

IN BRIEF:
This perspective highlights the significance of chronic pain, its underpinning biology and the need for composite biomarker signatures alongside self-report. Combining evidence from animal and human research spanning genetics through to neuroimaging, we highlight a few key areas of promise.
What is Pain?
Pain is our body’s alarm system, warning us either of danger in the environment, injury or the presence of disease. It is necessarily intrinsically unpleasant (hurts) which enables protection by driving immediate attention, action and adaptive learning. To be effective, protective pain must be so overpowering that we cannot ignore it. Consequently, it is intimately linked to negative emotions. Physiologically, it is triggered by the activation of high threshold primary sensory neurons – the noxious stimulus detecting nociceptors, by specific transduction machinery in their peripheral terminals (Woolf and Ma, 2007). This is the nociceptive pain evoked by pin prick, touching something too hot or any potentially tissue damaging chemical, thermal or mechanical stimulus. Lack of the capacity to experience nociceptive pain is catastrophic, tips of fingers and toes are damaged, lips and tongues are mutilated, and life expectancy is reduced, as witnessed in individuals with congenital insensitivity to pain due to rare recessive gene mutations resulting either in loss of nociceptors or their functional disruption (Bennett and Woods, 2014).
Post injury or infection, adaptive processes are engaged both in the peripheral and central nervous systems (PNS and CNS, respectively) that manifest as an amplification of noxious inputs resulting in an exaggeration and prolongation of pain (hyperalgesia) due to peripheral and central sensitization (Hucho and Levine, 2007). Additionally, there is the generation of hypersensitivity such that normally innocuous inputs now produce pain – the phenomenon of allodynia - a warm shower becomes painful after sun burn or a skin cut is tender to touch – this helps drive guarded protection of the injured area until it heals (Latremoliere and Woolf, 2009). Both nociceptive and acute inflammatory pain and the neurobiological mechanisms responsible are key then to survival. Treatments that block nociceptive and inflammatory pain need, therefore, to be used with caution. Elimination of the protective elements of acute pain are only required temporarily and in a highly controlled fashion, as for surgery, obstetrics or dentistry. Control of inflammatory pain needs to be a tight balance between reducing suffering and enabling healing, such as postoperatively or after trauma. There is a particular challenge though for pain reduction in arthritis patients using either non-steroidal anti-inflammatory drugs or a promising new treatment with anti-nerve growth factor antibodies, where pain needs to be managed such that the underlying joint disease is not worsened by overuse of pain-free but still damaged joints (Denk et al., 2017).
Chronic pathological pain is entirely different from acute nociceptive pain. Chronic pain is defined clinically as pain that persists beyond normal tissue healing time – normally 3–4 months. This includes chronic inflammatory pain, such as rheumatoid arthritis, neuropathic pain arising from injury to or disease of the nervous system, such as diabetic painful neuropathy or post-herpetic neuralgia, and dysfunctional or idiopathic pain, such as fibromyalgia or irritable bowel syndrome (https://www.iasp-pain.org/terminology). Chronic pathological pain rarely may also be the consequence of gain-of-function mutations in voltage-gated sodium ion channels expressed in nociceptors (Bennett and Woods, 2014; Dib-Hajj et al., 2015). Pathological pain includes conditions where the pain is not signaling the presence of an ongoing noxious stimulus (i.e. it is not nociceptive) nor ones which are enabling healing (i.e. not acute inflammatory pain), the pain here is not protective and is instead pathological. Persistent maladaptive pains are major sources of disability globally with 1/5 adults fulfilling this definition and a consequential high socioeconomic burden and cost (~$600 billion per annum in the USA)(IOM, 2011). The opioid crisis in the USA is a stark reminder of the suffering and a need for better non-addictive treatments (HEAL).
Such clinical pain has a complex biology and pathophysiology with multiple diverse pathways affected. These include abnormal peripheral drivers such as ectopic activity in injured axons, alterations in transmission and processing in the spinal cord and higher brain centers due to sensitization, amplification and disinhibition, through to modifications in perception. Exactly what is responsible for the transition of acute to chronic pain and why some individuals are more susceptible than others is an area of active research; mitigating the development of persistent pathological pain by targeting the responsible mechanisms being the goal.
Figure 1a, b provides a simplified overview of acute and chronic pain pathways and some mechanisms.
Figure 1a, b. Mechanisms of acute and chronic pain:


The normal physiological response to an acute noxious stimulus is depicted in black with the involvement of Aδ, and C fiber nociceptors transducing the input from the periphery to the superficial laminae of the dorsal horn of the spinal cord where they can be modulated. From the spinal cord, signals are relayed via regions in the brain stem to the brain where pain emerges as a perception and the sensory and emotional context and learning is applied to interpret and aid future avoidance of the stimulus. The major classes of chronic pain and the processes which are believed to lead to chronic pain in susceptible individuals are depicted in red. During inflammation, while the stimulus e.g. activated immune cells or a skin incision is present, there exists a status of peripheral sensitization (PS) characterized by erythema and tenderness to innocuous stimuli, typically heat. PS goes away once the peripheral pathology resolves. Stimuli activating nociceptors that are noxious, repeated and sustained e.g. following nerve injury, induce the process of central sensitization (CS) in the dorsal horn spinal cord. Initially CS is protective and enables the organism to avoid further injury due to a heightened awareness of its surroundings but at some point CS becomes pathological. CS produces pain in non-inflamed tissue by co-opting novel inputs e.g. Aβ fibers, thus mechanical pain is typical of CS and heat pain is more typical of peripheral sensitization. Recently it has been shown that a subset of corticospinal neurons (CSNs) known to originate in the primary and secondary somatosensory cortex and to directly innervate the spinal dorsal horn via CST axons can directly modulate normal and pathological tactile sensory processing in the spinal cord. Facilitation and descending inhibition are processes that occur due to different regions of the brain and brainstem inhibiting or activating (or even disinhibiting) nociceptive inputs to the spinal cord. The effect can be seen on both mechanical and heat sensations in different forms of chronic pain and an imbalance in this system (less inhibition, more facilitation) is a key mechanism, as are changes in the brain’s neurochemistry, structure and functional activity. Shown in green are the current methodologies that are used to define biomarkers at the particular levels of nociceptive and pain processing that apply to acute and chronic pain
Chronic Pain as a Symptom or Disease
The recognition that most chronic pain is maladaptive and mechanistically quite different from acute protective pain has been a major conceptual breakthrough within the pain field. Nevertheless it is hard to manage chronic pain effectively because the processes in the nervous system driving the pain are not easily identified and targeted for treatment. Pain in these circumstances is not a simply a symptom of some distinct disease pathology but rather the expression of a pathologically functioning nervous system. Of course there are some conditions, such as osteoarthritis, where there is a primary peripheral pathological driver and replacement of the joint can ameliorate the chronic pain. However, even here the response to such interventions is not universally beneficial, as the pain might become centralized in some patients (De Oliveira Silva et al., 2018; Soni et al., 2018). Furthermore, most effective treatment of pain (especially its prevention) needs to be not simply symptom suppressing (switching off the sensation of pain) but rather targeted at the underlying pathophysiological mechanisms within the nervous system that drive the chronicity, and are, in consequence, disease-modifying. For example, within the PNS, a local anesthetic that temporalily silences ectopic activity in an injured nerve is symptom suppressing while one that reduces expression of those ion channels that drive this pacemaker like activity would be disease modifying. Within the CNS a short lived inhibition of activity in nociceptive projection neurons in the spinal cord would be symptom suppressing while one that prevented long lasting changes in synaptic input strength would be disease modifying. The International Association for the Study of Pain has recently classified chronic pain for the International Classification of Diseases (ICD-11) as a disease or symptom, with either chronic primary pain, such as fibromyalgia where there is no known trigger, or chronic secondary pain, such as chronic neuropathic pain that develops after a lesion to the nervous system (Treede et al., 2019). This classification is welcome as it recognizes chronic pain as a disease in its own right.
So, although we use a single word ‘pain’, multiple distinct mechanisms contribute to the generation and maintenance of this complex, multidimensional and subjective experience. The ACTTION-American Pain Society Pain Taxonomy (AAPT) provides an evidence-based and multi-dimensional (or composite) approach to classifying chronic pain that attempts to address the need for mechanism based assessment and treatment (Fillingim et al., 2014). We similarly argue for an approach where pain is defined by the primary pathophysiological drivers responsible (Vardeh et al., 2016), as this will help better identify targets for successful intervention. However, to do this well we need biomarkers of mechanisms.
Composite Biomarker Signatures for Pain
Similar to other complex neurological diseases like Alzheimer’s, it is highly unlikely that one biomarker will be able to capture ‘pain’ in its entirety. Exploiting advanced analytical tools like neural networks, artificial intelligence or machine learning algorithms to combine multiple objective biomarkers into ‘composite pain biomarker signatures’ is more likely to be successful for understanding pain and developing new treatments (Baskin et al., 2016). We need to abandon the view that pain categorized as mild, moderate and severe determines therapeutic choice, to a model where we recognize that the pain patient’s experience is the consequence of multiple diverse neurobiological processes each of which offer an opportunity to be managed differently. We recognize that this is not trivial to achieve. We must develop therapies (drug, surgical, physical, psychological) that selectively target these mechanisms, and we must develop objective ways to assess the presence of the mechanisms and the efficacy of any modulation of them. We need to follow oncology where there has been a shift from general cytotoxic agents to treatments aimed specifically at the unique features of the cancer in an individual patient.
In this perspective, we ask the question: are objective biomarkers of pain and the mechanisms that drive it possible? However, we first need to interrogate why relying solely on subjective ratings is inadequate for pain assessment and treatment.
The Challenge of Measuring Pain
The International Association for the Study of Pain currently defines pain as “An unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” This definition does not capture the fact that pain may be both protective and pathological and also misses key features of clinical pain conditions; that pain may arise either in the absence of any stimulus or in response to a stimulus that would normally only evoke an innocuous sensation. Finally, it doesn’t readily serve non-verbal individuals as it requires the experience to be described.
At its simplest, pain comprises two core dimensions of intensity (magnitude) and unpleasantness (affect). When it comes to clinical pain there are many subjective variants in how the sensation is described; stabbing, burning, throbbing, pulsing, grinding and shock-like are but a few examples. Although an enormous amount of effort has been devoted to examining these pain descriptors, looking, for example, to see if the subjective sense of burning specifically reflects neuropathic pain, the results are, to date, generally not clear-cut. Much of how we describe pain is learned, conditioned by our experiences or shaped by our language and society or clinicians (Bourke, 2014). While report of the presence of pain is critical, as in duration, periodicity, location and intensity, descriptors are unlikely to share a simple one-to-one mapping with any underpinning mechanism.
Nonetheless, the standard way to assess the presence and magnitude of pain is to ask a person to describe it using rating scales and symptom-based questionnaires. For rating scales the challenge is how variable is an individual’s assessment of their pain – if you rate your pain at 7 out of 10 today how accurately can you gauge whether your pain tomorrow is identical or different? Is your worst pain, moreover, the same as someone with a different genetic, cultural, or environmental upbringing? Pain experiences are also malleable to various factors, such as mood, context and cognition, making the consequential pain report both complex and variable even to the same nociceptive input. Neuroimaging (discussed below) has shown that these factors influence the pain experience and consequent self-report via different mechanisms to the ones driving the underlying pathology. So, while these factors are a key part of the multidimensional pain experience their contribution needs to be distentangled and mechanistically understood; particularly if we want to know what our therapy is targeting ‘underneath’ the pain experience that is a mixed represention of all these inputs and influences.
Figure 2a illustrates the common tools we have to measure pain. Figure 2b illustrates how we measure pain in children and adults ranging from the awake and cognitively able to the deeply sedated/anaesthetized and cognitively impaired, and Figure 2c illustrates how pain is measured in animals.
Figure 2a.
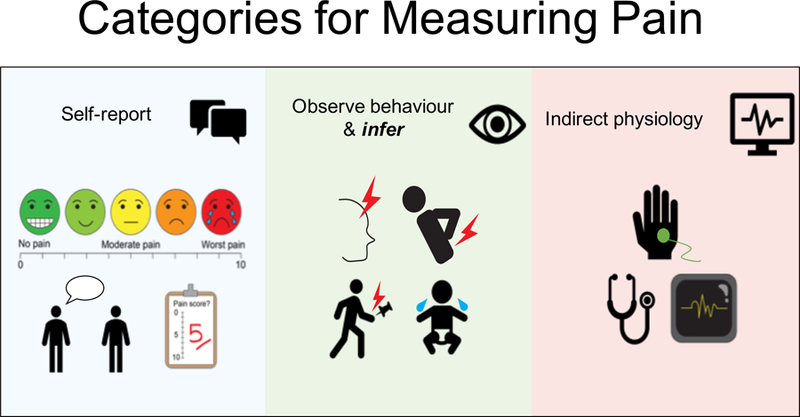
Categories for Measuring Pain.
Figure 2b. How we currently ‘measure pain’ in humans:

This falls into the following broad categories: (i) self-reports using rating scales/descriptors/questionnaires to exogenous stimuli or any ongoing and spontaneous pain; (ii) observed measures of pain-like behavior; (iii) indirect measures of physiology/autonomic changes. (ii) currently is subjective and may suffer from cultural and social biases/influences as well as a lack of sensitivity and specificity but artificial intelligence/machine learning methods may remove the subjectivity and identify more sensitive components, (iii) are indirect assessments and make significant assumptions when relating these physiological measures to the underlying subjective state.
Figure 2c. How we currently ‘measure pain’ in animals:

Preclinical pain-related categories are shown with increasing complexity from top to bottom. The vast majority of testing still involves use of exogenously applied thermal and mechanical stimuli (typically to the plantar surface of the hind paws of rodents) and reflexive output measures. Other measures of behavior based on choice (avoidance of an activity or avoidance of an aversive condition) and complex measures of physiological function are being increasingly introduced. Finally, we anticipate that human observation on animal behavior will be supplanted by computer-based machine-learning approaches to identify specific pain-related behaviors, such as facial grimace.
Current attempts to measure pain - Ratings:
For an awake, conscious and cognitively able individual the commonest way to measure pain is by rating intensity using a visual analogue (VAS), numeric rating (NRS) or verbal rating scale (VRS) but the sensitivity and robustness/reliability of these measures is low, even with training (Smith et al., 2016). Subjective ratings alert us to the patient’s report of the presence of pain – but it can be challenging to verify if the report is genuine or not. Capturing pain qualities on a range of single-item descriptors is also difficult; therefore, multidimensional outcome measures are used to try to reveal the mix of sensory and affective pain components, but they are empirically based and reveal little about the specific nature or degree of the pain. Helpful disease specific scales have also been developed (e.g. DN4 and PainDetect for neuropathic pain) and their specificity, sensitivity and reliability is being validated (Bouhassira et al., 2005; Freynhagen et al., 2006; Haanpaa et al., 2011).
The subjective assessment of pain – whether acute or chronic - can capture if pain is present, some degree of its severity and duration, and probably most reliably, where anatomically it feels to be occurring. Additional features of the pain experience can also be captured, such as if it is spontaneous or evoked, intermittent or continuous, deep or superficial. However, the presence of pain as a common endpoint tells us very little or arguably nothing about the presence of the multiple distinct pathologies that can drive pain. One other important issue with ratings is that high variance leads to reduced assay sensitivity i.e. the ability of a randomized control trial to detect an active treatment. First identified by Harris and colleagues (Harris et al., 2005) in a trial studying the effect of milnacipran on fibromyalgia pain ratings, the effect of high pre-treatment pain variability was found to significantly predict a treatment response specifically in the placebo arm. Farrar and colleagues (Farrar et al., 2014) also found the same phenomenon when they analysed clinical trials involving diabetic neuropathy (n=1226) and peripheral herpetic neuralgia (n=1514) patients. Both groups suggested that this problem be mitigated by excluding patients that showed the highest baseline pain rating variability (the top 20–25%) from the trial. It was made clear that exclusions must be performed prior to randomization into the treatments to avoid violating internal validity. However, overall, even with these recommendations on how to manage the variability of pain ratings, we need to recognize that there is something problematic and too insensitive with how we’re currently measuring pain and relief. As patient self-report remains the gold-standard, development of more sensitive rating scales and even ‘composite’ scales combining rating with physical functioning show promise in this regard (Dworkin et al., 2012; Patel et al., 2018; Smith et al., 2016). However, it’s time we argue, for an additional strategy.
What if you cannot rate pain - indirect measures to assess pain:
Obviously animals cannot self-report pain, but neither can a baby, a demented elderly person, an anesthetised or comatose patient. Crying, grimacing and guarding are taken to reflect pain in non-verbal awake individuals and in animals, but the extent to which they reflect the intensity of the pain is not clear and specificity is low – babies cry also because they are hungry or uncomfortable. A baby’s inability to verbally report pain poses an obvious barrier to pain management, and assessing pain in the elderly demented is equally problematic (Corbett et al., 2012). Responses to noxious stimulation can be quantified through a variety of behavioural (e.g. reflex withdrawal) and physiological (e.g. rise in heart rate, fall in oxygen saturation) changes (Moultrie et al., 2017). While such surrogate measures can guide treatment options, several studies indicate that these underestimate likely pain experience in infants and possibly contributes to an under-recognition and under-treatment of infant pain (Jones et al., 2017; Slater et al., 2008). Similarly, using an isolated forearm test during anaesthesia has led to the recognition that a minority of anesthetized patients are ‘aware’ and responsive (Pandit et al., 2014) – alerting us to the need for better measures to assess if these patients are also in pain (Mhuircheartaigh et al., 2013). Autonomic/physiological changes are commonly used as pain surrogates including heart rate, pupil dilation, blood pressure, galvanic skin response (GSR) changes and breathing rate. However, GSR and heart rate variability can, for example, be modified without changing nociceptive input, simply by altering the threat value or context of the experience (Leknes et al., 2013). As pain interferes with daily activities, tools thought to measure some aspect of pain indirectly in awake individuals, include functionality, mobility, frailty, emotional state and quality of life (Dworkin et al., 2005); however, they may also be measuring factors such as anxiety or sleep deprivation, so, as with GSR measures, interpretation requires caution.
In animal research or veterinary practice indirect behavioral or physiological metrics are the only option to test for presence of pain. Enhanced behaviors, such as: facial grimacing (Langford et al., 2010 ), licking/guarding paws/flinches and reactions to previously non-noxious stimuli; inhibited behaviors, such as: burrowing (Andrews et al., 2012), nest building (Jirkof et al., 2013), wheel running (Cobos et al., 2012); conditioned place preference (King et al., 2009); rearing activity (Matson et al., 2007); gnawing (Rohrs et al., 2018); gait (Angeby Moller et al., 2018)) and EEG (LeBlanc et al., 2016) are all used. However, they are at best surrogates of and not direct measures of pain.
For all these assessments, absence of signs does not necessarily mean absence of pain; similarly, their presence may also not reflect the experience of pain. Some measures in animals are closely related to the ethogram of the animal and others are surrogates of human clinical measures. For translational success preclinical to man, it is important to have metrics that are also translatable. This is where composite pain biomarkers can help us.
The growing need for biomarkers
Figure 3 illustrates situations where objective biomarkers for pain would benefit society.
Figure 3. A need for pain biomarkers:
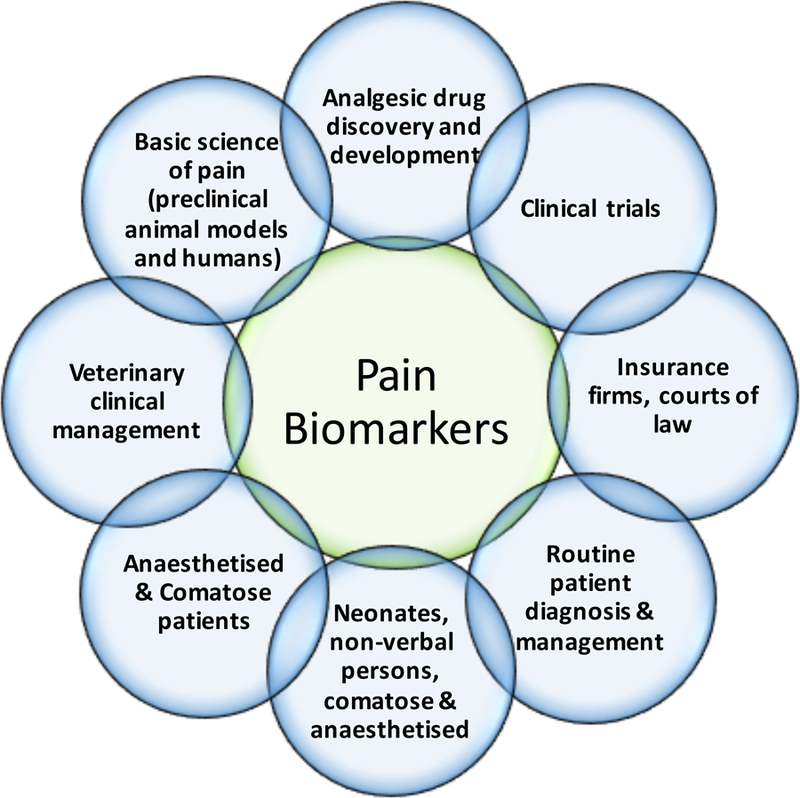
There are numerous situations that would benefit from the availability of specific, sensitive and accurate biomarkers for pain. Each of these are in themselves complex areas of biological science and medicine that require different combinations of the features of biomarkers listed in Table 1 relative to each other. For example, while routine patient management would benefit from use of safety and monitoring biomarkers, it would arguably be of greater benefit for non-verbal patients and neonates for a pharmacodynamic response biomarker. The consequences of false negative findings from a pain biomarker, include: (1) trust issues between doctor-patient, employee-patient or family-patient; (2) denial of medical treatment; (3) mental health, stress, spousal/family issues; (4) financial/insurance and employment issues; (5) privacy/legal (medical malpractice issues). The consequences of false positive findings, include: (1) unnecessary, costly, harmful analgesic treatment in non-communicative patients; (2) human, infrastructure, financial and time resources; (3) misunderstanding as a substitute for self-report. These are serious issues to be considered in the development of any pain biomarker, as has been discussed (Davis et al., 2017).
Let’s take the development of new analgesics as an illustrative example. Existing approved analgesics provide 50% or more pain relief in far less than a third of patients (Breivik et al., 2013). Many promising new compounds fail to reach the market as analgesics because they are discarded in early drug development due to a lack of significant reduction in pain in randomised placebo-controlled trials (Hewitt et al., 2009). Pain relief in the placebo arm— which is often large— can confound, therefore, potentially valuable, mechanistic, and pharmacodynamically active analgesic effects of the study drug (Chizh et al., 2009). Negative expectation may drive a self-report of no analgesia even if the drug has effective action on a key underpinning mechanism (e.g. attenuating ectopic firing of peripheral nerve, down-regulating central sensitization, etc) (Bingel et al., 2011). Therefore, there is a clear need for objective outcome measures of pharmacodynamic efficacy that demonstrate target engagement and modulation of pain related mechanisms in early patient drug studies. They provide early confidence that a compound has reached the tissue, for example the brain and receptor being targeted, and ideally can be used both in preclinical and clinical studies. Failure of efficacy in the face of proof of target engagement is a better filter for eliminating targets. We rely on preclinical mechanistic target validation to guide us on the analgesic potential of novel targets, but we need to note that sometimes the wrong mechanism is targeted. For example, a program that developed both mechanism-based and target engagement biomarkers for a compound with some preclinical efficacy yet delivered a major clinical trial failure, is that of a fatty acid amide hydrolase (FAAH) inhibitor (Huggins et al., 2012). This showed complete lack of analgesia in a phase 2 clinical trial for osteoarthritis, despite clear mechanism-based increases in endogenous cannabinoids. Was the wrong mechanism targeted? Contrasting that is the development of antibody therapies targeting NGF and its use as a mechanistic based biomarker, as illustrated in Figure 4. This provides an excellent example of successful translation from preclinical to patients with a treatment that targets a mechanism relevant to the pathogenesis of chronic pain (Aloe et al., 1992; Indo, 2001; Lewin et al., 1993; Lowe et al., 1997; Sevcik et al., 2005). Interestingly, although an immunoaffinity LC–MS/MS assay exists that can measure NGF in healthy volunteers and also in patients (Neubert et al., 2013), the final piece of the NGF story that is missing is the formal association of NGF levels in a patient population treated with or without a blocking antibody and assessment of pain.
Figure 4. Cross species validation of biomarker evaluation:
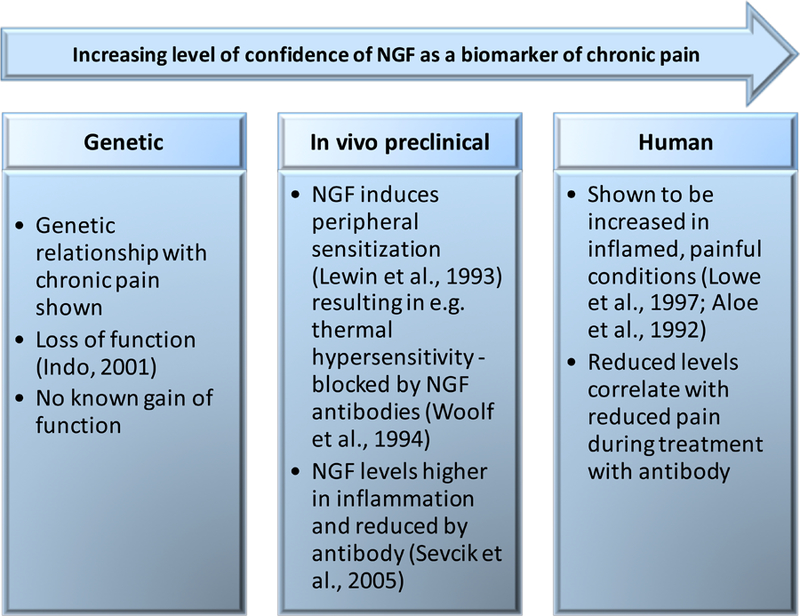
Nerve growth factor (NGF) has potential as a mechanistic pain biomarker in inflammatory conditions since its relationship to chronic inflammatory pain translates both across species and from genetics to therapy. However, for NGF to constitute such a biomarker its specificity and sensitivity will need to be evaluated and met.
What is a biomarker?
A joint FDA-NIH working group (Biomarkers, Endpoints and other Tools – BEST) identified seven distinct biomarker categories that could be applied across the whole spectrum of biological research (BEST, 2016). Further, the FDA categorized 4 types of biomarkers relevant to the development of drugs and biologics: (1) diagnostic, (2) prognostic, (3) predictive and (4) pharmacodynamic. Therefore, a biomarker is a characteristic that can be objectively measured and evaluated as an indicator of a normal biological process, a pathologic process, or pharmacological (and non-pharmacological) response to a therapeutic intervention. In Table 1, we summarize biomarker definitions and detail situations where they might be used. Currently there are no clinical biomarkers for pain approved by the FDA or European Medicines Agency (EMA) for use in analgesic drug development trials.
Table 1.
Biomarker Definitions with Present and Future Examples for Pain
| Type of Biomarker | Definition | Pain Examples (present; future) |
|---|---|---|
| Diagnostic | To detect or confirm the presence of a disease or condition. | QST; EEG; intra-epidermal nerve fibre density; |
| microneurography; neuroimaging, Genetics | ||
| Monitoring | To assess status of a disease or condition or effect of a medical product by any biomarker that is measured serially. | QST; compound levels in plasma, CSF; |
| Neuroimaging; EEG; intra-epidermal nerve fibre density | ||
| Pharmacodynamic/Response | To show that a biological response occurs in an individual exposed to a medical product. | QST; neuroimaging; EEG; Changes in cytokines |
| Specific mechanistic/biochemical pain drivers; intra-epidermal nerve fibre density | ||
| Predictive | To identify individuals more likely than individuals without the biomarker to experience a favourable or unfavourable effect from exposure to a medical product. | Genetics |
| Neuroimaging; EEG; intra-epidermal nerve fibre density | ||
| Prognostic | To identify likelihood of a clinical event, disease recurrence or progression in patients with disease of interest. | Genetics |
| Neuroimaging; EEG; intra-epidermal nerve fibre density | ||
| Safety | Measured before or after an exposure to a medical product to indicate likelihood, presence or extent of toxicity. | Treatment related e.g. sedation, tolerance, constipation, respiratory depression |
| Neuroimaging; EEG | ||
| Susceptibility/Risk | Potential for developing a disease or medical condition | Genetics |
| Neuroimaging; EEG |
Adapted from “BEST (Biomarkers, Endpoints, and other Tools) Resource”, a publication produced by the joint FDA-NIH Biomarker Working Group, December, 2016 (BEST, 2016)
In this perspective, we will not review and assess all potential pain mechanism biomarkers. Several recent and excellent reviews summarize the possibilities for immunohistochemistry/skin biopsy (transduction), microneurography (transmission), quantitative sensory testing (QST) and electroencephalography (EEG)/neuroimaging (perception) as potential pain biomarkers (Davis et al., 2017; Gasparotti et al., 2017; Smith et al., 2017). Instead, we focus on a few promising biomarker opportunities and discuss how they might be deployed as part of a future pain biomarker composite signature.
Neuroimaging Based Biomarkers
Probably the area that has caught most attention as a pain biomarker is neuroimaging. There is something appealing about supposedly ‘seeing’ pain, which lends itself to being thought of as an ideal objective biomarker; however, the standard biomarker criteria must be rigorously applied. Brain imaging is not a surrogate of self-report. In fact, it reveals far more than just the neural processing underpinning one dimension (e.g. intensity) of pain often captured in a self-report; it is an activity-dependent correlate of the entire experience generating countless possible signatures (Tracey and Mantyh, 2007). One early approach by neuroimagers has been to deconstruct the complex signal of the pain experience into constituent elements, whether as activity within a network or particular set of brain regions for subsequent selective diagnostic monitoring or targeting (Ploghaus et al., 1999). Chronic pain is often accompanied by co-morbidities such as depression, anxiety, catastrophizing, fatigue, sleep disturbance and poor cognition – and these all contribute to the pain experience. The relationship, therefore, between a patients’ self-report of pain and their concurrent regional brain activity is complex, and studies mostly using acute pain stimuli in healthy volunteers have shown how these factors profoundly alter the neural processing of nociceptive inputs – almost acting as central neural amplifiers or attenuators of the experience (Berna et al., 2018; Berna et al., 2010; Eippert et al., 2009a; Ploghaus et al., 2001; Sprenger et al., 2012; Tracey et al., 2002; Vachon-Presseau et al., 2016a; Wiech and Tracey, 2009). Related, even the analgesic effect measured by self-report, of a defined concentration of an intravenous dose of the fast-acting mu-opioid, remifentanil, can be enhanced by positive expectation and obliterated by negative expectation (Bingel et al., 2011). Such experiments reveal that subjective pain reports are highly malleable and that the processing of nociceptive inputs is modulated by factors like expectation, anxiety and mood. We now know that as pain becomes chronic, these influences alongside neural expression of ongoing, spontaneous or evoked pain in chronic pain patients become yet more important and complex (Harper et al., 2018a; Hashmi et al., 2013; Vachon-Presseau et al., 2016b). If we can identify the presence of such modulating influences this would greatly help assessment of the value of self-report, for example in analgesic efficacy trials.
Neuroimaging Tools in Pain Research
An array of neuroimaging tools are used in the study of acute and chronic pain: functional magnetic resonance imaging (fMRI) and associated advanced magnetic resonance methods, such as: spectroscopy, quantitative cerebral blood flow, diffusion imaging for white matter tract delineation, structural MRI and voxel based morphometric analysis; positron emission tomography (PET), electroencephalography (EEG) and magnetoencephalography (MEG). A growing body of work using EEG/MEG to characterize nociceptive and pain signals exists but is not discussed further here (see review (Ploner and May, 2017)). Most functional brain imaging studies use Blood Oxygen Level Dependent (BOLD) imaging and require a phasic stimulus to elicit a measurable signal. Measuring the brain’s functional response to an applied noxious stimulus or provoking a clinical symptom, like brush evoked allodynia or a painful joint squeeze, while producing a painful experience also recruit regions of the brain contributing to the entire multidimensional experience including attention, fear and anxiety. While the whole network is a reflection of the multidimensional experience – giving a drug or providing an intervention that produces changes only in a component of the experience - may not target those elements related to nociceptive processing or pathogenic mechanisms. For example, midazolam - a drug targeting anxiety - attenuates brain activity during pain but this should not be interpreted therefore as ‘analgesic’. A novel paradigm design that identified brain regions related to pain anticipation/anxiety rather than receipt of nociceptive afferents (Ploghaus et al., 1999) revealed it was the anticipatory/anxiety related and not nociceptive brain regions that were preferentially modulated by the drug (Wise et al., 2007). Other research has further dissected the particular roles brain regions subserve in the multidimensional pain experience (Woo et al., 2014; Woo et al., 2017). A particular challenge for brain imagers is that the smallest unit of analysis in fMRI, the voxel, integrates activity across hundreds of thousands to millions of neurons that have potentially distinct functional properties. Therefore, there is a “many-to-one” problem when mapping any sensory percept or psychological process to activity in a particular voxel or cluster. Failure to grasp this subtlety, coupled with over-reliance on ‘reverse-inference’ (i.e. inferring the meaning of brain activity in a pain study based upon what is found from another study – pain related or not) is a real problem and can lead to a lack of apparent reproducibility and interpretative issues. Nonetheless, meta-analyses reveal consistent results regarding structure and activity for a range of chronic pain disorders (Dehghan et al., 2016; Huang et al., 2016). An alternative approach is to relate the entire interconnected network of brain activity, indirectly assessed using BOLD resting state data (i.e. no exogenous stimulus), as a global marker of acute and chronic pain, and this might even include response to treatment (Harris et al., 2013; Kucyi and Davis, 2015; Napadow et al., 2012).
Other neuroimaging areas being developed with potential as biomarkers, include: functional neuroimaging of the spinal cord (Eippert et al., 2017; Sprenger et al., 2012; Stroman et al., 2014); and infant pain biomarkers of pain and analgesic efficacy (Moultrie et al., 2017; Slater et al., 2010). These latter studies demonstrate that pain-related behavioural measures may underestimate pain experience in infants (Jones et al., 2017).
Here, we will discuss two approaches for developing imaging-based biomarkers: one aimed at capturing pain perception using a single measurement, and the other aimed at developing a suite of biomarkers of pathogenic mechanisms, including risk factors for developing chronic pain (i.e. vulnerability and prognostic). While all these metrics are promising and have revealed a lot about the neural basis of acute and chronic pain, a concerted effort to combine these neuroimaging based metrics in disciplined ways for better diagnosis, prognosis, prediction and pharmacodynamics is now needed.
Machine-learning to generate biomarkers of pain perception.
Machine learning (ML) exploits the ability of computers to learn from and subsequently make predictions about data and are being used to identify what pattern or set of regions predict whether a person experienced pain or not. This approach has the advantage that it removes the ‘expert’ brain imaging scientist from making an interpretation, which often relies on the reverse-inference problem detailed above. Remember though, we use reverse-inference when interpreting a patient’s pain behavior, so it is not a unique problem to brain imaging data. ML is also useful for solving the problem of overlapping activity within a single voxel or cluster, because it identifies in spatially separable patterns, the different responses elicited by different stimuli. What might appear in normal univariate analysis as one ‘blob’ common to different stimuli or tasks can now be seen using Multi-Variate Pattern Analyses (MVPA) as having a unique and identifiable spatial pattern ‘under the blob’ for a specific stimulus (Haynes and Rees, 2006). In the pain field, for example, this has helped resolve whether somatic, social or empathic/vicarious pain occurs within overlapping brain regions – which they do not (Krishnan et al., 2016; Woo et al., 2014).
When applied to neuroimaging (functional imaging as well as EEG), MVPA can be used to detect response patterns associated with a particular subjective feature or experimental variable (e.g. pain intensity or relief). The data need to be split into: ‘training’ groups, used to obtain a provisional brain marker and ‘test’ groups, which evaluate predictive accuracy. Early machine-learning studies in pain relied on single datasets (Brodersen et al., 2012; Marquand et al., 2010; O’Muircheartaigh et al., 2015), but more recent ones have drawn on multiple datasets that include a range of pain experiences both from healthy individuals (Wager et al., 2013) and those subjected to experimental models of pain (e.g. the capsaicin model of secondary hyperalgesia) and from patients (Duff et al., 2015). Robust regional networks modulated by analgesics can be identified using this approach from multiple studies and can be tested for utility as diagnostic, prognostic, predictive and pharmacodynamic biomarkers. They could also be applied in situations where there is no self-report (e.g. anesthesia and babies) in conjunction with indirect measures, such as heart rate, galvanic skin response, pupil dilation, to determine what combination of measures best reflects the signal revealed by imaging as pain related.
Signatures of pain perception?
Two main networks identified by MVPA are the Neurological Pain Signature (NPS) and Pain-Analgesic Network (Duff et al., 2015; Wager et al., 2013). For acute nociceptive pain, the NPS is a distributed pattern of regions that matches commonly reported ‘pain-processing’ regions in humans and animals and tracks well with the magnitude of pain. For example, when 2 stimuli are experienced as different in pain intensity by 2 points on a 10-point numeric rating scale, NPS activity predicts which stimulus is more painful with >90% accuracy. In addition, the NPS has sensitivity and specificity in distinguishing between somatic pain and non-painful warmth, pain anticipation, pain recall, or emotionally evocative images, but is unable to distinguish placebo analgesia (Zunhammer et al., 2018). As this signature is generated from healthy volunteers experiencing similar and very brief phasic pain experiences, we need to examine the applicability of the NPS across distinct types of other acute pain (e.g., heat, cold, mechanical, inflammatory), and research is underway to generate new signatures reflective of different chronic pain conditions, such as fibromyalgia (Lopez-Sola et al., 2017).
Another classification-based protocol has utilized machine learning analysis of several FMRI analgesic clinical trials to derive neural signatures of pain and analgesic experiences (Duff et al., 2015). The advantage of these signatures is that they are based on data across a range of drug classes from chronic pain patients, that from healthy volunteer experimental models of pain mechanisms and symptoms (central sensitization) and acute pain in healthy controls. The protocol detects pharmacodynamic effects where a compound shows consistent effects on brain responses across individuals, and efficacy where the compound’s effects correspond to an analgesic signature from the classification database. The database can be used therefore, for assessing novel analgesics. For example, in an independent FMRI data set (n=24) from a double-blind, randomized, placebo-controlled, three-way crossover study in a healthy volunteer model of central sensitization (not used in the database and signature generation), the reduction in the brain response to painful stimuli by gabapentin could be correctly distinguished from placebo in 79% of individuals (p=0.003) using the signature. In contrast, responses following ibuprofen (non-efficacious in this model and so a negative control) could not be distinguished from placebo (45%, p=0.72). In the assessment for analgesic efficacy, the classifier correctly identified the gabapentin arm in 17 of 24 subjects (p=0.03), whereas for ibuprofen discrimination was below chance (p=0.92). Specific analgesic compounds may have moreover, distinct signatures; a classifier trained on an independent study of gabapentin reliably identified gabapentin (p=0.03) while classifiers trained on studies of the opioid remifentanil failed to identify gabapentin (p=0.5), while successfully identifying the effects of remifentanil (Wanigasekera, 2018). Such studies highlight the potential for this approach to aid analgesic drug development in an unbiased way. Figure 5 illustrates the approach.
Figure 5. Machine learning in biomarker development.
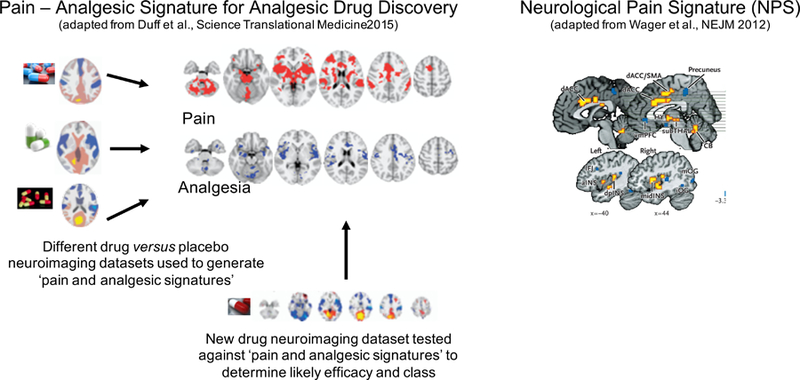
Illustration of Neurological Pain Signature and Pain-Analgesia Signature for Analgesic drug development as ‘biomarkers’ currently being developed using machine learning tools and data from human neuroimaging studies.
Cautionary note about machine-learning approaches in pain:
These approaches, while promising, rely on the self-report to train the classifier (e.g. pain or no pain) and additional ways to classify training data sets without self-report would be of interest. If a patient complains of pain and a neuroimaging biomarker confirms its presence, then this needs to be regarded seriously even if a cause for the pain cannot be defined (e.g. idiopathic pain). Furthermore, emotional pain is as relevant to the patient and should not be treated as ‘second class’ pain despite being challenging to understand and diagnose. Machine learning methods that generate emotional pain signatures/biomarkers, provided they have specificity and sensitivity, would be helpful diagnostically in this regard (Krishnan et al., 2016; Woo et al., 2014). However, the brain decoding of thoughts, feelings and perceptions, while offering unprecedented opportunities scientifically (Kragel et al., 2018), might have serious societal and personal/legal consequences and caution is advised to avoid misuse of such data. For example what to do if there is no neuroimaging pain biomarker/signature but the patient complains of pain – should the subjective self-report usurp the imaging data or vice versa? These precise issues have been discussed by a recent task-force (Davis et al., 2017).
Neuroimaging based biomarkers of pathogenic mechanisms
Neuroimaging can generate a suite of potential biomarkers of mechanisms driving pain as verified in preclinical models. Structural, functional and neurochemical changes, particularly when combined, all have potential as pain biomarkers.
Anatomical changes:
changes in gray matter volume (voxel based morphometric increases and decreases) and abnormalities in white matter and brain connectivity are observed in individuals with different chronic pain conditions (Apkarian et al., 2004; Tatu et al., 2018; Wartolowska et al., 2012), with some predictive of the conversion from acute to chronic back pain (Vachon-Presseau et al., 2016b). However, these anatomical changes reverse upon resolution of the pain condition, so might relate to co-morbid features that also resolve upon cessation of pain (Gwilym et al., 2010; Rodriguez-Raecke et al., 2009). The lack of current specificity of these anatomical changes and lack of any cellular understanding of what these changes in voxel-based morphometry or white matter connectivity mean (Sampaio-Baptista and Johansen-Berg, 2017) makes their utility as biomarkers, at this stage, limited.
Resting networks:
Resting state networks are a hallmark of normal brain function (Mitra and Raichle, 2016) and changes in these might provide markers reflective of a specific pain state or treatment response, as has been shown for a range of conditions (Becerra et al., 2014; Ceko et al., 2015; Harper et al., 2018a; Harris et al., 2013; Kucyi and Davis, 2015; Loggia et al., 2013; Napadow et al., 2012). Nevertheless, it is still unclear how specific these changes are to the underpinning mechanisms of spontaneous pain.
Neurochemical changes:
Proton Magnetic Resonance Spectroscopy (1H-MRS) reveals increased levels of the excitatory neurotransmitter glutamate and decreased concentrations of the inhibitory neurotransmitter GABA in the posterior insula of fibromyalgia patients compared to healthy controls (Foerster et al., 2012). They suggest the posterior insula may be a pain promoting region whose overactivity contributes to the pain syndrome. A neuropathic pain model in the rat generated similar changes, while increasing insular glutamate and decreasing GABA levels in non-injured rats resulted in mechanical allodynia (Watson, 2016). Further, pregabalin reduces aberrant posterior insula glutamate/glutamine signals in patients with fibromyalgia (Harris et al., 2013).
Imaging tonic pain to reveal potential biomarkers?:
Much of the work described above highlights a dominant role for the insula in pain. Involvement of the dorsal posterior insula in ongoing pain is supported by the sustained changes in its activity, whose magnitude correlates with nociceptive input and pain intensity, detected using a novel neuroimaging technique (arterial spin labeling) that allows for measurement of neural events lasting tens of minutes (Segerdahl et al., 2015b). Also using an ASL approach, Loggia and colleagues found an association between the magnitude of self-reported clinical pain and connectivity between the insula, among other regions, and the default mode resting state network in chronic pain (Loggia et al., 2013). Discussion of the posterior insula’s role in pain by the literature illustrates nicely some of the challenges in defining regional activity using univariate analyses and how new techniques, such as arterial spin labelling, offer different opportunities to understand how tonic pain might be represented in the brain – giving us potentially new biomarker signatures (Davis et al., 2015; Loggia, 2019; Segerdahl et al., 2015a). While we don’t suggest this region alone encodes pain – recent animal work highlighting a dominant role for the amygdala in pain unpleasantness, for example, reminds us of pain’s complexity and requirement to activate many brain regions (Corder et al., 2019) – several results suggest that this region provides potential as a possible biomarker of nociceptive drive and pain intensity – especially given: the failure to activate it, unlike most other pain-related brain regions, by empathy, hypnosis, or recalled pain (Fairhurst et al., 2012; Raij et al., 2005; Wager et al., 2013); it is part of the NPS and Pain-Analgesic network; predictive coding identifies its role encoding stimulus intensity (Geuter et al., 2017); it is not encoding saliency (Horing, 2018); and there is alteration of pain upon posterior insula modulation (acute and tonic) in animals and humans (Dimov et al., 2018; Garcia-Larrea and Mauguiere, 2018; Han et al., 2015; Lin et al., 2017).
Descending Pain Modulatory System – brainstem potential biomarker?:
The descending pain modulatory system (DPMS) is a brainstem-subcortical-cortical network that modulates nociceptive processing in the dorsal horn in a brainstem driven anti- and pro- nociceptive manner to control nociceptive input to the brain (Basbaum and Fields, 1984). Preclinical studies reveal that the anti-nociceptive component of the system is protective against neuropathic pain and the pro-nociceptive arm as contributing to spontaneous pain and tactile hypersensitivity (De Felice et al., 2011; Wang et al., 2013), including a direct input from the somatosensory cortex to the dorsal horn (Liu et al., 2018). Neuroimaging in human volunteers has identified regions of the brainstem that become active during the development and maintenance of capsaicin-induced central sensitisation and secondary mechanical hyperalgesia, during somatic and visceral pain, and during post-opioid hyperalgesia (Lee et al., 2008; Zambreanu et al., 2005). Such altered activity (less inhibition and more facilitation) has been verified in multiple patient cohorts, such as migraine, fibromyalgia, osteoarthritic hip and knee pain and most recently diabetic painful neuropathy (Coulombe et al., 2017; Gwilym et al., 2009; Harper et al., 2018b; Mainero et al., 2011; Marciszewski et al., 2018; Segerdahl et al., 2018; Soni et al., 2018). Stratifying patients based upon the presence or absence of descending pain modulatory system involvement may be a potential biomarker for predicting outcome to treatment/surgery. For example, osteoarthritis pain patients prior to joint replacement surgery who score high on neuropathic pain measures show increased facilitation within the brainstem compared to those whose clinical presentation is more indicative of nociceptive pain – and this was predictive of surgical outcome (Soni et al., 2018). In our view, the brainstem offers a potentially interesting biomarker of pathogenic pain and for use in drug development.
Biomarker readouts of pharmacodynamic efficacy versus placebo
Pharmacological neuroimaging is a viable tool in terms of reproducibility, sensitivity and ability to deliver relevant pharmacodynamic measurements (Wanigasekera et al., 2016; Wise et al., 2004). It is also used to predict efficacy and treatment response (Harris et al., 2013; Wanigasekera et al., 2012) and determine whether a drug has action on defined pathologic mechanisms. Gabapentin, for example, significantly modulates the development of central sensitisation compared to placebo (Iannetti et al., 2005). Activity in the brainstem during central sensitisation can differentiate gabapentin versus ibuprofen or placebo with greater sensitivity than analgesic scores (Wanigasekera et al., 2016). A range of brain and spinal cord fMRI neuroimaging experiments have identified that placebo analgesia utilizes the inhibitory arm of the descending pain modulatory system (Eippert et al., 2009a; Eippert et al., 2009b; Tracey, 2010). Expectation-driven placebo effects are not easy to eliminate since it is a normal component of any active therapy – as are nocebo (side) effects whose neurobiology is similarly being elucidated by neuroimaging (Bingel et al., 2011; Tinnermann et al., 2017). Such effects highlight the problem of measuring pharmacodynamic efficacy solely by behavioural analgesia. Further, the assumption that expectation-driven placebo analgesic effects are non-specific and equal in both the drug and placebo arms is increasingly being questioned (Colagiuri, 2010; Kirsch, 2000), not least because the neural mechanism driving placebo analgesia might be interfered with by centrally acting drugs. In a recent study of post-traumatic neuropathic pain patients treated chronically with pregabalin, tramadol and placebo, we were able to differentiate each arm using neuroimaging alone; verify that the placebo mechanism was recruited during the placebo arm by patients and show that the placebo mechanisms was interfered with during the two drug arms (Wanigasekera et al., 2018). A study in osteoarthritis similarly verifies imaging’s ability to infer distinct mechanisms in the absence of subjective differences (Tetreault et al., 2018).
It is clear that by using neuroimaging, placebo-related analgesic mechanisms can be distinguished from pharmacological-related analgesic mechanisms and, as such, might be useful in go/no-go decision making during analgesic drug discovery. A further advantage is that matching studies can be performed cross-species (Becerra et al., 2013; Upadhyay et al., 2013).
Cautionary note - the Bayesian Brain and Perception
Our assessment on how perceptions are constructed and updated in the brain are beginning to change. Sensory perception, vision, pain, or touch for example, is an amalgam of inputs and priors (an internal probalistic model that is updated by neural processing of sensory information in a manner approximating Bayesian probability) (Edwards et al., 2012; Parr et al., 2018). A prior might bias the perceptual outcome irrespective of current sensory inflow – (i.e. model is not updated, normally via a prediction error, by the nociceptive information). This concept holds true for pain perception (Wiech, 2016; Wiech et al., 2014). This is important for pain biomarkers because in the presence of pathogenic signal inputs to the brain, priors might powerfully influence the final perception. So, it is possible that a drug or treatment might have high efficacy in terms of targeted change in a pathogenic mechanism, but a subject’s priors might be such that it takes a while for a shift in their pain perception - creating a discrepancy between efficacy against a mechanism and the patient’s report. So, subjective ratings might be misleading and decoupled in time from modulation of underpinning mechanisms. Clearly, this has huge relevance for clinical trial design.
Biobank, Imaging Derived Phenotypes and Polyphenic Imaging for Composite Biomarker Development:
Combining multiple neuroimaging-based measures of the various pathogenic mechanisms that each contribute to the overall pain experience will likely provide a ‘composite pain biomarker’ with greater sensitivity and specificity. This is what is needed and work has begun. A study used machine learning-based predictions of clinical pain with two types of neuroimaging (resting state and arterial spin labeling) plus autonomic metrics was recently published (Lee et al., 2018b). When all three multimodal parameters were combined patient classification between lower and higher clinical pain intensity states had the best performance, illustrating the power of a machine learning based composite approach.
In psychiatric conditions and Alzheimer’s disease, combining measures from multiple modalities (e.g. imaging, genetics, biochemistry, psychological measures) increases predictive value. For example, combining advanced MRI measures and CSF protein biomarkers in patients with mild cognitive impairment had 91% accuracy, 85% sensitivity, 96% specificity for predicting conversion to Alzheimer’s disease (Douaud et al., 2013) and a recent study identified distinct multivariate brain morphological patterns that increased in predictive value when combined with cognitive and polygenic risk scores in schizophrenia and bipolar disorders (Doan et al., 2017). Pioneering work predicting acute to chronic pain conversion is in its infancy due to the challenges and costs of capturing patients early and following them longitudinally. So far it is limited to patients already in the acute phase of pain and before converting to chronic pain, or not, but the findings (structural, functional, genetic) are very promising (Baliki et al., 2012; Vachon-Presseau et al., 2016b).
However, to test hypotheses regarding what brain networks might reflect if a person is vulnerable or resilient to developing chronic pain (for example, an imbalance in the brainstem descending pain modulatory system (Denk et al., 2014)), we need to gather data well before tissue injury, onset of diabetes or exposure to chemotherapy (all of which might lead to chronic pain) then follow the subjects over time. UK Biobank (imaging component) aims to address this precise problem for many diseases by acquiring high-quality imaging data from 100,000 predominantly healthy participants whose health outcomes will be tracked over several decades. Structural, diffusion and functional imaging modalities are being collected from the brain, alongside body and cardiac imaging, genetics, lifestyle measures, biological phenotyping and health records. Discovery of imaging markers at an early stage of a broad range of diseases, as well as better insight into disease mechanisms, is the goal and even after the first 5000 participants’ data was released, an impressive range of associations between imaging derived brain phenotypes (IDPs) and other measures was realised (Miller et al., 2016). More recently, genome-wide association studies of 3,144 functional and structural brain imaging phenotypes show that many of these IDPs are heritable (Elliott et al., 2018). More than a 100 areas of the human genome were identified that influence the brain. As the genetic basis of brain structure is largely unknown, this newly identified and rich set of genetic effects will greatly help our understanding of the mechanisms by which the brain develops, functions, becomes damaged by disease and may heal itself or may enable persistent pathological functioning.
The potential of Biobank is huge, with clear potential to combine polyphenic imaging derived pain phenotypes with advanced polygenetic biomarkers of pain.
Genetic biomarkers of pain
Although there was, as for most diseases, great excitement in the pain field after the completion of the full human genome sequence in the early 2000’s, for the potential to discover genetic drivers of pain, progress in identifying such “pain genes” has been slow and generally disappointing. This, despite the relatively large heritable component (40–60%) revealed by multiple twin studies both of pain sensitivity in healthy individuals (Norbury et al., 2007) and of the pain experienced by patients (Burri et al., 2018; Momi et al., 2015; Vehof et al., 2014; Visscher et al., 2018). Identification of those genetic variants that contribute to enhanced pain could potentially both identify/validate novel analgesic targets and constitute a pain biomarker. What would such a genetic biomarker look like? An estimate of the degree/nature/extent/duration of the pain likely to be experienced by a healthy individual in response to a noxious stimulus, the risk of developing pain after a nerve injury, exposure to cancer chemotherapeutic agents or with diabetes, and its severity, type and persistence? As well as the likelihood of an analgesic response to defined therapeutic interventions?
The evaluation of genetic contributors to pain is severely handicapped by the lack of objective biomarkers of pain, as with all clinical investigations in the field – creating variable subjective rating phenotypes with which to try to associate gene variants, the generally small cohorts studied so far, and the difficulty in assessing a familial aspect of clinical pain if many people in a given family are not exposed to the trigger – e.g. traumatic nerve injury. Neuroimaging based endophenotypes might help bridge the gap from genetics to self report (Lee and Tracey, 2013). There has been some success in identifying rare genetic causes of pain occurring in absence of any pathology, as with gain-of-function mutations in voltage-gated sodium channels that produce conditions like inherited erythromyalgia and paroxysmal extreme pain disorder (Kapetis et al., 2017) or in CACNA1A in hemiplegic migraine, but there is nothing comparable for common pain conditions.
It well may be that different gene variants contribute to the pain sensitivity occurring in response to different classes of stimuli (mechanical/thermal/chemical), to the risk of developing spontaneous pain and/or allodynia, and in the conversion of acute to chronic pain. None of these features are currently captured in large cohorts like the UK Biobank, though that is changing with an enhanced series of questionnaires relating to chronic pain being released to the 350,000 Biobank participants who have consented to be contacted via electronic media. This phenotyping is designed to capture a wide range of conditions including headache, chronic widespread pain/chronic fatigue syndrome, post-surgical pain, musculoskeletal pain and neuropathic pain.
Perhaps one major contributor to the slow progress in identifying pain-driving genetic variants has been the assumption that pain risk for different types of pain (e.g. headache, temporomandibular joint pain, fibromyalgia, diabetic neuropathy) will be monogenic largely driven by variants in a single gene. While single gene causes of pain do occur, such mendelian mutations are extremely rare. In contrast while many common single gene variant contributors to pain have been identified in multiple typically underpowered GWAS studies (Veluchamy et al., 2018; Zorina-Lichtenwalter et al., 2016), although a few recent studies are at last beginning to use larger cohorts (Suri et al., 2018), the effect size is generally small, and many candidates are non-replicable. Interestingly though, replicated variants do not seem restricted to one anatomical site or type of pathology (Meloto et al., 2018).
Perhaps we have been looking for the wrong thing? Maybe pain is not driven by variants in a single gene but rather by a polygenic combinatorial effect of common variants in multiple differing genes and intronic sites, each with an individually small effect? Such a recognition is occurring for many common diseases where polygenic risk score analyses are identifying individuals at high risk of developing type 2 diabetes (Mahajan et al., 2018) and schizophrenia (Alloza et al., 2018), as well as breast cancer, inflammatory bowel disease and coronary artery disease where the polygenic risk score has a prevalence 20-fold higher than rare monogenic mutations with a comparable risk (Khera et al., 2018). This is a game changer – combinations of many genetic variants can be found that influence susceptibility to disease in many individuals. One paper so far has defined a polygenic contribution to migraine (Gormley et al., 2018) and another to chronic pain (McIntosh et al., 2016). Identification of polygenic risk scores for pain will require very large cohorts (many 100’s of thousands) and very well phenotyped patients – but the investment is going to be well worth it. Furthermore, application of machine learning/deep neural network algorithms to interrogate gene variants across the whole genome (Sundaram et al., 2018) will enable identification of complex pain risk “DNA fingerprints” likely to comprise tens of thousands of variants. We need to embrace this complexity as we move away from just seeking single base variant causes of pain to the identification of the totality of genetic contributors and modifiers of pain, which will constitute a true genetic pain biomarker. One that will in due course embrace epigenetic elements such as DNA methylation from blood, CSF and tissue samples and of course neuroimaging, and may enable us to identify who is at risk and for what, how badly and how long.
Such a polygenetic approach will complement efforts described above from UK Biobank with the next steps to link ‘polyphenic’ imaging with polygenetic information to better understand complex behavior.
Patient derived neurons as pain biomarkers
There are growing opportunities arising from phenotyping neurons differentiated from patient derived stem cells (induced pluripotent stem cells, iPSCs) for modeling disease, screening for treatment options, detecting biomarkers or even for acting as biomarkers of disease risk in their own right (Gendron et al., 2017; Schrenk-Siemens et al., 2018; Vadodaria et al., 2018; Wainger et al., 2014; Xu et al., 2018). iPSCs contain of course the genetic background of the patients but not the epigenetic marks derived from environmental interaction or the effects of aging. It is now possible to differentiate iPSCs into many defined types of neurons e.g. nociceptors, cortical and motor neurons although a limitation is that typically they remain immature. For pain, the capacity to generate human nociceptors from stem cell is certainly most promising (Chambers et al., 2012; Eberhardt et al., 2015; Wainger et al., 2015) especially for studying channelopathies like those arising from Nav1.7 mutations (Yang et al., 2018) where the hyperexcitability phenotype is easy to capture either electrophysiologically or with calcium reporter readouts. More complex three-dimensional multicell organoids for modelling cortex (Madhavan et al., 2018; Sloan et al., 2018) and skin (Lee et al., 2018a) are being developed and offer exciting new ways to study complex non-cell autonomous phenotypes.
Tremendous effort is being devoted to identifying and measuring phenotypes in stem cell derived neurons that are relevant to disease. For pain this could include membrane excitability, intracellular calcium levels, gene expression, membrane trafficking, mitochondrial function, axonal growth, susceptibility to axon terminal withdrawal, metabolic activity, post-translational modifications, sensitization of TRP channels and response to stressors (chemotherapeutic agents/axonal damage/hyperglycemia).
Biomarkers of pain risk could include phenotypic changes indicative of developing inflammatory pain, painful diabetic neuropathy, chemotherapy induced painful neuropathy or post-surgical neuropathic pain. For example, if patient derived nociceptors displayed increased relative excitability to axonal injury or a greater degree of TRPV1 responsiveness on exposure to inflammatory mediators that may be associated with a risk of neuropathic pain or peripheral sensitization respectively. Biomarkers of treatment response could include sensitivity to sodium channel blockers in lines with Nav1.7 gain of function mutations. Ultimately it may be possible to reconstruct dorsal horn circuits to test relative propensity for the synaptic mechanisms underlying central sensitization, synaptic facilitation and disinhibition, as well as strength of descending modulatory inputs. As this space is explored we need to keep in mind line and batch variations, culture-based artefacts due to karyotypic abnormalities, the possibility that large numbers of lines will be required to define disease relevant phenotypes and the advantages of using robotics to minimize variance.
Nevertheless, the future looks promising as we can begin to explore disease mechanisms in human cells and from patients with defined conditions. This will likely contribute to the identification of new targets and the capacity to conduct phenotypic screens for analgesic treatments as well as clinical trials in a dish. If the technology becomes sensitive enough this could be a major feature of precision pain medicine e.g. screen for the risk of an individual developing chemotherapy-induced neuropathy on exposure to chemotherapeutic agents and identify the best treatment options for a patient by measuring response of injured neurons to range of therapeutics alone or in combination. In the end though, success will of course also depend on how well the patient from whom the iPSC line was generated, was phenotyped and for that we need to harness the potential of the mechanism-based biomarkers discussed in this perspective, as well as improved ways of capturing self-report.
Conclusion:
In this perspective, our aim was to alert the reader to the problem of pain and the complexity in its measurement across a range of circumstances. Figure 6 summarises our perspective and suggestions regarding the development of composite pain biomarkers. We challenge the notion that self-report using various rating tools is a robust measure, particularly in chronic pain conditions. As a consequence, objective biomarkers that reflect key underpinning mechanisms are desperately needed if we are to do things better in terms of analgesic drug development, diagnosis, patient stratification, and treatment targeting. Fortunately, there are a range of possible pain biomarkers being developed that span species as well as spatial and temporal scales. Going forwards consortia that bring these different biomarker groups together are needed alongside advanced analytical methods that combine the biomarkers in a supervised manner so that they are diagnostic, prognostic, predictive and pharmacodynamic. If successful, this truly will herald a new era for determining the presence of pain, assessing its intensity and underlying pathophysiology and most importantly treating it more effectively.
Figure 6.
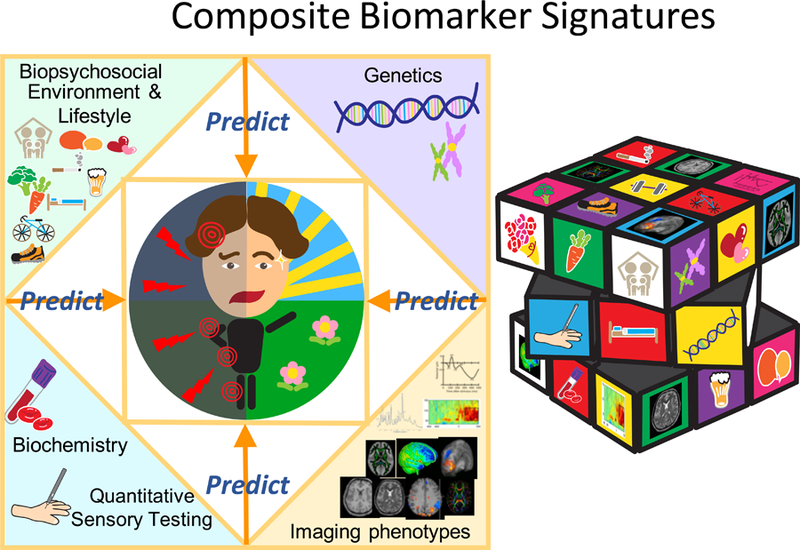
A Composite Biomarker Signatures for Pain.
KEY POINTS:
Pain is a subjective sensory experience that can, mostly, be reported, but cannot be directly measured or quantified. Nevertheless, a suite of biomarkers related to mechanisms, neural activity and susceptibility, offer the possibility - especially when used in combination - to produce objective pain-related indicators with the specificity and sensitivity required for diagnosis, evaluation of risk of developing pain and of analgesic efficacy. Such composite biomarkers will also provide improved understanding of pain pathophysiology.
Acknowledgements:
IT is supported by the Wellcome Trust and Nuffield Benefaction. CJW and NAA were supported by grant NS105076 from the NIH. We wish to thank Dr Idy Hiu Ting Ho for expert help generating the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
Clifford Woolf is on SAB of Biogen and is a founder of Nocion Therapeutics.
Nick Andrews has founding shares in Nocion Therapeutics.
Irene Tracey is on the Neuroscience SAB of Amgen and is part of Innovative Medicines Initiative PainCare-Biopain. She has a patent on depth of anaesthesia monitoring.
References:
- Alloza C, Cox SR, Blesa Cabez M, Redmond P, Whalley HC, Ritchie SJ, Munoz Maniega S, Valdes Hernandez MDC, Tucker-Drob EM, Lawrie SM, et al. (2018). Polygenic risk score for schizophrenia and structural brain connectivity in older age: A longitudinal connectome and tractography study. Neuroimage 183, 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloe L, Tuveri MA, Carcassi U, and Levi-Montalcini R (1992). Nerve growth factor in the synovial fluid of patients with chronic arthritis. Arthritis Rheum 35, 351–355. [DOI] [PubMed] [Google Scholar]
- Andrews N, Legg E, Lisak D, Issop Y, Richardson D, Harper S, Pheby T, Huang W, Burgess G, Machin I, and Rice AS. (2012). Spontaneous burrowing behaviour in the rat is reduced by peripheral nerve injury or inflammation associated pain. Eur J Pain 16, 485–495. [DOI] [PubMed] [Google Scholar]
- Angeby Moller K, Svard H, Suominen A, Immonen J, Holappa J, and Stenfors C. (2018). Gait analysis and weight bearing in pre-clinical joint pain research. J Neurosci Methods 300, 92–102. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, and Gitelman DR. (2004). Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 24, 10410–10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, and Apkarian AV. (2012). Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci 15, 1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, and Fields HL. (1984). Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci 7, 309–338. [DOI] [PubMed] [Google Scholar]
- Baskin II, Winkler D, and Tetko IV. (2016). A renaissance of neural networks in drug discovery. Expert Opin Drug Discov 11, 785–795. [DOI] [PubMed] [Google Scholar]
- Becerra L, Sava S, Simons LE, Drosos AM, Sethna N, Berde C, Lebel AA, and Borsook D. (2014). Intrinsic brain networks normalize with treatment in pediatric complex regional pain syndrome. Neuroimage Clin 6, 347–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Upadhyay J, Chang PC, Bishop J, Anderson J, Baumgartner R, Schwarz AJ, Coimbra A, Wallin D, Nutile L, et al. (2013). Parallel buprenorphine phMRI responses in conscious rodents and healthy human subjects. J Pharmacol Exp Ther 345, 41–51. [DOI] [PubMed] [Google Scholar]
- Bennett DL, and Woods CG. (2014). Painful and painless channelopathies. Lancet Neurol 13, 587–599. [DOI] [PubMed] [Google Scholar]
- Berna C, Leknes S, Ahmad AH, Mhuircheartaigh RN, Goodwin GM, and Tracey I. (2018). Opioid-Independent and Opioid-Mediated Modes of Pain Modulation. J Neurosci 38, 9047–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna C, Leknes S, Holmes EA, Edwards RR, Goodwin GM, and Tracey I. (2010). Induction of Depressed Mood Disrupts Emotion Regulation Neurocircuitry and Enhances Pain Unpleasantness. Biol Psychiat 67, 1083–1090. [DOI] [PubMed] [Google Scholar]
- BEST (2016). In BEST (Biomarkers, EndpointS, and other Tools) Resource (Silver Spring; (MD: )). [Google Scholar]
- Bingel U, Wanigasekera V, Wiech K, Mhuircheartaigh RN, Lee MC, Ploner M, and Tracey I. (2011). The Effect of Treatment Expectation on Drug Efficacy: Imaging the Analgesic Benefit of the Opioid Remifentanil. Sci Transl Med 3. [DOI] [PubMed] [Google Scholar]
- Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, Cunin G, Fermanian J, Ginies P, Grun-Overdyking A, et al. (2005). Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 114, 29–36. [DOI] [PubMed] [Google Scholar]
- Bourke J. (2014). The Story of Pain (Oxford University Press; ). [Google Scholar]
- Breivik H, Eisenberg E, O’Brien T, and Openminds (2013). The individual and societal burden of chronic pain in Europe: the case for strategic prioritisation and action to improve knowledge and availability of appropriate care. BMC Public Health 13, 1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen KH, Wiech K, Lomakina EI, Lin CS, Buhmann JM, Bingel U, Ploner M, Stephan KE, and Tracey I. (2012). Decoding the perception of pain from fMRI using multivariate pattern analysis. Neuroimage 63, 1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri A, Ogata S, Rice D, and Williams FMK. (2018). Twelve-year follow-up of chronic pain in twins: Changes in environmental and genetic influence over time. Eur J Pain [DOI] [PubMed]
- Ceko M, Shir Y, Ouellet JA, Ware MA, Stone LS, and Seminowicz DA. (2015). Partial recovery of abnormal insula and dorsolateral prefrontal connectivity to cognitive networks in chronic low back pain after treatment. Hum Brain Mapp 36, 2075–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, et al. (2012). Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizh BA, Priestley T, Rowbotham M, and Schaffler K. (2009). Predicting therapeutic efficacy - experimental pain in human subjects. Brain Res Rev 60, 243–254. [DOI] [PubMed] [Google Scholar]
- Cobos EJ, Ghasemlou N, Araldi D, Segal D, Duong K, and Woolf CJ. (2012). Inflammation-induced decrease in voluntary wheel running in mice: a nonreflexive test for evaluating inflammatory pain and analgesia. Pain 153, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colagiuri B. (2010). Participant expectancies in double-blind randomized placebo-controlled trials: potential limitations to trial validity. Clin Trials 7, 246–255. [DOI] [PubMed] [Google Scholar]
- Corbett A, Husebo B, Malcangio M, Staniland A, Cohen-Mansfield J, Aarsland D, and Ballard C. (2012). Assessment and treatment of pain in people with dementia. Nat Rev Neurol 8, 264–274. [DOI] [PubMed] [Google Scholar]
- Corder G, Ahanonu B, Grewe BF, Wang D, Schnitzer MJ, and Scherrer G. (2019). An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe MA, Lawrence KS, Moulin DE, Morley-Forster P, Shokouhi M, Nielson WR, and Davis KD. (2017). Lower Functional Connectivity of the Periaqueductal Gray Is Related to Negative Affect and Clinical Manifestations of Fibromyalgia. Front Neuroanat 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KD, Bushnell MC, Iannetti GD, Lawrence K St, and Coghill R. (2015). Evidence against pain specificity in the dorsal posterior insula. F1000Res 4, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KD, Flor H, Greely HT, Iannetti GD, Mackey S, Ploner M, Pustilnik A, Tracey I, Treede RD, and Wager TD. (2017). Brain imaging tests for chronic pain: medical, legal and ethical issues and recommendations. Nat Rev Neurol 13, 624–638. [DOI] [PubMed] [Google Scholar]
- De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, Ossipov MH, Vanderah TW, Lai J, Dussor GO, et al. (2011). Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain 152, 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira Silva D, Rathleff MS, Petersen K, Azevedo FM, and Barton CJ. (2018). Manifestations of Pain Sensitization Across Different Painful Knee Disorders: A Systematic Review Including Meta-analysis and Metaregression. Pain Med [DOI] [PubMed]
- Dehghan M, Schmidt-Wilcke T, Pfleiderer B, Eickhoff SB, Petzke F, Harris RE, Montoya P, and Burgmer M. (2016). Coordinate-based (ALE) meta-analysis of brain activation in patients with fibromyalgia. Hum Brain Mapp 37, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, Bennett DL, and McMahon SB. (2017). Nerve Growth Factor and Pain Mechanisms. Annu Rev Neurosci 40, 307–325. [DOI] [PubMed] [Google Scholar]
- Denk F, McMahon SB, and Tracey I. (2014). Pain vulnerability: a neurobiological perspective. Nat Neurosci 17, 192–200. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, and Waxman SG. (2015). NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci 16, 511–519. [DOI] [PubMed] [Google Scholar]
- Dimov LF, Toniolo EF, Alonso-Matielo H, de Andrade DC, Garcia-Larrea L, Ballester G, Teixeira MJ, and Dale CS. (2018). Electrical stimulation of the insular cortex as a novel target for the relief of refractory pain: An experimental approach in rodents. Behav Brain Res 346, 86–95. [DOI] [PubMed] [Google Scholar]
- Doan NT, Kaufmann T, Bettella F, Jorgensen KN, Brandt CL, Moberget T, Alnaes D, Douaud G, Duff E, Djurovic S, et al. (2017). Distinct multivariate brain morphological patterns and their added predictive value with cognitive and polygenic risk scores in mental disorders. Neuroimage Clin 15, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud G, Menke RA, Gass A, Monsch AU, Rao A, Whitcher B, Zamboni G, Matthews PM, Sollberger M, and Smith S. (2013). Brain microstructure reveals early abnormalities more than two years prior to clinical progression from mild cognitive impairment to Alzheimer’s disease. J Neurosci 33, 2147–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff EP, Vennart W, Wise RG, Howard MA, Harris RE, Lee M, Wartolowska K, Wanigasekera V, Wilson FJ, Whitlock M, et al. (2015). Learning to identify CNS drug action and efficacy using multistudy fMRI data. Sci Transl Med 7. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, et al. (2005). Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 113, 9–19. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, Turk DC, Peirce-Sandner S, Burke LB, Farrar JT, Gilron I, Jensen MP, Katz NP, Raja SN, Rappaport BA, et al. (2012). Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. Pain 153, 1148–1158. [DOI] [PubMed] [Google Scholar]
- Eberhardt E, Havlicek S, Schmidt D, Link AS, Neacsu C, Kohl Z, Hampl M, Kist AM, Klinger A, Nau C, et al. (2015). Pattern of Functional TTX-Resistant Sodium Channels Reveals a Developmental Stage of Human iPSC- and ESC-Derived Nociceptors. Stem Cell Reports 5, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MJ, Adams RA, Brown H, Parees I, and Friston KJ. (2012). A Bayesian account of ‘hysteria’. Brain 135, 3495–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eippert F, Bingel U, Schoell ED, Yacubian J, Klinger R, Lorenz J, and Buchel C. (2009a). Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron 63, 533–543. [DOI] [PubMed] [Google Scholar]
- Eippert F, Finsterbusch J, Bingel U, and Buchel C. (2009b). Direct evidence for spinal cord involvement in placebo analgesia. Science 326, 404. [DOI] [PubMed] [Google Scholar]
- Eippert F, Kong YZ, Winkler AM, Andersson JL, Finsterbusch J, Buchel C, Brooks JCW, and Tracey I. (2017). Investigating resting-state functional connectivity in the cervical spinal cord at 3 T. Neuroimage 147, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, Marchini J, and Smith SM. (2018). Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhurst M, Fairhurst K, Berna C, and Tracey I. (2012). An fMRI Study Exploring the Overlap and Differences between Neural Representations of Physical and Recalled Pain. Plos One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JT, Troxel AB, Haynes K, Gilron I, Kerns RD, Katz NP, Rappaport BA, Rowbotham MC, Tierney AM, Turk DC, and Dworkin RH. (2014). Effect of variability in the 7-day baseline pain diary on the assay sensitivity of neuropathic pain randomized clinical trials: an ACTTION study. Pain 155, 1622–1631. [DOI] [PubMed] [Google Scholar]
- Fillingim RB, Bruehl S, Dworkin RH, Dworkin SF, Loeser JD, Turk DC, Widerstrom-Noga E, Arnold L, Bennett R, Edwards RR, et al. (2014). The ACTTION-American Pain Society Pain Taxonomy (AAPT): an evidence-based and multidimensional approach to classifying chronic pain conditions. J Pain 15, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerster BR, Petrou M, Edden RA, Sundgren PC, Schmidt-Wilcke T, Lowe SE, Harte SE, Clauw DJ, and Harris RE. (2012). Reduced insular gamma-aminobutyric acid in fibromyalgia. Arthritis Rheum 64, 579–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freynhagen R, Baron R, Gockel U, and Tolle TR. (2006). painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 22, 1911–1920. [DOI] [PubMed] [Google Scholar]
- Garcia-Larrea L, and Mauguiere F. (2018). Pain syndromes and the parietal lobe. Handb Clin Neurol 151, 207–223. [DOI] [PubMed] [Google Scholar]
- Gasparotti R, Padua L, Briani C, and Lauria G. (2017). New technologies for the assessment of neuropathies. Nat Rev Neurol 13, 203–216. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, Carlomagno Y, Daughrity LM, Jansen-West K, Perkerson EA, et al. (2017). Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuter S, Boll S, Eippert F, and Buchel C. (2017). Functional dissociation of stimulus intensity encoding and predictive coding of pain in the insula. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley P, Kurki MI, Hiekkala ME, Veerapen K, Happola P, Mitchell AA, Lal D, Palta P, Surakka I, Kaunisto MA, et al. (2018). Common Variant Burden Contributes to the Familial Aggregation of Migraine in 1,589 Families. Neuron 98, 743–753 e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwilym SE, Filippini N, Douaud G, Carr AJ, and Tracey I. (2010). Thalamic Atrophy Associated With Painful Osteoarthritis of the Hip Is Reversible After Arthroplasty A Longitudinal Voxel-Based Morphometric Study. Arthritis Rheum-Us 62, 2930–2940. [DOI] [PubMed] [Google Scholar]
- Gwilym SE, Keltner JR, Warnaby CE, Carr AJ, Chizh B, Chessell I, and Tracey I. (2009). Psychophysical and Functional Imaging Evidence Supporting the Presence of Central Sensitization in a Cohort of Osteoarthritis Patients. Arthrit Rheum-Arthr 61, 1226–1234. [DOI] [PubMed] [Google Scholar]
- Haanpaa M, Attal N, Backonja M, Baron R, Bennett M, Bouhassira D, Cruccu G, Hansson P, Haythornthwaite JA, Iannetti GD, et al. (2011). NeuPSIG guidelines on neuropathic pain assessment. Pain 152, 14–27. [DOI] [PubMed] [Google Scholar]
- Han J, Kwon M, Cha M, Tanioka M, Hong SK, Bai SJ, and Lee BH. (2015). Plasticity-Related PKMzeta Signaling in the Insular Cortex Is Involved in the Modulation of Neuropathic Pain after Nerve Injury. Neural Plast 2015, 601767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DE, Ichesco E, Schrepf A, Halvorson M, Puiu T, Clauw DJ, Harris RE, Harte SE, and Network MR. (2018a). Relationships between brain metabolite levels, functional connectivity, and negative mood in urologic chronic pelvic pain syndrome patients compared to controls: A MAPP research network study. Neuroimage Clin 17, 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DE, Ichesco E, Schrepf A, Hampson JP, Clauw DJ, Schmidt-Wilcke T, Harris RE, and Harte SE. (2018b). Resting Functional Connectivity of the Periaqueductal Gray Is Associated With Normal Inhibition and Pathological Facilitation in Conditioned Pain Modulation. J Pain 19, 635 e631–635 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RE, Napadow V, Huggins JP, Pauer L, Kim J, Hampson J, Sundgren PC, Foerster B, Petrou M, Schmidt-Wilcke T, and Clauw DJ. (2013). Pregabalin rectifies aberrant brain chemistry, connectivity, and functional response in chronic pain patients. Anesthesiology 119, 1453–1464. [DOI] [PubMed] [Google Scholar]
- Harris RE, Williams DA, McLean SA, Sen A, Hufford M, Gendreau RM, Gracely RH, and Clauw DJ. (2005). Characterization and consequences of pain variability in individuals with fibromyalgia. Arthritis Rheum 52, 3670–3674. [DOI] [PubMed] [Google Scholar]
- Hashmi JA, Baliki MN, Huang L, Baria AT, Torbey S, Hermann KM, Schnitzer TJ, and Apkarian AV. (2013). Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain 136, 2751–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes JD, and Rees G. (2006). Decoding mental states from brain activity in humans. Nat Rev Neurosci 7, 523–534. [DOI] [PubMed] [Google Scholar]
- HEAL https://www.nih.gov/research-training/medical-research-initiatives/heal-initiative.
- Hewitt DJ, Hargreaves RJ, Curtis SP, and Michelson D. (2009). Challenges in analgesic drug development. Clin Pharmacol Ther 86, 447–450. [DOI] [PubMed] [Google Scholar]
- Horing B, Sprenger C, Buchel C. (2018). Dorsal Posterior Insula Responses are Related to Pain and not Salience when Using Painful and Non-Painful Heat ans Salience-Matched Sound Stimuli In IASP World Congress on Pain (Boston, USA: ). https://www.iasp-pain.org/terminology. [Google Scholar]
- Huang L, Kutch JJ, Ellingson BM, Martucci KT, Harris RE, Clauw DJ, Mackey S, Mayer EA, Schaeffer AJ, Apkarian AV, and Farmer MA. (2016). Brain white matter changes associated with urological chronic pelvic pain syndrome: multisite neuroimaging from a MAPP case-control study. Pain 157, 2782–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, and Levine JD. (2007). Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 55, 365–376. [DOI] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, and Young T. (2012). An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153, 1837–1846. [DOI] [PubMed] [Google Scholar]
- Iannetti GD, Zambreanu L, Wise RG, Buchanan TJ, Huggins JP, Smart TS, Vennart W, and Tracey I. (2005). Pharmacological modulation of pain-related brain activity during normal and central sensitization states in humans. P Natl Acad Sci USA 102, 18195–18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo Y. (2001). Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum Mutat 18, 462–471. [DOI] [PubMed] [Google Scholar]
- IOM (2011). In Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research (Washington (DC)). [PubMed] [Google Scholar]
- Jirkof P, Fleischmann T, Cesarovic N, Rettich A, Vogel J, and Arras M. (2013). Assessment of postsurgical distress and pain in laboratory mice by nest complexity scoring. Lab Anim 47, 153–161. [DOI] [PubMed] [Google Scholar]
- Jones L, Fabrizi L, Laudiano-Dray M, Whitehead K, Meek J, Verriotis M, and Fitzgerald M. (2017). Nociceptive Cortical Activity Is Dissociated from Nociceptive Behavior in Newborn Human Infants under Stress. Curr Biol 27, 3846–3851 e3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetis D, Sassone J, Yang Y, Galbardi B, Xenakis MN, Westra RL, Szklarczyk R, Lindsey P, Faber CG, Gerrits M, et al. (2017). Network topology of NaV1.7 mutations in sodium channel-related painful disorders. BMC Syst Biol 11, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, and Kathiresan S. (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, and Porreca F. (2009). Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 12, 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch I. (2000). Are drug and placebo effects in depression additive? Biol Psychiatry 47, 733–735. [DOI] [PubMed] [Google Scholar]
- Kragel PA, Koban L, Barrett LF, and Wager TD. (2018). Representation, Pattern Information, and Brain Signatures: From Neurons to Neuroimaging. Neuron 99, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Woo CW, Chang LJ, Ruzic L, Gu X, Lopez-Sola M, Jackson PL, Pujol J, Fan J, and Wager TD. (2016). Somatic and vicarious pain are represented by dissociable multivariate brain patterns. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucyi A, and Davis KD. (2015). The dynamic pain connectome. Trends Neurosci 38, 86–95. [DOI] [PubMed] [Google Scholar]
- Langford DJ, Bailey AL, Chanda ML, Clarke SE, Drummond TE, Echols S, Glick S, Ingrao J, Klassen-Ross T, Lacroix-Fralish ML, et al. (2010). Coding of facial expressions of pain in the laboratory mouse. Nat Methods 7, 447–449. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, and Woolf CJ. (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10, 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc BW, Bowary PM, Chao YC, Lii TR, and Saab CY. (2016). Electroencephalographic signatures of pain and analgesia in rats. Pain 157, 2330–2340. [DOI] [PubMed] [Google Scholar]
- Lee J, Bscke R, Tang PC, Hartman BH, Heller S, and Koehler KR. (2018a). Hair Follicle Development in Mouse Pluripotent Stem Cell-Derived Skin Organoids. Cell Rep 22, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Mawla I, Kim J, Loggia ML, Ortiz A, Jung C, Chan ST, Gerber J, Schmithorst VJ, Edwards RR, et al. (2018b). Machine learning-based prediction of clinical pain using multimodal neuroimaging and autonomic metrics. Pain. [DOI] [PMC free article] [PubMed]
- Lee M, and Tracey I. (2013). Neuro-genetics of persistent pain. Curr Opin Neurobiol 23, 127–132. [DOI] [PubMed] [Google Scholar]
- Lee MC, Zambreanu L, Menon DK, and Tracey I. (2008). Identifying Brain Activity Specifically Related to the Maintenance and Perceptual Consequence of Central Sensitization in Humans. J Neurosci 28, 11642–11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leknes S, Berna C, Lee MC, Snyder GD, Biele G, and Tracey I. (2013). The importance of context: When relative relief renders pain pleasant. Pain 154, 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, and Mendell LM. (1993). Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci 13, 2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RL, Douaud G, Filippini N, Okell TW, Stagg CJ, and Tracey I. (2017). Structural Connectivity Variances Underlie Functional and Behavioral Changes During Pain Relief Induced by Neuromodulation. Sci Rep-Uk 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Latremoliere A, Li X, Zhang Z, Chen M, Wang X, Fang C, Zhu J, Alexandre C, Gao Z, et al. (2018). Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loggia ML, Kim J, Gollub RL, Vangel MG, Kirsch I, Kong J, Wasan AD, and Napadow V. (2013). Default mode network connectivity encodes clinical pain: an arterial spin labeling study. Pain 154, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loggia ML, Segerdahl AR, Howard MA, Tracey I. (2019). Imaging Clinically Relevant Pain States using Arterial Spin Labeling. Pain Reports. [DOI] [PMC free article] [PubMed]
- Lopez-Sola M, Woo CW, Pujol J, Deus J, Harrison BJ, Monfort J, and Wager TD. (2017). Towards a neurophysiological signature for fibromyalgia. Pain 158, 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe EM, Anand P, Terenghi G, Williams-Chestnut RE, Sinicropi DV, and Osborne JL. (1997). Increased nerve growth factor levels in the urinary bladder of women with idiopathic sensory urgency and interstitial cystitis. Br J Urol 79, 572–577. [DOI] [PubMed] [Google Scholar]
- Madhavan M, Nevin ZS, Shick HE, Garrison E, Clarkson-Paredes C, Karl M, Clayton BLL, Factor DC, Allan KC, Barbar L, et al. (2018). Induction of myelinating oligodendrocytes in human cortical spheroids. Nat Methods 15, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainero C, Boshyan J, and Hadjikhani N. (2011). Altered functional magnetic resonance imaging resting-state connectivity in periaqueductal gray networks in migraine. Ann Neurol 70, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciszewski KK, Meylakh N, Di Pietro F, Mills EP, Macefield VG, Macey PM, and Henderson LA. (2018). Changes in brainstem pain modulation circuitry function over the migraine cycle. J Neurosci [DOI] [PMC free article] [PubMed]
- Marquand A, Howard M, Brammer M, Chu C, Coen S, and Mourao-Miranda J. (2010). Quantitative prediction of subjective pain intensity from whole-brain fMRI data using Gaussian processes. Neuroimage 49, 2178–2189. [DOI] [PubMed] [Google Scholar]
- Matson DJ, Broom DC, Carson SR, Baldassari J, Kehne J, and Cortright DN. (2007). Inflammation-induced reduction of spontaneous activity by adjuvant: A novel model to study the effect of analgesics in rats. J Pharmacol Exp Ther 320, 194–201. [DOI] [PubMed] [Google Scholar]
- McIntosh AM, Hall LS, Zeng Y, Adams MJ, Gibson J, Wigmore E, Hagenaars SP, Davies G, Fernandez-Pujals AM, Campbell AI, et al. (2016). Genetic and Environmental Risk for Chronic Pain and the Contribution of Risk Variants for Major Depressive Disorder: A Family-Based Mixed-Model Analysis. PLoS Med 13, e1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloto CB, Benavides R, Lichtenwalter RN, Wen X, Tugarinov N, Zorina-Lichtenwalter K, Chabot-Dore AJ, Piltonen MH, Cattaneo S, Verma V, et al. (2018). Human pain genetics database: a resource dedicated to human pain genetics research. Pain 159, 749–763. [DOI] [PubMed] [Google Scholar]
- Mhuircheartaigh RN, Warnaby C, Rogers R, Jbabdi S, and Tracey I. (2013). Slow-Wave Activity Saturation and Thalamocortical Isolation During Propofol Anesthesia in Humans. Sci Transl Med 5. [DOI] [PubMed] [Google Scholar]
- Miller KL, Alfaro-Almagro F, Bangerter NK, Thomas DL, Yacoub E, Xu J, Bartsch AJ, Jbabdi S, Sotiropoulos SN, Andersson JL, et al. (2016). Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat Neurosci 19, 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, and Raichle ME. (2016). How networks communicate: propagation patterns in spontaneous brain activity. Philos Trans R Soc Lond B Biol Sci 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momi SK, Fabiane SM, Lachance G, Livshits G, and Williams FM. (2015). Neuropathic pain as part of chronic widespread pain: environmental and genetic influences. Pain 156, 2100–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moultrie F, Slater R, and Hartley C. (2017). Improving the treatment of infant pain. Curr Opin Support Palliat Care 11, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napadow V, Kim J, Clauw DJ, and Harris RE. (2012). Decreased intrinsic brain connectivity is associated with reduced clinical pain in fibromyalgia. Arthritis Rheum 64, 2398–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert H, Muirhead D, Kabir M, Grace C, Cleton A, and Arends R. (2013). Sequential protein and peptide immunoaffinity capture for mass spectrometry-based quantification of total human beta-nerve growth factor. Anal Chem 85, 1719–1726. [DOI] [PubMed] [Google Scholar]
- Norbury TA, MacGregor AJ, Urwin J, Spector TD, and McMahon SB. (2007). Heritability of responses to painful stimuli in women: a classical twin study. Brain 130, 3041–3049. [DOI] [PubMed] [Google Scholar]
- O’Muircheartaigh J, Marquand A, Hodkinson DJ, Krause K, Khawaja N, Renton TF, Huggins JP, Vennart W, Williams SC, and Howard MA. (2015). Multivariate decoding of cerebral blood flow measures in a clinical model of on-going postsurgical pain. Hum Brain Mapp 36, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit JJ, Andrade J, Bogod DG, Hitchman JM, Jonker WR, Lucas N, Mackay JH, Nimmo AF, O’Connor K, O’Sullivan EP, et al. (2014). 5th National Audit Project (NAP5) on accidental awareness during general anaesthesia: summary of main findings and risk factors. Br J Anaesth 113, 549–559. [DOI] [PubMed] [Google Scholar]
- Parr T, Rees G, and Friston KJ. (2018). Computational Neuropsychology and Bayesian Inference. Front Hum Neurosci 12, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KV, Allen R, Burke L, Farrar JT, Gewandter JS, Gilron I, Katz NP, Markman JD, Marshall SF, Resnick M, et al. (2018). Evaluation of composite responder outcomes of pain intensity and physical function in neuropathic pain clinical trials: an ACTTION individual patient data analysis. Pain 159, 2245–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploghaus A, Narain C, Beckmann CF, Clare S, Bantick S, Wise R, Matthews PM, Rawlins JNP, and Tracey I. (2001). Exacerbation of pain by anxiety is associated with activity in a hippocampal network. J Neurosci 21, 9896–9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploghaus A, Tracey I, Gati JS, Clare S, Menon RS, Matthews PM, and Rawlins JNP. (1999). Dissociating pain from its anticipation in the human brain. Science 284, 1979–1981. [DOI] [PubMed] [Google Scholar]
- Ploner M, and May ES. (2017). EEG and MEG in pain research - Current state and future perspectives. Pain. [DOI] [PubMed]
- Raij TT, Numminen J, Narvanen S, Hiltunen J, and Hari R. (2005). Brain correlates of subjective reality of physically and psychologically induced pain. Proc Natl Acad Sci U S A 102, 2147–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, and May A. (2009). Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J Neurosci 29, 13746–13750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrs EL, Neubert JK, Caudle RM, and Allen KD. (2018). Behavioral characteristics of capsaicin mediated cutaneous, myogenic, and arthrogenic orofacial nociception in rats. Arch Oral Biol 92, 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio-Baptista C, and Johansen-Berg H. (2017). White Matter Plasticity in the Adult Brain. Neuron 96, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrenk-Siemens K, Rosseler C, and Lampert A. (2018). Translational Model Systems for Complex Sodium Channel Pathophysiology in Pain. Handb Exp Pharmacol 246, 355–369. [DOI] [PubMed] [Google Scholar]
- Segerdahl AR, Mezue M, Okell TW, Farrar JT, and Tracey I. (2015a). The dorsal posterior insula is not an island in pain but subserves a fundamental role - Response to: “Evidence against pain specificity in the dorsal posterior insula” by Davis et al. F1000Res 4, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerdahl AR, Mezue M, Okell TW, Farrar JT, and Tracey I. (2015b). The dorsal posterior insula subserves a fundamental role in human pain. Nat Neurosci 18, 499–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerdahl AR, Themistocleous AC, Fido D, Bennett DL, and Tracey I. (2018). A brain-based pain facilitation mechanism contributes to painful diabetic polyneuropathy. Brain 141, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevcik MA, Ghilardi JR, Peters CM, Lindsay TH, Halvorson KG, Jonas BM, Kubota K, Kuskowski MA, Boustany L, Shelton DL, and Mantyh PW. (2005). Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain 115, 128–141. [DOI] [PubMed] [Google Scholar]
- Slater R, Cantarella A, Franck L, Meek J, and Fitzgerald M. (2008). How well do clinical pain assessment tools reflect pain in infants? PLoS Med 5, e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater R, Cornelissen L, Fabrizi L, Patten D, Yoxen J, Worley A, Boyd S, Meek J, and Fitzgerald M. (2010). Oral sucrose as an analgesic drug for procedural pain in newborn infants: a randomised controlled trial. Lancet 376, 1225–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, Andersen J, Pasca AM, Birey F, and Pasca SP. (2018). Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc 13, 2062–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Amtmann D, Askew RL, Gewandter JS, Hunsinger M, Jensen MP, McDermott MP, Patel KV, Williams M, Bacci ED, et al. (2016). Pain intensity rating training: results from an exploratory study of the ACTTION PROTECCT system. Pain 157, 1056–1064. [DOI] [PubMed] [Google Scholar]
- Smith SM, Dworkin RH, Turk DC, Baron R, Polydefkis M, Tracey I, Borsook D, Edwards RR, Harris RE, Wager TD, et al. (2017). The Potential Role of Sensory Testing, Skin Biopsy, and Functional Brain Imaging as Biomarkers in Chronic Pain Clinical Trials: IMMPACT Considerations. J Pain 18, 757–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni A, Wanigasekera V, Mezue M, Cooper C, Javaid MK, Price AJ, and Tracey I. (2018). Central sensitisation in knee osteoarthritis: Relating pre-surgical brainstem neuroimaging and PainDETECT based patient stratification to arthroplasty outcome. Arthritis Rheumatol. [DOI] [PMC free article] [PubMed]
- Sprenger C, Eippert F, Finsterbusch J, Bingel U, Rose M, and Buchel C. (2012). Attention modulates spinal cord responses to pain. Curr Biol 22, 1019–1022. [DOI] [PubMed] [Google Scholar]
- Stroman PW, Wheeler-Kingshott C, Bacon M, Schwab JM, Bosma R, Brooks J, Cadotte D, Carlstedt T, Ciccarelli O, Cohen-Adad J, et al. (2014). The current state-of-the-art of spinal cord imaging: Methods. Neuroimage 84, 1070–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram L, Gao H, Padigepati SR, McRae JF, Li Y, Kosmicki JA, Fritzilas N, Hakenberg J, Dutta A, Shon J, et al. (2018). Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50, 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri P, Palmer MR, Tsepilov YA, Freidin MB, Boer CG, Yau MS, Evans DS, Gelemanovic A, Bartz TM, Nethander M, et al. (2018). Genome-wide meta-analysis of 158,000 individuals of European ancestry identifies three loci associated with chronic back pain. PLoS Genet 14, e1007601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatu K, Costa T, Nani A, Diano M, Quarta DG, Duca S, Apkarian AV, Fox PT, and Cauda F. (2018). How do morphological alterations caused by chronic pain distribute across the brain? A meta-analytic co-alteration study. Neuroimage Clin 18, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault P, Baliki MN, Baria AT, Bauer WR, Schnitzer TJ, and Apkarian AV. (2018). Inferring distinct mechanisms in the absence of subjective differences: Placebo and centrally acting analgesic underlie unique brain adaptations. Hum Brain Mapp 39, 2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinnermann A, Geuter S, Sprenger C, Finsterbusch J, and Buchel C. (2017). Interactions between brain and spinal cord mediate value effects in nocebo hyperalgesia. Science 358, 105–108. [DOI] [PubMed] [Google Scholar]
- Tracey I. (2010). Getting the pain you expect: mechanisms of placebo, nocebo and reappraisal effects in humans. Nat Med 16, 1277–1283. [DOI] [PubMed] [Google Scholar]
- Tracey I, and Mantyh PW. (2007). The cerebral signature and its modulation for pain perception. Neuron 55, 377–391. [DOI] [PubMed] [Google Scholar]
- Tracey I, Ploghaus A, Gati JS, Clare S, Smith S, Menon RS, and Matthews PM. (2002). Imaging attentional modulation of pain in the periaqueductal gray in humans. J Neurosci 22, 2748–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB, et al. (2019). Chronic pain as a symptom or a disease: the IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 160, 19–27. [DOI] [PubMed] [Google Scholar]
- Upadhyay J, Baker SJ, Rajagovindan R, Hart M, Chandran P, Hooker BA, Cassar S, Mikusa JP, Tovcimak A, Wald MJ, et al. (2013). Pharmacological modulation of brain activity in a preclinical model of osteoarthritis. Neuroimage 64, 341–355. [DOI] [PubMed] [Google Scholar]
- Vachon-Presseau E, Centeno MV, Ren W, Berger SE, Tetreault P, Ghantous M, Baria A, Farmer M, Baliki MN, Schnitzer TJ, and Apkarian AV. (2016a). The Emotional Brain as a Predictor and Amplifier of Chronic Pain. J Dent Res 95, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon-Presseau E, Tetreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW, et al. (2016b). Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 139, 1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadodaria KC, Amatya DN, Marchetto MC, and Gage FH. (2018). Modeling psychiatric disorders using patient stem cell-derived neurons: a way forward. Genome Med 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardeh D, Mannion RJ, and Woolf CJ. (2016). Toward a Mechanism-Based Approach to Pain Diagnosis. J Pain 17, T50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehof J, Zavos HM, Lachance G, Hammond CJ, and Williams FM. (2014). Shared genetic factors underlie chronic pain syndromes. Pain 155, 1562–1568. [DOI] [PubMed] [Google Scholar]
- Veluchamy A, Hebert HL, Meng W, Palmer CNA, and Smith BH. (2018). Systematic review and meta-analysis of genetic risk factors for neuropathic pain. Pain 159, 825–848. [DOI] [PubMed] [Google Scholar]
- Visscher CM, Schouten MJ, Ligthart L, van Houtem CM, de Jongh A, and Boomsma DI. (2018). Shared Genetics of Temporomandibular Disorder Pain and Neck Pain: Results of a Twin Study. J Oral Facial Pain Headache 32, 107–112. [DOI] [PubMed] [Google Scholar]
- Wager TD, Atlas LY, Lindquist MA, Roy M, Woo CW, and Kross E. (2013). An fMRI-based neurologic signature of physical pain. N Engl J Med 368, 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, Buttermore ED, Oliveira JT, Mellin C, Lee S, Saber WA, Wang AJ, Ichida JK, Chiu IM, Barrett L, et al. (2015). Modeling pain in vitro using nociceptor neurons reprogrammed from fibroblasts. Nat Neurosci 18, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, King T, De Felice M, Guo W, Ossipov MH, and Porreca F. (2013). Descending facilitation maintains long-term spontaneous neuropathic pain. J Pain 14, 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanigasekera V, Duff E, Tracey I. (2018). Validation of an FMRI Based Classification Pipeline for Detecting Analgesic Efficacy from Neuroimaging Data In IASP World Congress on Pain (Boston, USA: ). [Google Scholar]
- Wanigasekera V, Lee MC, Rogers R, Kong YZ, Leknes S, Andersson J, and Tracey I. (2012). Baseline reward circuitry activity and trait reward responsiveness predict expression of opioid analgesia in healthy subjects. P Natl Acad Sci USA 109, 17705–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanigasekera V, Mezue M, Andersson J, Kong YZ, and Tracey I. (2016). Disambiguating Pharmacodynamic Efficacy from Behavior with Neuroimaging Implications for Analgesic Drug Development. Anesthesiology 124, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanigasekera V, Wartolowska K, Huggins JP, Duff EP, Vennart W, Whitlock M, Massat N, Pauer L, Rogers P, Hoggart B, and Tracey I. (2018). Disambiguating pharmacological mechanisms from placebo in neuropathic pain using functional neuroimaging. Brit J Anaesth 120, 299–307. [DOI] [PubMed] [Google Scholar]
- Wartolowska K, Hough MG, Jenkinson M, Andersson J, Wordsworth BP, and Tracey I. (2012). Structural changes of the brain in rheumatoid arthritis. Arthritis Rheum-Us 64, 371–379. [DOI] [PubMed] [Google Scholar]
- Watson CJ. (2016). Insular balance of glutamatergic and GABAergic signaling modulates pain processing. Pain 157, 2194–2207. [DOI] [PubMed] [Google Scholar]
- Wiech K. (2016). Deconstructing the sensation of pain: The influence of cognitive processes on pain perception. Science 354, 584–587. [DOI] [PubMed] [Google Scholar]
- Wiech K, and Tracey I. (2009). The influence of negative emotions on pain: Behavioral effects and neural mechanisms. Neuroimage 47, 987–994. [DOI] [PubMed] [Google Scholar]
- Wiech K, Vandekerckhove J, Zaman J, Tuerlinckx F, Vlaeyen JWS, and Tracey I. (2014). Influence of prior information on pain involves biased perceptual decision-making. Curr Biol 24, R679–R681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RG, Lujan BJ, Schweinhardt P, Peskett GD, Rogers R, and Tracey I. (2007). The anxiolytic effects of midazolam during anticipation to pain revealed using fMRI. Magn Reson Imaging 25, 801–810. [DOI] [PubMed] [Google Scholar]
- Wise RG, Williams P, and Tracey I. (2004). Using fMRI to quantify the time dependence of remifentanil an algesia in the human brain. Neuropsychopharmacol 29, 626–635. [DOI] [PubMed] [Google Scholar]
- Woo CW, Koban L, Kross E, Lindquist MA, Banich MT, Ruzic L, Andrews-Hanna JR, and Wager TD. (2014). Separate neural representations for physical pain and social rejection. Nat Commun 5, 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CW, Schmidt L, Krishnan A, Jepma M, Roy M, Lindquist MA, Atlas LY, and Wager TD. (2017). Quantifying cerebral contributions to pain beyond nociception. Nat Commun 8, 14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, and Ma Q. (2007). Nociceptors--noxious stimulus detectors. Neuron 55, 353–364. [DOI] [PubMed] [Google Scholar]
- Xu B, Magli A, Anugrah Y, Koester SJ, Perlingeiro RCR, and Shen W. (2018). Nanotopography-responsive myotube alignment and orientation as a sensitive phenotypic biomarker for Duchenne Muscular Dystrophy. Biomaterials 183, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mis MA, Estacion M, Dib-Hajj SD, and Waxman SG. (2018). NaV1.7 as a Pharmacogenomic Target for Pain: Moving Toward Precision Medicine. Trends Pharmacol Sci 39, 258–275. [DOI] [PubMed] [Google Scholar]
- Zambreanu L, Wise RG, Brooks JCW, Iannetti GD, and Tracey I. (2005). A role for the brainstem in central sensitisation in humans. Evidence from functional magnetic resonance imaging. Pain 114, 397–407. [DOI] [PubMed] [Google Scholar]
- Zorina-Lichtenwalter K, Meloto CB, Khoury S, and Diatchenko L. (2016). Genetic predictors of human chronic pain conditions. Neuroscience 338, 36–62. [DOI] [PubMed] [Google Scholar]
- Zunhammer M, Bingel U, Wager TD, and Placebo Imaging C (2018). Placebo Effects on the Neurologic Pain Signature: A Meta-analysis of Individual Participant Functional Magnetic Resonance Imaging Data. JAMA Neurol. [DOI] [PMC free article] [PubMed]