

SUMMARY
The cyclin-dependent kinases Cdk4 and Cdk6 form complexes with D-type cyclins to drive cell proliferation. A well-known target of cyclin D-Cdk4,6 is the retinoblastoma protein Rb, which inhibits cell-cycle progression until its inactivation by phosphorylation. However, the role of Rb phosphorylation by cyclin D-Cdk4,6 in cell-cycle progression is unclear because Rb can be phosphorylated by other cyclin-Cdks, and cyclin D-Cdk4,6 has other targets involved in cell division. Here, we show that cyclin D-Cdk4,6 docks one side of an alpha-helix in the Rb C terminus, which is not recognized by cyclins E, A, and B. This helix-based docking mechanism is shared by the p107 and p130 Rb-family members across metazoans. Mutation of the Rb C-terminal helix prevents its phosphorylation, promotes G1 arrest, and enhances Rb’s tumor suppressive function. Our work conclusively demonstrates that the cyclin D-Rb interaction drives cell division and expands the diversity of known cyclin-based protein docking mechanisms.
In Brief
Precise timing of cell-cycle transitions relies on regulation of the activity and specificity of cyclin-dependent kinases. Topacio et al. show that the G1 cyclin-Cdk complex cyclin D-Cdk4,6 targets its well-known substrate, the retinoblastoma protein Rb, through recognition of a C-terminal alpha-helix and demonstrate that this specific cyclin-Cdk-substrate interaction drives cell proliferation.
Graphical Abstract

INTRODUCTION
The cyclin-dependent kinases Cdk4 and Cdk6 (Cdk4,6) are activated by the D-type cyclins D1, D2, and D3 (cyclin D) to drive cell-cycle progression from G1 to S phase (Morgan, 1997; Sherr and Roberts, 2004). One important target of Cdk4,6 is the retinoblastoma tumor suppressor protein Rb, which binds and inhibits the activating E2F transcription factors. Rb phosphorylation promotes its dissociation from E2Fs and thereby drives the expression of E2F-target genes that initiate DNA replication (Bertoli et al., 2013; Dick and Rubin, 2013; Sherr and McCormick, 2002). The importance of Rb phosphorylation and the frequent observation of increased Cdk4,6 activity in cancer has contributed to the consensus model that these kinases, activated by their D-type cyclin partners, phosphorylate and inhibit Rb to drive cell-cycle progression (Burkhart and Sage, 2008; Lundberg and Weinberg, 1998).
In the current model, cyclin D-Cdk4,6 activity gradually increases until it triggers a positive feedback loop that commits cells to passing the restriction point just prior to the G1/S transition (Merrick et al., 2011; Pardee, 1974; Schwarz et al., 2018). As cells progress through G1, cyclin D-Cdk4,6 gradually phosphorylates Rb and triggers the onset of E2F-dependent expression of cyclins E and A (Bertoli et al., 2013). Cyclins E and A then bind Cdk1 and Cdk2 to form complexes that continue to phosphorylate Rb (Merrick et al., 2011; Morgan, 2007; Narasimha et al., 2014). In addition, cyclin E- and A-dependent Cdk complexes phosphorylate and inhibit the E3 ubiquitin ligase APC/C activating subunit Cdh1, stabilizing APC/CCdh1 substrates, including cyclin A (Di Fiore et al., 2015; Jaspersen et al., 1999; Kramer et al., 2000; Zachariae et al., 1998). The sequential activation of these interconnected positive feedback loops progressively drives commitment to cell division in the face of exposure to anti-proliferative conditions (Cappell et al., 2016, 2018; Pardee, 1974; Schwarz et al., 2018; Yung et al., 2007; Zetterberg and Larsson, 1985).
However, in opposition to the prevailing model of gradually increasing cyclin D-Cdk4,6 activity triggering G1/S, cyclin D levels are nearly constant through G1 (Hitomi and Stacey, 1999). Moreover, Rb is mono-phosphorylated during early- to mid-G1, suggesting that cyclin D-Cdk4,6 activity does not gradually increase through G1 (Narasimha et al., 2014). E2F-dependent transcription increases at the same time that Cdk2 activity increases in late G1, implying that cyclin D-dependent mono-phosphorylated Rb is still capable of interacting with E2F transcription factors to inhibit transcription. This raises the possibility that Rb inactivation in late G1 is due to hyper-phosphorylation by Cdk2 kinase complexes, and cyclin D-Cdk4,6 promotes the G1/S transition through a different mechanism (Narasimha et al., 2014).
If not Rb, what could be the main target of cyclin D-Cdk4,6 driving cell-cycle progression? Possible substrates, whose phosphorylation promotes cell-cycle progression, include a mediator of antiproliferative transforming growth factor β (TGF-β) signaling Smad3 (Matsuura et al., 2004), an APC/C co-activator Cdh1 (The et al., 2015), and a cell-cycle transcription factor FOXM1 (Anders et al., 2011). Further supporting an oncogenic role for Cdk4,6, there is a growing body of literature showing that Cdks directly phosphorylate metabolic enzymes to regulate metabolism in yeast and human cells (Ewald, 2018; Ewald et al., 2016; Salazar-Roa and Malumbres, 2017; Zhao et al., 2016). More specifically, in mammalian cells, cyclin D3-Cdk6 kinase complexes phosphorylate and inactivate the key glycolytic enzymes PFKP and PKM2 to shunt glycolytic intermediates toward NADPH and GSH production, which mitigates ROS accumulation to promote cell survival (Wang et al., 2017). These Rb-independent roles for cyclin D-Cdk4,6 in promoting cell proliferation raise the question as to what is the in vivo function of the cyclin D-Cdk4,6-Rb interaction.
To determine the function of Rb phosphorylation by cyclin D-Cdk4,6, we sought to generate variants of Rb that could no longer interact with cyclin D-Cdk4,6 while preserving all the other interactions with other cyclin-Cdk complexes. The specificity of substrate binding and phosphorylation by cyclin-Cdk complexes is generally determined by the ability of the cyclin to recognize docking sites on substrates (Morgan, 2007). Previously identified docking sites on substrates are short linear amino acid motifs. In budding yeast, the G1 cyclin Cln2 recognizes an LP docking motif and the S phase cyclins Clb5 and Clb3 recognize substrates with RxL docking motifs (Bhaduri and Pryciak, 2011; Cross and Jacobson, 2000; Kõivomägi et al., 2011; Loog and Morgan, 2005). In animal cells, cyclin A-Cdk2 and cyclin E-Cdk2 complexes also utilize RxL-based docking through their α1 helix hydrophobic patches to bind and phosphorylate substrate proteins including Rb, p107, p27, and Cdc6 (Adams et al., 1999; Hirschi et al., 2010; Russo et al., 1996; Schulman et al., 1998; Takeda et al., 2001; Wohlschlegel et al., 2001). While cyclin D appears to have a hydrophobic patch on its α1 helix similar to cyclins E and A, it has not been shown if this patch also recognizes RxL motifs. Cyclin D has an N-terminal LxCxE motif, which binds Rb’s LxCxE cleft (Dick and Rubin, 2013; Dowdy et al., 1993). However, mutation of this LxCxE cleft has only a modest effect in vivo suggesting that either the cyclin D-Rb interaction is of limited importance or that there exist additional docking interactions. Supporting the existence of an additional cyclin D-Rb docking interaction, truncation of the Rb C terminus disrupts phosphorylation by cyclin D1-Cdk4 in vitro, and this truncated Rb variant slows division and inhibits E2F-dependent gene expression in cultured cells (Gorges et al., 2008; Pan et al., 2001; Wallace and Ball, 2004).
In this study, we analyzed the docking interactions between Rb and cyclin D-Cdk4,6 complexes. We found that cyclin D-Cdk4,6 targets the Rb family of proteins for phosphorylation primarily by docking a C-terminal alpha-helix. Importantly, this Rb C-terminal helix is not recognized by the other major cell-cycle cyclin-Cdk complexes cyclin E-Cdk2, cyclin A-Cdk2, and cyclin B-Cdk1. Thus, mutation of this helix disrupts cyclin D’s ability to phosphorylate Rb and preserves Rb regulation by other cyclin-Cdk complexes. Disruption of helix-based docking reduced Rb phosphorylation, induced G1 cell-cycle arrest in cell lines, and slowed tumor growth in vivo. Taken together, our results show that cyclin D-Cdk4,6 phosphorylates and inhibits Rb via a C-terminal helix and that this interaction is a major driver of cell proliferation.
RESULTS
RxL- and LxCxE-Based Docking Mutations Broadly Affect Cyclin-Cdk Complexes
To determine the function of Rb phosphorylation by cyclin D-Cdk4,6, we sought to generate an Rb variant that does not interact with cyclin D-Cdk4,6, but does interact with other cyclin-Cdk complexes (Figures 1A and 1B). To identify specific cyclin D-Rb interactions, we performed in vitro kinase assays on Rb protein variants with a panel of purified cyclin-Cdk complexes (Schwarz et al., 2018) (Figure S1). In contrast to other studies of Rb phosphorylation that used Rb fragments, we were able to purify full-length Rb protein from bacteria for our study. In these kinase assay experiments, mutation of Rb residues required for docking interactions manifest as reduced kinase activity toward Rb. Compared with other cyclin-Cdk complexes, cyclin D-Cdk4,6 exhibited no detectable kinase activity toward the model Cdk substrate histone H1 (Matsushime et al., 1994) but was capable of phosphorylating Rb (Figures 1C–1E).
Figure 1. Cyclin D-Cdk4,6 Complexes Target Rb for Phosphorylation by Docking a C-Terminal Helix.

(A) Schematic of the cyclin-Cdk docking sites and the 14 accessible Cdk phosphorylation sites on Rb.
(B) Reported interactions and mutations of cyclin-Cdk docking sites on Rb.
(C) In vitro kinase assays using the denoted cyclin-Cdk complexes and histone H1 or variants of Rb. RbRxLmut lacks all C-terminal RxL sequences that dock cyclin hydrophobic patches. RbLxCxE cleft mut lacks the LxCxE docking cleft. RbΔC-term lacks the C-terminal amino acids 893–928. The Coomassie-stained gels showing equal amounts of substrate used in each reaction are placed below each autoradiograph.
(D and E) Quantification of H1 kinase assays (D) and Rb kinase assays (E) using the denoted cyclin-Cdk complexes. Data are mean ± SEM; n = 2.
(F) The ratio of kinase activity of the denoted cyclin-Cdk complexes toward Rb and RbΔC-term. Data are mean ± SEM; n = 2.
(G) Helical wheel projection of the predicted Rb C-terminal helix. The black and gray arrows indicate the beginning and end of the helix, respectively. Circles represent hydrophilic residues, diamonds represent hydrophobic residues, triangles represent potentially negatively charged residues, and pentagons represent potentially positively charged residues. Colored amino acids correspond to alanine or proline substitutions in (H).
(H) In vitro kinase assays of Rb protein variants by cyclin D1-Cdk4. ΔCdk denotes Rb lacking the 14 Cdk phosphorylation sites. ΔHelix denotes Rb lacking the C-terminal helix amino acids 895–915. Helix mut. denotes an Rb variant where the predicted docking interface residues F897, L901, and R908 are substituted with alanines. Data are mean ± SEM; n = 2.
(I) Immunoblot analysis of a GST-pull-down binding assay. The GST-tagged Rb bait proteins were incubated with 3X FLAG-tagged cyclin D1 prey proteins. * Denotes a degradation product below full-length GST-Rb.
(J) Quantification cyclin D1-Cdk4 pulled down in (F). Data are mean ± SEM; n = 3.
(K and L) Quantification of in vitro kinase assays of the denoted Rb variant with (K) cyclin D1-Cdk4 or (L) cyclin E1-Cdk2. Data are mean ± SEM; n = 3.
(M) Comparison of the docking mechanisms recognized by cyclin D-Cdk4,6 and cyclin E-Cdk2.
To test the effect of mutating known cyclin D-Rb docking interactions, we first removed the LxCxE binding cleft in the Rb pocket domain that interacts with proteins containing the LxCxE motif, such as viral oncoproteins and cyclin D (Dick et al., 2000; Dowdy et al., 1993; Markey et al., 2007). This resulted in only a 1.7-fold ± 0.3-fold reduction in phosphorylation by cyclin D kinase complexes (Figures 1C and S1L), consistent with reports that the LxCxE-docking interaction alone is weak and its removal has modest effects in cells (Dick et al., 2000; Guiley et al., 2015). Moreover, we observed that previously reported LxCxE cleft mutations similarly affected cyclin E-, A-, and B-dependent phosphorylation of Rb in vitro and therefore may not be specific for cyclin D (Figures 1C and S1L).
Next, we tested the effect of mutating the RxL motifs on Rb that are reported to interact with the hydrophobic patch region of the S phase cyclins E and A (Adams et al., 1999; Hirschi et al., 2010; Schulman et al., 1998). We generated an Rb variant lacking all 5 RxL sequences in its unstructured regions (Figure 1A and 1B). Compared to wild type Rb, this RxL variant of Rb was phosphorylated 2.0-fold ± 0.1-fold less by cyclin E-Cdk2 and 2.9- fold ± 0.2-fold less by cyclin A-Cdk2 (Figures 1C and S1L). This implies that, while cyclins E and A use RxL docking as previously reported, this is not the only mechanism they use to identify their substrates. Cyclin B-Cdk1 phosphorylation of Rb was unchanged by mutating the RxL motifs, suggesting that cyclin B does not use its hydrophobic patch to phosphorylate Rb. While it has not been studied extensively, cyclin D has an α1 helix hydrophobic patch as cyclins E, A, and B, but it is composed of different amino acid residues (Morgan, 2007). We observed that the Rb variant lacking RxL motifs was phosphorylated 4.1- fold ± 0.4-fold less by cyclin D-Cdk4,6, suggesting that cyclin D recognizes RxL motifs as other cyclins (Figures 1C and S1L). While the RxL docking is clearly important for cyclin D, it is shared with other cyclins so that its mutation would not specifically disrupt the cyclin D-Rb interaction.
Cyclin D-Cdk4,6 Complexes Target Rb for Phosphorylation by Docking a C-Terminal Helix
To generate an Rb protein variant that does not interact with cyclin D-Cdk4,6, but does interact with other cyclin-Cdk complexes, we next examined the effect of truncating the final 37 amino acids of Rb. Truncation of the Rb C terminus, which included these 37 amino acids, was previously shown to reduce Rb phosphorylation in vitro and promote Rb’s ability to arrest cells in G1 (Gorges et al., 2008; Pan et al., 2001; Wallace and Ball, 2004) (Figure S2). C-terminal truncation of Rb reduced phosphorylation by cyclin D1-Cdk4,6 20-fold ± 10-fold (Figures 1C and S1L) and increased the Michaelis-Menten constant KM beyond our measurement limit of ~5 μM (Figure S2G), indicating the presence of a docking interaction in the final 37 amino acids of the Rb C terminus. Importantly, this C-terminal truncation is specific for cyclin D-Cdk4,6 complexes and does not affect the phosphorylation of Rb by cyclin E-Cdk2, cyclin A-Cdk2, and cyclin B-Cdk1 complexes (Figures 1C, 1F, and S2H).
We next sought to determine how the Rb C terminus promotes phosphorylation by cyclin D-Cdk4,6. Cyclin substrate docking has previously been shown to arise from short linear motifs of a few amino acids in intrinsically disordered regions on the target proteins, a common mechanism for evolution of protein-protein interactions (Bloom and Cross, 2007; Davey et al., 2015). However, such a short linear motif model is unlikely to explain the interaction of cyclin D with the Rb C terminus because in vitro Rb phosphorylation by cyclin D-Cdk4 is affected by mutations over a large range of amino acids (Wallace and Ball, 2004). While the Rb C terminus is intrinsically disordered, it is known to adopt structure when bound to other proteins (Rubin et al., 2005). We therefore examined the Rb C terminus for potential secondary structure and found a stretch of 21 amino acids with alpha-helix propensity (Rb 895–915; Figures 1G and S3). Deletion of this potential helix (ΔHelix) or disruption of this helix by proline substitution (Q899P or A902P) drastically reduced Rb phosphorylation by cyclin D-Cdk4,6, which was comparable to the phosphorylation of an Rb variant (ΔCdk) lacking all 14 accessible Cdk phosphorylation sites (Figures 1G, 1H, S2A, and S2B).
The Rb C-terminal helix is predicted to have one face composed primarily of hydrophobic residues (Figure 1G). We tested whether these residues could form an interface between the Rb C-terminal helix and cyclin D-Cdk4,6 by measuring phosphorylation of alanine substitution mutants by cyclin D-Cdk4. Rb phosphorylation was disrupted when individual predicted interface residues were mutated but was unaffected when individual adjacent residues were mutated (Figure S2C). For example, the impact of arginine-to-alanine substitutions depended on which face of the helix it occurred on (Figures 1G and 1H; compare R908A versus R910A). Combined alanine substitution of three of the core interface residues (F897, L901, and R908), denoted as RbHelix mut, disrupted phosphorylation of Rb by cyclin D-Cdk4,6 similar to that of RbΔHelix (Figures 1G, 1H, and S2D). To test whether the orientation of the helix was important for phosphorylation, we reversed the primary sequence of the helix within the C terminus of full-length Rb. This variant (rev. Helix) was poorly phosphorylated by cyclin D-Cdk4, suggesting that the orientation of the interface is important for docking in addition to the polarity and acid-base properties of the interface residues (Figure S2D).
Because alanine has high helical propensity (Pace and Scholtz, 1998), these substitutions likely disrupt the recognition of Rb by cyclin D-Cdk4,6 without affecting the C-terminal helix structure. We found that alanine substitution of the core interface residues did not change alpha-helix propensity (Figure S3B) and confirmed that the core interface mutations disrupted cyclin D-Cdk4,6 binding to Rb with GST-pull-down assays (Figures 1I and 1J).
Finally, we examined the effect of combining mutations on Rb and found that LxCxE, RxL, and helix mutations were additive (Figures 1K, 1L, and S2G). This suggests that these docking sites interact with different parts of cyclin D, but it is unclear why disruption of only two of three available cyclin D docking mechanisms can render Rb unphosphorylatable in vitro. Taken together, our analysis reveals a mechanism of helix-based docking for cyclin D-Cdk4,6 within a diverse set of poorly understood docking interactions distinct for each cyclin (Figure 1M).
Cyclin D1-Cdk4,6 Phosphorylates Synthetic Substrates Fused to the Rb C-Terminal Helix
To further test the helix-based docking model, we sought to confer increased cyclin D activity to poor Cdk4,6 substrates such as an Rb peptide containing a single Cdk consensus phosphorylation site (Grafström et al., 1999; Kitagawa et al., 1996). To do this, we generated a fusion protein containing a GST tag, Rb amino acids 775–790 that contain a single Cdk site, a Gly-Ser linker, and the Rb C-terminal helix (Figures 2A and S4; Table S1). Fusing the helix to this previously poor substrate led to a 13.9 fold ± 0.1-fold increase in phosphorylation by cyclin D1-Cdk6 but did not affect phosphorylation by cyclin E1-Cdk2 (Figures 2B and 2C). We also observed a similar increase in activity toward fusion proteins containing single Cdk sites on peptides derived from Histone H1 (Matsushime et al., 1994) or Rb amino acids 790–805 (Figures S4G and S4H). Cyclin D1-Cdk6 did not phosphorylate synthetic substrates containing the Rb C-terminal helix with the predicted core interface residues substituted with alanines (Figures 2B, S4C, S4F, and S4G). We again tested the impact of the orientation of these interface residues on docking by generating a synthetic substrate containing a reversed Rb C-terminal helix. Our observation that this synthetic substrate was poorly phosphorylated by cyclin D1-Cdk6 is consistent with our data suggesting that the orientation of the C-terminal helix is critical for docking of full-length Rb (Figures 2A, 2B, S2C, S4C, and S4F). Moreover, fusion of the Rb C-terminal helix did not affect the phosphorylation of these synthetic substrates by cyclin E1-Cdk2, further supporting that the Rb C-terminal helix is specific for cyclin D-Cdk4,6 complexes (Figures 2C–2E, S4D, S4F, and S4H). Taken together, these data show that the Rb C-terminal helix is sufficient to direct cyclin D-Cdk4,6 complexes for phosphorylation, regardless of intrinsic preferences for particular Cdk sites since it enhances phosphorylation for both the relatively weak and strong Cdk phosphorylation sites that we tested (Figure S4).
Figure 2. The Rb C-Terminal Helix Is Sufficient to Recruit Cyclin D1-Cdk4,6 Complexes for Phosphorylation.

(A) Schematic of the engineered GST-Cdk phosphorylation site fusion protein containing a GST tag and the Rb amino acids 775–787 containing a single Cdk site fused to either no docking site, the Rb C-terminal helix docking site (+Helix), the Rb C-terminal helix docking site with the three interface residues substituted with alanines (+Helix mut.), or the Rb C-terminal helix docking site in reverse (+Helix rev.).
(B and C) In vitro kinase assays of the indicated engineered GST-Cdk phosphorylation site fusion protein by (B) cyclin D1-Cdk6 or(C) cyclin E1-Cdk2. Data are mean ± SEM; n = 3.
(D and E) Schematic of docking interaction between cyclin D-Cdk4,6 complexes and (D) the engineered GST-Cdk phosphorylation site fused to the Rb C-terminal helix docking site in comparison to (E) mutant versions of the engineered GST-Cdk phosphorylation site fusion protein.
D-Type Cyclins Recognize the Rb C-Terminal Helix
To determine the subunit of the cyclin D-Cdk4,6 complex that interacts with the Rb C-terminal helix, we generated the remaining D-type cyclin-Cdk complexes formed by cyclins D2 and D3 with Cdk4 and Cdk6. All six cyclin D-Cdk4,6 complexes target Rb for phosphorylation through helix-based docking (Figure 3A). Because all three D-type cyclins have the α1 helix hydrophobic patch, we sought to determine what the cyclin D hydrophobic patch recognized. To do this, we purified Cdk6 in complex with a cyclin D1 variant in which the residues that form the hydrophobic patch were substituted with alanines (cyclin D1HP mut.). Compared to wild type cyclin D1-Cdk6, cyclin D1HP mut-Cdk6 exhibited weaker kinase activity toward Rb consistent with a function for the hydrophobic patch (Figure 3B). However, cyclin D1Hp mut.-Cdk6 did not phosphorylate an Rb variant where the C-terminal helix interface residues were substituted with alanines. This implies that the hydrophobic patch is not responsible for recognizing the Rb C-terminal helix (Figure 3B).
Figure 3. D-Type Cyclins Recognize the Rb C-Terminal Helix.

(A) In vitro kinase assays with all six cyclin D-Cdk4,6 complexes and the denoted Rb variants. The Coomassie-stained gels showing equal amounts of substrate used in each reaction are placed below each autoradiograph. Fold changes ± SEM of wild type to Helix mutant phosphorylation are: 30 ±10for cyclin D1-Cdk4; 19 ± 3 for cyclin D2-Cdk4; 90 ± 20 for cyclin D3-Cdk4; 17 ± 6 for cyclin D1-Cdk6; 19 ± 8 for cyclin D2-Cdk6; 30 ± 20 for cyclin D3-Cdk6. Representative experiments shown (out of two independent experiments).
(B) In vitro kinase assays of the denoted Rb variant with cyclin D1-Cdk6 or cyclin D1HP mut.-Cdk6. The Coomassie-stained gels showing equal amounts of substrate used in each reaction are placed below each autoradiograph. Representative experiments shown (out of two independent experiments).
(C) In vitro kinase assays using the denoted cyclin D1-Cdk6 complexes with GST-Rb775–787(S780)+Helix docking or GST-Rb775–787(S780)+RxL docking. Cyclin D1HPmut.- denotes mutation of the hydrophobic patch on cyclin D1. Cyclin D1LxCxE mut denotes mutation of the LxCxE motif on cyclin D1. The Coomassie-stained gels showing equal amounts of substrate used in each reaction are placed below each autoradiograph. Representative experiments shown (out of three independent experiments).
(D and E) Quantification of the phosphorylation of the engineered GST-Cdk phosphorylation site fusion protein containing helixdocking from Rb (+Helix) or RxL docking from Cdc6 (+RxL) by wild type cyclin D1-Cdk6 versus (D) cyclin D1HP mut -Cdk6 or (E) cyclin D1LxCxE mut -Cdk6. Data are mean ± SEM; n = 3.
(F) In vitro kinase assays with cyclin D1 fused to Cdk2. GST-Rb775–787(S780) without docking or GST-Rb775–787(S780)+Helix docking were used as substrates. The Coomassie-stained gel showing equal amounts of substrate used in each reaction is placed below the autoradiograph. Representative experiments shown (out of two independent experiments).
(G) Schematic of the docking interactions between cyclin D1, D2, and D3 complexes and their substrates. Cyclins D1–D3 are capable of recognizing substrate proteins through helix-based docking, independent of the Cdk present in the complex.
To test if cyclin D’s hydrophobic patch docks RxL sequences similar to cyclin A and E (Adams et al., 1999; Hirschi et al., 2010; Schulman et al., 1998; Takeda et al., 2001), we compared the ability of cyclin D1-Cdk6 and cyclin D1HP mut.-Cdk6 to phosphorylate synthetic substrates containing an RxL motif. Mutation of the cyclin D hydrophobic patch removed the ability of cyclin D1-Cdk6 to phosphorylate a fusion protein containing a GST tag, the Rb amino acids 775–790 containing a single Cdk site, and a peptide containing an RxL sequence derived from the known Cdk substrate Cdc6 (Takeda et al., 2001) (Figures 3C, 3D, and S4E). However, the cyclin D hydrophobic patch mutation did not affect the ability of cyclin D1-Cdk6 to phosphorylate a fusion protein where the RxL docking sequence was replaced by the Rb C-terminal helix (Figures 3C and 3D). As expected, mutation of the LxCxE sequence in cyclin D did not affect phosphorylation of any of these substrates (Figures 3C and 3E). Taken together, these data suggest that the hydrophobic patch on cyclin D recognizes linear RxL sequences, including those on Rb.
That cyclin D’s hydrophobic recognized RxL sequences but not the Rb C-terminal helix raised the question if cyclin D or Cdk4,6 was responsible for helix docking. To test if cyclin D was responsible for helix-based docking, we purified cyclin D1-Cdk2 complexes and observed that cyclin D1-Cdk2 complexes phosphorylate synthetic substrates containing the C-terminal helix (Figure 3F). Because cyclin E1-Cdk2 complexes do not use helix docking but cyclin D1-Cdk2 complexes can use helix-based docking, our experiments suggest that the helix-docking site likely lies on cyclin D rather than Cdk4,6. Thus, the hydrophobic patch of cyclin D likely recognizes RxL sequences, while a different region on cyclin D is likely responsible for recognizing the Rb C-terminal helix (Figure 3G).
A C-Terminal Docking Helix Is Present across the Metazoan Rb Protein Family
We next examined if other cyclin D-Cdk4,6 substrates used helix-based docking. As few Cdk4,6 substrates are known (Anders et al., 2011; Malumbres and Barbacid, 2005), we initially examined the Rb family members, p107 and p130, which are similar in sequence and function (Dick and Rubin, 2013). We hypothesized that all human and mouse Rb family proteins, based on their sequence, have C-terminal docking helices and found that they were targeted by cyclin D-Cdk4,6 in vitro (Figures 4A–4D and S5). Alanine substitutions at residues we predict to be the docking interface for p107 and p130 also disrupted phosphorylation by cyclin D1-Cdk4 (Figures 4A–4D). We observed a similar requirement for helix-based docking with mouse Rb (Figures S5K and S5L). The presence of helix-based docking across the Rb family led us to search for the conserved sequence motif, which we identified by aligning 682 C-terminal sequences of metazoan Rb family members (Medina et al., 2016) and generating a consensus sequence motif (Figure 4E). We did not find such C-terminal helices in Rb sequences outside metazoans, suggesting that helix-based docking is a metazoan innovation (Figure 4F). Taken together, our results suggest that cyclin D-Cdk4,6 targets the Rb family through a similar mechanism across metazoans. A motif search through the human proteome looking for such docking helices on proteins containing Cdk consensus phosphorylation sites suggests as many as 70 proteins may use this mechanism (Table S2).
Figure 4. A C-Terminal Docking Helix Is Present in the Metazoan Rb Protein Family.
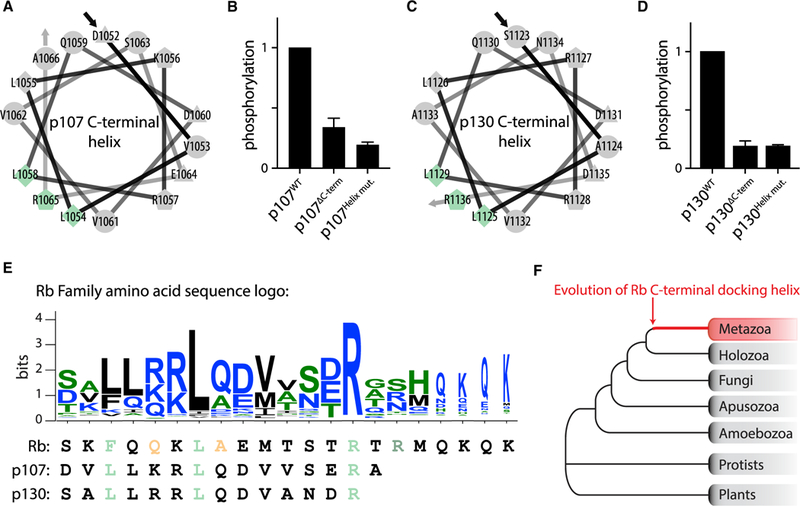
(A–D) Helical wheel projection of the predicted C-terminal helix for human (A) p107 and (C) p130, and in vitro kinase assays of human (B) p107 and (D) p130 variants by cyclin D1-Cdk4. DC-term denotes truncating p107 after amino acid position 1014 and p130 after amino acid position 1122. Helix mut. denotes alanine substitution of the predicted docking interface residues indicated in blue and green in (A) and (C). Data are mean ± SEM; n = 2.
(E) C-terminal helix sequence motif generated using 682 sequences of metazoan Rb family members. In the logo, blue denotes hydrophilic residues, green denotes neutral amino acids, and black denotes hydrophobic amino acids. Below the logo, C-terminal helix residues for Rb, p107, and p130 are aligned. The C-terminal helix residues that were tested for importance to cyclin D-docking are colored according to Figure 1G and (A) and (C).
(F) A C-terminal Rb helix was not found outside metazoan proteome sequences.
The Cyclin D-Rb Interaction Promotes the G1/S Transition, Rb Dissociation from Chromatin, and E2F1 Activation
The identification of the Rb C-terminal helix allowed us to test the function of Rb phosphorylation by cyclin D-Cdk4,6 in cell-cycle control. This is because mutation of Rb’s C-terminal helix disrupts phosphorylation by cyclin D-Cdk4,6, but not other cell-cycle-dependent cyclin-Cdk complexes so that the introduction of Rb C-terminal helix mutations specifically tests the function of the cyclin D-Rb interaction.
To test the function of the cyclin D-Rb interaction, we examined immortalized human mammary epithelial cells (HMECs) expressing doxycycline-inducible Rb variant proteins fused to Clover fluorescent protein and 3FLAG affinity tags (Figures 5A, 5B, S6A and S6B). We chose to examine HMECs because they are a non-transformed cell line previously used to study cell growth and proliferation (Sack et al., 2018). Expression of exogenous wild type Rb in HMECs had a minor effect on cell-cycle progression and cell size, whereas expression of RbHelix mut., which lacks helix-based docking, increased the fraction of cells in G1 from 50% to 75%, and increased cell size by 55% (Figures 5C and 5D). Expression of RbHelix mut. had a similar effect to exposing cells to 500 nM palbociclib, a Cdk4,6 inhibitor (Figure 5C). Expression of the double mutant RbLxCxE cleft+Helix mut. protein, which lacks LxCxE- and helix-based docking mechanisms, resulted in a dramatic G1 cell-cycle arrest and a cell size increase similar to the effect of expressing an RbΔCdk protein lacking all 14 Cdk phosphorylation sites (Figures 5C and 5D). The cell size and cell-cycle phase phenotypes correlated with the amount of chromatin-bound Clover-3FLAG-Rb in G1 phase cells for each Rb variant (Figure 5E). More specifically, following a low salt wash (Håland et al., 2015; Lundberg and Weinberg, 1998), there was 11.5-fold more RbΔCdk than wild type Rb that remained bound to chromatin in G1 in a representative experiment. These effects were not due to differences in Rb protein expression as measured using a quantitative immunoblot (Figures S6A and S6B). We also examined Rb variant expression in an Rb-deficient U-2 OS cell line and observed that both U-2 OS and U-2 OS RB1−/− cell lines responded to exogenous Rb expression similarly to HMECs. In these cell lines, RbHelix mut. caused a greater increase in the G1 fraction of cells than wild type Rb (Figure S6C).
Figure 5. The Cyclin D-Cdk4,6-Rb Interaction Promotes the G1/S Transition, Rb Dissociation from Chromatin, and E2F1 Activation.

(A) Map of PiggyBac integration constructs containing doxycycline-inducible human RB1 gene fused to fluorescent Clover and 3X FLAG affinity tag sequences.
(B) Composite phase contrast and Clover fluorescence images showing expression of Clover-3X FLAG-Rb in HMECs with and without 500 ng/mL doxycycline.
(C-E) Cell-cycle analysis by EdU incorporation and DAPI staining (data are mean ± SEM; n = 2) (C), cell size in G1 (representative experiments shown, out of three independent experiments) (D), and amount of chromatin-bound Rb in G1 (representative experiments shown, out of three independent experiments) (E) in HMECs expressing Clover-3FLAG alone (empty), wild type Rb, or the indicated mutant Rb following a 48-h induction with doxycycline (500 ng/mL). RbLxCxE cleft mut. lacks the LxCxE docking cleft. RbHellx mut. denotes an Rb variant where the predicted docking interface residues F897, L901, and R908 are substituted with alanines. RbLxCxE cleft+Helix mut. contains both indicated mutations. RbΔCdk lacks the 14 Cdk phosphorylation sites. Listed in the upper right of each histogram are median FSC for (D) and Clover-3FLAG-Rb for (E).
(F) Schematic of Rb phosphorylation time course experiment. T98G cells were arrested by serum starvation (−FBS), Rb was induced (+dox), and samples were collected at 0, 4, 8, 12, and 24 h after release (+10% FBS, +dox).
(G) Immunoblot analysis of lysates from Rb phosphorylation time course described in (F) with the denoted antibodies. Representative experiments shown (out of three independent experiments).
(H and I) Quantification of (H) phospho-Rb(807/811)over total Clover-3FLAG-Rband (I) E2F1 from Rb phosphorylation time course in (G). Data are mean ± SEM; n = 3.
Next, we examined the phosphorylation of exogenously expressed wild type Rb and RbHelix mut. in T98G cells that were arrested and synchronously released into the cell cycle following serum starvation (Figure 5F). Cells expressing RbHelix mut. displayed less phosphorylated Rb and had weaker E2F1 expression, suggesting that cyclin D-Cdk4,6-dependent phosphorylation during G1 is important for E2F1-dependent transcription and cell-cycle entry (Figure 5G–5l). We chose to examine E2F1 expression because it is a target of activating E2F transcription factors (Johnson et al., 1994). Consistent with helix-based docking being important for activating E2F-dependent gene expression, we observed lower E2F1 protein levels in cells expressing the RbHelix mut. variant compared to wild type Rb (Figure 5I). Taken together, these experiments are consistent with our biochemical results and show the additive effect of disrupting LxCxE- and helix-based docking mechanisms. Moreover, our analysis strongly supports the role of the cyclin D-Rb interaction as a critical driver of cell-cycle entry at the G1/S transition.
Disruption of the Cyclin D-Rb Interaction Slows Tumor Growth
We next sought to test our model that cyclin D docks the Rb C-terminal helix to promote cell-cycle progression in an animal model. To determine if the Rb variant lacking helix-based docking, RbHellx mut., is a more potent tumor suppressor than wild type Rb, we integrated a vector containing doxycycline-inducible Rb alleles into a Kras+/G12D; Trp53−/− mouse pancreatic ductal adenocarcinoma cell line (Mazur et al., 2015) (Figures 6A, S6D, and S6E). We then allografted these cell lines into NSG mice by subcutaneous implantation. Tumors were allowed to engraft and grow for 5 days before we induced expression of the exogenous Rb alleles (Figure 6A). The tumor suppressor function of Rb was enhanced in the RbHelix mut. variant lacking helix-based docking and was more enhanced by the removal of the Cdk sites as in RbΔCdk (Figure 6B; see Figure S7 for fold changes and p values for all genotype comparisons). At the end of a biological replicate experiment, tumors were extracted and weighed, further demonstrating the difference between the tumor suppressor potency of wild type Rb and RbHelix mut. (Figure 6C). Interestingly, we did not find an additive effect when an LxCxE cleft mutation, RbLxCxE cleft mut., was combined with either wild type Rb or RbHelix mut. (Figure S7), consistent with the previously reported mild phenotype of this mutation (Dick et al., 2000). Our results here support the model in which the cyclin D-Rb helix docking interaction drives Rb inactivation and cell-cycle progression in vivo.
Figure 6. Disruption of the Cyclin D-Cdk4,6-Rb Interaction Slows Tumor Growth.
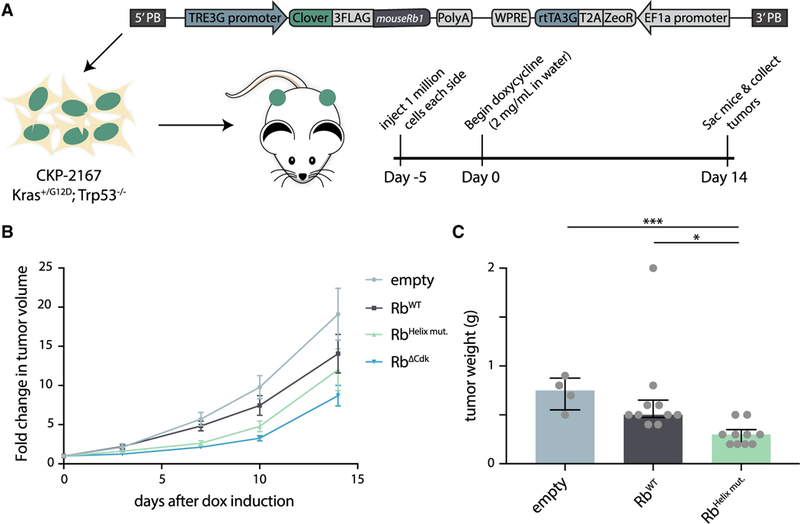
(A) Schematic of mouse experiment. PiggyBac integration constructs containing doxycycline-inducible mouse Rb1 fused to fluorescent Clover and 3FLAG affinity tag sequences were transfected into Kras+/G12D; Trp53−/− mouse pancreatic ductal adenocarcinoma cells (CKP-2167). Approximately 1 million CKP-2167 cells expressing variants of doxycycline-inducible mouse Rb were allografted by subcutaneous implantation. After 5 days of engraftment and growth, mice were given water supplemented with doxycycline (2 mg/mL) for 2 weeks.
(B) Fold change in tumor volume compared to day 0 were calculated from caliper measurements. Data are mean ± SEM. A table of p values for all fold-change comparisons can be found in Figure S7B. n = 12 for empty, RbWT, RbHelix mut., and RbΔCdk.
(C) Median tumor weight and interquartile ranges 15 days after doxycycline induction of either empty vector, wild type Rb, or RbHelix mut. n.s., *p > 0.05, **p ≤ 0.05, and ***p ≤ 0.001. n = 4 for empty, n = 10 for RbWT, and n = 10 for RbHelix mut.
DISCUSSION
Taken together, our work demonstrates the importance of helix-based cyclin D docking on Rb to promote phosphorylation, drive cell-cycle progression, and inhibit Rb’s tumor suppressive function. This study was motivated by the apparent conflict between the long-standing model of the G1/S transition and recent reports that cyclin D-Cdk4,6 may drive cell proliferation and survival through Rb-independent mechanisms. We identified a C-terminal helix in Rb that specifically docks cyclin D (Figure 7A). Mutation of the Rb C-terminal helix disrupts its interaction with cyclin D but preserves the ability of other cyclin-Cdk complexes to phosphorylate Rb. Expressing Rb protein variants lacking the C-terminal helix arrested cells in G1 and slowed tumor growth even though cyclin D-Cdk4,6 could still interact with all of its other targets. A model where cyclin D inhibits Rb indirectly through downstream cyclins is therefore unlikely because these Rb variants lacking the docking helix are readily targeted by downstream cyclins E and A. Thus, Rb phosphorylation by cyclin D-Cdk4,6 is a crucial first step in driving the G1/S transition.
Figure 7. Cyclin D-Cdk4,6 Inactivates Rb through Multiple, Specific Docking Interactions to Drive Cell-Cycle Progression.

(A) Schematic of the multiple docking interactions between cyclin D and Rb. The LxCxE cleft in the Rb pocket interacts with an LxCxE motif at the N terminus of cyclin D. The RxL motifs on the C terminus of Rb interact with the hydrophobic patch (HP) of cyclin D. The C-terminal helix of Rb interacts with an unknown part of cyclin D. Together, these interactions contribute to how the Cdk4 and Cdk6 active sites target the 14 accessible Cdk sites on Rb.
(B) A model for the major functions of cyclin D-Cdk4,6 complexes. Cyclin D-Cdk4,6 docks, phosphorylates, and inactivates Rb to promote S Phase. Cyclin D-Cdk4,6 also promotes cell proliferation and survival through phosphorylation of other substrate proteins.
While Rb is clearly an important target for cyclin D to promote cell proliferation, it may not be the only such target. Although we believe the major cell-cycle function of cyclin D-Cdk4,6 is to inactivate and phosphorylate Rb through helix-based docking, the complex also independently influences cell survival by phosphorylating and inactivating metabolic enzymes. Thus, cell proliferation in vivo may be enhanced by increasing cell survival and by driving cell division through distinct cyclin D-Cdk4,6 targets (Figure 7B). By examining Rb-docking helices across metazoans, we identified a consensus helix sequence motif, which we then used to generate a list of potential substrates in the human proteome (Table S2). This list may provide more insight into non-canonical substrates and functions of cyclin D-Cdk4,6 that could promote proliferation.
In general, progression through the eukaryotic cell cycle is characterized by a monotonic increase in cyclin-dependent kinase activity (Stern and Nurse, 1996). However, not all cell-cycle-dependent substrates are phosphorylated at the same time (Swaffer et al., 2016, 2018). Early substrates are typically targets of early cyclin-Cdk complexes via specific docking mechanisms (Loog and Morgan, 2005). To prevent later substrates from being phosphorylated by the early activated cyclin-Cdk complexes, these early complexes are typically characterized by lower intrinsic kinase activity. For example, in budding yeast, peptides containing a single Cdk site derived from histone H1 were phosphorylated at progressively higher rates by G1-phase, S-phase, and mitotic cyclins (Kõivomägi et al., 2011). Here, we report a similar progressive increase in intrinsic kinase activity toward histone H1 by animal cyclin-Cdk complexes. By far, the lowest activity toward H1 was found in cyclin D-Cdk4,6 complexes. Thus, the progression of the cell cycle from high-specificity cyclin-Cdk complexes through to high-activity complexes is likely a conserved feature of eukaryotic cell-cycle control.
The low activity of cyclin D-Cdk4,6 complexes may be compensated for by the specificity of their docking mechanisms. In contrast to previously identified cyclin-Cdk docking motifs, which are short linear motifs in intrinsically disordered regions of proteins, Rb helix-based docking is unique in its structural requirement. While short linear motifs are easy to evolve (Davey et al., 2015), the addition of a structural requirement, such as a helix, likely makes it much more difficult. This is because both the helix and the appropriate interface residues must align. The difficulty of evolving helix-based docking could therefore underlie the observation that cyclin D-Cdk4,6 appears to have relatively few substrates compared to other cyclin-Cdk complexes (Anders et al., 2011; Malumbres and Barbacid, 2005). We speculate that this low-activity, high-docking-specificity kinase has few substrates so it may adequately phosphorylate key targets, such as Rb.
Cyclin D-Cdk4,6 complexes clearly play a role in cancer, and a series of cancer drugs targeting the ATP pockets of Cdk4,6 are successfully emerging from clinical trials (Sherr et al., 2016). However, while Cdk4,6 inhibitors such as palbociclib have been observed to limit disease progression, the differences in overall survival compared to standard of care are not statistically significant (Finn et al., 2016; Turner et al., 2018). That Cdk4,6 inhibitors have significant off-target activities (Hafner et al., 2017) raises the possibility that these cancer therapies can be improved by a new class of drugs targeting cyclin substrate recognition and helix-based docking.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan M. Skotheim (skotheim@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Transfection
Immortalized human mammary epithelial cells HMEC-hTERT1 cells were cultured in MEGM Mammary Epithelial Cell Growth Medium (Lonza CC-3150) (Sack et al., 2018). T98G, CKP-2167, and U-2 OS cells were cultured in DMEM supplemented with 10% FBS, 4.5g/L Glucose, 2 mM L-glutamine, and Sodium Pyruvate (Mazur et al., 2015). Cell lines stably expressing doxycycline-inducible Rb variants were generated by transfecting cells plated into individual wells of a 6 well dish with 2.2 mg of doxycycline-inducible PiggyBac integration plasmid and 1.1 mg of PiggyBac Transposase plasmid (Ding et al., 2005) using the FuGene HD reagent (Promega E2311). Zeocin (400ug/mL) selection began two days after transfection. Zeocin resistant cells were maintained as polyclonal cell lines. Prior to flow cytometry and immunoblot analysis, cells were grown in the presence of doxycycline (500 ng/mL) for two days.
Doxycycline-inducible PiggyBac integration vector construction
We cloned an Rb expression cassette, driven by the TRE3G doxycycline-inducible promoter (Clontech 631168), into a PiggyBac integration plasmid containing 5′ and 3′ PiggyBac homology arms (Ding et al., 2005; Shariati et al., 2018). The expression cassette contains the human RB1 or mouse Rb1 genes fused to fluorescent Clover and 3FLAG affinity tag sequences, a zeocin resistance gene, and a Tet-On 3G transactivator gene driven by the Ef1α promoter.
Subcutaneous tumor implantation
CKP-2167 expressing variants of doxycycline-inducible mouse Rb were allografted by subcutaneous implantation in NSG mice. For each implantation, approximately 1 million cells were suspended in 100 μL of 1X PBS and mixed with 100 μL of Matrigel Basement Membrane Matrix (Corning 356237). This mixture was injected into the left and right flanks of each mouse. After five days of engraftment and growth, mice were given water supplemented with doxycycline (2 mg/mL) for two weeks. Tumor lengths and widths were measured with calipers and volumes were estimated with the formula V= (4/3) × π × r3, where r is half of the average tumor diameter or (length + width) ÷ 4. At the end of the experiment, mice were sacrificed, and tumors were harvested. Animal studies were done in compliance with the Stanford Administrative Panel on Laboratory Animal Care Protocol 13565.
METHOD DETAILS
Protein expression and purification
Full-length, N-terminally glutathione S-transferase-tagged (GST-tagged), Rb family proteins were expressed in the E. coli strain BL21, purified by glutathione-agarose affinity chromatography (Sigma-Aldrich G4510), and eluted with 50mM Tris pH 8.0; 100mM KOAc; 25mM MgOAc; 10% glycerol; 15mM Glutathione (Sigma-Aldrich G4251).
GST-Cdk phosphorylation site fusion proteins (Figure S4; Table S1) containing a GST tag, a TEV-protease cleavage site, and a single Cdk phosphorylation site peptide were expressed and purified as described above. The three different Cdk phosphorylation sites that we used were (1) a Cdk site patterned after a region of similar sequence found in histone H1 protein (Hagopian et al., 2001; Kõivomägi et al., 2011) (H1 site: PKTPKKAKKL), (2) an S780 Cdk site from Rb (Kitagawa et al., 1996) (Rb 775–787: RPPTLSPIPHIPR), and (3) an S795 Cdk site from Rb (Grafström et al., 1999) (Rb 790–805: YKFPSSPLRIPGGNIY). To test docking specificity of the cyclin-Cdk complexes, we also fused these GST-Cdk site fusion proteins to the Rb C-terminal Helix (Rb 895–915: SKFQQKLAEMTSTRTRMQKQK) or the Cdc6 RxL docking motif (Takeda et al., 2001) (Cdc6 89–103: HTLKGRRLVFDNQLT) using a G4S glycine-serine linker (GGGGS).
Human cyclin-Cdk fusion complexes were purified from budding yeast cells (Schwarz et al., 2018) using a 3X FLAG affinity purification method, modified from a previous protocol used for HA-tag purification (McCusker et al., 2007). Briefly, N-terminally tagged cyclin-Cdk fusions were cloned into 2-micron vectors using a glycine-serine linker (Rao et al., 1999) and overexpressed from the GAL1 budding yeast promoter. The overexpressed 3FLAG-tagged cyclin-Cdk complexes were then purified by immunoaffinity chromatography using ANTI-FLAG M2 affinity agarose beads (Sigma-Aldrich A2220) and eluted with 0.2 mg/mL 3X FLAG peptide (Sigma-Aldrich F4799). We note that similar cyclin-Cdk fusions have previously been able to restore wild-type function in vivo (Chytil et al., 2004).
In vitro kinase assays
For all experiments, equal amounts of substrate and purified kinase complexes were used. Substrate concentrations were kept in the range of 1–5 mM for different experiments, but did not vary within any experiment. Reaction aliquots were taken at two time points (8 and 16 minutes) and the reaction was stopped with SDS-PAGE sample buffer. The basal composition of the assay mixture contained 50 mM HEPES pH 7.4, 20 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl2, 10 mM MgOAc, 40 mM KOAc, 6 mM glutathione, 0.2 mg/ml 3X FLAG peptide, 6% glycerol, 3 mM EGTA, 0.2 mg/ml BSA and 500 μM ATP (with 2 μCi of [γ−32P] ATP added per reaction; PerkinElmer BLU502Z250UC). Histone H1 protein used as a general substrate for Cdk was purchased from EMD Millipore (14–155). Phosphorylated proteins were separated on 10% SDS-PAGE gels. Phosphorylation of substrate proteins was visualized using autoradiography (Typhoon 9210; GE Healthcare Life Sciences). Autoradiographs were quantified with the ImageQuant TL Software. We note that phosphorylation of H1 and Rb by our cyclin-Cdk fusions occurs in the linear range for the 8- and 16-minute time-points (Figures S1D–S1K). Within every experiment and quantification, the reactions have been conducted at the same time, run on the same gel, and images were taken of the same exposures. The few cases in which the gels were cropped (Figure S1B) were done to rearrange the order of lanes to increase figure clarity.
Glutathione S-transferase (GST) pull down assay
GST-tagged Rb proteins were dialyzed to remove glutathione using a buffer containing 50mM Tris pH 8.0, 100mM KOAc, 25mM MgOAc, and 10% glycerol. Next, GST-Rb proteins were bound to glutathione-agarose beads (Sigma-Aldrich G4510) for 1 hour at 4°C in a binding buffer containing 50 mM Tris pH 8.0,150 mM NaCl, 1% Triton X-100, and 1 mM DTT. This bead-GST-Rb mixture was washed with this binding buffer and incubated with an equimolar amount of 3FLAG-tagged cyclin-Cdk complexes for 2–3 hours at 4°C. Beads were then washed with binding buffer and eluted with 2X SDS-PAGE sample buffer. Input and pulldown samples were then analyzed by immunoblotting.
Circular dichroism
Circular dichroism (CD) spectra were recorded using a Jasco J-1500 CD spectrometer. Samples contained 30 μM recombinant Rb 890–920 in a buffer containing 25 mM sodium phosphate and 100 mM NaCl (pH 6.1). The data were fit by calculating a weighted average of reference spectra measured for poly-L-lysine, which adopts known different secondary structures depending on pH and temperature (Greenfield and Fasman, 1969).
Helix prediction, bioinformatics analysis, and helix motif search
Secondary structure predictions were carried out on the PSIPRED protein structure prediction server using the PSIPRED v3.3 Secondary Structure prediction method (Buchan et al., 2013) (http://bioinf.cs.ucl.ac.uk/psipred/). Helical wheel projections of predicted C-terminal helices were generated using the “Helical Wheel Projections” tool (http://rzlab.ucr.edu/scripts/wheel/wheel.cgi).
The full-length sequences of Rb family members (Medina et al., 2016) were aligned by MAFFT-L-INS-I (Katoh et al., 2017). A profile Hidden-Markov model (HMM) was generated using the HMMER3 web service (Finn et al., 2015). JackHMMER used to iteratively search for Rb homologs based on the profile HMM, using a stringent E-value cutoff of 1e-10 against UniProt Reference Proteomes. Of the total 1072 eukaryotic Rb homologs retrieved, 682 were metazoan. All sequences were re-aligned using MAFFT-L-INS-i (maxitr = 1000). The aligned metazoan sequences were then trimmed to focus on sequence positions occupied by human Rb. Membership in RB1, RBL1, or RBL2 sub-families were determined by manual examination of the phylogenetic tree generated using Fast Tree (Price et al., 2009). Non-metazoan Rb sequence from taxa closely related to metazoan were examined manually.
Aligned sequences from the helix region of metazoan RB family members were used to generate a position specific weight matrix (PSSM). The PSSM was used to score protein sequences in the UniProtKB/Swiss-Prot reviewed human proteome (UP000005640). PSIPRED was used perform helicity prediction on sequences with PSSM scores above 20, which corresponds to 5 standard deviation above the mean random score distribution). The sequences were further filtered by the presence of CDK substrate sites ([S/T]P) within the full-length protein. All analysis was done using BioPython (Cock et al., 2009). Results from helix motif search are listed in Table S2.
Flow cytometry analysis
Flow cytometry analysis was performed on BD LSRII.UV cytometer. Live cells were prepared for flow cytometry by washing with 1X PBS, trypsinizing, and resuspending in 1X PBS. For live cells, we measured forward scatter area as a readout for cell size. For measurement of newly synthesized DNA by nucleoside analog incorporation, cells were incubated with 10 μM EdUfor30 minutes at 37°C, fixed in 3% formaldehyde for 10 minutes at 37°C, and permeabilized with 90% ethanol for 30 minutes on ice. EdU was detected using the Click-iT Plus EdU Alexa Fluor 594 Flow Cytometry Assay Kit (C10646) and cells were stained with 3 μM DAPI for 10 minutes at room temperature. For these fixed cells, we measured EdU, DAPI fluorescence as a readout for DNA content, Clover green protein fluorescence as a readout for the amount of tagged exogenous Rb, and forward scatter area as a readout for cell size. To measure the amount of DNA-associated Rb, we used a soluble protein extraction method (Håland et al., 2015). Briefly, the cells were harvested by trypsinization and pelleted by centrifugation. The cell pellet was then resuspended in ice-cold low salt extraction buffer (0.1% Igepal CA-630,10 mM NaCl, 5 mM MgCl2, 0.1 mM PMSF, 10 mM Potassium phosphate buffer pH 7.4) and incubated on ice for 1 minute. Then, the cells were fixed by adding paraformaldehyde to a final concentration of 3% and incubating on ice for 1 hour. Fixed cells were washed once with 1×PBS and then stained with 20 μM Hoechst 33342 DNA dye for 10 minutes at 37°C in 1×PBS. DNA content and DNA-bound Clover-tagged Rb amounts were measured with a BD LSRII.UV flow cytometer.
Immunoblotting
Portions of harvested tumors were resuspended at 100 mg/mL in RIPA buffer supplemented with protease and phosphatase inhibitors. Next, we homogenized these tumor samples with pestles in tubes, and then sonicated the samples for 10 s at 50% amplitude on a Fisherbrand Model 120 Sonic Dismembrator. Cells cultured on dishes were collected by scraping in 1× PBS and lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Proteins from lysates were separated on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane using the iBlot 2 dry blotting system (Invitrogen IB21001).
Membranes were incubated overnight at 4°C with the following antibodies: Phospho-Rb (Ser807/811) (D20B12) XP Rabbit mAb (Cell Signaling Technology #8516), Purified Mouse Anti-Human Retinoblastoma Protein Clone G3–245 (BD Biosciences 554136), Monoclonal ANTI-FLAG M2 antibody produced in mouse (Sigma-Aldrich F1804), E2F-1 Antibody (Cell Signaling Technology #3742), and mouse monoclonal β-Actin Antibody N-21 (Santa Cruz Biotechnology sc-130656). The primary antibodies were detected using the fluorescently labeled secondary antibodies IRDye 680LT Goat anti-Mouse IgG (LI-COR 926–68020) and IRDye 800CW Goat anti-Rabbit IgG (LI-COR 926–32211). Membranes were imaged on a LI-COR Odyssey CLx and analyzed with LI-COR ImageStudio software.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values were expressed as means ± SEM except for tumor weight, which was expressed as medians and interquartile ranges. Statistical analyses were performed by Student’s t test (two-tailed) using GraphPad Prism 7. P values < 0.05 are considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
Original images of autoradiographs and immunoblots are available at Mendeley Data under the following link: https://doi.org/10.17632/xy3xv64×2d.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Rb (Ser807/811) (D20B12) XP Rabbit mAb | Cell Signaling Technology | Cat#8516; RRID: AB_11178658 |
| Purified Mouse Anti-Human Retinoblastoma Protein Clone G3–245 | BD Biosciences | Cat#554136; RRID: AB_395259 |
| Monoclonal ANTI-FLAG M2 antibody produced in mouse | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| E2F-1 Antibody | Cell Signaling Technology | Cat#3742; RRID: AB_2096936 |
| Mouse monoclonal β-Actin Antibody N-21 | Santa Cruz Biotechnology | Cat#sc-130656; RRID: AB_2223228 |
| IRDye 680LT Goat anti-Mouse IgG | LI-COR | Cat#926–68020; RRID: AB_10706161 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR | Cat#926–32211; RRID: AB_621843 |
| ANTI-FLAG M2 affinity agarose beads | Sigma-Aldrich | Cat#A2220; RRID: AB_10063035 |
| Bacterial and Virus Strains | ||
| E. coli BL21 | Agilent | Cat#230245 |
| E. coli DH5α | Agilent | Cat#200231 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Palbociclib | Santa Cruz Biotechnology | Cat#sc-478943 |
| FuGene HD reagent | Promega | Cat#E2311 |
| Zeocin Selection Reagent | GIBCO | Cat#R25001 |
| Doxycycline, Hyclate, CAS 24390–14-5, | Calbiochem | Cat#324385–1GM |
| Matrigel Basement Membrane Matrix | Corning | Cat#356237 |
| L-Glutathione reduced | Sigma-Aldrich | Cat#G4251 |
| Glutathione-Agarose | Sigma-Aldrich | Cat#G4510 |
| 3X FLAG peptide | Sigma-Aldrich | Cat#F4799 |
| [γ−32P] ATP | PerkinElmer | Cat# BLU502Z250UC |
| HistoneH1 protein | EMD Millipore | Cat#14–155 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dilactate) | Invitrogen | Cat#D3571; RRID: AB_2307445 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Invitrogen | Cat#H1399 |
| Critical Commercial Assays | ||
| Click-iT Plus EdU Alexa Fluor 594 Flow Cytometry Assay Kit | Invitrogen | Cat#C10646 |
| Deposited Data | ||
| Raw data and images | Mendeley Data | https://doi.org/10.17632/xy3xv64x2d.1 |
| Experimental Models: Cell Lines | ||
| Human: HMEC-hTERT1 | Dr. S. Elledge | Sack et al., 2018 |
| Human: T98G | Dr. C. Shwarz | Schwarz et al., 2018 |
| Human: U-2 OS | A.C. Chaikovsky | N/A (unpublished) |
| Human: U-2 OS RB1−/− | A.C. Chaikovsky | N/A (unpublished) |
| Mouse: CKP-2167 | Dr. P. K. Mazur | Mazur et al., 2015 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NSG | The Jackson Laboratory via Dr. M. Winslow | Cat#005557; RRID: IMSR_JAX:005557 |
| Budding yeast: yBT000 W303 MATa bar1::HISG cdc28:: cdc28-as1 sic1Δ::URA3 | Dr. M. Kõivomägi | Kõivomägi et al., 2011 |
| Budding yeast: yBT004 W303 MATa bar1::HISG cdc28:: cdc28-as1 sic1Δ::URA3 LexA-ER-haB112::HIS3 | Dr. M. Kõivomägi | Kõivomägi et al., 2011 |
| Recombinant DNA | ||
| Tet-On 3G Inducible Expression System (pCMV-Tet3G Regulator Plasmid & pTRE3G Response Plasmid) | Clontech Laboratories via Dr. A. Shariati | Cat#631168 |
| pGEX-4T-1 | GE Healthcare | Cat#28–9545-49 |
| PBSKHRbc | Dr. R. Weinberg via Addgene | Cat#1761; RRID: Addgene_1761 |
| pCMV HA hRb delta CDK | Dr. S. Dowdy via Addgene | Cat#58906; RRID: Addgene_58906 |
| See Table S3 for all plasmids generated for this study | N/A | N/A |
| Other | ||
| MEGM Mammary Epithelial Cell Growth Medium | Lonza | Cat#CC-3150 |
| DMEM With 4.5g/L Glucose, Sodium Pyruvate; Without L-Glutamine, Phenol Red | Corning | Cat#17205CV |
| Fetal Bovine Serum, qualified, heat inactivated, USDA- approved regions | GIBCO | Cat#10438026 |
| Penicillin Streptomycin 100X Solution | HyClone | Cat#SV30010 |
| L-Glutamine, 200mM Solution | HyClone | Cat#SH30034.02 |
Highlights.
Cyclin D, but not other cyclins, targets a C-terminal alphahelix docking motif on Rb
Helix-based docking is shared by the p107 and p130 Rb-family members across metazoans
Helix-based docking is a major driver of Rb phosphorylation and the G1/S transition
ACKNOWLEDGMENTS
We thank Nick Dyson, Ioannis Sanidas, and Joe Lipsick for their thoughtful comments on the manuscript, Mart Loog for discussions on cyclin docking, Nick Buchler for discussions on Rb evolution, and Amy Tarangelo and Matthew Swaffer for assistance with graphical abstract and figure design. We thank Ali Shariati fordox-inducible PiggyBac reagents, Andrea Chaikovsky for U-2 OS and U-2 OS RB1−/− cells, Martha Cyert for GST binding assay reagents, Monte Winslow for NSG mice, Garry Coles for technical help with mice, Steve Elledge for HMEC-hTERT1 cells, and Robert Weinberg and Steve Dowdy for Rb plasmids. Flow cytometry data were collected on an instrument in the Stanford Shared FACS Facility obtained using the NIH S10 Shared Instrument Grant S10RR027431. This work was supported by the NIGMS (R01 GM092925 and R01 GM115479) and the NCI through the Cancer Biology Training Grant (T32 CA009302).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.03.020.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Li X, Sellers WR, Baker KB, Leng X, Harper JW, Taya Y, and Kaelin WG Jr. (1999). Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol. 19, 1068–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, and Sicinski P (2011). A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, and de Bruin RAM (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri S, and Pryciak PM (2011). Cyclin-specific docking motifs promote phosphorylation of yeast signaling proteins by G1/S Cdk complexes. Curr. Biol. 21, 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J, and Cross FR (2007). Multiple levels of cyclin specificity in cell-cycle control. Nat. Rev. Mol. Cell Biol. 8, 149–160. [DOI] [PubMed] [Google Scholar]
- Buchan DWA, Minneci F, Nugent TCO, Bryson K, and Jones DT (2013). Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart DL, and Sage J (2008). Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, and Meyer T (2016). Irreversible APC(Cdh1) inactivation underlies the point of no return for cell-cycle entry. Cell 166, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Mark KG, Garbett D, Pack LR, Rape M, and Meyer T (2018). EMI1 switches from being a substrate to an inhibitor of APC/CCDH1 to start the cell cycle. Nature 558, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chytil A, Waltner-Law M, West R, Friedman D, Aakre M, Barker D, and Law B (2004). Construction of a cyclin D1-Cdk2 fusion protein to model the biological functions of cyclin D1-Cdk2 complexes. J. Biol. Chem. 279, 47688–47698. [DOI] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, and de Hoon MJ (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR, and Jacobson MD (2000). Conservation and function of a potential substrate-binding domain in the yeast Clb5 B-type cyclin. Mol. Cell. Biol. 20, 4782–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Cyert MS, and Moses AM (2015). Short linear motifs - ex nihilo evolution of protein regulation. Cell Commun. Signal. 13, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B, Davey NE, Hagting A, Izawa D, Mansfeld J, Gibson TJ, and Pines J (2015). The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev. Cell 32, 358–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, and Rubin SM (2013). Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Sailhamer E, and Dyson NJ (2000). Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20, 3715–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, and Xu T (2005). Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473–83. [DOI] [PubMed] [Google Scholar]
- Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, and Weinberg RA (1993). Physical interaction of the retinoblastoma protein with human D cyclins. Cell 73, 499–511. [DOI] [PubMed] [Google Scholar]
- Ewald JC (2018). How yeast coordinates metabolism, growth and division. Curr. Opin. Microbiol. 45, 1–7. [DOI] [PubMed] [Google Scholar]
- Ewald JC, Kuehne A, Zamboni N, and Skotheim JM (2016). The yeast cyclin-dependent kinase routes carbon fluxes to fuel cell cycle progression. Mol.Cell 62, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, and Eddy SR (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43 (W1), W30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al. (2016). Palbociclib and Letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936. [DOI] [PubMed] [Google Scholar]
- Gorges LL, Lents NH, and Baldassare JJ (2008). The extreme COOH terminus of the retinoblastoma tumor suppressor protein pRb is required for phosphorylation on Thr-373 and activation of E2F. Am. J. Physiol. Cell Physiol. 295, C1151–C1160. [DOI] [PubMed] [Google Scholar]
- Grafström RH, Pan W, and Hoess RH (1999). Defining the substrate specificity of cdk4 kinase-cyclin D1 complex. Carcinogenesis 20, 193–198. [DOI] [PubMed] [Google Scholar]
- Greenfield N, and Fasman GD (1969). Computed circulardichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116. [DOI] [PubMed] [Google Scholar]
- Guiley KZ, Liban TJ, Felthousen JG, Ramanan P, Litovchick L, and Rubin SM (2015). Structural mechanisms of DREAM complex assembly and regulation. Genes Dev. 29, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Mills CE, Subramanian K, Chen C, Chung M, Boswell SA, Everley RA, Walmsley CS, Juric D, and Sorger P (2017). Therapeutically advantageous secondary targets of abemaciclib identified by multi-omics profiling of CDK4/6 inhibitors. bioRxiv. 10.1101/211680. [DOI] [Google Scholar]
- Hagopian JC, Kirtley MP, Stevenson LM, Gergis RM, Russo AA, Pavletich NP, Parsons SM, and Lew J (2001). Kinetic basis for activation of CDK2/cyclin A by phosphorylation. J. Biol. Chem. 276, 275–280. [DOI] [PubMed] [Google Scholar]
- Håland TW, Boye E, Stokke T, Grallert B, and Syljuåsen RG (2015). Simultaneous measurement of passage through the restriction point and MCM loading in single cells. Nucleic Acids Res. 43, e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi A, Cecchini M, Steinhardt RC, Schamber MR, Dick FA, and Rubin SM (2010). An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 17, 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi M, and Stacey DW (1999). Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr. Biol. 9, 1075–1084. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, and Morgan DO (1999). Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 9, 227–236. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Ohtani K, and Nevins JR (1994). Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 8, 1514–1525. [DOI] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, and Yamada KD (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. Published online September 6, 2017. 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, and Taya Y (1996). The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 75, 7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Kõivomägi M, Valk E, Venta R, lofik A, Lepiku M, Morgan DO, and Loog M (2011). Dynamics of Cdk1 substrate specificity during the cell cycle. Mol.Cell 42,610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, and Peters JM (2000). Mitotic regulation of the APC activatorproteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loog M, and Morgan DO (2005). Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature 434, 104–108. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, and Weinberg RA (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2005). Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30, 630–641. [DOI] [PubMed] [Google Scholar]
- Markey MP, Bergseid J, Bosco EE, Stengel K, Xu H, Mayhew CN, Schwemberger SJ, Braden WA, Jiang Y, Babcock GF, et al. (2007). Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 26, 6307–6318. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, and Kato JY (1994). D-typecyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 14, 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura l., Denissova NG, Wang G, He D, Long J, and Liu F (2004). Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 430, 226–231. [DOI] [PubMed] [Google Scholar]
- Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA, Vangala D, et al. (2015). Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapyfor pancreatic ductal adenocarcinoma. Nat. Med. 21, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker D, Denison C, Anderson S, Egelhofer TA, Yates JR 3rd, Gygi SP, and Kellogg DR (2007). Cdk1 coordinates cell-surface growth with the cell cycle. Nat. Cell Biol. 9, 506–515. [DOI] [PubMed] [Google Scholar]
- Medina EM, Turner JJ, Gordan R, Skotheim JM, and Buchler NE (2016). Punctuated evolution and transitional hybrid network in an ancestral cell cycle of fungi. eLife 5, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick KA, Wohlbold L, Zhang C, Allen JJ, Horiuchi D, Huskey NE, Goga A, Shokat KM, and Fisher RP (2011). Switching Cdk2 on oroffwith small molecules to reveal requirements in human cell proliferation. Mol. Cell 42, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO (1997). Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291. [DOI] [PubMed] [Google Scholar]
- Morgan DO (2007). The Cell Cycle (New Science Press). [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by monophosphorylation. eLife 3, 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, and Scholtz JM (1998). A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Cox S, Hoess RH, and Grafström RH (2001).Acyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 61, 2885–2891. [PubMed] [Google Scholar]
- Pardee AB (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RN, Stamm NB, Otto K, Kovacevic S, Watkins SA, Rutherford P, Lemke S, Cocke K, Beckmann RP, Houck K, et al. (1999). Conditional transformation of rat embryo fibroblast cells by a cyclin D1-cdk4 fusion gene. Oncogene 18, 6343–6356. [DOI] [PubMed] [Google Scholar]
- Rubin SM, Gall A-L, Zheng N, and Pavletich NP (2005). Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106. [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, and Pavletich NP (1996). Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 382, 325–331. [DOI] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. (2018). Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 173, 499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Roa M, and Malumbres M (2017). Fueling the cell division cycle. Trends Cell Biol. 27, 69–81. [DOI] [PubMed] [Google Scholar]
- Schulman BA, Lindstrom DL, and Harlow E (1998). Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. USA 95, 10453–10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz C, Johnson A, Koivomagi M, Zatulovskiy E, Kravitz CJ, Doncic A, and Skotheim JM (2018). A precise Cdk activity threshold determines passage through the restriction point. Mol. Cell 69, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariati A, Dominguez A, Wernig M, Qi S, and Skotheim J (2018). Reversible inhibition of specific transcription factor-DNA interactions using CRISPR. bioRxiv. 10.1101/282681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, and McCormick F (2002). The RB and p53 pathways in cancer. Cancer Cell 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, and Roberts JM (2004). Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18, 2699–2711. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Beach D, and Shapiro GI (2016). Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern B, and Nurse P (1996). A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 12, 345–350. [PubMed] [Google Scholar]
- Swaffer MP, Jones AW, Flynn HR, Snijders AP, and Nurse P (2016). CDK substrate phosphorylation and ordering the cell cycle. Cell 167, 1750–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaffer MP, Jones AW, Flynn HR, Snijders AP, and Nurse P (2018). Quantitative phosphoproteomics reveals the signaling dynamics of cell-cycle kinases in the fission yeast Schizosaccharomyces pombe. Cell Rep. 24, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Wohlschlegel JA, and Dutta A (2001). A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 276, 1993–1997. [DOI] [PubMed] [Google Scholar]
- The I, Ruijtenberg S, Bouchet BP, Cristobal A, Prinsen MBW, van Mourik T, Koreth J, Xu H, Heck AJR, Akhmanova A, et al. (2015). Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat. Commun. 6, 5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Slamon DJ, Ro J, Bondarenko I, Im S-A, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, et al. (2018). Overall survival with Palbociclib and Fulvestrant in advanced breast cancer. N. Engl. J. Med. 379, 1926–1936. [DOI] [PubMed] [Google Scholar]
- Wallace M, and Ball KL (2004). Docking-dependent regulation of the Rb tumor suppressor protein by Cdk4. Mol. Cell. Biol. 24, 5606–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, Suski JM, et al. (2017). The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlegel JA, Dwyer BT, Takeda DY, and Dutta A (2001). Mutational analysis of the Cy motif from p21 reveals sequence degeneracy and specificity for different cyclin-dependent kinases. Mol. Cell. Biol. 21, 4868–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung Y, Walker JL, Roberts JM, and Assoian RK (2007). A Skp2 auto-induction loop and restriction point control. J. Cell Biol. 178, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, and Seufert W (1998). Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282, 1721–1724. [DOI] [PubMed] [Google Scholar]
- Zetterberg A, and Larsson O (1985). Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 82, 5365–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Chen Y, Carey L, and Futcher B (2016). Cyclin-dependent kinase co-ordinates carbohydrate metabolism and cell cycle in S. cerevisiae. Mol. Cell 62, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original images of autoradiographs and immunoblots are available at Mendeley Data under the following link: https://doi.org/10.17632/xy3xv64×2d.1.