

Abstract
By using chronic lymphocytic leukemia as target for discovery in cancer pathogenesis we discovered that the great majority of CLLs (75-85%) carry a deletion of miR-15a and miR-16-1 at 13q14. We also discovered that miR-15/16 are negative regulators of the BCL2 oncogene. Thus the loss of the two negative regulators causes BCL2 overexpression and leukemia. A corollary of this is that CLL is very sensitive to the anti BCL2 drug venetoclax that can induce complete remission in CLL patients. Since leukemia patients may carry billions of leukemia cells, it is quite likely that some (few) of the leukemic cells are resistant to venetoclax. Thus, since microRNAs have multiple targets, we looked for other proteins that may be overexpressed in CLL because of the low of miR-15/16. We discovered that ROR1 an embryonal antigen expressed on most (~ 90%) CLL, but not on normal B cell, is also regulated by miR-15/16. Thus CLL cells are also sensitive to monoclonal antibodies against ROR1. Venetoclax and monoclonal antibodies against ROR1 act synergistically in killing CLL cells.
Keywords: CLL, BCL2, ROR1, miR-15/16, tsRNA, tRNA derived fragments
13q deletions in chronic lymphocytic leukemia, miR-15/16 and BCL2.
Chronic lymphocytic leukemia (CLL) is the most common leukemia in humans. Quite often CLL patients can live normally showing mild symptoms and do not need treatment for a number of years (Sgambati et al., 2001). CLL is a complex disease in which a rare population of CD5 positive B-cells is expanded (Bullrich and Croce, 2001; Sgambati et al., 2001). Most often CLL patients with high expression of ZAP-70 (Zeta-chain-associated protein kinase 70) and unmutated IgH VH gene show a clinically aggressive disease, while patients expressing low levels of ZAP-70 and mutated IgH VH do not need immediate treatment (Bullrich and Croce, 2001; Sgambati et al., 2001). The common chromosomal aberrations in CLL cells include deletions at 11q (18%), 17p (8%), 13q (60%), and trisomy 12 (12%−16%) (Dohner et al., 2000; Edelmann et al., 2012).
Multiple publications showed that 17p deletions result in inactivation of TP53 and miR-3676 - miR-4521 cluster (miR-3676 targets TCL1 an import and driver in aggressive CLL) (Balatti et al., 2015; Palamarchuk et al., 2012); trisomy 12 CLLs are associated with NOTCH1 (Notch homolog 1, translocation-associated (Drosophila)) gain of function mutations (Balatti et al., 2012); while ATM (ataxia-telangiectasia mutated) gene may be inactivated by 11q deletions (Bullrich et al., 1999). Interestingly, CLL patients with 11q and 17p deletions usually require immediate treatment while patients with 13q deletions often have indolent disease (Dohner et al., 2000).
The discovery of miR-15/16 as a target of 13q deletions in CLL and role its role in targeting BCL2 were reviewed multiple times. Briefly, in 2000-2002 we thought that 13q14 region deleted in most CLLs contains an important tumor suppressor gene. Despite the extensive effort by us and several other laboratories no protein coding gene inactivated in CLL was found (Bullrich et al., 2001; Mertens et al., 2002; Migliazza et al., 2001; Rondeau et al., 2001). At that time we identified several interesting CLL cases, one with a small 30 kb deletion, and another one with a translocation within this deletion. At that time since genomic databases were frequently updated and using one of these and we determined that a cluster of two microRNA genes, miR-15a and miR-16-1, was located in the 30 kb region (Calin et al., 2002), and miR-15/16 cluster was the only gene in that region. Subsequent studies revealed that miR-15/16 was the target of 13q deletions in CLL and that was the first example that a microRNA gene was involved in cancer (Calin et al., 2002).
Since miR-15/16 is a target of 13q14 deletions we thought that miR-15/16 could target important oncogene(s) in B-cells. We searched available databases and identified predicted miR-15/16 targets. One of the top predicted targets was BCL2, a critical oncogene overexpressed in most CLL cases (Cimmino et al., 2005). BCL2 has a very important role in the pathogenesis of solid cancers as well as lymphoid malignancies. Located mostly in mitochondria, BCL2 protein indices survival and decreases cell death by inhibiting the release of Cytochrome C into the cytoplasm (Grabow et al., 2012; Sanchez-Beato et al., 2003). By analyzing homology between miR-15/16 and the BCL2 mRNA we found that both miR-15 and miR-16 potentially target bases 3287 to 3279 in the 3’end of the BCL2 cDNA (Cimmino et al., 2005). By studying the expression levels of miR-15, miR-16 and BCL2 in CLL we found that expression of miR-15/16 was lowest in the samples expressing high levels of BCL2; and expression of miR-15/16 was highest in the samples expressing low levels of BCL2. We then confirmed that miR-15/16 directly targeted BCL2 by experiments and concluded that BCL2 overexpression in CLL is due the loss of miR-15/16 (Cimmino et al., 2005).
MicroRNAs can not be easily delivered into cancer cells, thus, at least at this time miR-15/16 is not suitable to use as a CLL drug. On the other hand, BCL2 is highly expressed in almost all CLL cells and thus represents a good target to develop anti cancer drugs. Recently, Abbott was able to develop a drug, an inhibitor of Bcl2 (Souers et al., 2013). Venetoclax (also called ABT-199) targets Bcl2 by inhibiting its protein-protein interactions. Venetoclax was used to treat previously treated relapsed patients showing 17p deletions, and even in these difficult to treat cases venetoclax had of 80% response rate (Croce and Reed, 2016).
ROR1, a new miR-15/16 target.
Dr. Kipps laboratory has discovered the receptor kinase like orphan receptor 1 (ROR1) and found that is an oncoembryonic antigen found on most CLL B-cells (approximately in 90% of CLL patients) and not on normal B-cells or normal adult tissues, except a small subgroup of B-cell precursors named hematogones (Baskar et al., 2008; Broome et al., 2011). Additionally, antibodies targeting ROR1 can inhibit ROR1 induced cell growth in cells expressing both ROR1 and Tcl1, such as human aggressive CLL (Widhopf et al., 2014). We analyzed the 3’ untranslated region (UTR) of the ROR1 gene with bioinformatic tools and found that ROR1 is a predicted target of miR-15/16. Thus, we carried out luciferase experiments and proved that miR-15/16 target the 3’ UTR of ROR1 (Rassenti et al., 2017).
Since previous studies have shown that approximately 90% of CLL patients are positive for ROR1 (Fukuda et al., 2008), but approximately 10% have low-to-negligible levels of ROR1, we investigated a cohort of ROR1 low and ROR1 high for the expression of microRNAs (Rassenti et al., 2017). These experiments demonstrated that the expression of ROR1 is inversely correlated with the expression of miR-15a and miR-16-1. ROR1 high and low cells had a significant difference in expression of miR-15a and miR-16-1 (Rassenti et al., 2017). By Western blotting, we investigated a Burkitt lymphoma cell line that does not express BCL2 (Raji) and a T-cell leukemic cell line that expresses BCL2 (Jurkat), and a panel of CLL cells from patients for the expression of ROR1 and Bcl2. The ROR1 high CLL cells expressed high levels of Bcl2, while ROR1 negative cells expressed very low levels of Bcl2 (Rassenti et al., 2017). At this point we quantitated the differentially expressed microRNA in the ROR1 low and high CLL samples shown a signature of 17 microRNAs can discriminate these two groups of CLL samples (Rassenti et al., 2017). The results of these experiments indicated that the determinants of the expression of ROR1 are miR-15a and miR-16-1. The miR-15/16 expression was low when ROR1 expression was high and miR-15/16 expression was high when ROR1 expression was low (Rassenti et al., 2017). At this point we wanted to establish the relationship between ROR1 and Bcl2 expressions and percentage of CLL B-cells harboring a 13q14 deletion. We analyzed an additional cohort of 35 cases of CLL for Bcl2 expression and percentage of CLL cells carrying the 13q deletion by FISH analysis. Our results showed that samples with low expression of ROR1 had low expression of intracellular Bcl2 and an average percentage of CLL B-cells carrying in 13q deletion of 22%; while CLL cases with high expression of ROR1 had higher expression of Bcl2 and had an average % of CLL B-cells with a 13q deletion of 55% (Rassenti et al., 2017).
The discovery that miR-15/16 target ROR1, in addition to BCL2, may initiate the development of new combination therapy by targeting the same leukemia cells through different biological pathways. Since normal adult tissues generally lack expression of ROR1 and only leukemia cells are express high levels of ROR1, due of the loss of the miR-15/16 expression, single agent treatment with anti-ROR1 monoclonal antibodies should not have negative side effects, and should target only the leukemia cells of most CLL patients. However, it is possible that some rare CLL cells may escape the drug effect due to loss or modulation of the expression of ROR1. Thus, a combination therapy with Venetoclax and anti-ROR1 antibodies (cirmtuzumab) should be able to eradicate CLL cells even in case of selection of a clone mutated for one of the targets that would lead to resistance to one of the two drugs (Rassenti et al., 2017).
Since loss of miR-15/16 is an initial event in CLL pathogenesis causing overexpression of Bcl2 and ROR1, we hypothesized that combination therapy with molecules targeting each of these proteins would have synergistic effect. To investigate if cirmtuzumab can kill CLL cells, we analyzed viability of CLL cells. We carried out these experimants with and without venetoclax to evaluate for the synergic activity of venetoclax and cirmtuzumab CLL cells expressing high levels of ROR1. After 16 hours of treatment, the combination of venetoclax and cirmtuzumab was significantly more cytotoxic than treatment with venetoclax alone, cirmtuzumab alone or in combination with control human antibody (Rassenti et al., 2017). At 16 hours of treatment cirmtuzumab alone did not significantly affect CLL cells viability. Treatment with venetoclax alone resulted in about 50% of cell death, while addition of cirmtuzumab resulted in about 75% of cell death, suggesting that venetoclax and cirmtuzumab have a synergic effect (Rassenti et al., 2017).
MiR-15/16 deletion causes leukemia in mice.
Tumor suppressor function of any coding or noncoding gene is never fully evidenced until their deletion in mice results in a tumor phenotype. To prove that miR-15/16 is bona fide tumor suppressor Klein at al in 2010 inactivated miR-15/16 in mice (Klein et al., 2010). They generated two knockout alleles: one of the alleles had small deletion of miR-15/16 only; another allele was called MDR (minimal deleted region) had miR-15/16 and the neighboring Dleu2 gene deleted (Klein et al., 2010). Both mouse strains developed CLL like disease at the age of 18 months. Malignant B-cells were CD5, CD19 and IgM positive, and penetrance was ~25% for miR-15/16 knockouts and ~40% for MDR strain. Interestingly, the knockout of MDR allele had more severe phenotype then that of miR-15/16 only implying that additional regulatory elements in MDR may also contribute to CLL pathogenesis (Klein et al., 2010).
There are two miR-15/16 clusters in humans, miR-15a/16-1 and miR-15b/16-2. MiR-15/16 and MDR mice described above were generated by deleting miR-15a/16-1 cluster. Very recently we generated a mouse expressing no miR-15/16 by crossing miR-15a/16-1 and miR-15b/16-2 knockouts (DKO) (Lovat et al., 2018). Northern Blot analysis confirmed complete absence of miR-15/16 expression in splenocytes of these mice. DKO mice showed a significant reduction of overall survival (65% at the age of 12 months). Additional examination determined that DKO mice developed ignificant splenomegaly (Lovat et al., 2018). We then carried out histological analysis and to our surprise 77% of DKO mice developed a disease very similar to human AML while 23% developed B-cell malignancies. 20% of DKO mice also developed extra-splenic AML related disease, which involved livers and lungs. Using flow cytometry analysis we were able to confirm AML phenotype. While CD11b/Gr1 double positive cells we 1% in spleens and 40% in bone marrow of control mice, DKO spleens contained 10-15% CD11b/Gr1 double positive cells while DKO bone marrow contained 60-70% such cells (Lovat et al., 2018).
TsRNAs, a new class of small noncoding RNAs involved in CLL.
TCL1 (T-cell leukemia/lymphoma 1) oncogene is a critical gene in the pathogenesis of aggressive CLL. We and others previously showed that transgenic mice overexpressing TCL1 in B-cells develop the aggressive form of CLL (Allegra et al., 2014; Bichi et al., 2002; Yan et al., 2006). TCL1 overexpression transforms B-cells by activating Akt and inhibiting AP-1, Dnmt3A and Dnmt3B (Palamarchuk et al., 2012; Pekarsky et al., 2000; Pekarsky et al., 2008). While studying the regulation of TCL1 expression, we identified the microRNA cluster miR-4521/3676 and determined that miR-3676 is a powerful regulator of TCL1 expression that targets three consecutive 28-bp repeats within the 3’ UTR of TCL1 (Balatti et al., 2015). Subsequently we were studying in detail the miR-4521/3676 cluster, because of its proximity to the TP53 gene (this region includes TP53 and miR-4521/3676 and is deleted in 7-10% of CLLs). We found that these two microRNAs are associated with tRNA sequences and represent a recently identified class of small noncoding RNAs, tsRNAs (Pekarsky et al., 2016). TsRNAs is a new class of small RNAs derived from tRNAs during tRNA processing (Haussecker et al., 2010). tRNAs are expressed from tRNA genes by RNA polymerase III. TsRNAs are molecules produced from the 3’ end of pre-tRNAs by endonuclease RNase Z, they are unique sequences starting at the 3’ ends of tRNAs and ending at the stop signal for RNA polymerase III (four consecutive T nucleotides) (Haussecker et al., 2010; Martens-Uzunova et al., 2013).
Since tsRNAs are physically similar to piRNAs (they are both single-stranded short RNAs containing no secondary structures), we investigated if the tsRNAs derived from this region (ts-3676 and ts-4521) can also function as piRNAs (Pekarsky et al., 2016). We carried out an RNA immunoprecipitation experiment and determined that ts-3676 and ts-4521 can bind PiwiL2. Indeed PiwiL2 complexes were enriched with ts-3676 and ts-4521 but not with control microRNAs. At the same time both, ts-3676 and ts-4521 and control microRNAs were present in Ago1 and Ago2 complexes as expected (Pekarsky et al., 2016). These results demonstrate that the miR-4521/3676 locus expresses two small RNAs, ts-3676 and ts-4521, which may function as microRNAs as well as piRNAs by binding to PiwiL2 (Pekarsky et al., 2016).
We then investigated expression of ts-3676 and ts-4521 in CLL and lung cancer. In CLL we found that ts-3676 and ts-4521 are down-regulated in all cytogenetic CLL groups (17p deleted, 11q deleted, 13q deleted and normal karyotype) (Pekarsky et al., 2016). To determine the expression of ts-3676 and ts-4521 in lung cancer, we used 17 lung cancer samples and matched normal lung tissues. Real-time RT-PCR experiments revealed a drastic down-regulation of ts-3676 and ts-4521 expression in lung cancer samples compared to matched normal lung controls. Sequencing analysis of ~500 CLL samples and ~300 lung cancer samples revealed that ts-3676 and ts-4521 are mutated in ~1% of CLLs and 2% of lung cancer samples (Pekarsky et al., 2016).
Thus, to our knowledge, we identified first two tsRNAs (produced from 3’ end of pre tRNAs) involved in cancer. Our data established that ts-3676 and ts-4521 do not originate from the processing that involves microRNAs (Drosha cleavage followed by Dicer cleavage), but derive from a Thr tRNA and a Ser tRNA respectively (Pekarsky et al., 2016). Similarly to miR-15/16, the first two microRNAs found altered in cancer and inactivated in 13q deleted CLL cases, ts-3676 and ts-4521 are inactivated in 17p deleted CLL cases (Balatti et al., 2015). While miR-15/16 target BCL2 and ROR1, critical genes in CLL pathogenesis, ts-3676 targets TCL1, an oncogene critical in aggressive CLL (Balatti et al., 2015).
Since ts-3676 and ts-4521 are down-regulated in CLL and lung cancer, we decided to determine if other tsRNAs can be differentially expressed in cancer. We retrieved all tRNA sequences from genomic databases, generated a list of all tsRNAs by isolating unique sequences starting from the 3’ end of tRNAs and ending with the four-T RNA Polymerase III stop signal. Our list contained ~120 unique tsRNA sequences (Pekarsky et al., 2016). Using this list we designed a microarray chip with a goal of studying expression patterns of tsRNAs in a variety of tumor tissues and normal controls. First we studied tsRNA expression signatures in CLL. We selected 11 indolent CLL patient samples, 12 aggressive CLL patient samples and 8 normal controls and carried out a microarray experiment using a tsRNA chip we designed. We found that 15 tsRNAs were significantly up- or down-regulated in indolent CLL versus aggressive CLL comparison. In addition, nine tsRNAs were significantly differentially expressed in aggressive CLL compared normal controls; and that 10 tsRNAs were dysregulated in indolent CLL versus normal controls (Pekarsky et al., 2016). To investigate tsRNA are signatures in lung cancer, we carried out a similar experiment using seven lung cancer samples and five normal paired lung tissue samples. This resulted in the identification of six tsRNAs dysregulated in lung cancer samples versus normal lung tissues (Pekarsky et al., 2016). Interestingly, ts-46 and ts-47 were significantly down-regulated in lung cancer samples versus normal lung tissues, and in aggressive CLL when compared to normal controls. This indicates that ts-46 and ts-47 could potentially have tumor suppressor functions (Pekarsky et al., 2016). To confirm these observations in another study we showed that both, ts-46 and ts-47 suppress colonies formation is lung cancer cell lines (Balatti et al., 2017). In a subsequent study we investigated 14 paired samples (tumor and normal surrounding tissue) from 7 colon adenoma patients and 16 paired samples (tumor and normal surrounding tissue) from 8 patients with colon adenocarcinoma. This resulted in a signature of 8 tsRNAs for colon adenoma and another signature of 7 tsRNAs for colon adenocarcinoma and a signature (Balatti et al., 2017). Interestingly ts-3676 and ts-4521 were inactivated in adenomas but not in adenocarcinomas. Both comparisons showed up- regulation of Ts-40 in colon cancer indicating possible oncogenic function for this tsRNA (Balatti et al., 2017). In another experiment we studied tsRNA signature in human lymphocytes with and without activation of c-MYC. Among 15 differentially expressed tsRNAs, ts-47 was the most down-regulated by c-MYC overexpression, indicating that c-MYC may be responsible for don-regulation of ts-47 in CLL and lung cancer (Balatti et al., 2017).
Figure 1. MiR-15/16, BCL2 and ROR1 in CLL.
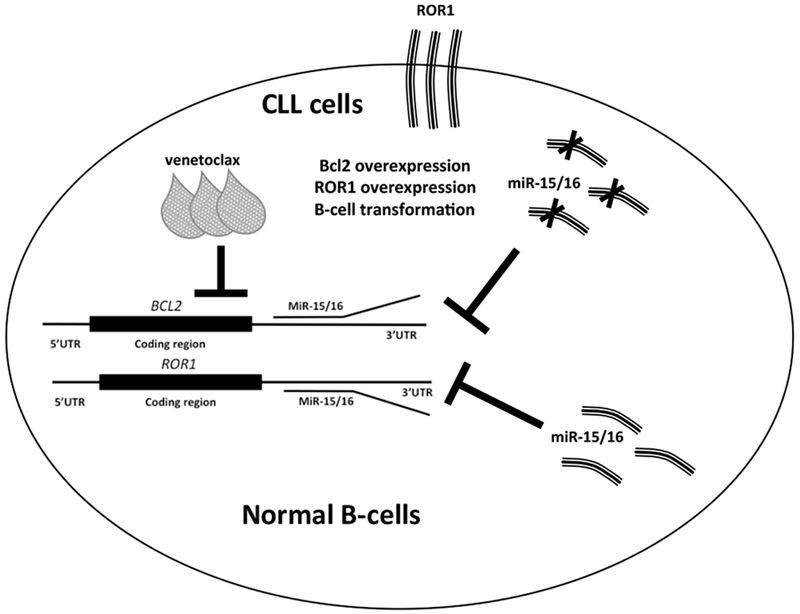
In normal B-cells miR-15/16 inhibit ROR1 and BCL2. In CLL cells miR-15/16 is deleted, ROR1 and BCL2 are overexpressed causing B-cell transformation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- Allegra D, Bilan V, Garding A, Dohner H, Stilgenbauer S, Kuchenbauer F, Mertens D, 2014. Defective DROSHA processing contributes to downregulation of MiR-15/−16 in chronic lymphocytic leukemia. Leukemia 28(1), 98–107. [DOI] [PubMed] [Google Scholar]
- Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, Pekarsky Y, Croce CM, 2012. NOTCH1 mutations in CLL associated with trisomy 12. Blood 119(2), 329–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatti V, Nigita G, Veneziano D, Drusco A, Stein GS, Messier TL, Farina NH, Lian JB, Tomasello L, Liu CG, Palamarchuk A, Hart JR, Bell C, Carosi M, Pescarmona E, Perracchio L, Diodoro M, Russo A, Antenucci A, Visca P, Ciardi A, Harris CC, Vogt PK, Pekarsky Y, Croce CM, 2017. tsRNA signatures in cancer. Proceedings of the National Academy of Sciences of the United States of America 114(30), 8071–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatti V, Rizzotto L, Miller C, Palamarchuk A, Fadda P, Pandolfo R, Rassenti LZ, Hertlein E, Ruppert AS, Lozanski A, Lozanski G, Kipps TJ, Byrd JC, Croce CM, Pekarsky Y, 2015. TCL1 targeting miR-3676 is codeleted with tumor protein p53 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America 112(7), 2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, Staudt LM, Wilson WH, Wiestner A, Rader C, 2008. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 14(2), 396–404. [DOI] [PubMed] [Google Scholar]
- Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, Russo G, Hardy RR, Croce CM, 2002. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A 99(10), 6955–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome HE, Rassenti LZ, Wang HY, Meyer LM, Kipps TJ, 2011. ROR1 is expressed on hematogones (non-neoplastic human B-lymphocyte precursors) and a minority of precursor-B acute lymphoblastic leukemia. Leukemia research 35(10), 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullrich F, Croce C, 2001. Molecular Biology of Chronic Lymphocytic Leukemia. Chronic Lymphocytic Leukemias, Second Edition, Revised and Expanded, Bruce Cheson , Ed. Dekker Marcel, Inc. New York, 9–32. [Google Scholar]
- Bullrich F, Fujii H, Calin G, Mabuchi H, Negrini M, Pekarsky Y, Rassenti L, Alder H, Reed JC, Keating MJ, Kipps TJ, Croce CM, 2001. Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer research 61(18), 6640–6648. [PubMed] [Google Scholar]
- Bullrich F, Rasio D, Kitada S, Starostik P, Kipps T, Keating M, Albitar M, Reed JC, Croce CM, 1999. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res 59(1), 24–27. [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM, 2002. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99(24), 15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM, 2005. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102(39), 13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM, Reed JC, 2016. Finally, An Apoptosis-Targeting Therapeutic for Cancer. Cancer research 76(20), 5914–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, Dohner K, Bentz M, Lichter P, 2000. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343(26), 1910–1916. [DOI] [PubMed] [Google Scholar]
- Edelmann J, Holzmann K, Miller F, Winkler D, Buhler A, Zenz T, Bullinger L, Kuhn MW, Gerhardinger A, Bloehdorn J, Radtke I, Su X, Ma J, Pounds S, Hallek M, Lichter P, Korbel J, Busch R, Mertens D, Downing JR, Stilgenbauer S, Dohner H, 2012. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 120(24), 4783–4794. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Chen L, Endo T, Tang L, Lu D, Castro JE, Widhopf GF 2nd, Rassenti LZ, Cantwell MJ, Prussak CE, Carson DA, Kipps TJ, 2008. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proceedings of the National Academy of Sciences of the United States of America 105(8), 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabow S, Waring P, Happo L, Cook M, Mason KD, Kelly PN, Strasser A, 2012. Pharmacological blockade of Bcl-2, Bcl-x(L) and Bcl-w by the BH3 mimetic ABT- 737 has only minor impact on tumour development in p53-deficient mice. Cell Death Differ 19(4), 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA, 2010. Human tRNA-derived small RNAs in the global regulation of RNA silencing. Rna 16(4), 673–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R, 2010. The DLEU2/miR- 15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17(1), 28–40. [DOI] [PubMed] [Google Scholar]
- Lovat F, Fassan M, Sacchi D, Ranganathan P, Palamarchuk A, Bill M, Karunasiri M, Gasparini P, Nigita G, Distefano R, Veneziano D, Dorrance AM, Garzon R, Croce CM, 2018. Knockout of both miR-15/16 loci induces acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens-Uzunova ES, Olvedy M, Jenster G, 2013. Beyond microRNA--novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer letters 340(2), 201–211. [DOI] [PubMed] [Google Scholar]
- Mertens D, Wolf S, Schroeter P, Schaffner C, Dohner H, Stilgenbauer S, Lichter P, 2002. Down-regulation of candidate tumor suppressor genes within chromosome band 13q14.3 is independent of the DNA methylation pattern in B-cell chronic lymphocytic leukemia. Blood 99(11), 4116–4121. [DOI] [PubMed] [Google Scholar]
- Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E, Guccione E, Qu X, Chien M, Murty VV, Gaidano G, Inghirami G, Zhang P, Fischer S, Kalachikov SM, Russo J, Edelman I, Efstratiadis A, Dalla-Favera R, 2001. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood 97(7), 2098–2104. [DOI] [PubMed] [Google Scholar]
- Palamarchuk A, Yan PS, Zanesi N, Wang L, Rodrigues B, Murphy M, Balatti V, Bottoni A, Nazaryan N, Alder H, Rassenti L, Kipps TJ, Freitas M, Croce CM, Pekarsky Y, 2012. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL). Proceedings of the National Academy of Sciences of the United States of America 109(7), 2555–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Balatti V, Palamarchuk A, Rizzotto L, Veneziano D, Nigita G, Rassenti LZ, Pass HI, Kipps TJ, Liu CG, Croce CM, 2016. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proceedings of the National Academy of Sciences of the United States of America 113(18), 5071–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, Russo G, Tsichlis P, Croce CM, 2000. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proceedings of the National Academy of Sciences of the United States of America 97(7), 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Palamarchuk A, Maximov V, Efanov A, Nazaryan N, Santanam U, Rassenti L, Kipps T, Croce CM, 2008. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Proceedings of the National Academy of Sciences of the United States of America 105(50), 19643–19648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassenti L, Balatti V, Ghia EM, Palamarchuk A, Tomasello L, Fadda P, Pekarsky Y, Widhopf Ii GF, Kipps T, Croce CM, 2017. MicroRNA dysregulation to identify therapeutic target combinations for Chronic Lymphocytic Leukemia. Proc Natl Acad Sci U S A 114(40), 10731–10736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondeau G, Moreau I, Bezieau S, Petit JL, Heilig R, Fernandez S, Pennarun E, Myers JS, Batzer MA, Moisan JP, Devilder MC, 2001. Comprehensive analysis of a large genomic sequence at the putative B-cell chronic lymphocytic leukaemia (B- CLL) tumour suppresser gene locus. Mutat Res 458(3–4), 55–70. [DOI] [PubMed] [Google Scholar]
- Sanchez-Beato M, Sanchez-Aguilera A, Piris MA, 2003. Cell cycle deregulation in B-cell lymphomas. Blood 101(4), 1220–1235. [DOI] [PubMed] [Google Scholar]
- Sgambati M, Linet M, Devesa S, 2001. Chronic Lymphocytic Leukemia, Epidemiological, Familial, and Genetic Aspects. Chronic Lymphocytic Leukemias, Second Edition, Revised and Expanded, Bruce Cheson , Ed. Dekker Marcel, Inc. New York, 33–62. [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW, 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine 19(2), 202–208. [DOI] [PubMed] [Google Scholar]
- Widhopf GF 2nd, Cui B, Ghia EM, Chen L, Messer K, Shen Z, Briggs SP, Croce CM, Kipps TJ, 2014. ROR1 can interact with TCL1 and enhance leukemogenesis in Emu-TCL1 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 111(2), 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XJ, Albesiano E, Zanesi N, Yancopoulos S, Sawyer A, Romano E, Petlickovski A, Efremov DG, Croce CM, Chiorazzi N, 2006. B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 103(31), 11713–11718. [DOI] [PMC free article] [PubMed] [Google Scholar]