

Abstract
The inappropriate expansion of self-reactive “bystander” T cells can contribute to autoimmune disease. In this issue of Immunity, Watanabe et al. (2014) demonstrate that the tumor suppressor p53 prevents the cytokine-dependent proliferation of T cells in the absence of cognate antigens.
The hallmark of an adaptive immune response is the activation and clonal expansion of antigen-specific T and B lymphocytes. The recruitment of a T cell into an immune response requires the coordinated delivery of three distinct signals. Signal 1 is delivered when the T cell receptor (TCR) engages cognate antigenic peptides presented by major histocompatibility complexes (MHC) displayed on the surface of an antigen-presenting cell (APC). Signal 2 is delivered upon the recognition of costimulatory ligands presented by APCs that have been exposed to proinflammatory stimuli. Lastly, signal 3 is delivered by combinations of cytokines that support the survival, proliferation, effector function, and differentiation of T cells that have received signals 1 and 2. In particular, cytokines that signal through the common gamma chain, such as interleukin-2 (IL-2), contribute to signal 3 by promoting the survival and expansion of T cells. This system is exquisitely selective, and for the most part ensures that nonspecific “bystander” lymphocytes do not participate in immune responses. The effectiveness of this safety mechanism is remarkable, given that the vast majority of the T cells present in secondary lymphoid organs are not specific for antigens derived from the offending pathogen and that many of these cells are capable of recognizing self-antigens. These potentially hazardous cells are exposed to the same milieu of costimulatory ligands and cytokines available to neighboring antigen-specific T cells. Consequently, the mechanisms that prevent the inappropriate expansion of bystander T cells might contribute to the avoidance of autoimmune disorders. In this issue of Immunity, Watanabe et al. (2014) provide insight into this process by demonstrating that the tumor suppressor p53 (TP53 in human; Trp53 in mouse) imposes the checkpoint that prevents the proliferation of bystander T cells exposed to signal 3, in the form of IL-2, and that this check-point is lifted by signals delivered through the TCR.
A tremendous amount of study has been devoted to the roles of p53 in cancer because most human cancers involve either inactivating mutations in p53 itself or in components of the p53 pathway (Green and Kroemer, 2009). Given the importance of p53 in cancer biology, it is somewhat surprising that so little is known about its functions in T cells. The best characterized role of p53 is that of a transcription factor that either halts the cell cycle or induces apoptotic cell death in response to DNA damage. Consequently, several earlier studies have addressed the roles of p53 in T cell development, during the recombination of genomic TCR loci. Consistent with its roles in cancer biology, p53 contributes to the suppression of thymocyte proliferation during the checkpoint associated with the recombination of the TCRβ locus and the subsequent formation of a functional pre-TCR. This process requires the introduction of double-strand DNA breaks and is expected to activate p53. Remarkably, the loss of p53 is sufficient to permit thymocytes that are incapable of generating a functional pre-TCR to progress from the coreceptor “double-negative” (DN) stage to the CD4+ CD8+ “double-positive” (DP) stage (Guidos et al. 1996; Haks et al. 1999). B cell development is similarly restored in recombinase-deficient mice lacking p53. The ability of activated B cells to undergo cycles of expansion and somatic hyper-mutation is also influenced by p53. In particular, BCL6, a transcriptional repressor that is a hallmark of germinal center B cells, attenuates Trp53 transcription (Phan and Dalla-Favera, 2004). Thus, these B cells are able to evade the cytostatic and proapoptotic effects that would normally be triggered by p53 in response to the mutations induced during somatic hypermutation by the activation-induced cytidine deaminase, AID.
In the current study, Watanabe et al. demonstrate that IL-2 signaling alone directed the upregulation of p53 and inhibited T cell proliferation and that the genetic ablation of Trp53 enabled the spontaneous proliferation of naive T cells exposed to IL-2 in the absence of antigen. They further showed that although stimulation via the TCR rapidly increases the amount of p53 protein, antigenic stimulation ultimately reduced the amounts of p53 protein and mRNA, thereby enabling proliferation in response to IL-2 (Figure 1). Watanabe et al. proceeded to confirm these observations in vivo by using TCR transgenic T cells in an adoptive-transfer model. In sum, their observations indicate that p53 imposes the checkpoint that prohibits the expansion of T cell clones in the absence of antigenic stimuli.
Figure 1. The Tumor Suppressor p53 Blocks Bystander T Cell Proliferation Triggered by IL-2.
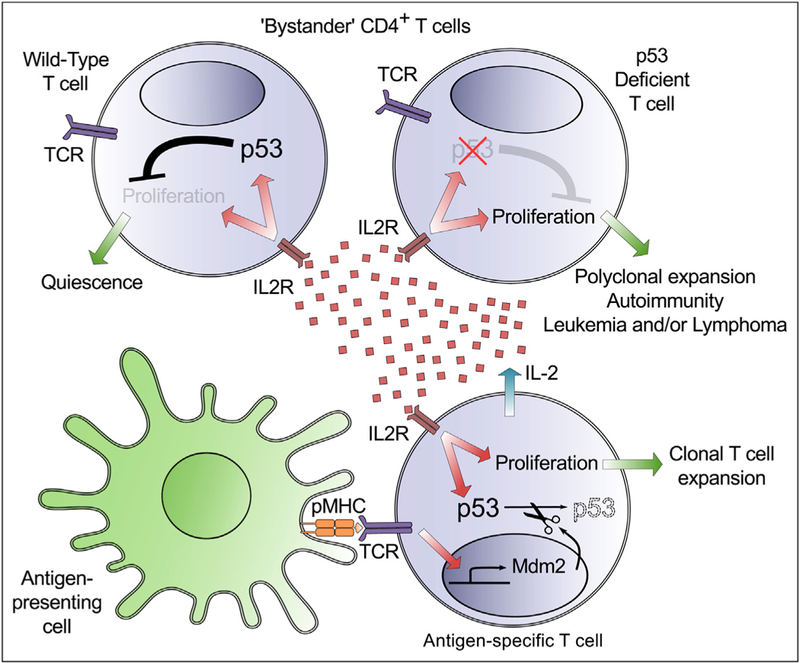
(upper left) In response to a pathogenic challenge, CD4+ T cells that are not specific for antigen are exposed to mitogenic cytokines such as IL-2. In normal animals, IL-2-dependent signals increase the abundance of p53 protein, blocking the proliferation of these “bystander” T cells.
(upper right) T cells that lack functional p53 proliferate when exposed to IL-2, even in the absence of TCR signaling, confirming the essential role of p53 in the enforcement of this checkpoint.
(bottom right) The specific recognition of antigen (pMHC) by the TCR increases the concentration of the mRNA encoding Mdm2, a ubiquitin ligase that targets p53 for proteasomal degradation. The ensuing degradation of p53 enables the expansion of antigen-specific T cell clones while ensuring that potentially self-reactive “bystander” cells do not participate in the immune response.
Because the activation of naive CD4+ T cells in the periphery is not associated with directed DNA rearrangement or mutation, it is not immediately obvious why p53 should have evolved to enforce this checkpoint. However, the transient upregulation of p53 following TCR ligation might serve a practical purpose. In many long-lived vertebrates, thymic involution significantly reduces the output of naive T cells in the adult, increasing the average age of the naive T cell pool. In humans, peripheral naive T cells can survive 6–10 years, providing ample opportunity for the accumulation of DNA damage (den Braber et al. 2012).
The transient induction of p53 following TCR ligation might delay the initiation of cell cycle to allow the resolution of this DNA damage. This mechanism is likely to play an important role in limiting the initiation of T cell lymphoma. It will be interesting to determine whether other cytokines that signal through the common gamma chain also increase the abundance of p53 expression. In particular, IL-7 promotes thymocyte survival and expansion prior to the checkpoints associated with the formation of a viable pre-TCR (Xiong et al. 2013; Singer et al. 2008). A logical extension of the current study would be to examine whether IL-7 terminates its role in thymocyte expansion by inducing p53 and whether signaling through newly formed pre-TCR complexes lifts the resulting developmental blockade.
Another prospect raised by this study is the possibility that the autoimmune disease observed in T cell-specific conditional Trp53 knockout animals might be caused, at least in part, by the inappropriate activation of self-reactive bystander T cells. Although the study that initially reported this autoimmune disorder (Kawashima et al. 2013) attributed the disease to a reduction in the numbers of regulatory T (Treg) cells, the few Treg cells observed in these animals are fully functional. It will be interesting to determine the extent to which each of these mechanisms contributes to the observed autoimmune pathology.
The authors also investigated the mechanism by which the amount of p53 is reduced following antigenic stimulation. They discovered that the abundance of the mRNA encoding Mdm2, a negative regulator of p53, is upregulated in cells exposed to cognate antigen-bearing APCs, but is unperturbed in cells exposed to IL-2 alone. Mdm2 is an E3 ubiquitin ligase that binds to and inhibits the transcriptional functions of p53, and that can mark p53 for proteasomal degradation (Figure 1; Kruse and Gu, 2009). To determine whether Mdm2 is responsible for the TCR-induced decrease in p53 abundance and for subsequent CD4+ T cell proliferation, Watanabe et al. used the small-molecule inhibitor Nutlin3a, which blocks the interaction between Mdm2 and p53 and leads to p53 stabilization and transcriptional activation. They found that the treatment of antigen-stimulated cells with Nutlin3a increased the abundance of p53 and abolished the proliferation of these cells. Although Nutlin3a also blocks the interaction of Mdm2 with the p53 family members p73 and p63, Nutlin3a did not have a significant effect on the proliferation of p53-deficient cells, indicating that p53 is the critical inhibitor of proliferation in this system.
Perhaps the most exciting aspect of the current study is that it opens up numerous avenues of research. Both p53 and Mdm2 are central points in complex webs of intersecting pathways. For example, the amount of p53 increased in the first 48 hr following antigen plus IL-2 stimulation, despite the fact that there was no increase in the abundance of the Trp53 mRNA. This clearly suggests some form of translational or posttranslational control. Future studies will need to determine the precise mechanisms by which p53 is stabilized by cytokine stimulation, as well as which downstream targets of p53 are responsible for blocking proliferation in cells exposed to IL-2 alone. Another question left unaddressed concerns the relative roles of TCR signaling and costimulatory signaling in the upregulation of Mdm2. Nevertheless, the study presented by Watanabe et al. has provided crucial insight into a previously unappreciated mechanism that suppresses the proliferation of bystander CD4+ T cells during adaptive immune responses. It will be important to further clarify how p53 influences Tcell activation, the initiation of autoimmunity, and the formation of T cell lymphomas. Perhaps this study will lift the checkpoint that has thus far limited the recruitment of “bystander” immunologists into studies of p53.
REFERENCES
- den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mögling R, de Boer AB, Willems N, Schrijver EHR, Spierenburg G, Gaiser K, et al. (2012). Immunity 36, 288–297. [DOI] [PubMed] [Google Scholar]
- Green DR, and Kroemer G. (2009). Nature 458, 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, and Danska JS (1996). Genes Dev. 10, 2038–2054. [DOI] [PubMed] [Google Scholar]
- Haks MC, Krimpenfort P, van den Brakel JH, and Kruisbeek AM (1999). Immunity 11, 91–101. [DOI] [PubMed] [Google Scholar]
- Kawashima H, Takatori H, Suzuki K, Iwata A, Yokota M, Suto A, Minamino T, Hirose K, and Nakajima H. (2013). J. Immunol 191, 3614–3623. [DOI] [PubMed] [Google Scholar]
- Kruse JP, and Gu W. (2009). Cell 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan RT, and Dalla-Favera R. (2004). Nature 432, 635–639. [DOI] [PubMed] [Google Scholar]
- Singer A, Adoro S, and Park JH (2008). Nat. Rev. Immunol 8, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Moon K,Vacchio MS, Hathcock KS, and Hodes RJ (2014). Immunity 40, this issue, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Parker BL, Dalheimer SL, and Yankee TM (2013). Immunology 138, 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]