

Abstract
To inform novel personalized medicine approaches for race and socioeconomic disparities in head and neck cancer.
We examined germ-line and somatic mutations, immune signatures, and epigenetic alterations linked to neighborhood determinants of health in Black and Non-Latino White (NLW) head and neck cancer patients
Cox proportional hazards revealed that Black HNSCC patients with PAX5 (p=0.06) and PAX1 (p=0.017) promoter methylation had worse survival than NLW patients, after controlling for education, zipcode and TNM stage (n=118). We also found that promoter methylation of PAX1 and PAX5 (n=78), was correlated with neighborhood characteristics at the zip code level (p<0.05). Analyses also showed differences in the frequency of TP53 mutations (n=32) and Tumor Infiltrating Lymphocyte (TIL) counts (n=24), and the presence of a specific C -> A germ-line mutation in JAK3, chr19:17954215 (protein P132T), in Black HNSCC (n=73) patients (p<0.05), when compared to NLW (n=37) patients. TIL counts are associated (p=0.035) with long-term (>5 years), when compared with short-term survival (< 2 years).
We show bio-social determinants of health associated with survival in Black HNSCC patients which, together with racial differences shown in germ-line mutations, somatic mutations and TIL counts, suggests that contextual factors may significantly inform precision oncology services for diverse populations.
Keywords: Bio-social determinants of health, JAK3 germ-line mutation and somatic mutations differences in Black head and neck squamous cell carcinoma (HNSCC) patients, social determinants of health linked to epigenetic changes in HNSCC, racial differences in Tumor Infiltrating Lymphocytes number in HNSCC patients, HNSCC disparities
Introduction
Molecular heterogeneity among patient populations plays an important role in determining cancer prognosis and can enable novel precision medicine applications [1–5]. Precision medicine is a powerful tool to overcome cancer disparities by taking into account. In cancer, solid tumors derive from microscopic, clonal cellular proliferations that come about in progressive stages by the acquisition of somatic alterations[1–3]. Accumulation of somatic mutations is associated with cellular divisions that progressively acquire stem cell characteristics. These characteristics are gained by replicative errors that arise in response to hereditary burden, environmental stressors and random error [6, 7]. Responses to environmental factors correlate closely with the acquisition of DNA methylation alterations and other DNA-based signatures, which may contribute to cancer heterogeneity [8–10]. When heterogeneity is examined, the impact of racial differences in somatic and germline mutations, promoter methylation and immuno-infiltration in cancer is largely unexplored [11]. Most of the genomic sequencing studies have been performed on genomes of European descent [12–14]. Only a handful of studies have reported racial and ethnic differences in the genomic landscape of triple negative breast cancer and head and neck patients [15–18].
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide and clinical outcomes vary throughout racial/ethnic and socioeconomic groups. Out of all of the racial/ethnic groups, during the last 40 years the group who has had the best survival, clinical outcome and earlier stage at diagnosis of HNSCC has been Non-Latino Whites (NLW) [19]. Regardless of race, ethnicity and other socio-demographic characteristics, novel molecular characterization tools and strategies based on the detection of genetic and epigenetic markers offer new hope for improved risk assessment, early cancer detection, and therapeutic intervention, as well as tumor surveillance in cancer for all patients [20]. Contrary to the goal of benefiting all patients, the impact of these strategies in reducing HNSCC survival disparities has been limited by an incomplete understanding of population dynamics in HNSCC heterogeneity, particularly in cancer’s early development stages, and subsequent clinical outcomes among racial/ethnic groups other than White, for whom social determinants continue to play critical roles in late-stage diagnosis, health care access, and worse clinical outcomes [21].
Genomic and epigenomic alterations that have been linked with HNSCC outcomes, such as TP53 mutations [22–27], loss of heterozygosity [28], microsatellite instability [29], and differential DNA methylation [30] are associated with environmental exposures, such as cigarette smoke and alcohol consumption [31]. Recent evidence points towards another exposure with differential survival, namely HPV+ and HPV- status in HNSCC. HPV status is possibly related to different pathway involvement in the initiation and progression of HPV–related disease [32]. Moreover, HPV+ HNSCC tumors mostly occur in the oropharynx, show superior responses to chemotherapy and radiotherapy [33, 34], and are less prevalent in Blacks than in NLW patients [35].
To ascertain the landscape of differences between Black and NLW HNSCC patients we surveyed genomic, epigenomic and immuno-oncology signatures in three separate cohorts: Cohort 1 – Racial Correlates of Genomic and Epigenomic differences; Cohort 2 – Social Correlates of Genomic and Epigenomic differences; Cohort 3 – Racial Correlates of Immuno-oncology differences.
To document frequency differences in HNSCC somatic mutations and epigenetic alterations between Black and NLW, we analyzed exome sequencing data on ~18,000 protein-encoding genes from 32 HNSCC cases, together with genome-wide DNA methylation array and genome-wide mRNA array data on these 32 patients and frequency matched 32 uvulopalatopharyngealplasty (UPPP) controls. The DNA methylation results were validated with genome-wide DNA methylation array and genome-wide mRNA array data from 279 HNSCC patients from the Cancer Genome Atlas (TCGA) project and Quantitative Methylation Specific PCR (QMSP) data in 76 HNSCC patients from Hopkins.
We then examined genome-wide DNA methylation and genome-wide mRNA alterations in a second Discovery cohort of 24 HNSCC patients and frequency matched 27 UPPP controls, 70% of which were Black. To verify the robustness of our findings we used fluorogenic, quantitative methylation specific PCR (qMSP) in a second Validation cohort of 118 HNSCC patients, 55% of which were Black. The qMSP resutls were used to perform social determinants of health analyses at the zipcode level, using publicly available Census data. We also performed targeted exome-sequencing in 16 Black HNSCC patients using a Hotspot Panel, which profiles 50 frequently mutated in human cancer genes. We used Sanger sequencing to confirm the mutation frequency of specific gene alterations. We also used droplet digital PCR to examine racial differences in the immune signatures of HNSCC patients in Discovery Cohort 3 (n=24) (Supplementary Figure 1).
Material and Methods:
Clinical samples
The head and neck cancer specimens collected for this study conform to the criteria we have previously used and to those established the NIH TCGA Biospecimen Selection Process. All samples were obtained following patient consent. The study was approved by Johns Hopkins Hospitals Institutional Review Board, and was performed in accordance with Health Insurance Portability and Accountability Act (HIPAA) guidelines to safeguard Protected Health Information and the US Common Rule. Unique patient identifiers for each sample were created at study entry, allowing blind annotation and tracking of demographic, clinical, and molecular data. Tumor samples were obtained at the time of initial tumor resection. Clinical data elements were carefully collected for each patient using criteria modeled after the currently updated TCGA clinical data requirements for other tumor types. Each tumor sample used in the study was obtained in accordance with the above protocol, and was subjected to pathologic review by a qualified pathologist in order to allow 1) confirmation of the diagnosis of squamous cell carcinoma; 2) confirmation that the representative tumor section contains greater than 80% tumor cell nuclei; and 3) confirmation that the sample is less than 40% necrotic. Forensic microsatellite repeat analysis and HLA genotyping was conducted on all tumor samples as well as the DNA obtained from normal tissue from the same patient in order to confirm matching tumor and normal sample identities. In addition to the above quality control measures, each DNA sample will be tested for efficiency of amplification using a stringent protocol developed in the laboratory. Any sample not meeting these criteria will not be included in the study. Summaries of all quality control analyses of samples used in this project will be available for databases containing the resulting sequencing data. The tumor DNA and RNA samples we will use for molecular studies are selected to be of high purity and quality in order to ensure sensitive detection of genomic alterations; these are critical parameters for the success of such high throughput DNA and RNA sequencing projects. Purity is defined as >80% tumor cells, and quality is defined by isolation of high molecular weight DNA and RNA, assayed by gel electrophoresis, PicoGreen quantification, Bioanalyzer profiles, and by a quantitative functional assay for PCR amplification.
Our Head and Neck Tumor Bank (HAND) stores over 70,000 specimens from over 14,000 patients recruited during the past 20 years. We are fortunate to have available a large collection of such high quality frozen sections of surgical samples of HNSCC, linked with clinical information. These samples are accompanied by paired normal tissue (lymphocyte DNA), which is critical for exome sequencing studies. This sample collection has been maintained and curated by Dr. Wayne Koch, Department of Otolaryngology-Head and Neck Surgery, Johns Hopkins University School of Medicine, with support from the Johns Hopkins HNSCC SPORE grant.
Patient characteristics
Patient cohorts for the different phases of this project were drawn from 248 Black and 248 NLW HNSCC patients treated in Johns Hopkins, for whom we had access to their medical record and had obtained consent to store tumor tissue. Their clinical and socio-demographic characteristics are listed in Table 1. We frequency matched the samples on sex (70.2% of Black and 80.6% of NLW are males); age (the median age for Black is 59.6 years and for NLW is 58.4 years); and site (25.8% of Black and 25% of NLW have oral cancer; 29% of Black and 32.3% of NLW have oropharyngeal cancer; 7.3% of Black and 8.1% of NLW have hypopharyngeal cancer; and 34.7% of Black and 32.3% of NLW patients have larynx cancer). A subset of oropharyngeal tumors was HPV+: 21% of Black and 49% of NLW. Furthermore, the were no differences in stage (10.5% of Black and 12.9% of NLW have Stage 1; 8.1% of Black and 11.3% of NLW have Stage 2; 13.7% of Black and 18.5% of NLW are Stage 3; and 57.3% of Black and 43.5% of NLW are Stage 4) or ethanol use (41.1% of Black and 31.5% of NLW were Heavy drinkers; 20.2% of Black and 25.8% of NLW were occasional drinkers; and 21.8% of Black and 14.5% of NLW did not drink). There were more active smokers among Black (71%) than NLW (50.8%). Consequently, there were more former smokers (NLW-17.7%; Black-9.7%) and non-smokers, (NLW-16.1%; Black- 4.8%), as listed in Table 1 (p=0.02). These are most likely a reflection of the racial differences and composition in our HNSCC tumor bank, by tumor subsite. We adjusted for smoking status as a possible confounder in our comparisons.
Table 1.
Clinical and socio-demographic characteristics
| Total | Black | Non-Latino Whites | |
|---|---|---|---|
| Total n | 496 | 248 | 248 |
| Sex n (%) | |||
| Female | 122 | 74 (29.8) | 48 (19.4) |
| Male | 374 | 174 (70.2) | 200 (80.6) |
| Age n (%) | |||
| Median (y) | 59.6 | 58.4 | |
| <40y | 24 | 12 (4.8) | 12 (4.8) |
| 40–55y | 152 | 74 (29.8) | 78 (31.5) |
| >55y | 318 | 160 (64.5) | 158 (63.7) |
| Site n (%) | |||
| Oral Cavity (%) | 124 (25) | 64 (25.8) | 60 (25) |
| Oropharynx (%) | 152 (30.6) | 72 (29) | 80 (32.3) |
| Nasopharynx (%) | 6 (1.2) | 2 (0.8) | 4 (1.6) |
| Hypopharynx (%) | 38 (7.7) | 18 (7.3) | 20 (8.1) |
| Larynx (%) | 166 (33.5) | 86 (34.7) | 80 (32.3) |
| Unknown (%) | 8 (1.6) | 6 (2.4) | 2 (0.8) |
| Stage n (%) | |||
| 1 | 58 (11.7) | 26 (10.5) | 32 (12.9) |
| 2 | 48 (9.7) | 20 (8.1) | 28 (11.3) |
| 3 | 80 (16.1) | 34 (13.7) | 46 (18.5) |
| 4 | 250 (50.4) | 142 (57.3) | 108 (43.5) |
| unknown | 60 (12.1) | 26 (10.5) | 34 (13.7) |
| Smoking n (%) | |||
| Active | 302 (60.9) | 176 (71) | 126 (50.8) |
| Past | 68 (13.7) | 24 (9.7) | 44 (17.7) |
| Non-smokers | 52 (10.5) | 12 (4.8) | 40 (16.1) |
| unknown | 74 (14.9) | 36 (14.5) | 38 (15.3) |
| ETOH n (%) | |||
| Heavy | 180 (36.3) | 102 (41.1) | 78 (31.5) |
| Occasional | 114 (23) | 50 (20.2) | 64 (25.8) |
| None | 90 (18.1) | 54 (21.8) | 36 (14.5) |
| unknown | 102 (2.6) | 42 (16.9) | 70 (28.2) |
Gene-set enrichment analysis.
Gene set enrichment analysis of functional themes was performed to capture biological processes over-represented in the various conditions under investigation using Analysis of Functional Annotation. A chi-squared test was applied to test whether each Functional Gene Set (FGS) was over-represented in any of the gene list associated with any of the investigated contrasts/conditions (e.g. gene associated with hypermethylated promoters in HNSCC). In the present study, individual, non-redundant genes, as annotated in the NCBI Entrez gene database (R/Bioconductor package org.Hs.eg.db version 2.4.6) were used as the total gene space, and contingency tables were used to identify gene sets over-represented in the investigated conditions.
Correction for multiple hypothesis testing was obtained separately for each FGS collection, by applying the Benjamini and Hochberg method[69–71] as implemented in the multtest R/Bioconductor package. Overall, this approach is analogous to Gene Set Enrichment Analysis (GSEA)[72], and has already been successfully applied in other studies[73, 74]. The heatmaps color bar represents the negative log10 False Discovery Rate (FDR). For each gene set collection, the sets for which at least one condition showed FDR < 0.01 were reported. The top 150 conditions were reported when too many gene sets where retrieved.
Social determinants of health
We used qMSP to compare the differences in promoter DNA methylation of four genes, NID2, EDNRB, PAX1, and PAX5, identified as biomarkers of HNSCC risk in prior publications. The unbiased discovery of promoter methylation biomarkers of HNSCC using genome-wide DNA methylation arrays and next-generation sequencing, as well the design and optimization of qMSP primers and probes to validate these findings in separate cohorts, is described in previous publications from our lab [75, 76].
Neighborhood-level variables
Median household income, home ownership, home vacancy and insurance coverage rates using corresponding (patient) 2010 zip code tabulation areas (ZCTAs) were provided by the US Census Bureau’s American Community Survey. ZCTAs are generalized areal representations of United States Postal Service (USPS) ZIP Code service areas [77, 78]. Based on this zip code data, the Census Bureau aggregates ZCTAs from addresses contained within each block. This aggregation of data allows a point-based data set (addresses) to be converted into an area feature dataset ZCTAs [79].
Statistical Analysis
Three methods were implemented to assess the relationship between molecular alterations, clinical factors and neighborhood zipcode-level interactions with HNSCC survival. First, we estimated HNSCC survival linked co-variables using the Kaplan-Meier method. Second, we used Cox proportional hazards models to conduct univariate and bivariate linear and logistic regression models to determine if there was a correlation between study variables and HNSCC survival was used to analyze the effects of co-variables on survival. Third, a geographically-weighted regression analysis was applied to evaluate if this correlation occurred after weighing for local neighbors.
Kaplan-Meier method
The log-rank test was used to test the significance of the association between survival and the following categorical variables: promoter methylation of EDNRB, NID2, PAX5, PAX1 and tumor stage; and the following determinants of health at the zip code level: high school diploma, median family income, health insurance, home ownership rate and home vacancy rate. Promoter methylation of EDNRB, NID2, PAX5, PAX1 categorical variables were recorded as follows: 1= Methylated; 2= Unmethylated. Data for high school diploma, median family income, health insurance, home ownership rate and home vacancy rates categorical variables are as follows: 1= Above the Median; 2= Below the Median.
Cox proportional hazard regression
Cox proportional hazard regression was used to test the significance of the association between survival and the following continuous variables: promoter methylation of EDNRB, NID2, PAX5, PAX1 and the following determinants of health at the zip code level: high school diploma, median family income, health insurance, home ownership rate and home vacancy rate. We then used Cox proportional hazard models for univariate and multivariate analyses to estimate hazard ratios (HRs) with 95% confidence intervals (CIs).
Multiple regression models were developed to describe the relationship between HNSCC survival and differential promoter methylation, clinical factors and neighborhood-level interactions within the entire study population and among racial strata. All statistical tests were two-sided, and p-values <0.05 were considered as statistically significant. Analyses were performed using STATA version 13.
Geographically-weighted regression (GWR) analyses
To assess spatial relationships of racial/ethnic disparities between EDNRB, NID2, PAX5, PAX1 promoter methylation, primary site, median family income, health insurance rate, home ownership rate and home vacancy rate, we conducted a geographically weighted regression (GWR). Due to the substantial geographic variation in the United States, GWR is performed to identifying spatial heterogeneities in regression models of geo-referenced data. Local regression coefficients and associated statistics (i.e. proportion of variance explained, correlation coefficients) can then be mapped to visualize how the explanatory power of covariates changes spatially.
Spatial autocorrelation was tested for and found within the residuals from linear regressions using both a local and a global Moran’s I. The global Moran’s I is an analysis on whether or not variables seem to be spatially correlated (i.e. high values are next to high values) or not spatially correlated (i.e. values don’t appear to affect one another). The closer the value is to −1/n-1 (−1/13) the more random the data. The closer to 1 the more correlated, the closer to −1 the more negatively correlated. Closeness was determined by Euclidean distance from the center of each zip code using threshold distance of 0.036529. To evaluate the strength of the correlation of racial/ethnic disparities between EDNRB, NID2, PAX5, PAX1 promoter methylation, primary site, median family income, health insurance rate, home ownership rate and home vacancy rate accounting for the local geographical weight around a focal point, geographically weighted regression models were constructed to weigh the neighborhoods using ArcGIS (Version 10.1). However, logistic regression models with geographical weighting could not converge to estimate model coefficients using the maximum likelihood approach.
Thus, only geographically-weighted linear regression was conducted in this study. The weighting scheme utilized in this study was the Gaussian kernel function [30]. The bandwidth or number of neighbors used for each local estimation, is perhaps the most important parameter for GWR and the optimal bandwidth was determined by minimizing Akaike Information Criterion (AICc). The Gaussian kernel bandwidth varies across space with the decay function of weighting neighborhoods. The kernel type was set up as adaptive to account for the density of spatial features and the optimal bandwidth was determined by minimizing Akaike Information Criterion (AICc) [31]. The parameter estimates were mapped in ArcInfo (Version 9.3).
Immuno-oncology determinants of survival
The QuanTILfy assay droplet digital PCR (ddPCR) assay quantifies T-lymphocytes using a multiplex ddPCR system, which amplifies rearranged TCRβ loci from genomic DNA using 45 forward primers, each specific to one or multiple functional TCR Vβ segments, and 13 reverse primers, each specific to a TCR Jβ segment. The multiplex reaction also includes in each well one of a series of 35 minor groove binder 6-carboxyfluorescein (FAM) TaqMan probes complementary to 52 different Vβ gene segments, and a VIC probe complementary to ribonuclease (RNase) P protein subunit p30 (RPP30) per well, which serves as a reference gene to permit normalized quantification. To identify clonal expansion of TCR genes, ddPCR assays were designed for eight Vβ gene segment subgroups, each of which contains the forward primers and TaqMan probes specific to a non-overlapping subset of Vβ gene segments. Each Vβ gene subgroup was combined with all 13 Jβ gene segment primers, as well as the RPP30 primers and probe, effectively creating a multiplex ddPCR assay for TCRβ rearrangement detection. Each of the 52 possible TCRβ gene segments is measured once in exactly one of the wells. Therefore, the sum of counts from all wells gives a precise digital quantification of the total number of rearranged TCRβs in the sample.
Single Nucleotide Variant in JAK3
Discovery with Next Generation Sequencing
We used the Ion AmpliSeq™Cancer Hotspot Panel v2 to profile 50 frequently mutated in human cancer genes in a discovery cohort of HNSCC Black patients. Libraries for the discovery cohort were generated using the Ion AmpliSeq Library kit 2.0 according to the manufacturer’s instructions (Life Technologies, Carlsbad, USA). Included in this panel were primers for 207 amplicons covering 2800 Catalog of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) of 50 genes with known cancer associations (ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, JAK2, JAK3, IDH2, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53 and VHL). 10ng DNA from the tumor samples was used as the template to prepare the library. Amplified libraries were quantified using the Qubit 2.0 Fluorometer and the High Sensitivity Qubit Assay Kit (Life Technologies). Amplified libraries were assessed for quality (size and concentration) using the Agilent 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA) following the Bioanalyzer standard protocol. The AmpliSeq libraries were clonally amplified on to Ion Sphere Particles (ISPs) using emulsion PCR following standard Ion Torrent protocols. ISP preparation was performed using the automated Ion Torrent OneTouch2 system following the manufacturer’s protocol (MAN0007220 Revision 4.0). The Qubit Fluorometer was used to assess ISP quality after ISP preparation but before ISP enrichment. Up to eight specimens were barcoded with Ion Xpress Barcode Adapters (Life Technologies), pooled, and run on a single Ion 318 chip. This includes multiple patient samples and one control, which we rotate among water, normal, and a mix of positive control cell lines.
Validation with Sanger sequencing
The results were validated with Sanger sequencing in a separate cohort of 76 HNSCC patients. The region surrounding the P132T amino-acid site of JAK3 were sequenced by sanger sequencing. Amplification of JAK3 region was performed by polymerase chain reaction (PCR) performed in 12.5 μl reactions containing 1× PCR Buffer (67 mM Tris- HCl, pH 8.8, 6.7 mM MgCl2, 16.6 mM NH4SO4, 10 mM 2-mercaptoethanol), 10 mM dNTPs (Invitrogen, San Diego, CA), 10 μM forward (5’-GTAAAACGACGGCCAGTCTGTGAGGCCTCCGCAGA-3’ and 10 μM reverse (5’-CAGGAAACAGCTATGACCGATTGCATGCCAGTCCTCA-3’) primers, 1.25 U Platinum Taq (Invitrogen, San Diego, CA) and 10 ng DNA. Reactions were performed in 364-well plates using ABI 7900 thermocycler (Applied Biosystems, Foster City, CA) as follows: 1 cycle of 96°C for 30 seconds; 40 cycles of 95°C for 30 sec, 66°C for 60 sec, 72°C for 60 sec;1 cycle of 72°C for 5 min. Templates have been purified by QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). Sanger sequencing was performed by GENEWIZ using ABI 3730xl DNA analyzer for capillary electrophoresis and fluorescent dye terminator detection.
Quantitative real-time reverse transcription PCR
HNSCC RNA from a subsample of Black patients were assessed for JAK3 and GAPDH expression levels using quantitative real-time reverse transcription (RT)-PCR (TaqMan). Reverse transcription was performed with random hexamer primers and Superscript II Reverse Transcriptase (Invitrogen Corp.) according to manufacturer’s instructions. Quantitative RT-PCR was then performed on the Applied Biosystems 7900 Sequence Detection Instrument (Applied Biosystems) using TaqMan expression assays (Life Technologies).
Results
HNSCC disparities in Somatic mutations and DNA methylation
We observed disparities in the frequency of mutated and methylated events in the PAX, NOTCH1 and TP53 pathways (Supplementary Table 1). We observed higher frequencies of TP53 and NOTCH1 mutations (NOTCH1mut) in Black HNSCC patients and no differences in PAX1 or PAX5 methylation across all tumor sites, and interestingly, these patterns differed when we stratified by anatomic subsite. Outside the oropharynx, Black patients have a higher frequency of PAX5 methylation (PAX5met) and TP53 mutations (p53mut) than NLW, and no NOTCH1mut. Inside the oropharynx, Black patients have a lower frequency of PAX5met when compared with NLW. Inside the oropharynx, NLW also had a higher frequency of combined p53mut or PAX5met, while Blacks had a higher frequency of combined NOTCH1mut or PAX1 methylation (PAX1met). Complex genetic and epigenetic interactions between PAX, NOTCH, and p53 pathways may be differentially driving HNSCC initiation and progression events in Black and NLW HNSCC patients.
Patients showed obvious differences in the genetic landscapes of HPV-associated and HPV-negative HNSCC. In the HPV-associated tumors compared to those tumors not related to HPV, far fewer genes were mutated per tumor (4.8 ± 3 versus 20.6 ± 16.7, p<0.05, Welch two sample t-test). These data are consistent with previous results on HNSCC, as well as on HPV-associated cervical cancers (10–12). More cancer-related mutations were identified in tumors from patients with a history of tobacco use compared to those from patients who did not use tobacco (21.6 ± 17.8 versus 9.5 ± 6.5, p<0.05, Welch two sample t-test).
We also observed gene-specific gain and loss of methylation events correlated with etiologic factors. PAX1 promoter is not methylated in HPV positive tumors, whereas PAX1 gains methylation in HPV negative tumors PAX1 promoter methylation is also observed in most patients with a history of tobacco exposure (71%), while only 33% of patients without tobacco exposure history exhibited methylation in the promoter region of PAX1. Most HPV negative tumors (83%) show promoter methylation of PAX5 compared to only 25% of HPV positive tumors. On the contrary, tumors from patients with a history of tobacco exposure (57%) had similar frequency of promoter methylation in PAX5 when compared with tumors from patients with no history of tobacco (67%). We also observed concurrent genomic and epigenomic associations with viral and tobacco exposures; patients with TP53 mutations also had PAX1 promoter methylation, a history of tobacco exposure, and were HPV negative.
Somatic mutation profile in Black compared to NLW HNSCC patients
We examined the mutational profile in a separate cohort of Black HNSCC patients to verify the findings observed in the Discovery cohort. Black HNSCC patients have different frequencies of somatic mutations in the 50 genes most commonly mutated in human cancers when compared to NLW HNSCC patients, as well as across anatomic sites. Black larynx cancer patients had a higher frequency of TP53, PIK3CA, JAK3, KIT, APC and MET somatic mutations. NOTCH1 had a higher frequency of somatic mutations in Black oropharyngeal cancer patients and CDKN2A had a higher frequency of somatic mutations in Black oral cancer patients (Supplementary Table 2).
Germ-line mutation in JAK3 in Black HNSCC patients
In addition to somatic mutations, we found a germline C - > A mutation in JAK3, chr19:17954215 (protein P132T) together with a paired polymorphism (at position chr19:17954149) in 25% of the samples in Discovery cohort, of which all happened to be males with larynx cancer. In an additional cohort of 76 HNSCC patients (39 Black and 37 NLW), we validated this finding with Sanger sequencing, where we found this same JAK3 mutation in 8% of the Black patients while none of the NLW patients had it. Black males with larynx cancer (67%) and Black females with cancer of the oropharynx (33%) had this JAK3 germ-line mutation. This is a known SNP (rs3212723) present in COSMIC (COSM34216) and the dbSNP databases. This SNP is present only in people from Africa or African descent in the 1000 genome project: Masai (MKK NA 21737), American from the Southwest (ASW NA 19834, NA19701) and Yoruba (YRI NA 18502, NA 18504, NA 19238). Further sequencing of the cohorts showed that Normal Black patients had the mutation at a higher proportion than Black HNSCC patients.
Gene expression signatures in Black HNSCC patients
We then examined genome-wide expression and DNA methylation array data from 56 HNSCC patients and 59 normal epithelium controls. Analysis in Black HNSCC patients had gene-set enrichment analysis show that promoter methylation correlates with downregulation in several genes (Figure 1), many of which are in immune related pathways: Antigen Processing; Chemokine Signaling; Citokine-Citokine Receptor interactions; Natural Killer Cell Cytotoxicity. The top altered pathways revealed by Gene Ontology (GO) clustering involved the loss of IRF4, IRF8, PAX5, PAX1, CXCL12, EBF1 and PARP15, as can be seen in Supplementary Table 3.
Figure 1.
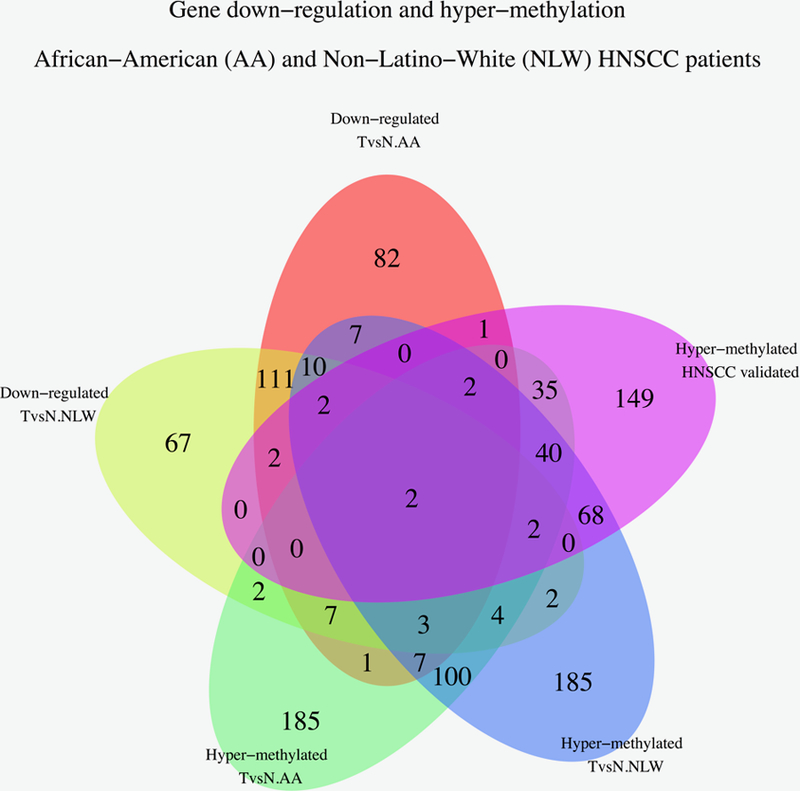
Venn diagrams that show the intersection of genes that in 49 AA and NLW HNSCC patients compared to 51 UPPP controls.
Social determinants of HNSCC survival disparities: Neighborhood effects that may impact genetic and epigenetic changes.
The overall majority of the HNSCC patients in our cohort reside in the state of Maryland (76.1%). Most of the remaining patients reside in the following states: The District of Columbia (DC) 1.7%, Delaware (DE) 8.5%, Pennsylvania (PA) 4.3%, Virginia (VA) 4.2%, and West Virginia (WVA) 4.2%. The proportion of patients residing in Maryland (MD) differed by race (p= 0.002): Black (61.8%) and NLW (38.2%). Most of the patients residing in the state of MD (51.7%) resided outside of Baltimore City, and most were NLW patients (73.5%). The majority of Black patients with MD residency lived in Baltimore City (62.3%).
There were notable racial differences in zipcode-level neighborhood factors across the six states. Median family income differed significantly between Black and NLW patients (p < 0.0001). NLW patients had a 38.2% higher zipcode-level median family income ($65,005) than Black patients ($47,040). The median zipcode-level health insurance rate also differed significantly between Black (87.8%) and NLW patients (89.8%) (P=0.001). The median zipcode-level homeownership rate was 5% lower for Black (86.9%) compared to NLW patients (91.9%) (P=0.02). NLW patients (8.1%) had a median home vacancy rate, which was 61.7% higher among Black (13.1%) than NLW (P= 0.032).
Choropleth thematic maps were created for every Baltimore City zip code in which our HNSCC patients resided, using the layer property in ArcGIS. Choropleth maps aggregate data in the form of counts across a defined geographic space (zip codes in this instance), indicating differences by shaded or colored areas [36]. The resulting Choropleth thematic maps geographically depicted the spatial distribution of PAX5 methylation (in quartiles) and home vacancy rates at the zip code level, for all the HNSCC patients in our cohort who reside in Baltimore. Darker colors represent a higher quartile PAX5 promoter methylation in the PAX5 methylation map (Figure 2A), or a higher percentage of home vacancies per zip code in the home vacancy map (Figure 2B). Choropleth thematic maps were labeled with zip-code level information for two zipcodes that represent opposite ends of the socio-economic spectrum in Baltimore: Roland Park (21210) and Madison/East End (21205). The residents of Roland Park have a median annual income of $90,492 (the highest in Baltimore City), a 3.4% unemployment rate, and a mean life expectancy of 83.1 years life expectancy rate. In contrast, the residents of Madison/East have a median annual income of $30,389 (in the lowest quartile), a 14.4% unemployment rate, and a mean life expectancy of 64.8 years life expectancy rate. HNSCC patients in our cohort who lived in the zip code with the highest median annual income had the lowest levels of PAX 5 methylation and home vacancy rates, after adjusting for race. Conversely, HNSCC patients in our cohort who lived in the zip code with some of the lowest median annual income, had the highest levels of PAX 5 methylation and home vacancy rates, after adjusting for race. Home vacancy is defined as the proportion of homes that are not occupied in a specific area and is a known social determinant of health of the build environment. This environmental measure is correlated with undesirable health outcomes. This information suggests that there may be exposures to multiple environmental hazards and social stressors at the zip-code level, which may contribute to differences in HNSC risk, indolence and aggressiveness. Home vacancy rates and PAX5 promoter methylation may be part of the complex mechanism by which the exposome becomes biology, which is reflected in the different patterns of somatic mutation and promoter methylation changes between Black and NLW HNSCC patients in our cohort.
Figure 2.
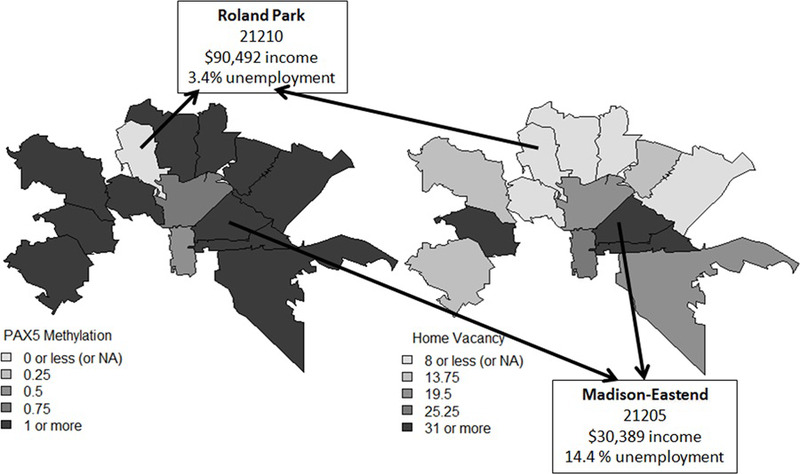
Choropleth maps geographically depicted the spatial distribution of selected variables at the zipcode level for all the HNSCC patients in our cohort who reside in Baltimore: (2A) PAX5 promoter methylation map (in quartiles); and (2B) home vacancy rates map (zip code level). Darker colors represent a higher quartile in the PAX5 promoter methylation map, or a higher percentage of home vacancies per zip code in the home vacancy map. Choropleth thematic maps were labeled with zip-code level information for two zipcodes that represent opposite ends of the socio-economic spectrum in Baltimore: Roland Park (21210) and Madison/East End (21205). The residents of Roland Park have a median annual income of $90,492 (the highest in Baltimore City), while the residents of Madison/ East End have a median annual income of $30,389.
We then examined if survival disparities in HNSCC are linked to neighborhood determinants of health. Using data from the US Census Bureau’s American Community Survey, we examined the relationships of social determinant of health variables at the neighborhood-level and promoter methylation of PAX5, PAX1, EDNRB and NID2 in the HNSCC patients residing in Baltimore. We examined five domains at the zip-code level that have previously been identified as social determinants of health: educational attainment (percentage of High school diplomas), median household income, home ownership, vacancy status and insurance coverage. Pooled logistic regression analysis showed an association between percentage of residents with High School Diploma at the zip-code level (HD) and PAX5 (p=0.09) and PAX1 (p=0.05) promoter methylation (Figure 3A). Survival analyses revealed that patients with PAX5 (p= 0.001) or NID2 (p=0.05) promoter methylation had a worse outcome than those without it. Cox proportional hazards regression multivariable analysis, revealed that Black patients with PAX5 (p=0.06) and PAX1 (p=0.017) methylation had worse survival than NLW, after controlling for HD, zipcode and TNM stage. (Figure 3B).
Figure 3.
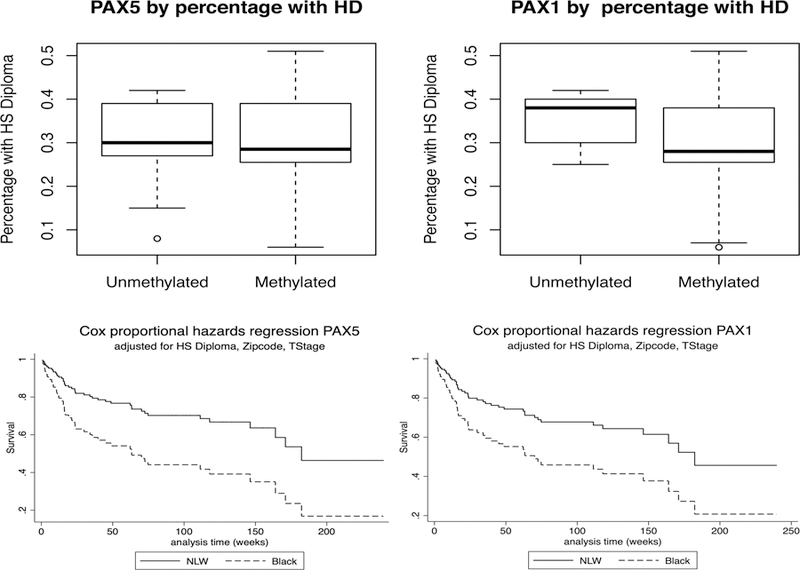
A) Pooled logistic regression analysis showed a marginally significant association between the percentage of residents with High School Diploma at the zip-code level (HD) with PAX5 (p=0.09) and PAX1 (p=0.05) promoter methylation. 3B) Multivariable Cox proportional hazards regression analysis shows that AA patients with PAX5 (p=0.06) and PAX1 (p=0.017) promoter methylation had worse survival than NLW, after controlling for HD, zipcode and TNM stage.
In subsequent multivariate Cox regression analyses of TCGA HNSCC data (n=279) we confirmed that patients with PAX5 methylation had worse survival than those without it (HR =1.63), after adjusting for smoking, surgical margin and p53 mutations. (p=0.03). This is a very similar hazard ratio (HR) that for current or previous smoking (HR=1.88; p=0.03) and positive surgical margins (HR=1.76; p=0.02). We also found that combined somatic TP53 mutations and PAX5 promoter methylation are linked to worse outcomes when compared to patients with either alteration alone, after adjusting for smoking, and surgical margin (HR=2.16; p<0.001) (Figure 4). This is a slightly higher ratio than for p53 mutation alone, after adjusting for smoking, surgical margin and PAX5 (HR=2.06; p=0.004) (Supplementary Table 4).
Figure 4.
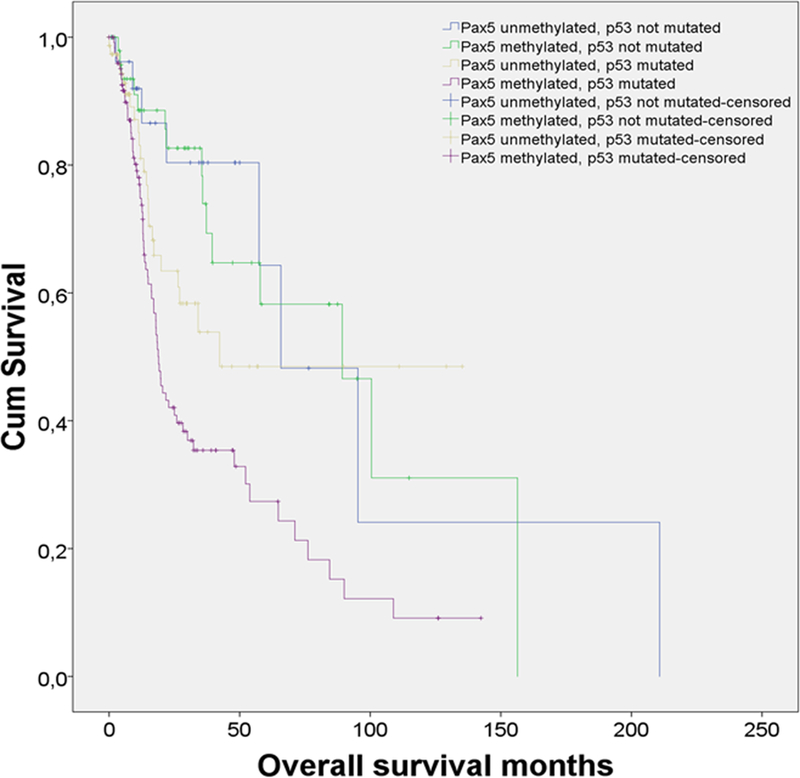
Results from a Kaplan Meier analysis of 279 HNSCC patients from TCGA. (A). Patients with promoter methylation of PAX5 have worse outcomes than patients without PAX5 methylation (p=0.026). (B) Patients with combined somatic TP53 mutations and PAX5 promoter methylation have poorer outcomes when compared to patients with TP53 mutations, who do not have PAX5 methylation (p<0.012).
Immuno-oncology determinants of survival in HNSCC.
When we used the QuanTILfy digital PCR assay[37] to examine TILs in HNSCC tumor tissue, the number of TILS was found to be increased in long-term survivors (>5yrs) compared to short-term survivors (<5yrs) (p <0.05) (Figure 5A). We also compared TIL counts by site and observed more in larynx cancers (Figure 5B). Cancers had larger TIL counts and clonality in Black patients, compared to NLWs (Figure 5C).
Figure 5.

Tumor Infiltrating lymphocytes (TILs) as immuno-oncology determinants in HNSCC, quantified with the QuanTILfy assay, a droplet digital PCR assay. Larger numbers of TILs are associated with survival (5A), larynx cancer (5B) and Black race (5C).
Bio-social markers for Personalized Medicine workflows
A Precision Medicine Index that integrates Contextual, Bio-Psychosocial and Molecular Data can be a useful tool for clinicians, patients, public health practitioners, behavioral therapists and policy makers. DNA methylation, the best-understood epigenomic mark, is a molecular marker that can summarize multiple external and internal factors that modulate health and disease. It is a tangible measurement of how the environment, social forces, emotions and psychological processes modulate gene expression, and eventually impinge upon normal and disrupted, intracellular and extracellular processes, via molecular pathways that are slowly being mapped and validated. In Figure 6A we depict how contextual, demographic, lifestyle and molecular markers can be useful markers for a variety of HNSCC endpoints: a) Molecular markers of clean surgical margins; Biosocial markers of outcome disparities; and Precision Medicine markers of therapeutic response. These markers can also help us to create a better biological understanding of the complex relationships and interactions between different biological systems and functions. For instance, Figure 6B shows that PAX5 methylation levels inversely correlate (r=−0.83) with TIL counts in HNSCC patients. These data is consistent with both, the inverse association between PAX5 methylation and survival and the direct association between TILS and survival we have observed. We have also observed that the HNSCC patients in our cohort who lived in the zip code with the lowest median annual income had the highest levels of PAX 5 methylation and home vacancy rates, after adjusting for race. Together these data suggests that PAX5 methylation may be a bio-social marker, linked to both adverse social determinants of health and poor survival outcomes in head and neck cancer. Figure 6C shows that JAK3 expression in Black HNSCC patients differs by anatomic location. Figure 6D shows that Notch1 mutations and PAX1 methylation levels also differ by anatomic location and race in HNSCC patients. Together these data suggests that Black HNSCC patients have different methylation and mutation profiles than NLW HNSCC, which may underlie some of the unexplained survival disparities in HNSCC. Notch1 mutations are higher in Black HNSCC, but they are only observed in the oropharynx, where Black HNSCC patients also show a higher frequency of PAX1 methylation.
Figure 6.

A. Molecular markers, combined with external and internal environment variables, can be used as biosocial markers of head and neck cancer outcome disparities, head and neck cancer markers and precision medicine markers for head and neck cancer patients. Figure 6B shows that PAX5 methylation levels inversely correlate (r=−0.83) with TIL counts in HNSCC patients. Figure 6C shows that JAK3 expression in Black HNSCC patients differs by anatomic location. Figure 6D shows that Notch1 mutations and PAX1 methylation levels also differ by anatomic location and race in HNSCC patients.
Discussion
This is the first study to examine molecular differences in NLW and Black HNSCC patients. We performed genome-wide bioinformatics analyses and validated the frequency of selected somatic mutations, germline mutations, promoter methylation, gene expression and TILs using conventional and droplet PCR primers and probes. We also examined the association of promoter methylation with well-understood social determinants of health, identified as neighborhood characteristics at the zipcode level. To our knowledge this is the most comprehensive characterization of molecular disparities in any tumor type.
Genomic and epigenomic inactivation of tumor suppressor genes may partly explain survival disparities in HNSCC [36, 38–40]. We show genetic, epigenetic and Tumor Infiltrating Lymphocytes (TILs) frequency differences between Black and NLW HNSCC patients. Black larynx cancer patients had a higher frequency of TP53, PIK3CA, JAK3, KIT, APC and MET somatic mutations. NOTCH1 had a higher frequency of somatic mutations in Black oropharyngeal cancer patients and CDKN2A had a higher frequency of somatic mutations in Black oral cancer patients. We also found racial differences in the frequency of a germline C - > A mutation in JAK3, chr19:17954215 (protein P132T) in larynx cancer. This is the first germline mutation associated to racial differences in HNSCC.
JAKs (or Janus kinases) are a family of tyrosine kinases that involved in cytokine receptor-mediated intracellular signal transduction. Upon receptor activation JAKs phosphorylate the transcription factors known as STATs and initiate the JAK-STAT signaling pathway. Four JAK family members have been identified (JAK1, JAK2, JAK3 and Tyk2), which share a similar protein domain structure: a kinase domain, a regulatory pseudo -kinase domain, a SH2 domain and a FERM domain. The FERM domain of JAK family members mediates the association of JAK with other enzymes and cytokine receptors.
JAK3 associates with the IL-2 receptor gamma-chain. It is predominantly expressed in immune cells and transduces a signal in response to its activation via tyrosine phosphorylation by interleukin receptors. Mutations in this gene are associated with autosomal SCID (severe combined immunodeficiency disease). JAK3 is a non-receptor tyrosine kinase involved in various processes such as cell growth, development, or differentiation. JAK3 mediates essential signaling events in both innate and adaptive immunity and plays a crucial role in hematopoiesis during T-cell development. In the cytoplasm, JAK3 plays a pivotal role in signal transduction via its association with type I receptors sharing the common subunit gamma such as IL2R, IL4R, IL7R, IL9R, IL15R and IL21R. Following ligand binding to cell surface receptors, JAK3 phosphorylates specific tyrosine residues on the cytoplasmic tails of the receptor, creating docking sites for STATs proteins. Subsequently, JAK3 phosphorylates the STATs proteins once they are recruited to the receptor. Phosphorylated STATs then form homodimer or heterodimers and translocate to the nucleus to activate gene transcription. For example, upon IL2R activation by IL2, JAK3 molecules bind to IL2R beta (IL2RB) and gamma chain (IL2RG) subunits inducing the tyrosine phosphorylation of both receptor subunits on their cytoplasmic domain. Then, STAT5A AND STAT5B are recruited, phosphorylated and activated by JAK3. Once activated, dimerized STAT5 translocates to the nucleus and promotes the transcription of specific target genes in a cytokine-specific fashion.
Survival analyses revealed that patients with PAX5 (p= 0.001) or NID2 (p=0.05) promoter methylation had a worse outcome than those without it. We also found an association of PAX5 and PAX1 promoter methylation with social determinants of health at the zipcode level. Cox proportional hazards regression multivariable analysis, revealed that Black HNSCC patients with PAX5 (p=0.06) and PAX1 (p=0.017) methylation had worse survival than NLW, after controlling for HD, zipcode and TNM stage.
PAX5 maintains cellular identity by repressing gene expression throughout B cell differentiation [41–43]. Dysregulated expression of PAX5 is involved in differentiation block [44], somatic hypermutation and immunoglobulin heavy chain class switch recombination [45], leukemogenesis [46], and positive regulation of c-Met transcription[47]. PAX5 association to different tumor types suggests it has diagnostic or prognostic utility in lung cancer[48], Hodgkin lymphoma[49], acute lymphocytic leukemia[50], breast cancer[50], oral cancer [51, 52], gastric cancer [53], head and neck cancer [54, 55], and esophageal cancer [55]. A recent study demonstrated that the lack of expression of PAX5 in lymphoid neoplasms is associated with promoter hypermethylation, leading to PAX5 silencing in cases characterized by poor clinical outcome [56]. Aberrant PAX5 promoter methylation is also associated with HNSCC [57]. PAX5 promoter methylation is also significantly associated with poor survival in gastric cancer. PAX5 can induce cell apoptosis through direct upregulation of TP53, impacting downstream targets in gastric cancer [58]. When expressed, PAX5 binds to the TP53 promoter, inducing TP53 expression, and consequently suppressing cell proliferation. But when PAX5 is methylated, this tumor suppressor function is abrogated. Spatial analyses identified survival differences and PAX5 methylation in Black and NLW HNSCC patients, after adjusting for socioeconomic characteristics at the zipcode level.
HNSCC disparities are partly mediated by differences in access to care, stage at diagnosis, insurance status, attitudes of health providers, as well as human papilloma virus (HPV) infection status [59, 60] and measures of economic advantage [61]. Variations in tumor-associated immune responses between Black and NLWs have also been implicated in cancer health disparities [62–64]. Molecular alterations linked to HNSCC survival, such as TP53 mutations[65], loss of heterozygosity[28], microsatellite instability[29], and differential DNA methylation[30] have been associated to HNSCC main risks factors: tobacco smoking, alcohol consumption and HPV status [31]. Most HPV+ tumors occur in the oropharynx, show superior responses to chemotherapy and radiotherapy[32–34], and are less prevalent in Black HNSCC patients [36, 61]. They are also characterized by Tumor Infiltrating Lymphocytes (TILs) of the stroma and tumor nests, suggesting HPV+ HNSCC tumors do not evade immune surveillance [66]. The results we obtained with the QuanTILfy assay support the notion that disparities in HNSCC may be related to differences in TIL’s quantity and clonality.
Our results suggest a complex interrelationship between molecular, clinical and social factors with racial differences in HNSCC survival. Our results underscore the importance of increasing the number of molecular and genomic studies that examine health disparities in HNSCC. HNSCC, like all solid tumors, is thought to be initiated and to progress through a series of clonal and sub-clonal genetic alterations. The frequency and timing of these alterations driving head and neck tumorigenesis in mostly NLW patients are the focus of several research groups’ efforts, including ours. But relatively few efforts have been devoted to study the genetic and epigenetic alterations critical to the development of HNSCC in Black or Latinos.
The canonical clonal hypothesis of cancer posits that a tumor is initiated by a driver mutation, defined as a genetic alteration that increases the ratio of cell birth to cell death. Once these initiated cells avoid death to expand, successive clonal expansions occur with each new driver gene mutation. While there is no full consensus, mathematical models predict that at least 3–5 driver mutations in the same cell are needed to initiate an oncogenic process. It is now understood there is heterogeneity throughout this process, with clonal bottlenecks appearing as some clones predominate over other clones during tumor initiation and progression. Therefore, at any given point in tumor development, there will be at least some heterogeneity within the tumor as a result of anatomic constraints coupled with competing clonal growth, and some due to other factors that are not well understood. These observations reinforce the idea that most somatic mutations found in common adult tumors have arisen as a result of serial sub-clonal developments, each characterized by a few potential drivers. These drivers can function as the genetic fingerprints of different cancerous tumors.
Together these results suggests that somatic and germ-line mutations, epigenetic alterations and TILs may provide a unique molecular fingerprint associated with the higher levels of exposure to biopsychosocial stressors that many racial and ethnically diverse groups are exposed to, mainly by living in disadvantaged neighborhoods. Molecular and social determinants of health at the neighborhood level can potentially inform precision oncology services for racial and ethnically diverse populations.
The main limitation of this study is sample size. Approximately 20% of patient samples in the head and neck tumor bank at Hopkins are from African American patients and some of these samples have been depleted throughout the years. Therefore, we were unable to measure all markers in all patients. We were also limited by the lack of funding for sequencing projects that aim to characterize racial and ethnic disparities in cancer in general and HNSCC specifically. This has certainly not been a priority as evidenced by the small frequency of TCGA samples from Black, Latino/Hispanic and Asian patients. Measurement of molecular alterations in these populations may increase our understanding of the role they play in cancer disparities, and their interplay with other molecular, psychosocial and clinical factors related to cancer indolence and aggressiveness across diverse populations.
This is a discovery project, reporting molecular differences that had never been identified in HNSCC racial disparities research. We are only showing associations, not cause and effect relationships. We did not set out to prove the molecular reasons for health disparities in HNSCC. This would require another study design with a much larger sample size to confirm the observed associations in this initial study, followed by work on patient derived tumorgrafts[67, 68]. Ultimately, molecular analysis in precision medicine may be helpful to better elucidate whether observed genomic and epigenomic alterations represent a distinct entity with clinical, immunophenotypic, and molecular characteristics or an incidental phenomenon during malignant transformation. Larger samples sizes will also provide the opportunity to systematically close the existing gap between exposure data and diagnostic/prognostic data. Data science tools can be set in place to integrate demographic, social, economic and behavioral data, at the individual and neighborhood levels, with molecular features, clinical information and health services organization data.
This challenge can be addressed with big data analytic strategies, which may include machine learning and computer algorithms to integrate patient demographic, psychosocial, clinical, pathology and molecular profiles with treatment recommendations, health insurance coverage, clinical and health information data at the medical center level, and publicly available geocoded socioeconomic, environmental, social determinants of health data at neighborhood levels. We show schematic representations of the Data Integration Modules and Data/Systems Modules (Supplementary Figure 2) that can be added to an existing or newly created biospecimen bank, using the Johns Hopkins Head and Neck tumor bank database (HAND) as an example. This integration can become the foundation for Precision Medicine platforms that track the health information and status of individual from cradle to grave, using the similar mathematical modeling used by air traffic controllers to follow the trajectory or flight path of an airplane flying from one airport to another. We show elements that highlight the similarities between both analogies, as well as a framework in which contextual, social, biological and psychological process can determine and in turn influence epigenomic changes in Supplementary Figure 3. In sum, we now have the understanding and capabilities of combining multiple big data streams into next generation precision medicine tools with machine learning and quantum entanglement capabilities, which will allow us to obtain precise depictions of molecular, clinical, psychosocial and contextual portraits to track health trajectories across the life-span. The markers described in this manuscript may be useful to improve our understanding of HNSCC survival, guide HNSCC treatment options and inform public health strategies designed to reduce HNSCC cancer disparities.
Supplementary Material
Acknowledgements:
Financial support: This research was supported by National Cancer Institute grants U01CA84986 and K01CA164092 and CA121113; National Institute of Dental and Craniofacial Research grants P50DE019032 Head and Neck Cancer SPORE, and RC2 DE20957.
Footnotes
Conflicts of Interests: There are no conflicts of interests
References
- 1.Wu ES, et al. , Effect of actionable somatic mutations on racial/ethnic disparities in head and neck cancer prognosis. Head Neck, 2016. 38(8): p. 1234–41. [DOI] [PubMed] [Google Scholar]
- 2.Bandera EV, et al. , Racial/Ethnic Disparities in Ovarian Cancer Treatment and Survival. Clin Cancer Res, 2016. [DOI] [PMC free article] [PubMed]
- 3.Jones S, et al. , Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med, 2015. 7(283): p. 283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dayem Ullah AZ, et al. , SNPnexus: assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res, 2018. 46(W1): p. W109–W113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finotti A, et al. , Liquid biopsy and PCR-free ultrasensitive detection systems in oncology (Review). Int J Oncol, 2018. 53(4): p. 1395–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stratton MR, Campbell PJ, and Futreal PA, The cancer genome. Nature, 2009. 458(7239): p. 719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin J, et al. , A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome Res, 2007. 17(9): p. 1304–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bozic I, et al. , Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A, 2010. 107(43): p. 18545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomasetti C and Vogelstein B, Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science, 2015. 347(6217): p. 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomasetti C, Li L, and Vogelstein B, Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science, 2017. 355(6331): p. 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan SH, Petrovics G, and Srivastava S, Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int J Mol Sci, 2018. 19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawan C, et al. , Epigenetic drivers and genetic passengers on the road to cancer. Mutat Res, 2008. 642(1–2): p. 1–13. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrov LB, et al. , Mutational signatures associated with tobacco smoking in human cancer. Science, 2016. 354(6312): p. 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spratt DE, et al. , Racial/Ethnic Disparities in Genomic Sequencing. JAMA Oncol, 2016. 2(8): p. 1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ademuyiwa FO, et al. , Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res Treat, 2017. 161(3): p. 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keenan T, et al. , Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. J Clin Oncol, 2015. 33(31): p. 3621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parada H Jr., et al. , Race-associated biological differences among luminal A and basal-like breast cancers in the Carolina Breast Cancer Study. Breast Cancer Res, 2017. 19(1): p. 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Souza G, et al. , Differences in the Prevalence of Human Papillomavirus (HPV) in Head and Neck Squamous Cell Cancers by Sex, Race, Anatomic Tumor Site, and HPV Detection Method. JAMA Oncol, 2016. [DOI] [PMC free article] [PubMed]
- 19.Popejoy AB and Fullerton SM, Genomics is failing on diversity. Nature, 2016. 538(7624): p. 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boden G, et al. , Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes, 2008. 57(9): p. 2438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green RC, et al. , Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine. Am J Hum Genet, 2016. 98(6): p. 1051–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodwin WJ, et al. , Unequal burden of head and neck cancer in the United States. Head Neck, 2008. 30(3): p. 358–71. [DOI] [PubMed] [Google Scholar]
- 23.Jiron J, et al. , Racial disparities in Human Papillomavirus (HPV) associated head and neck cancer. Am J Otolaryngol, 2014. 35(2): p. 147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu GF, et al. , Racial parities in outcomes after radiotherapy for head and neck cancer. Cancer, 2014. 120(2): p. 244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina MA, et al. , African American and poor patients have a dramatically worse prognosis for head and neck cancer: an examination of 20,915 patients. Cancer, 2008. 113(10): p. 2797–806. [DOI] [PubMed] [Google Scholar]
- 26.Schrank TP, et al. , Case-matching analysis of head and neck squamous cell carcinoma in racial and ethnic minorities in the United States--possible role for human papillomavirus in survival disparities. Head Neck, 2011. 33(1): p. 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worsham MJ, et al. , Disparate molecular, histopathology, and clinical factors in head and neck squamous cell carcinoma racial groups. Otolaryngol Head Neck Surg, 2012. 147(2): p. 281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poeta ML, et al. , TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med, 2007. 357(25): p. 2552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan D, et al. , Definition of a region of loss of heterozygosity at chromosome 11q23.3–25 in head and neck squamous cell carcinoma using laser capture microdissection technique. Diagn Mol Pathol, 2004. 13(1): p. 33–40. [DOI] [PubMed] [Google Scholar]
- 30.Zuo C, et al. , Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg, 2009. 141(4): p. 484–90. [DOI] [PubMed] [Google Scholar]
- 31.Tan HK, et al. , Quantitative methylation analyses of resection margins predict local recurrences and disease-specific deaths in patients with head and neck squamous cell carcinomas. Br J Cancer, 2008. 99(2): p. 357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Namazie A, et al. , Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope, 2002. 112(3): p. 472–81. [DOI] [PubMed] [Google Scholar]
- 33.Marsit CJ, et al. , Hypermethylation of E-cadherin is an independent predictor of improved survival in head and neck squamous cell carcinoma. Cancer, 2008. 113(7): p. 1566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanbery L, et al. , High SEPT9_v1 Expression Is Associated with Poor Clinical Outcomes in Head and Neck Squamous Cell Carcinoma. Transl Oncol 3(4): p. 239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fakhry C, et al. , Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst, 2008. 100(4): p. 261–9. [DOI] [PubMed] [Google Scholar]
- 36.Settle K, et al. , Racial survival disparity in head and neck cancer results from low prevalence of human papillomavirus infection in black oropharyngeal cancer patients. Cancer Prev Res (Phila), 2009. 2(9): p. 776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronson RE, et al. , Neighborhood mapping and evaluation: a methodology for participatory community health initiatives. Matern Child Health J, 2007. 11(4): p. 373–83. [DOI] [PubMed] [Google Scholar]
- 38.Arbes SJ Jr., et al. , Factors contributing to the poorer survival of black Americans diagnosed with oral cancer (United States). Cancer Causes Control, 1999. 10(6): p. 513–23. [DOI] [PubMed] [Google Scholar]
- 39.Murdock JM and Gluckman JL, African-American and white head and neck carcinoma patients in a university medical center setting. Are treatments provided and are outcomes similar or disparate? Cancer, 2001. 91(1 Suppl): p. 279–83. [DOI] [PubMed] [Google Scholar]
- 40.Gourin CG and Podolsky RH, Racial disparities in patients with head and neck squamous cell carcinoma. Laryngoscope, 2006. 116(7): p. 1093–106. [DOI] [PubMed] [Google Scholar]
- 41.Carotta S, et al. , Pax5 maintains cellular identity by repressing gene expression throughout B cell differentiation. Cell Cycle, 2006. 5(21): p. 2452–6. [DOI] [PubMed] [Google Scholar]
- 42.Holmes ML, Pridans C, and Nutt SL, The regulation of the B-cell gene expression programme by Pax5. Immunol Cell Biol, 2008. 86(1): p. 47–53. [DOI] [PubMed] [Google Scholar]
- 43.Holmes ML, et al. , Repression of Flt3 by Pax5 is crucial for B-cell lineage commitment. Genes Dev, 2006. 20(8): p. 933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Douglas WG, et al. , Development of head and neck squamous cell carcinoma is associated with altered cytokine responsiveness. Mol Cancer Res, 2004. 2(10): p. 585–93. [PubMed] [Google Scholar]
- 45.Dege C and Hagman J, Activation of Aicda gene transcription by Pax5 in plasmacytoma cells. Immunol Res, 2013. 55(1–3): p. 155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medvedovic J, et al. , Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol, 2011. 111: p. 179–206. [DOI] [PubMed] [Google Scholar]
- 47.Kanteti R, et al. , PAX5 is expressed in small-cell lung cancer and positively regulates c-Met transcription. Lab Invest, 2009. 89(3): p. 301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren Y, et al. , Diagnostic utility of PAX2 and PAX5 in distinguishing non-small cell lung cancer from small cell lung cancer. Int J Clin Exp Pathol, 2015. 8(11): p. 14709–16. [PMC free article] [PubMed] [Google Scholar]
- 49.Ranuhardy D, et al. , CD30, CD15, CD50, and PAX5 Expressions as Diagnostic Markers for Hodgkin Lymphoma (HL) and Systemic Anaplastic Large Cell Lymphoma (sALCL). Acta Med Indones, 2018. 50(2): p. 104–109. [PubMed] [Google Scholar]
- 50.Moafi A, et al. , The correlation between Pax5 deletion and patients survival in Iranian children with precursor B-cell acute lymphocytic leukemia. Cell Mol Biol (Noisy-le-grand), 2017. 63(8): p. 19–22. [DOI] [PubMed] [Google Scholar]
- 51.Ribeiro IP, et al. , WT1, MSH6, GATA5 and PAX5 as epigenetic oral squamous cell carcinoma biomarkers - a short report. Cell Oncol (Dordr), 2016. 39(6): p. 573–582. [DOI] [PubMed] [Google Scholar]
- 52.Ribeiro IP, et al. , Genomic and epigenetic signatures associated with survival rate in oral squamous cell carcinoma patients. J Cancer, 2018. 9(11): p. 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng J, et al. , Applicability of the methylated CpG sites of paired box 5 (PAX5) promoter for prediction the prognosis of gastric cancer. Oncotarget, 2014. 5(17): p. 7420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayashi M, et al. , Paired box 5 methylation detection by droplet digital PCR for ultra-sensitive deep surgical margins analysis of head and neck squamous cell carcinoma. Cancer Prev Res (Phila), 2015. 8(11): p. 1017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayashi M, et al. , Innovative rapid gene methylation analysis of surgical margin tissues in head and neck cancer. Ann Surg Oncol, 2014. 21(9): p. 3124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazzi S, et al. , Rare lymphoid neoplasms coexpressing B- and T-cell antigens. The role of PAX-5 gene methylation in their pathogenesis. Hum Pathol, 2009. 40(9): p. 1252–61. [DOI] [PubMed] [Google Scholar]
- 57.Guerrero-Preston R, et al. , Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics, 2014. 9(7): p. 1031–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X, et al. , Epigenetic inactivation of paired box gene 5, a novel tumor suppressor gene, through direct upregulation of p53 is associated with prognosis in gastric cancer patients. Oncogene, 2012. 31(29): p. 3419–30. [DOI] [PubMed] [Google Scholar]
- 59.Robins HS, et al. , Digital genomic quantification of tumor-infiltrating lymphocytes. Sci Transl Med, 2013. 5(214): p. 214ra169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peterson CE, et al. , Racial differences in head and neck squamous cell carcinomas among non-Hispanic black and white males identified through the National Cancer Database (1998–2012). J Cancer Res Clin Oncol, 2016. 142(8): p. 1715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peterson CE, et al. , Measures of economic advantage associated with HPV-positive head and neck cancers among non-Hispanic black and white males identified through the National Cancer Database. Cancer Epidemiol, 2017. 48: p. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitworth A, New research suggests access, genetic differences play role in high minority cancer death rate. J Natl Cancer Inst, 2006. 98(10): p. 669. [DOI] [PubMed] [Google Scholar]
- 63.Lippman SM and Lee JJ, Reducing the “risk” of chemoprevention: defining and targeting high risk−-2005 AACR Cancer Research and Prevention Foundation Award Lecture. Cancer Res, 2006. 66(6): p. 2893–903. [DOI] [PubMed] [Google Scholar]
- 64.Guerrero-Preston R, et al. , Bumphunting methylation sequencing identifies a critical link between PAX, NOTCH, and TP53 pathways associated to poor survival in Head and Neck Squamous Cell Carcinoma. . Epigenetics, submitted.
- 65.Yang HC, et al. , Genome-wide association study of young-onset hypertension in the Han Chinese population of Taiwan. PLoS One, 2009. 4(5): p. e5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Westra WH, The changing face of head and neck cancer in the 21st century: the impact of HPV on the epidemiology and pathology of oral cancer. Head Neck Pathol, 2009. 3(1): p. 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Izumchenko E, et al. , Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol, 2017. 28(10): p. 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ben-David U, et al. , Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet, 2017. 49(11): p. 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reichert TE, et al. , Absent or low expression of the zeta chain in T cells at the tumor site correlates with poor survival in patients with oral carcinoma. Cancer Res, 1998. 58(23): p. 5344–7. [PubMed] [Google Scholar]
- 70.Watanabe Y, et al. , Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod, 2010. 109(5): p. 744–52. [DOI] [PubMed] [Google Scholar]
- 71.Badoual C, et al. , PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res, 2013. 73(1): p. 128–38. [DOI] [PubMed] [Google Scholar]
- 72.Benjamini Y, et al. , Controlling the false discovery rate in behavior genetics research. Behav Brain Res, 2001. 125(1–2): p. 279–84. [DOI] [PubMed] [Google Scholar]
- 73.Kim SY and Volsky DJ, PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics, 2005. 6: p. 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mootha VK, et al. , Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A, 2003. 100(2): p. 605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ross AE, et al. , Gene expression pathways of high grade localized prostate cancer. Prostate, 2011. [DOI] [PubMed]
- 76.Tyekucheva S, et al. , Integrating diverse genomic data using gene sets. Genome Biol, 2011. 12(10): p. R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Michailidi C, et al. , Genome-wide and gene-specific epigenomic platforms for hepatocellular carcinoma biomarker development trials. Gastroenterol Res Pract, 2014. 2014: p. 597164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guerrero-Preston R, et al. , NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev Res (Phila), 2011. 4(7): p. 1061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bureau, U.S.C., American FactFinder, Glossary. Census 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.