factor TEAD4, Biochem. J 475 (12) (2018) 2043–2055. [DOI] [PubMed] [Google Scholar]
- [39].Gibault F, Coevoet M, Sturbaut M, Farce A, Renault N, Allemand F, Guichou JF, Drucbert AS, Foulon C, Magnez R, Thuru X, Corvaisier M, Huet G, Chavatte P, Melnyk P, Bailly F, Cotelle P, Toward the discovery of a novel class of YAP(-)TEAD interaction inhibitors by virtual screening approach targeting YAP (-)TEAD Protein(-)Protein interface, Cancers 10 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Smith S, Sessions RB, Shoemark D, Williams C, Ebrahimighaei R, McNeill M, Crump MP, McKay T, Harris G, Newby AC, Bond M, Anti-proliferative and anti-migratory effects of a novel YAP-TEAD interaction inhibitor identified using in silico molecular docking, J. Med. Chem. 62 (3) (2019) 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fromond C, Chene L, Soude A, Barth M, Montalbetti C, Broqua P, A rational approach for discovery of inhibitors of YAP-TEAD interaction, Cancer Res. 75 (2015). [Google Scholar]
- [42].Inventiva, New Compounds Inhibitors of the Yap/taz-Tead Interaction and Their Use in the Treatment of Malignant Mesothelioma, (2017) W02017064277.
- [43].Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, Deng H, Pan D, Luo X, Wu X, Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway, Nat. Chem. Biol 12 (4) (2016) 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, Dey A, Hannoush RN, Fairbrother WJ, Cunningham CN, Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling, Structure 24 (1) (2016) 179–186. [DOI] [PubMed] [Google Scholar]
- [45].Mesrouze Y, Meyerhofer M, Bokhovchuk F, Fontana P, Zimmermann C, Martin T, Delaunay C, Izaac A, Kallen J, Schmelzle T, Erdmann D, Chene P, Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ, Protein Sci. 26 (12) (2017) 2399–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].T.G.H. Corporation, TEAD Transcription Factor Autopalmitoylation Inhibitors, (2017) W02017053706.
- [47].Bum-Erdene K, Zhou D, Gonzalez-Gutierrez G, Ghozayel MK, Si Y, Xu D, Shannon HE, Bailey BJ, Corson TW, Pollok KE, Wells CD, Meroueh SO, Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEADYap protein-protein interaction, Cell Chem. Biol 26 (3) (2019) 378–389 e13. [DOI] [PubMed] [Google Scholar]
- [48].Gibault F, Coevoet M, Sturbaut M, Farce A, Renault N, Allemand F, Guichou JF, Drucbert AS, Foulon C, Magnez R, Thuru X, Corvaisier M, Huet G, Chavatte P, Melnyk P, Bailly F, Cotelle P, Toward the discovery of a novel class of YAP-TEAD interaction inhibitors by virtual screening approach targeting YAP-TEAD protein-protein interface, Cancers 10 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhou Z, Hu T, Xu Z, Lin Z, Zhang Z, Feng T, Zhu L, Rong Y, Shen H, Luk JM, Zhang X, Qin N, Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides, FASEB J. 29 (2) (2015) 724–732. [DOI] [PubMed] [Google Scholar]
- [50].Nasir MS, Jolley ME, Fluorescence polarization: an analytical tool for immunoassay and drug discovery, Comb. Chem. High Throughput Screen 2 (4) (1999) 177–190. [PubMed] [Google Scholar]
- [51].Zhu MR, Du DH, Hu JC, Li LC, Liu JQ, Ding H, Kong XQ, Jiang HL, Chen KX, Luo C, Development of a high-throughput fluorescence polarization assay for the discovery of EZH2-EED interaction inhibitors, Acta Pharmacol. Sin 39 (2) (2018) 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Moerke NJ, Fluorescence polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding, Curr. Protoc. Chem. Biol 1 (1) (2009) 1–15. [DOI] [PubMed] [Google Scholar]
- [53].Cheng HC, The power issue: determination of KB or Ki from IC50. A closer look at the Cheng-Prusoff equation, the Schild plot and related power equations, J. Pharmacol. Toxicol. Methods 46 (2) (2001) 61–71. [DOI] [PubMed] [Google Scholar]
- [54].Blanchard N, Richert L, Coassolo P, Lave T, Qualitative and quantitative assessment of drug-drug interaction potential in man, based on Ki, IC50 and inhibitor concentration, Curr. Drug Metabol 5 (2) (2004) 147–156. [DOI] [PubMed] [Google Scholar]
- [55].Cer RZ, Mudunuri U, Stephens R, Lebeda FJ, IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding, Nucleic Acids Res. 37 (2009) W441–W445 Web Server issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang JH, Chung TDY, Oldenburg KR, A simple statistical parameter for use in evaluation and validation of high throughput screening assays, J. Biomol. Screen 4 (2) (1999) 67–73. [DOI] [PubMed] [Google Scholar]
- [57].Hall MD, Yasgar A, Peryea T, Braisted JC, Jadhav A, Simeonov A, Coussens NP, Fluorescence polarization assays in high-throughput screening and drug discovery: a review, Methods Appl. Fluoresc 4 (2) (2016) 022001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Arkin MR, Glicksman MA, Fu H, Havel JJ, Du Y, Inhibition of protein-protein interactions: non-cellular assay formats, in: Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Inglese J, Iversen PW, Kahl SD, Kales SC, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr, Weidner JR, Wildey MJ, Xia M, Xu X (Eds.), Assay Guidance Manual, 2004. Bethesda (MD). [Google Scholar]
- [59].Huang X, Fluorescence polarization competition assay: the range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand, J. Biomol. Screen 8 (1) (2003) 34–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.