

Abstract
The NLRP3 inflammasome is activated in the cytoplasm of cells and its products such as IL-1β are exported through a non-classical ER-Golgi pathway. Several mechanistically distinct models including exocytosis of secretory lysosomes, microvesicles (MVs) and extracellular vehicles (EVs) have been proposed for their release. In this study, we hypothesized that the NLRP3 inflammasome product, IL-1β in response to exogenously administrated and endogenously produced D-ribose stimulation is released via extracellular vesicles including EVs via a sphingolipid-mediated molecular mechanisms controlling lysosome and multivesicular body (MVB) interaction. First, we demonstrated that both endogenous and exogenous D-ribose induced NLRP3 inflammasome activation to produce IL-1β, which was released via EVs in podocytes. Then, we found that colocalization of marker MVB marker VPS16 with IL-1β within podocytes increased upon D-ribose stimulation, which was accompanied by decreased colocalization of lysosome marker Lamp-1 and VPS16, suggesting decrease in MVB inclusion of IL-1β due to reduced lysosome and MVB interaction. All these changes were mimicked and accelerated by lysosome v-ATPase inhibitor, bafilomycin. Moreover, ceramide in podocytes was found elevated upon D-ribose stimulation, and prior treatments of podocyte with acid sphingomyelinase (Asm) inhibitor, amitriptyline, acid ceramidase (AC) inducer, genistein, or AC CRISPR/cas9 activation plasmids were found to decrease D-ribose-induced ceramide accumulation, EVs release and IL-1β secretion due to reduced interactions of lysosome with MVBs. These results suggest that inflammasome-derived products such as IL-1β during D-ribose stimulation are released via EVs, in which lysosomal sphingolipid-mediated regulation of lysosome function plays an important role.
Keywords: Extracellular vehicles, Inflammasome, Ceramide, D-Ribose, IL-1β, Podocytes, Glomeruli
1. Introduction
D-Ribose is a naturally occurring monosaccharide known as “molecular currency” because of its role in intracellular energy transfer [1]. D-Ribose has been used as supplemental therapy for replenishing ATP levels in certain pathologic conditions such as, chronic fatigue syndrome [2] fibromyalgia and myocardial dysfunction [3]. Recent studies demonstrated D-ribose induced intracellular ROS production and NF-κB activation leading to endothelial dysfunction and cognitive impairment, in particular of diabetic encephalopathy [4]. Our studies in mice reported that D-ribose-induced mesangial cell damage and renal dysfunction caused nephropathy mediated via RAGE-dependent NF-κB signaling pathway [5]. Prolonged administration of D-ribose in mice induces NLRP3 inflammasome activation in podocytes, contributing to podocyte injury and consequent glomerular sclerosis via AGE-RAGE signaling pathway. In this regard various pathological stimulations initiates formation and activation of Nlrp3 inflammasome leading to the autocatalysis and activation of caspase-1, which in turn lead to maturation of pro-inflammatory cytokines, such as bioactive interleukin-1β (IL-1β) and interleukin-18 (IL-18). This NLRP3 inflammasome activation has been considered as an intracellular machinery to trigger inflammatory response to lead to chronic inflammation or degenerative injury in a variety of tissues and organs, such as kidney [6] and liver [7]. Among inflammasome products, IL-1β is a primary proin- flammatory cytokine mainly involved in inflammatory processes during various pathological conditions, such as pyroptosis [8], and IL-1β was often used as a common prototype product in studies on NLRP3 inflammasomes [9]. The mechanistic pathway for the secretion of active IL-1β produced by D-ribose-induced NLRP3 inflammasome in podocytes to result in glomerular injury, which may be a crucial step for triggering D-ribose-mediated nephropathy during diabetes.
Over the last two decades, it has become clear that cells can release vesicles that harbor and deliver functional molecules to recipient cells, in particular, extracellular vehicles (EVs) that are originated from the luminal membrane of multivesicular bodies (MVBs) [10–12]. Earlier in 1990, Rubartelli et al. in human monocytes showed that IL-1β is actively secreted via intracellular vesicles involving translocation of intracellular membranes which is different from classical endoplasmic reticulum-Golgi route [13]. After a decade, exocytosis of proIL-1β containing vesicles was reported for the transport of cytosolic IL-1β out of the cell, a secretory route regulated by various factors like ATP and osmotic conditions [14]. However, further studies are needed to clarify whether the secretion of IL-1β produced via NLRP3 inflammasome activation is via enhanced EVs release under pathological conditions. It is now well known that EVs are small vesicles (30–100 nm) of endosomal origin delimited by a lipid bilayer, and their secretion maintains cellular homeostasis in its producing cells by exporting various unnecessary or harmful materials [15], and functions as an alternate disposal pathway to lysosomes [16]. EVs were recognized to be an important medium for cellular communication. EVs regulate various pathological and physiological processes by delivering mRNAs, miRNAs, and a variety of proteins to receptor cells [17]. The biogenesis of EVs and their release are regulated by various factors. One of important regulators in sphingolipid signaling pathway is ceramide, which was associated with MVBs formation and EVs release [18,19]. Ceramide has also been reported to initiate NLRP3 inflammasome formation and activation [20,21], and promote NLRP3-dependent IL-1β production [22]. Based on these results, we wondered whether the sphingolipid metabolism or ceramide is involved in EVs formation or release, in particular, under different pathological conditions.
To answer this question, the present study hypothesized that sphingolipid metabolism regulates lysosome trafficking and fusion with MVBs and thereby controls EVs release. IL-1β produced by D-ribose-induced NLRP3 inflammasome activation was released via EVs, which is regulated by lysosomal sphingolipid-mediated signaling pathway. We first examined whether both exogenously administrated D-ribose and endogenously produced D-ribose-induced NLRP3 inflammasome activation and accompanied increases in EVs release in podocytes. Then, we observed whether D-ribose enhanced EVs release is due to dysregulation of AC-ceramide mediated lysosome function, in particular, the interaction of lysosome with MVBs. Finally, we performed in vivo experiments to confirm the involvement of enhanced IL-1β release due to decreases in lysosome-MBVs interactions in podocyte dysfunction and glomerular injury. Our results together demonstrated that IL-1β produced by NLRP3 inflammasome activation in podocytes upon D-ribose stimulation is indeed increasingly released via EVs that is controlled by lysosomal sphingolipid pathway.
2. Materials and methods
2.1. Animals
Eight-week-old, male C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) were intraperitoneally (i.p.) injected vehicle or D-ribose (dissolved in 0.9% saline) at a dose of 2 g/kg BW, once a day, for 30 days. Then, mice were sacrificed and their kidneys were harvested. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the Virginia Commonwealth University.
2.2. Cell culture
A conditionally immortalized mouse podocyte cell line (Graciously provided by Dr. P. E. Klotman, Division of Nephrology, Department of Medicine, Mount Sinai School of medicine, New York, NY), was cultured and maintained as described previously [23]. For all experiments, culture medium was replaced with serum-free medium prior to treatments. Podocytes were incubated with D-ribose (25 mM, Sigma, USA) or phosphonoacetic acid (1.0 mM, Sigma Aldrich, USA) for 24 h. Acid ceramidase (AC), AC inducer genistein (genis, 20 μM, Sigma Aldrich, USA) and acid sphingomyelinase (Asm) inhibitor, amitriptyline (Ami, 20 μM, Sigma, St. Louis, MO, USA) were used 30 min prior to treatments [24,25].
2.3. Western blot analysis
Equal amount of protein was resolved on SDS-PAGE gels and transferred to PVDF membrane. After blocking, membranes were incubated with primary antibodies rabbit anti ASC (1:1000, cell signaling technology, USA), rabbit anti-Cle-Caspase-1 (1:1000, cell signaling technology, USA), rabbit anti-pro-Caspase-1 (1:1000, Abcam, Cambridge, MA, USA), rabbit anti-acid ceramidase antibody (1:1000, LSBio, USA) and rabbit anti-β-actin (1:10000, Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4 °C. After overnight incubation, membranes were washed and incubated with donkey anti-rabbit-HRP IgG (1:5000, Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at room temperature. Finally, bands were detected by chemiluminescence technique using LI-COR Odyssey Fc and the band intensity of target proteins were normalized to β-actin and calculated with Image J software version 1.44p (NIH, Bethesda, MD, USA).
2.4. Assays of Caspase-1 activity and IL-1β production
Cell culture medium was used to measure Caspase-1 activity using a commercial colorimetric assay kit (Biovision, Mountain View, CA), and IL-1β production was detected with mouse IL-1β ELISA kit (R&D systems, USA) according to manufacturer’s instructions.
2.5. CRISPR/Cas9 activation plasmid transfection
An AC CRISPR/Cas9 activation plasmid was purchased from Santa Cruz Biotechnology, Dallas, TX, USA. Plasmid transfection was performed with the Polyethylenimine (Polyscience Inc., Eppelheim, Germany) according to manufacturer’s instructions.
2.6. Immunofluorescence microscopy
After treatments, kidney slides and podocyte culture coverslips (cell density is 1000/well in 24-well-plate) were fixed, blocked and incubated with primary antibodies against NLRP3 (1:100, Abcam, Cambridge, MA, USA), cleaved-caspase-1 (1:200, Santa Cruz Biotechnology, Dallas, TX, USA), VPS16 (1:100, Proteintech Group, Chicago, IL, USA), IL-1β (1:200, R&D systems, USA), Lamp-1(1:100, Abcam, Cambridge, MA, USA) at 4 °C overnight. Then samples were incubated with corresponding secondary antibodies with either Alexa- 488- or Alexa-555-labeled (Invitrogen) for 1 h at room temperature in the dark room. Finally, samples were mounted with mounting medium containing DAPI sealed with nail polish and pictures were taken under confocal laser scanning microscope (Fluoview FV1000; Olympus, Tokyo, Japan). There were 5–6 slides in every group, and 3 to 5 frames were chosen in every sample at random to show the characteristics of cell statues. Co-localization coefficient was analyzed with Image Pro Plus 6.0 software (Media Cybernetics, Bethesda, MD) and expressed as Pearson’s correlation coefficient (PCC) [26].
2.7. Immunohistochemistry
The kidneys were dissected from the mice, fixed in 10% buffered formalin for 24 h, embedded in paraffin and were cut into 5 μm slices to make tissue slides. After deparrafinization followed by heat-induced antigen retrieval, slides were incubated with primary antibodies against Annexin-II (1:100, Santa Cruz Biotechnology, Dallas, TX, USA), CD63 (1:100, Santa Cruz Biotechnology, Dallas, TX, USA) and alkaline phosphatase (1:100, Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4 °C, and then incubated with biotinylated secondary antibodies and a streptavidin peroxidase complex (PK-7800, Vector Laboratories, Burlingame, CA, USA). Then slides were incubated with DAB and counterstained with hematoxylin. Followed by washing, dehydration, and finally slides were mounted with permount DPX and observed under microscopy as described previously [27]. The area percentage of the positive staining was calculated with Image Pro Plus 6.0 software [27].
2.8. EVs isolation from cultured podocytes
Culture medium was collected from the podocytes, centrifuged at 1200g for 15 min at 4 °C to remove the debris. Supernatant was transferred into sterile Eppendorf tubes and centrifuged at 10,000g for 30 min at 4 °C. 0.22 μm filter was used to remove microvesicles from the supernatant, and then centrifuged again at 100,000g for 90 min at 4 °C. Finally, supernatant was discarded and the remaining EVs pellet was resuspended in 50 μl cold PBS for further analysis [28].
2.9. Nanoparticle tracking analysis (NTA)
NTA measurements were performed with a NanoSight NTA3.2 Dev Build 3.2.16 (Malvern Instruments Ltd., UK), equipped with a sample chamber with a 638-nm laser and a Viton fluoroelastomer O-ring. The samples were injected in the sample chamber with sterile syringes (BD, New Jersey, USA) until the liquid reached the tip of the nozzle. All measurements were performed at room temperature. The screen gain and camera level was 10 and 13 respectively. Each sample was measured at standard measurement, 30 s with manual shutter and gain adjustments. Three measurements of the each sample was performed. 3D figures were exported from the software. Particles sized between 50 and 100 nm were calculated [29].
2.10. High performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) analysis of ceramides, sphingosine, and sphingosine-1- phosphate
Separation, identification and quantitation of ceramides, sphingosine, and sphingosine-1-phosphate were performed by HPLC-MS/MS. Internal standard solution containing 10 ng each of ceramide C12, sphingosine C17, and sphingosine-1-phosphate C17 was added to each cell sample and calibrator. Ceramides, SPH, and S1P were extracted using the Bligh-Dyer method under acidic conditions [30]. Briefly, 2:1 methanol: chloroform (v/v) and 1 N HCl was added to all samples and calibrators. Cell samples were sonicated for 30 s and centrifuged at 13,200 rpm for 5 min. Calibrators were mixed for 5 min then and centrifuged at 13,200 rpm for 5 min. The organic phases were collected, and the samples and calibrators were extracted again with chloroform. The organic phases were combined and evaporated to dryness using nitrogen gas, reconstituted in 100 μl ethanol, and placed in autosampler vials for HPLC-MS/MS analysis on a Sciex 6500+ QTRAP System with an IonDrive Turbo V source for TurbolonSpray® (Ontario, Canada) attached to a Shimadzu Nexera X2 UPLC system (Kyoto, Japan) controlled by Analyst software. Chromatographic separation was performed on a Restek Force Biphenyl column 100 ×3 mm, 5 μm (Bellefonte, PA). The acquisition mode used was multiple reactions monitoring (MRM).
2.11. Cell activity and cell damage assay
Cell activity was assessed by CCK-8 assay kit (Dojindo, Japan). Cells were incubated with 10% CCK-8 solution at 37 °C for 1 h. The absorbance was measured with a microplate reader at 450 nm wavelength. Lactate Dehydrogenase (LDH) level in culture supernatant can be detected to reflect cell damage. After treatment, the supernatant was collected and analyzed with LDH assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to manufacturer’s instructions.
2.12. Statistical analysis
Data are presented as means ± SE. The significant differences between and within multiple groups were examined using one way or two way ANOVA, followed by Duncan’s multiple-range test. P < 0.05 was considered statistically significant.
3. Results
3.1. Exogenous and endogenous D-ribose induced inflammasome formation in podocytes
As demonstrated in our previous study, exogenous D-ribose treatment induced formation and activation of NLRP3 inflammasome in podocytes. The present study confirmed whether endogenous D-ribose also induced the formation and activation of NLRP3 inflammasome by inhibition of ribokinase, an enzyme that catalyzes the formation of D-ribose-5-phosphate from D-ribose [31]. We observed that phosphonoacetic acid (PAA), a ribokinase inhibitor, induced NLRP3 inflammasome formation and activation in podocytes. As depicted in Fig. 1A, both D-ribose and PAA treatment significantly increased the colocalization of NLRP3 (green) and caspase-l(red). The Pearson correlation coefficient is shown in summarized bar Fig. 1B. By western blot analysis, both D-ribose and PAA treatment was found to remarkably increase the expression of cle-caspsae-1 as shown in Fig. 1C, D. In addition, biochemical analyses showed the caspase-1 activity was significantly increased in D-ribose and PAA treatment groups than vehicle (Fig. 1E). These results demonstrated that both exogenous and endogenous D-ribose induced formation and activation NLRP3 inflammasome in podocytes.
Fig. 1.
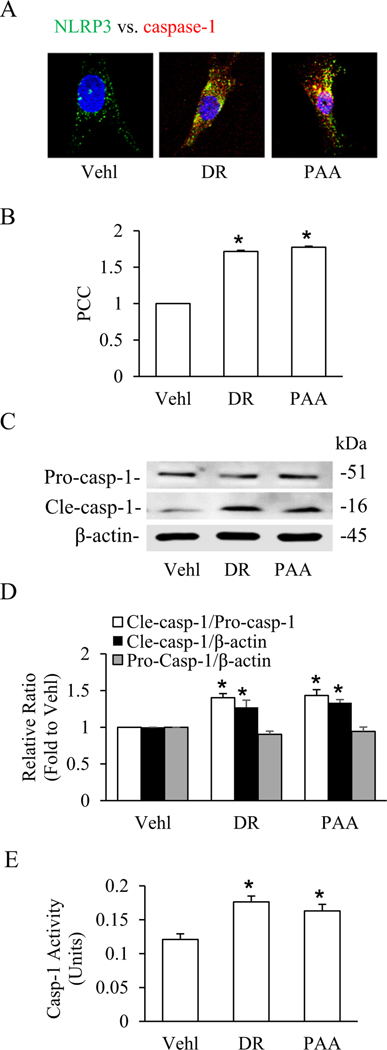
Exogenous and endogenous D-ribose-induced inflammasome formation and activation in podocytes.
A. Representative confocal microscopic images of NLRP3 (green) and caspase-1 (red) (n=4). B. Bar graph shows summarized data depicting co-localization of NLRP3 vs. caspase-1 in podocytes treated with D-ribose and PAA (n=4). C. Representative western blot gel of Pro-caspase-1 and Cle-caspase-1 in podocytes treated with D-ribose and PAA (n=4). D. Bar graph shows summarized data of Pro-caspase-1 and Cle-caspase-1 in podocytes treated with D-ribose and PAA (n=4). E. Caspase-1 activity in podocytes treated with D-ribose and PAA (n=5). *P < 0.05 versus Vehl group. Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid.
3.2. D-Ribose-induced IL-1β release through EVs secretion by lysosome dysfunction in podocytes
Literature cites that NLRP3 inflammasome formation and activation leads to production of mature IL-1β, IL-18. To explore how IL-1β, a proinflammatory cytokine is secreted out of cells, we assessed the contribution of EVs as IL-1β release mechanisms in an experimental cell model of podocytes. We confirmed EVs with its marker CD63 by western blot (Supplementary Fig. 1A). Using a nanoparticle tracking analysis system, we found that secretion of EVs (50–100 nm) from podocytes markedly increased by both D-ribose and PAA, as shown by representative 3-D histograms, which were further increased with prior treatment of bafilomycin A1 (Baf), a lysosome v-ATPase inhibitor (Fig. 2A). Fig. 2B showed summarized data of the particle number sized between 50 and 100 nm, and the curve of particle number vs. particle size < 200 nm was shown in Supplementary Fig. 1B. By ELISA, we observed that IL-1β production in EVs isolated from supernatant of D-ribose and PAA treatment groups were significantly higher than vehicle group which were further increased by pretreatment with bafilomycin A1 as shown in Fig. 2C. Moreover, the expression of cle-caspase-1/procaspase-1 and ASC in EVs were also found to be up-regulated after DR or PAA treatment (Supplementary Fig. 2). Using confocal microscopy, in Baf treated podocytes, we observed that both D-ribose and PAA markedly increased colocalization of MVBs (VPS16, green) and IL-1β (red) resulting in the secretion of EVs loaded with IL-1β in podocytes (Fig. 2D) Furthermore, we observed that Baf-induced lysosomal dysfunction caused more decreased co-localization of MVBs (VPS16, green) vs. lysosomes (Lamp-1, red) in both D-ribose and PAA treated podocytes which indicates loss of MVBs interactions or even fusion with lysosomes causing secretion of EVs (Fig. 2F). The Pearson colocalization coefficient (PCC) of (VPS16, green) vs. (IL-1β, red) was clearly increased while as PCC of VPS16 and Lamp-1 was decreased by both D-ribose and PAA treatment in Baf treated podocytes as shown in Fig. 2E, G. Together these results concluded that D-ribose also caused lysosomal dysfunction, indicating that D-ribose and bafilomycin might share similar mechanistic pathway in the secretion of extracellular vehicles carrying inflammasome products like IL-1β.
Fig. 2.

IL-1β release through EVs secretion mediated by lysosome dysfunction in D-ribose-treated podocytes.
A. Representative 3D graphs depicting number of EVs isolated from cell supernatant with different treatments (n = 6). B. Summarized bar graph shows changes in EVs number with different treatments (n = 6). C. Bar graph shows IL-1β levels in EVs isolated from cell supernatant with different treatments (n = 6). D. Representative confocal microscopic images showing the co-localization of VPS 16 (green) and IL-1β (red) and E. co-localization of VPS 16 (green) and Lamp-1 (red) in podocytes with different treatments (n = 5). Summarized data depicting co-localization of F. VPS 16 (green) and IL-1β (red) and G. co-localization of VPS 16 (green) and Lamp-1 (red) in podocytes with different treatments (n = 5). *P < 0.05 versus Ctrl-Vehl group, #P < 0.05 versus Ctrl group. Ctrl, control; Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid; Baf, bafilomycin A1.
3.3. D-Ribose-induced ceramide formation in podocytes regulated by lysosomal-ceramide sphingolipid pathway
HPLC-MS/MS analysis was performed to determine if exogenous and endogenous D-ribose-induced ceramide formation in podocytes. Fig. 3A shows a representative chromatogram of sphingosine (SPH), sphingosine-1-phosphate (S1P) and ceramide from podocytes with different treatments. Clear peaks corresponding to S1P C17, S1P, SPH C17, SPH, ceramides C12, C14, C16, C18, C20, C22 and C24 were detected. We observed that both D-ribose and PAA caused a significant increase in C16 production as compared to vehicle Ctrl group. Next, we tested if modulation of lysosomal-ceramide sphingolipid pathway regulated D-ribose-induced ceramide formation. For this podocytes were treated with amitriptyline (an Asm inhibitor), genistein (an AC activator) and acid ceramidase (AC) CRISPR activation plasmid. We found that amitriptyline, genistein and AC CRISPR activation plasmid significantly decreased both D-ribose and PAA-induced C16 production in podocytes. The summarized data is shown in Fig. 3B. However, there is no remarkable change in the sphingosine production with D-ribose and PAA treatment as compared to the vehicle. Interestingly, amitriptyline had no significant effect on sphingosine levels, while genistein and AC CRISPR activation plasmid treatment significantly increased sphingosine levels as compared to vehicle Ctrl group (Fig. 3C). These data suggest that lysosomal-ceramide sphingolipid pathway play an important role in D-ribose-induced ceramide formation in podocytes.
Fig. 3.

Effects of Asm inhibition and AC activation on ceramide C16 and sphingosine production in podocytes.
A. Representative chromatogram of ceramide and sphingosine (SPH) analyzed by UPLC-MS/MS analysis (n = 5). Summarized bar shows B. ceramide C16 and C. sphingosine production in podocytes with different treatments (n = 5). *P < 0.05 versus Ctrl-Vehl group, #P < 0.05 versus Ctrl group. Ctrl, control; Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid; Ami, amiftiptyline; Genis, genistein; CRISPR, acid ceramidase CRISPR activation plasmid; cps, Counts per second.
3.4. Asm inhibition blocked D-ribose-induced IL-1β secretion by blocking EVs release in podocytes
To investigate the role of the sphingolipid salvage pathway in D-ribose-induced IL-1β secretion by EVs release, podocytes were pretreated with amitriptyline, an Asm inhibitor. Using a nanoparticle tracking analysis system, we found that amitriptyline blocked secretion of EVs (50–100 nm) in both D-ribose and PAA treated podocytes as shown in (Fig. 4A). Next, by ELISA we observed that D-ribose and PAA-induced IL-1β production in EVs was significantly decreased by amitriptyline treatment (Fig. 4B), which was confirmed by decreased of colocalization of MVBs (VPS16, green) and IL-1β (red) (Fig. 4C). The colocalization coefficient (PCC) of both markers in D-ribose and PAA-treated podocytes was clearly reduced by amitriptyline as shown in bar graph (Fig. 4E). Furthermore, we found that amitriptyline significantly increased colocalization of MVBs (VPS16, green) vs. lysosomes (Lamp-1, red) in all treatment groups as shown Fig. 4D. The PCC analysis showed in the bar graph (Fig. 4F). These results indicate that lysosomal-ceramide pathway can modulate the IL-1β production by blocking the EVs release in podocytes which in turn can prevent the glomerular injury.
Fig. 4.

Effects of acid sphingomyelinase (Asm) inhibition on IL-1β release and EVs secretion.
A. Summarized data of EVs number isolated from cell supernatant with different treatments, as determined by Nanosight (n = 6). B. IL-1β levels in EVs isolated from cell supernatant with different treatments (n = 6). Representative confocal microscopic images showing the co-localization of C. VPS 16 (green) and IL-1β (red) and D. co-localization of VPS 16 (green) and Lamp-1 (red) with bafilomycin treatment in D-ribose and PAA- treated podocytes. Summarized bar graph depicting co-localization of E. VPS 16 (green) and IL-1β (red) and F. VPS 16 (green) and Lamp-1 (red) (n = 5). *P < 0.05 versus Ctrl-Vehl group, #P < 0.05 versus Ctrl group. Ctrl, control; Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid; Ami, amitriptyline.
3.5. Pharmacological and genetic activation of acid ceramidase decreased D-ribose-induced IL-1β secretion by blocking EVs release in podocytes
To further investigate the role of the lysosomal-ceramide sphingolipid pathway in both exogenous and endogenous D-ribose-induced IL-1β secretion by EVs release, podocytes were pretreated with genistein, an acid ceramidase (AC) activator. We observed that both EVs release (particle number) and IL-1β production was significantly increased by exogenous and endogenous D-ribose, whileas genistein treatment blocked this D-ribose-induced increase as shown in Fig. 5A, B. Confocal microscopy showed that genistein significantly decreased both exogenous and endogenous D-ribose-induced increased colocalization of MVBs (VPS16, green) vs. (IL-1β, red) (Fig. 5C), whileas colocalization of MVBs (VPS16, green) vs. lysosomes (Lamp-1, red) was significantly increased (Fig. 5D). The PCC analysis showed in the bar graph (Fig. 5E, F).
Fig. 5.

Effects of acid ceramidase (AC) activation on D-ribose-induced IL-1β release and EVs secretion.
A. Bar graph shows summarized data of EVs number isolated from cell supernatant with different treatments, as determined by Nanosight (n=6). B. IL-1β levels in EVs isolated from cell supernatant with different treatments (n=6). Representative confocal microscopic images showing the co-localization of C. VPS 16 (green) and IL-1β (red) and D. co-localization of VPS 16 (green) and Lamp-1 (red) with genestein treatment in D-ribose and PAA-treated podocytes (n=5). Summarized data depicting co-localization of E. VPS 16 (green) and IL-1β (red) and F. VPS 16 (green) and Lamp-1 (red) (n=5). *P < 0.05 versus Ctrl-Vehl group, #P < 0.05 versus Ctrl group. Ctrl, control; Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid; Genis, genistein.
To further confirm the role of AC enzyme, AC CRISPR activation plasmid was transfected into podocytes prior to treatment of D-ribose or Vehl. As shown in Fig. 6A and B, we observed a remarkable increase in AC expression with the treatment of AC CRISPR activation plasmid, indicating the good transfecting efficiency. Similar to genistein, an AC activator, AC CRISPR activator plasmid significantly decreased the D-ribose-induced increase in EVs number and IL-1β production (Fig. 6C and D). Also, colocalization of MVBs (VPS16, green) vs. (IL-1β, red) (Fig. 6E) was significantly decreased but colocalization of MVBs (VPS16, green) vs. lysosomes (Lamp-1, red) was significantly increased (Fig. 6F) by AC CRISPR activator plasmid by both exogenous and endogenous D-ribose in podocytes. The PCC analysis showed in the bar graph (Fig. 6G, H). Both pharmacological and genetic manipulations indicated that lysosomal-ceramide pathway in particular AC play an important role in D-ribose-induced IL-1β secretion via increased EVs release.
Fig. 6.

Effect of acid ceramidase CRISPR/Cas9 activation plasmid on D-ribose-induced IL-1β release and EVs secretion.
A. Representative western blot gel document showing the effect of acid ceramidase CRISPR activation plasmid on the expression of acid ceramidase (AC) in podocytes (n = 4). B. Summarized bar graph shows change in AC expression using AC CRISPR activation plasmid. C. Summarized data of EVs number isolated from cell supernatant treated with AC CRISPR activation plasmid in D-ribose and PAA-treated podocytes (n = 6). D. IL-1β levels in EVs isolated from cell supernatant treated with AC CRISPR activation plasmid in D-ribose and PAA-treated podocytes (n = 6). Representative confocal microscopic images showing the co-localization of E. VPS 16 (green) and IL-1β (red) and F. co-localization of VPS 16 (green) and Lamp-1 (red) treated with AC CRISPR activation plasmid in D-ribose and PAA-treated podocytes (n = 5). Summarized data depicting co-localization of G. VPS 16 (green) and IL-1β (red) and H. VPS 16 (green) and Lamp-1 (red) (n = 5). *P < 0.05 versus Ctrl-Vehl group, #P < 0.05 versus Ctrl group. Ctrl, control; Vehl, Vehicle; DR, D-ribose; PAA, Phosphonoacetic acid; CRISPR, acid ceramidase CRISPR activation plasmid.
3.6. D-Ribose-induced IL-1β release through EVs secretion by reduced lysosome-MVBs interactions in the glomeruli of mice
To validate our in vitro results whether D-ribose induce IL-1β release through EVs secretion in glomeruli in vivo, 8-week-old, male C57BL/6J mice were intraperitoneally (i.p.) injected with vehicle or D-ribose. We first found that after 30 days treatment of D-ribose, the plasma levels of IL-1β in mice was 348.71 ng/l, significantly up-regulated than Vehl group (326.87 ng/l). By immunohistochemistry we found that D-ribose significantly increased the expression of EVs markers like Annexin-II, CD63 and ALP in glomeruli as compared to vehicle treated mice as shown in the Fig. 7A. Furthermore, consistent to our in vitro data, D-ribose significantly decreased the colocalization of MVBs (VPS16, green) vs. lysosomes (Lamp-1, red) in glomeruli (Fig. 7B) whereas as colocalization of MVBs (VPS16, green) vs. (IL-1β, red) was significantly increased as shown in Fig. 7C. Moreover, D-ribose down-regulated the colocalization of nephrin (green) and VPS16 (red) in glomeruli (Fig. 7D). These results indicate that D-ribose result in reduced lysosome-MVB interactions, increasing fusion of MVBs with plasma membrane and enhanced IL-1β release through EVs secretion.
Fig. 7.

D-Ribose-induced IL-1β release and EVs secretion in the glomeruli of mice.
A. Representative images and summarized bar graph shows immunostaining of glomeruli for EVs markers like Annexin-II, CD63 and alkaline phosphatase (ALP) in the glomeruli of mice treated with D-ribose (n = 5). B. Representative confocal images and summarized bar diagram shows co-localization of VPS 16 (green) vs. Lamp-1 (red) in the glomeruli of mice treated with D-ribose (n = 5). C. Representative confocal images and summarized bar diagram shows co-localization of VPS 16 (green) vs. IL-1β (red) in the glomeruli of mice treated with D-ribose (n = 5). D. Representative confocal images and summarized bar diagram shows co-localization of Nephrin (green) vs. VPS16 (red) in the glomeruli of mice treated with D-ribose (n = 5). *P < 0.05 versus Vehl group. Vehl, Vehicle; DR, D-ribose.
4. Discussion
In the present study, we examined whether lysosomal-ceramide metabolism via AC regulates IL-1β secretion through extracellular vesicles during D-ribose-induced NLRP3 inflammasome activation in podocytes. Firstly, we demonstrated that both exogenous and endogenous D-ribose induced formation and activation of NLRP3 inflammasome in podocytes. Endogenous D-ribose formation was induced by phosphonoacetic acid (PAA), a ribokinase inhibitor, an enzyme that catalyzes the formation of D-ribose-5-phosphate from D-ribose. Both D-ribose and PAA was shown to increase the secretion of EVs, and also IL-1β levels in these isolated EVs, which were further increased by prior treatment of bafilomycin A1, a lysosome v-ATPase inhibitor. Increased colocalization of MVBs marker (VPS16) and IL-1β was observed. These results indicate that the secretion ofIL-1β containing EVs was increased in both D-ribose and PAA-treated podocytes. By HPLC-MS/MS analysis, we showed that D-ribose and PAA caused a significant increase in C16-ceramide production, which can be blocked by genetic and pharmacologic activation of AC expression and activity, but enhanced by inhibition of AC expression and activity. It is clear that AC is a critical enzyme that control ceramide levels in lysosomes and thereby may contribute to the control of lysosome function and consequent EVs release in podocytes during D-ribose stimulation.
Furthermore, we observed that amitriptyline, an Asm inhibitor, blocked D-ribose and PAA-induced secretion of EVs and their IL-1β levels from podocytes, which was followed by increased inclusion of IL-1β as shown by colocalization of VPS16 vs. IL-1β. However, the colocalization of MVBs (VPS16) vs. lysosomes (Lamp-1) was decreased, suggesting reduction of IL-1β containing MVBs degradation via lysosome that leads to increases in EVs release. We also demonstrated that genistein, an AC gene expression inducer and AC CRISPR/Cas9 activation plasmid attenuation of enhanced EVs release induced by both exogeneous and endogenous D-ribose in podocytes, which were associated with reduced lysosome-MVB interactions and increased IL-1β containing MVBs. These results suggest that lysosomal AC -mediated lysosomal ceramide metabolism plays a crucial role in IL-1β release via EVs during NLRP3 inflammasome activation induced by D-ribose.
D-Ribose has been recently emerging as a novel pathogenic factor for organ damages during diabetes mellitus. However, the molecular mechanism mediating the action of D-ribose remains poorly understood. In the present study, we first demonstrated that both exogenous and endogenous D-ribose induced the formation and activation of NLRP3 inflammasome in podocytes, confirming that NLRP3 inflammasome activation may be a critical mechanism for D-ribose induced-podocyte injury and glomerular sclerosis. NLRP3 inflammasome products such as IL-1β were released via EVs out of podocytes in response to either exogenously administrated or endogenously produced D-ribose, which was mimicked lysosome dysfunction induced by bafilomycin A1. We also found that IL-1β was also up-regulated after D-glucose treatment, which can be further increased with Bafilomycin (Supplementary Fig. 3A). Furthermore, we found that EVs isolated from vehicle-treated podocytes had no significant effects on mesangial cell activity. However, EVs isolated from D-ribose treated podocytes decreased mesangial cell activity by 10.5% compared to Vehl-treated cells (Supplementary Fig. 3B and C). To our knowledge, this is the first experimental evidence showing the products of NRLP3 inflammasome activation in podocytes upon D-ribose stimulation are released via EVs, thereby triggering the inflammatory response in glomeruli and leading to chronic sterile glomerular inflammation and ultimate sclerosis There is growing evidence that EVs are a molecular signaling package for some specific target cells [10,32] and are used by the originating cell to dispose unnecessary or harmful materials [15,33–35]. In the context of inflammation, some reports have shown that extracellular ATP stimulation in murine macrophages lead to P2X7R-induced formation of multivesicular bodies (MVBs), which contain IL-1β, and these inflammasome loaded intraluminal vesicles (ILVs) are released by exocytosis [36]. In human THP-1 monocytes and microglia cells, MVs shedding is predicted as a general mechanism for secretion IL-1β [37,38]. However, other studies in activated human monocytes also revealed that both pro-IL-1β and caspase-1 are entrapped in the vesicles, which as an endolysosomal compartment undergo exocytosis in the form of secretory lysosomes [14,39], but not via EVs. Dupont et al. also showed that autophagy via Atg5-dependent export pathway has a positive contribution to the secretion of the proinflammatory cytokine IL-1β in different mammalian cells [40]. It seems that different cells under different conditions may use different mechanisms to release IL-1β. Our results indicate that enhanced EVs secretion may be a crucial mechanism mediating the release of NLRP3 inflammasome produced IL-1β in podocytes upon D-ribose stimulation.
This activating mechanism instigating the inflammatory response in podocytes and glomeruli may also contribute to the development of diabetic nephropathy because D-ribose has now been emerging as a novel pathogenic factor for organ damages during diabetes mellitus in addition to elevated blood glucose levels. Therefore, blockade of NLRP3 inflammasome activation or inhibition of the release of NRLP3 inflammasome products may be a therapeutic strategy for prevent or treatment of diabetic nephropathy (DN), which is characterized by hyperfiltration, albuminuria, and decline in glomerular filtration rate (GFR), leading to progressive renal fibrosis or sclerosis [41].
In animal experiments, we also observed that mice on the high D-ribose diet had increased EVs in their glomeruli, as shown by largely increased levels of EVs markers CD63 and ALP in glomeruli. It was also found that inclusion of NRLP3 inflammasome product, IL-1β in MVB was increased, while the interaction of lysosome with MVBs was reduced in glomerular podocytes of these mice receiving the high D-ribose diet. These results indicate that D-ribose activated inflammasome in podocytes of mice may trigger consequent inflammatory response in glomeruli via EVs-mediated release of IL-1β or other products, which ultimately leads to podocyte dysfunction, glomerular injury and the development of glomerular sclerosis, as shown by previous studies [42–44]. Together, all data suggested that lysosomal sphingolipid pathway plays a crucial role in NLRP3 inflammasome-dependent IL-1β release via EVs leading to glomeruli podocyte injury. The present study did attempt to explore the mechanism by which EVs-derived inflammasome products such as IL-1β cause glomerular injury and ultimate sclerosis, recent studies have indicated that EVs produced and secreted in the kidney are not only an important biomarker to depict kidney function or disease, but also serve as an critical mechanism mediating intra-renal signaling, in particular, cell-to-cell communication, which may participate in the development of different kidney diseases [45–47]. There is evidence that podocyte-derived EVs increased in diabetic mice even before onset of albuminuria [48]. Increased EVs may serve as a signaling vesicle to trigger phenotype changes in neighbor cells, to produce podocyte dysfunction and to promote fibrogenesis in glomeruli or other tissues in the kidney [45–47], resulting in the development of albuminuria [49].
Another important finding of the present study is that IL-1β release via EVs is regulated by ceramide sphingolipid metabolism via lysosomal AC and Asm. Ceramide is released from the hydrolysis of membrane sphingomyelin by various sphingomyelinases such as Asm, serine palmitoyltransferase, and ceramide synthase [50]. Lysosomal ceramide is mainly metabolized into sphingosine by AC.
Studies have shown that ceramide may be importantly involved in activation of kidney tissue inflammation and thereby leads to chronic renal diseases under different pathological conditions such as obesity, hyperhomocysteinemia, diabetes mellitus and other renal inflammatory diseases. With respect to ceramide regulation of EVs biogenesis and secretion, there is report that SMPD3-derived ceramide triggers budding of EVs vesicles into multivesicular endosomes [19,32], suggesting that ceramide is important sphingolipid required for EVs formation, secretion and function [51], The present study demonstrated that D-ribose increased the ceramide levels in podocytes, which were decreased by both Asm inhibition with amitriptyline or AC activation by AC gene inducer, genestein or AC CRISPR/Cas9 activation plasmid. This suggests that increased D-ribose in the kidney may act on podocytes to not only induce NLRP3 inflammasome activation, but also increase ceramide level in lysosomes to promote EVs secretion, thereby transporting NLRP3 inflammosome products out of podocyte to exert their role in triggering glomerular inflammatory response.
In summary, the present study demonstrated that NLRP3 inflammasome products such as IL-1β were increasingly released via EVs secreted from podocytes upon D-ribose stimulation, which may lead to glomerular podocyte injury. This EVs-mediated release of NLRP3 inflammasome products is controlled by lysosomal ceramide-mediated signaling. Targeting this EVs-mediated release of NLRP3 inflammosome products regulated by ceramide signaling may represent a novel strategy for prevention and treatment of diabetic nephropathy given the important role of D-ribose in diabetic organ damage.
Supplementary Material
Acknowledgments
This study was supported by grants DK054927, HL075316, and P30DA033934 from National Institutes of Health of United States and grants 81573763 and 81530099 from National Natural Science Foundation of China. We also acknowledge the support from China Scholarship Council (CSC) (CSC Grant No. 201706010324).
Footnotes
Transparency document
The Transparency document associated with this article can be found, in online version.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbamcr.2019.02.007.
Conflict of interest
We declare that there is no conflict of interest to disclose.
References
- [1].Mahoney DE, Hiebert JB, Thimmesch A, Pierce JT, Vacek JL, Clancy RL, Sauer AJ, Pierce JD, Understanding D-ribose and mitochondrial function, Adv. Biosci. Clin. Med 6 (2018) 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jones K, Probst Y, Role of dietary modification in alleviating chronic fatigue syndrome symptoms: a systematic review, Aust. N. Z. J. Public Health 41 (2017) 338–344. [DOI] [PubMed] [Google Scholar]
- [3].Thompson J, Neutel J, Homer K, Tempero K, Shah A, Khankari R, Evaluation of D-ribose pharmacokinetics, dose proportionality, food effect, and pharmacodynamics after oral solution administration in healthy male and female subjects, J. Clin. Pharmacol 54 (2014) 546–554. [DOI] [PubMed] [Google Scholar]
- [4].Iannuzzi C, Borriello M, Carafa V, Altucci L, Vitiello M, Balestrieri ML, Ricci G, Irace G, Sirangelo I, D-Ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts, Biochim. Biophys. Acta 1862 (2016) 93–104. [DOI] [PubMed] [Google Scholar]
- [5].Hong JN, Wang XM, Zhang N, Fu H, Li WW, D-Ribose induces nephropathy through RAGE-dependent NF-kappa B inflammation, Arch. Pharm. Res 41 (2018) 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Conley SM, Abais JM, Boini KM, Li PL, Inflammasome activation in chronic glomerular diseases, Curr. Drug Targets 18 (2017) 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen Y, He X, Yuan X, Hong J, Bhat O, Li G, Li PL, Guo J, NLRP3 inflammasome formation and activation in nonalcoholic steatohepatitis: therapeutic target for antimetabolic syndrome remedy FTZ, Oxidative Med. Cell. Longev 2018 (2018) 2901871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang S, Yuan YH, Chen NH, Wang HB, The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease, Int. ImmunoPharmacol 67 (2019) 458–464. [DOI] [PubMed] [Google Scholar]
- [9].Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, Li PL, Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia, Redox Biol 13 (2017) 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tkach M, Thery C, Communication by extracellular vesicles: where we are and where we need to go, Cell 164 (2016) 1226–1232. [DOI] [PubMed] [Google Scholar]
- [11].Colombo M, Raposo G, Thery C, Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles, Annu. Rev. Cell Dev. Biol 30 (2014) 255–289. [DOI] [PubMed] [Google Scholar]
- [12].Schorey JS, Harding CV, Extracellular vesicles and infectious diseases: new complexity to an old story, J. Clin. Invest 126 (2016) 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rubartelli A, Cozzolino F, Talio M, Sitia R, A novel secretory pathway for interleukin-1-beta, a protein lacking a signal sequence, EMBO J. 9 (1990) 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A, The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolyso-some-related vesicles, Mol. Biol. Cell 10 (1999) 1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, Hara E, Exosomes maintain cellular homeostasis by excreting harmful DNA from cells, Nat. Commun 8 (2017) 15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Eitan E, Suire C, Zhang S, Mattson MP, Impact of lysosome status on extracellular vesicle content and release, Ageing Res. Rev 32 (2016) 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Balkom BW, Pisitkun T, Verhaar MC, Knepper MA, Exosomes and the kidney: prospects for diagnosis and therapy of renal diseases, Kidney Int 80 (2011) 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Llorente A, Skotland T, Sylvanne T, Kauhanen D, Rog T, Orlowski A, Vattulainen I, Ekroos K, Sandvig K, Molecular lipidomics of exosomes released by PC-3 prostate cancer cells, Biochim. Biophys. Acta 1831 (2013) 1302–1309. [DOI] [PubMed] [Google Scholar]
- [19].Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M, Ceramide triggers budding of exosome vesicles into multi-vesicular endosomes, Science 319 (2008) 1244–1247. [DOI] [PubMed] [Google Scholar]
- [20].Kolliputi N, Galam L, Parthasarathy PT, Tipparaju SM, Lockey RF, NALP-3 inflammasome silencing attenuates ceramide-induced transepithelial permeability, J. Cell. Physiol 227 (2012) 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD, The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance, Nat. Med 17 (2011) 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Boini KM, Xia M, Abais JM, Li G, Pitzer AL, Gehr TW, Zhang Y, Li PL, Activation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencing, Biochim. Biophys. Acta 1843 (2014) 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL, NADPH oxidasemediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia, Antioxid. Redox Signal. 18 (2013) 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dhar A, Dhar I, Bhat A, Desai KM, Alagebrium attenuates methylglyoxal induced oxidative stress and AGE formation in H9C2 cardiac myocytes, Life Sci 146 (2016) 8–14. [DOI] [PubMed] [Google Scholar]
- [25].Chowdhury P, Aminoguanidine (AG) induces induced both pro- and antioxidant effect in AR42J cells, a rat pancreatic tumor cell line, Ann. Clin. Lab. Sci 47 (2017) 572–580. [PubMed] [Google Scholar]
- [26].Yuan X, Wang L, Bhat OM, Lohner H, Li PL, Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: antioxidant action of butyrate, Redox Biol 16 (2018) 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li G, Chen Z, Bhat OM, Zhang Q, Abais-Battad JM, Conley SM, Ritter JK, Li PL, NLRP3 inflammasome as a novel target for docosahexaenoic acid metabolites to abrogate glomerular injury, J. Lipid Res 58 (2017) 1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Klymiuk MC, Balz N, Elashry MI, Heimann M, Wenisch S, Arnhold S, Exosomes isolation and identification from equine mesenchymal stem cells, BMC Vet Res 15 (2019) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Antes TJ, Middleton RC, Luther KM, Ijichi T, Peck KA, Liu WJ, Valle J, Echavez AK, Marban E, Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display, J. Nanobiotechnol. 16 (2018) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scherer M, Schmitz G, Liebisch G, High-throughput analysis of sphingosine 1-phosphate, sphinganine 1-phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography-tandem mass spectrometry, Clin. Chem 55 (2009) 1218–1222. [DOI] [PubMed] [Google Scholar]
- [31].Park J, van Koeverden P, Singh B, Gupta RS, Identification and characterization of human ribokinase and comparison of its properties with E-coli ribokinase and human adenosine kinase, FEBS Lett. 581 (2007) 3211–3216. [DOI] [PubMed] [Google Scholar]
- [32].Couzin J, Cell biology: the ins and outs of exosomes, Science 308 (2005) 1862–1863. [DOI] [PubMed] [Google Scholar]
- [33].Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K, Alzheimer’s disease beta-amyloid peptides are released in association with exosomes, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 11172–11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF, Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease, J. Biol. Chem 287 (2012) 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G, Cells release prions in association with exosomes, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qu Y, Franchi L, Nunez G, Dubyak GR, Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages, J. Immunol 179 (2007) 1913–1925. [DOI] [PubMed] [Google Scholar]
- [37].MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A, Rapid secretion of interleukin-1beta by microvesicle shedding, Immunity 15 (2001) 825–835. [DOI] [PubMed] [Google Scholar]
- [38].Bianco F, Pravettoni E, Colombo A, Schenk U, Moller T, Matteoli M, Verderio C, Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia, J. Immunol 174 (2005) 7268–7277. [DOI] [PubMed] [Google Scholar]
- [39].Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A, Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 9745–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V, Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta, EMBO J. 30 (2011) 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH, Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury, Kidney Int 65 (2004) 116–128. [DOI] [PubMed] [Google Scholar]
- [42].Ma CH, Kang LL, Ren HM, Zhang DM, Kong LD, Simiao pill ameliorates renal glomerular injury via increasing Sirt1 expression and suppressing NF-kappaB/NLRP3 inflammasome activation in high fructose-fed rats, J. EthnoPharmacol 172 (2015) 108–117. [DOI] [PubMed] [Google Scholar]
- [43].Wang W, Ding XQ, Gu TT, Song L, Li JM, Xue QC, Kong LD, Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microRNA-377, Free Radic. Biol. Med 83 (2015) 214–226. [DOI] [PubMed] [Google Scholar]
- [44].Xiong J, Wang Y, Shao N, Gao P, Tang H, Su H, Zhang C, Meng XF, The expression and significance of NLRP3 inflammasome in patients with primary glomerular diseases, Kidney Blood Press. Res 40 (2015) 344–354. [DOI] [PubMed] [Google Scholar]
- [45].Bruno S, Porta S, Bussolati B, Extracellular vesicles in renal tissue damage and regeneration, Eur. J. Pharmacol 790 (2016) 83–91. [DOI] [PubMed] [Google Scholar]
- [46].Erdbrugger U, Le TH, Extracellular vesicles in renal diseases: more than novel biomarkers? J. Am. Soc. Nephrol 27 (2016) 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pomatto MAC, Gai C, Bussolati B, Camussi G, Extracellular vesicles in renal pathophysiology, Front. Mol. BioSci 4 (2017) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhou H, Kajiyama H, Tsuji T, Hu X, Leelahavanichkul A, Vento S, Frank R, Kopp JB, Trachtman H, Star RA, Yuen PS, Urinary exosomal Wilms’ tumor-1 as a potential biomarker for podocyte injury, Am. J. Physiol. Ren. Physiol 305 (2013) F553–F559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Castrop H, Schiessl IM, Novel routes of albumin passage across the glomerular filtration barrier, Acta Physiol (Oxford) 219 (2017) 544–553. [DOI] [PubMed] [Google Scholar]
- [50].Boini KM, Xia M, Li CX, Zhang C, Payne LP, Abais JM, Poklis JL, Hylemon PB, Li PL, Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemia-induced glomerular injury in mice, Am. J. Pathol 179 (2011) 2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Elsherbini A, Bieberich E, Ceramide and exosomes: a novel target in cancer biology and therapy, Adv. Cancer Res 140 (2018) 121–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.