

Abstract
Mitochondria are capable of detecting cellular insults and orchestrating inflammatory responses. Mitochondrial reactive oxygen species (mtROS) are intermediates that trigger inflammatory signaling cascades in response to our newly proposed conditional damage associated molecular patterns (DAMP). We recently reported that increased proton leak regulates mtROS generation and thereby exert physiological and pathological activation of endothelial cells. Herein, we report the recent progress in determining the roles of proton leak in regulating mtROS, and highlight several important findings: 1) The majority of mtROS are generated in the complexes I and III of electron transport chain (ETC); 2) Inducible proton leak and mtROS production are mutually regulated; 3) ATP synthase-uncoupled ETC activity and mtROS regulate both physiologica006C and pathological endothelial cell activation and inflammation initiation; 4) Mitochondrial Ca2+ uniporter and exchanger proteins have an impact on proton leak and mtROS generation; 5) MtROS connect signaling pathways between conditional DAMP-regulated immunometabolism and histone post-translational modifications (PTM) and gene expression. Continuous improvement of our understanding in this aspect of mitochondrial function would provide novel insights and generate novel therapeutic targets for the treatment of sterile inflammatory disorders such as metabolic diseases, cardiovascular diseases and cancers.
Keywords: Proton leak, Mitochondrial reactive oxygen species (mtROS), Endothelial cell activation, Electron transport chain (ETC) uncoupling, Cardiovascular diseases
1. Introduction - mitochondria are “sentinel” organelles capable of detecting cellular insults and orchestrating inflammatory responses
Historically considered as merely cellular “powerhouses” that manufacture ATP and other metabolites, mitochondria are increasingly being recognized as “sentinel” organelles, which are capable of detecting cellular insults and orchestrating inflammatory responses [1]. Mitochondria are complex organelles, which contain their own DNA and are composed of a double membrane; outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). This double membrane gives rise to two compartments; 1) the intermembrane space (IMS) located between OMM and IMM; and 2) the matrix located in the space created by the IMM itself.
For the proper function and maintenance, mitochondria require coordination between their own genome and the nuclear genome within the cell. A complex cellular signaling network exists to mediate the crosstalk between the mitochondria and the nucleus,and also to elicit mitochondrial responses to cellular stresses [2,3]. Moreover, mitochondria are morphologically dynamic organelles where their shape is tightly regulated by mitochondrial biogenesis processes such as fusion, fission, mitophagy, and endoplasmic reticulum (ER) -mitochondria tethering/juxtaposition [4–7]. Many metabolic and cellular signals play a role in determining mitochondrial morphology, thus affecting its function [8].
The function of mitochondria is not confined to performing oxidative phosphorylation and supplying the ATP to perform cellular processes. There is ample evidence of mitochondria being involved in diverse functions ranging from regulating cellular metabolism to sterile inflammation [9]. From the past publications, it is apparent that mitochondria are important components in innate immunity. The innate immunity responds to infection and injury by recognizing pathogen associated molecular patterns (PAMP) or damage associated molecular patterns (DAMP) [10]. These PAMP and DAMP are recognized by PRR (pattern recognition receptors). Most extensively studied PRR types are TLR (Toll like receptors) and NLR (NOD like receptors). Additionally, RLR (RIG-I like receptors), AIM-2 (absent in melanoma), RAGE (receptor for advanced glycation end products), P2Y receptors and P2X receptors were identified as PRRs [11–14]. MAVS (mitochondrial antiviral signaling proteins) located on OMM are necessary adaptors for activation of RLR in response to viral infections [15–17]. Moreover, mitochondria play a central role in mediating signals of TLR and NLR [18–20].
Recently, our lab demonstrated that mitochondria are essential components in signaling mediated by conditional DAMP such as LPC (lysophosphatidylcholine) [21]. LPC is a member of lysophospholipids (LPL) family. Moreover, LPC is a bioactive lipid and is the major phospholipid component of oxidized low-density lipoproteins [22]. It is believed to be an important mediator in inflammatory disorders [23]. Previously, by using LPL family as a prototype, we identified the metabolites that elicit physiological functions at normal concentration but activate inflammatory responses at elevated concentrations as conditional DAMP [24,25]. We further reported that mitochondria act as sensors, which relay information to the nucleus to modulate the gene expression,and generate appropriate responses to overcome the insults caused by conditional DAMP [3,21]. Also, the ability of mitochondrial constituents to act as DAMP in response to cellular stresses is well established [26,27]. Therefore, mitochondria are important organelles that participate in various signaling pathways associated with triggering innate immunity and inflammation.
2. The majority of mitochondrial reactive oxygen species (mtROS) are generated in the complexes I and III of electron transport chain (ETC)
As an important part of cellular reactive oxygen species system [28], mtROS has been identified as an intermediate that trigger inflammatory signaling cascade in response to DAMP and PAMP [18,29,30]. Despite ROS (reactive oxygen species) being identified as a toxin for its high reactivity with lipids, proteins and nucleic acids, recent studies have suggested its important role in mediating physiological cellular signaling during homeostasis [1,31]. Mitochondria are a significant source of ROS in cells because more than 90% of cellular oxygen is consumed in mitochondria [32,33]; and ROS is produced as consequences of mitochondrial electron transport chain (ETC) activity during oxidative phosphorylation.
Oxidative phosphorylation is an essential cellular process that generate ATP, which is the main energy source in cells. During oxidative phosphorylation, the electrons removed from biological fuels such as glucose and fatty acids by electron donors reduced NADH and FADH2, go through a series of electron carrier system located in IMM, which is widely known as the ETC [34]. The mitochondrial ETC is comprised of a series of redox carriers named: Complex I: NADH dehydrogenase (ubiquinone), complex II (succinate dehydrogenase), complex III (ubiquinol-cytochrome c reductase) and complex IV (cytochrome oxidase). The stepwise transfer of electrons from complex I-IV ultimately reduce respiratory oxygen to water at complex IV [35].
Electron transport from complex I to IV is an exergonic process; and this energy is utilized to establish a proton gradient by pumping positively charged protons from the matrix to IMS. The proton gradient that is created across the IMM is named as proton-motive force (ΔP). Also, this process establishes a chemical gradient named mitochondrial membrane potential (ΔΨm) by increasing the positive charge and the negative charge in IMS and matrix, respectively [36]. This ΔP allows membrane potential-dependent ATP-synthase (complex V) to generate ATP from ADP (adenosine di-phosphate) when protons re-enter to the matrix through complex V [37]. Therefore, the proton gradient couple respiratory oxygen to ADP phosphorylation/ATP generation. However, during the stepwise transmission of electrons through the ETC, there is a chance for the electron to exit the ETC before being reduced to water at complex IV. This process is termed as electron leak, and result in generation of superoxide. Generally, superoxide generation is attributed to complexes I and III, however, other mitochondrial proteins such as flavin adenine dinucleotide (FAD)-linked pyruvate and α-ketoglutarate dehydrogenase complexes were also reported to generate mitochondrial superoxide [38]. A comprehensive review on the generation of mtROS and targeting mtROS as a therapy for inflammatory diseases and cancers had been previously published by our lab [1].
3. Inducible proton leak and mtROS production are mutually regulated
Lipid membranes show high conductance to protons; therefore, protons migrate to the matrix of the mitochondria across IMM independent of complex V. This process is termed as “proton leak”. During proton leak, energy is dissipated as heat instead of being used for ATP synthesis [39]. As proton leak depicts the protons that migrate into the matrix without producing ATP, it makes the coupling of substrate oxygen and ATP generation incomplete. However, proton leak is the principal, but not the only mechanism that incompletely couples substrate oxygen to ATP generation. Even though the contribution is insignificant, a phenomenon called “electron slip”, as the second mechanism, is also attributed to incomplete coupling of ATP generation and substrate oxygen as well. Electron slip refers to the process where electrons are transported via ETC without pumping protons to IMS and without contribution to ΔP. Therefore, electron slip result in disproportionate increase in oxygen consumption at high ΔP [40,41]. The exact mechanism of how proton leak takes place is not fully known [42].
Based on many publications, it is evident though “seemingly unproductive”, proton leak plays an important role in regulating the viability of the cells during normal physiological status as well as in pathology. Surprisingly, mitochondrial proton leak accounts for approximately 26% of the oxygen consumption rate in isolated hepatocytes [43]. As proton leak and ATP production both compete for ΔP, theoretically one can expect to observe less proton leak in the presence of high cellular energy demand. However, proton leak is responsible for almost 34% of oxygen consumption rate in skeletal muscles subjected to maximal tetanic contraction. Therefore, it is safe to conclude that proton leak contributes largely to maintaining the cellular metabolic rate, and therefore is not a futile process that occurs in the cells [44].
The physiological regulation of proton leak is categorized into two types; 1) basal/constitutive proton leak, and 2) regulated/inducible proton leak. Basal proton conductance is generally unregulated, and largely depends on the fatty acyl composition of the inner membrane phospholipids [45,46]. However, the proton conductance through the lipid bilayer of the IMM accounts for only 5% of the proton leak. Therefore, this suggests that majority of basal conductance is regulated via mitochondrial inner membrane proteins. Approximately, two-third of the basal proton conductance is correlated to the abundance of adenine nucleotide translocase (ANT), which is a mitochondrial protein found in the IMM[47].
Inducible proton leak is catalyzed by specific mitochondrial inner membrane proteins such as uncoupling proteins (UCPs) [48]. UCPs are a subfamily of the mitochondrial solute carrier family proteins that mediate transport of various metabolites across the IMM. There are five types of UCPs found in mammals, named as UCP1–5. Out of the five UCPs, UCP-1 was the first to be identified in brown adipose tissue (BAT) and has been extensively studied. UCP-1 is responsible for converting mitochondrial energy potential to heat, thus mainly regulate adaptive thermogenesis. Several studies have demonstrated that UCP-1 positive cells in white adipose tissue (WAT) show similar properties as brown adipocytes [49–51]. Furthermore, increased expression of UCP-1 in white adipocytes augmented the energy consumption to a similar level to that of brown adipocytes [52–54]. Therefore, it is clearly evident that the proton leak regulated by UCP-1 in BAT plays an essential role in thermogenesis. Even though, UCP-2 and UCP-3 reduce the mitochondrial coupling efficiency, their roles are largely focused on regulating reactive oxygen species (ROS) levels rather than regulating thermogenesis [29,55]. UCPs-mediated proton conductance is regulated at various levels such as molecular, transcriptional, translational, and proteolytic [56]. Moreover, many publications have demonstrated the involvement of UCPs in development of various pathologies such as cardiovascular diseases, oxidative stress, immune response, and type 2 diabetes mellitus. Table 1 summarizes some of the publications that demonstrated the involvement of proton leak or UCPs in the development of cardiovascular diseases and metabolic diseases. However, it is not clear whether the proton leak is directly involved at the molecular level in some of the studies that showed that UCPs are involved in disease progression. The effects that are mediated by UCPs might be independent to uncoupled respiration.
Table 1.
The roles of UCPs and proton leak in the development and the progression of cardiovcascular and metabolic diseases.
| Pathology | Findings and references |
|---|---|
| Obesity | Reduced proton leak and decreased expression of UCP-3 in skeletal muscles of diet resistant obese women [84]. |
| Lower rate of proton leak in primary muscles cells in diet resistant obese human subjects [85]. | |
| Diabetes mellitus | Ucp-2 preserves the endothelial function in diet induced obese mice [86]. |
| Hyperglycemia induced UCP-2 acts as a sensor and a negative regulator of mtROS production in HUVEC [66]. | |
| Cardiac IR injury | Ucp-2 and Ucp-3 increase the ischemic tolerance in rats [87]. |
| Proton leak is elevated after IR injury in isolated rat mitochondria. This increase is largely mediated by ANT [88]. | |
| Ucp-3 deletion exaggerates apoptosis and result in larger infarct size in mice [89]. | |
| Ucp-3 mediates the cardio-protection of H2O2 mediated preconditioning against IR injury by preserving the mitochondrial function [90]. | |
| Hypertension | Proton leak is reduced in maternally inherited hypertension attributed to the mutation in mitochondrial tRNAAla to G at 5665th position [91]. |
| Absence of Ucp2 accelerated the development of pulmonary hypertension in mice when exposed to intermittent hypoxia [92]. | |
| Atherosclerosis | Absence of Ucp-2 increased the development of unstable atherosclerotic plaques [65]. |
| Ucp-2 expression gradually decreased with increased lipid deposition in the aorta in ApoE−/− mice [93]. | |
| Proton leak maintains mitochondrial integrity in endothelial cells treated with low dose of LPC [21]. |
There are contradictory reports that link proton leak to ROS generation. Some reports provide evidence that the dissipation of ΔP [57–59] or presence of ADP decrease the mtROS production, while a few claim that uncoupling enhances the ROS production [21,60,61]. This contradictory evidence might be due to the differences in biological and experimental settings. However, the view that uncoupling decreases mtROS production prevails. Previous publications have demonstrated that uncoupling may play a protective role by mitigating ROS production in cells. This cyto-protective role of uncoupling was specifically observed in heart under conditions of oxidative stress such as ischemia reperfusion (IR) injury [62], aging [63] and diabetes [64]. Most prominently, expression of UCP1 and UCP2, and also chemical uncouplers offer protection against IR injury. Similarly, it was observed that uncoupling can exert a profound protective effect against toxicity produced in the presence of hyperglycemia in endothelial cells. In addition, protective role of uncoupling against atherosclerosis was reported [65,66].
On the other hand, increased level of ROS seems to cause an increase in proton leak [57]. It was observed that peroxynitrite, which is a potent inducer of lipid oxidation, increases the proton leak in isolated brain mitochondria [67]. Additionally, superoxide can also enhance the electron leak to a similar extent as peroxynitrite, and was shown to activate UCPs 1, 2 & 3 in rat BAT, kidney and skeletal muscles [68]. The UCPs and ANTs are attributed to enhanced proton conductance in the presence of ROS. Therefore, the presence of a protective feedback loop has been suggested, where increased ROS generation activates the mechanisms that promote proton leak; and the enhanced proton leak in turn reduce the ROS production limiting further damage to mitochondrial function [57,68].
However, it was also reported that increased mtROS is required to activate UCP-1 during thermogenesis. A recent study implied that mtROS production is elevated during thermogenesis in BAT. This increase in ROS generation was accompanied by elevated UCP-1 dependent respiration [69]. Furthermore, it was reported that mice rely exclusively on alternative mechanisms of thermogenesis in the absence of UCP-1 [70]. Unlike in UCP-1 intact experimental systems, quenching of mtROS did not inhibit thermogenesis in UCP-1 KO mice. This is strong evidence that mtROS is essential for UCP-1 dependent thermogenesis [71]. UCP-1 in normal cellular environment, remains in a purine-nucleotide bound state, which is its inactive form [72,73]. Current evidence suggests that rather than the expression level, activation status of UCP-1 is important for uncoupled respiration during thermogenesis. MtROS itself was identified as a mediator that activates not only UCP-1, but also UCP-2 and UCP-3 [68]. Detailed discussion about the most recent findings on the relationship between mtROS and UCP-1 mediated thermogenesis is well reviewed elsewhere [71].
4. ATP synthase-uncoupled ETC activity and mtROS regulate both physiological and pathological endothelial cell activation and inflammation initiation
Recently, our lab published the effects of conditional DAMP LPC on inducing endothelial cell activation [21]. Endothelial activation is the initial step of the inflammatory process that initiate the circulating immune cells to adhere and migrate across the endothelium that ultimately lead to progression of atherosclerosis. Recently, we proposed two types of endothelial activation: 1) Physiological endothelial cell activation, and 2) Pathological endothelial cell activation [74]. We described physiological endothelial cell activation as a mechanism that endothelial cells utilize to induce low-grade activation to recruit patrolling immune cells to maintain the homeostasis and good health of the endothelium and conduct immunosurveillance in tissues and vessels. On the other hand, we recognized pathological endothelial cell activation as a mechanism that is triggered by constant exposure of the endothelium to cardiovascular disease risk factor-derived stresses. Continuous bombardment of PAMP and DAMP may trigger prolonged activation of endothelial cells that ultimately lead to recruitment of pathological immune cells to the endothelium leading to development of a continuous inflammatory process that is difficult to resolve [75].
Our data showed that low dose of LPC treatment (10 μM) in endothelial cells generated ROS at a low level and promoted endothelial activation without compromising mitochondrial integrity, ATP generation and cell viability. In contrast, high dose of LPC (40 μM) treatment generated high level of ROS and activated apoptosis of the cells. This data shows that mitochondria have the ability to sense the level of insult caused by conditional DAMP and other cellular stresses, and mediate appropriate responses. Also, we hypothesize that mtROS is an important intermediate that enables mitochondria to fine tune between eliciting physiological transient low-grade inflammation and prolonged full-blown inflammatory responses depending on the intensity of the insult. Further, our data indicated that the levels of ROS generated mediate activation of differential signaling mechanisms in the cells such as site-specific histone 3 lysine 14 acetylation [21,76] and determine the homeostatic and pathological responses.
Interestingly, we observed that a low dose of LPC had a profound effect on oxidative phosphorylation system in the mitochondria. LPC (10 μM) treatment increased the mitochondrial oxygen reduction rate, maximal respiration rate while having no impact on the spare respiratory rate of the mitochondria. This indicated to us that the basal respiration was increased, thus the ETC activity was increased in endothelial cells treated with low dose of LPC. Despite the ETC activity being induced, we did not observe an increase in ATP generation in our experimental system. It was evident to us that the mitochondrial ETC activity was augmented not to increase energy production, but to mediate a different response that is uncoupled to ATP synthesis. Simultaneously, with the augmented ATP synthase-uncoupled ETC activity, we observed an increase in proton leak without dissipation of ΔΨm in the mitochondria. Therefore, it was evident that the ETC activity was increased in order to maintain a steady proton gradient, and thereby to stabilize ΔΨm and mitochondrial integrity in endothelial cells treated with low dose of LPC [21].
5. Mitochondrial Ca2+ uniporter and exchanger proteins have an impact on proton leak and mtROS generation
LPC acts through G-protein coupled receptors (GPCR). Our data indicated that low dose of LPC treatment significantly induced cytosolic and mitochondrial Ca2+ influx. So far, Ca2+ is one of the most important intracellular messengers that was extensively studied in various pathologies. Myriad of proteins are known to change their conformation and charge due to Ca2+ binding, therefore, the intrusion and extrusion of Ca2+ are tightly controlled at a cost of high energy consumption [77]. Just like ER, mitochondria are one of the important organelles that sequester Ca2+ in the cells. However, mitochondria utilize distinct mechanisms to that of ER to regulate Ca2+. Mitochondrial outer membrane is highly permeable to Ca2+ but the inner membrane is more selective and utilizes specific transporters and channels that allow Ca2+ influx [77].
During the maintenance of mitochondrial Ca2+ homeostasis, tight regulation of both influx and efflux of Ca2+ are essential. Ca2+ influx requires intact ΔΨm, suggesting that the maintenance of the negativity of the mitochondrial matrix is important for the flux [78,79]. While Ca2+ ions enter in to the mitochondria through mitochondrial Ca2+ uniporter (MCU), there are other mitochondrial exchanger proteins that are responsible for Ca2+ efflux [80]. In our experimental system, we observed that use of ruthenium red (RR - an inhibitor of Ca2+ transporters including MCU) with LPC attenuated the proton leak and also mtROS generation, suggesting that Ca2+ influx is the initial response to low intensity stress [21].
We hypothesize that LPC mediated induction of proton leak we observed was due to elevated activity of exchangers that are responsible for regulating Ca2+ homeostasis (Fig. 1). Previously it was shown that mitochondrial ion exchangers are linked with movement of protons across the IMM [81]. Mostly, mitochondrial Na+/Ca2+ exchanger (NCX) is the predominant antiporter that is involved in extrusion of Ca2+ [82]. The movement of protons via mitochondrial Na+/H+ exchanger (NHE), which is also an antiporter is indirectly related to the function of NCX by extruding Na+ that entered via NCX. In addition, there are reports confirming the presence of Ca2+ and proton antiporters, where Ca2+ is extruded at the expense of proton intrusion. Molecular identity of this Ca2+/H+ exchanger is still in debate. So far, Letm1 protein is the potential candidate that was recognized as this unidentified exchanger [77]. Regardless we did not quantify the activity of these exchangers in our experimental system, as mentioned above, we did observe an increase in Ca2+ movement and proton leak in response to LPC treatment in endothelial cells. Therefore, we hypothesize that the increased proton leak we observed was due to increased activity of exchangers that are responsible for mediating Ca2+ efflux, and hence we term this type of proton leak as “ion-mediated proton leak”. Therefore, we further postulate that increased proton conductance is a vital physiological process that is necessary to regulate Ca2+ while maintaining the mitochondrial integrity by stabilizing ΔΨm during propagation of low grade inflammation due to exposure to conditional DAMP.
Fig. 1. Proton leak is physiologically important to maintain the mitochondrial integrity when responding to low grade inflammation:
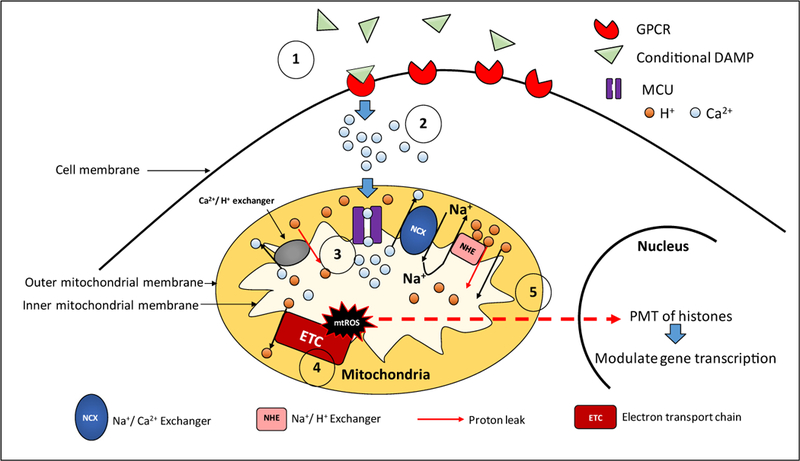
1) Conditional DAMP LPC acts through its receptors and increases Ca2+ influx in the cytosol.
2) Mitochondrial Ca2+ influx is mediated by MCU (Mitochondrial Ca2+ uniporter).
3) Ion mediated proton leak is increased to maintain the membrane potential in the presence of elevated mitochondrial Ca2+ influx. A) Indirect increase of proton leak: Na+/ Ca2+ exchanger (NCX) mediates Ca2+ extrusion while allowing Na+ intrusion. NHE (Na+/H+ exchanger) indirectly facilitates the function of NCX by allowing Na+ extrusion and increased proton leak to the matrix. B) Direct increase of proton leak: Ca2+/H+ exchanger mediates the efflux of Ca2+ at the expense of increased H+ influx in to the mitochondrial matrix.
4) Increased ATP uncoupled ETC (electron transport chain) activity. To maintain the mitochondrial membrane potential in the presence of increased proton leak, ATP uncoupled ETC activity is augmented. This leads to increased mtROS production.
5) MtROS mediated post-translational modification (PTM) of proteins. Through downstream targets that are yet to be identified, mtROS relay signaling to the nucleus and modulate histone PTM. Changes in the histone acetylation modulate gene expression and exert appropriate responses to induce low grade inflammation.
6. MtROS connects signaling pathways between conditional DAMP regulated immunometabolism and histone posttranslational modifications (PTM) and gene expression
Our data implied that increased Ca2+ in the cytosol and mitochondria are responsible for mtROS production, which in turn induce PTM of histones in the nucleus. We hypothesize that increased activity of ATP synthase uncoupled ETC activity that result due to augmented ion mediated proton leak is responsible for increased mtROS production. Despite the prevailing notion that increased proton leak mitigates mtROS production, there are reports that demonstrate increased mtROS production in the presence of enhanced proton leak, which is similar to our findings [69]. We specifically demonstrated that acetylation at lysine 14 residue of histone 3 protein (H3K14Ac) is responsible for induction of ICAM-1 (intercellular adhesion molecule −1) gene expression in LPC treated endothelial cells. ICAM-1 act as an adhesion molecule in endothelial cells and facilitate the recruitment of immune cells in to the endothelium. Previously, mtROS induced PTM in proteins that regulate mitochondrial fission was reported [83]. However, to the best of our knowledge, our report was the first to describe the ability of mtROS to elicit PTM of histone proteins, thus modifying the gene expression patterns. Future studies are required to identify the molecules that mitochondria utilize to relay signaling to other compartments that result in PTM of proteins.
In summary, the novel working model we propose is as follows (Fig. 1). In response to various stresses such as conditional DAMP LPC, GPCR gets activated and triggers Ca2+ influx in both cytosol and in the mitochondria. In order to maintain the mitochondrial integrity and propagate low-grade inflammatory response, ion mediated-proton leak is induced due to increased activity of ion exchangers. To avoid dissipation of ΔΨm, ATP synthase-uncoupled ETC activity is increased to propel the protons back to IMS from the matrix. Increased ETC activity leads to generation of mtROS that promote PTM of proteins via signaling partners that are yet to be elucidated. These mtROS-mediated PTM in proteins enable the cells to generate low-grade inflammation and activate responses to resolve the inflammation. The intensity of the stimuli and the duration of the insult may play a vital role in determining the type of response that mitochondria may elicit. However, we cannot negate the fact that uncoupling proteins may also contribute to the maintenance of ΔΨm during cellular insults that may alter ion flux in mitochondria. In our system we did not observe a significant modulation of uncoupling proteins. However, we did observe a slight increase in ANT, which might also contribute to the increased proton leak we observed. Therefore, proton leak whether it is ion dependent or protein dependent, gives the ability to the mitochondria to respond to low intensity cellular insults without compromising ΔΨm.
Acknowledgements
Funding
This work was supported by a NIH grant to XY (Grant No. RO1 - HL132399-01A1).
Abbreviations
- AIM-2
Absent in melanoma-2
- ANT
Adenine nucleotide transferase
- BAT
Brown adipose tissue
- DAMP
Damage associated molecular patterns
- ER
Endoplasmic reticulum
- ETC
Electron transport chain
- GPCR
G-protein coupled receptors
- HUVEC
Human umbilical vein cells
- ICAM-1
Intercellular adhesion molecule − 1
- IMM
Inner mitochondrial membrane
- IMS
Intermembrane space
- IR
Ischemia reperfusion
- LPC
Lysophosphatidylcholine
- LPL
Lysophospholipids
- MAVS
Mitochondrial antiviral signaling proteins
- MCU
Mitochondrial Ca2+ uniporter
- mtROS
Mitochondrial ROS
- NCX
Na+/Ca+ exchanger
- NHE
Na+/H+ exchanger
- NLR
NOD like receptors
- OMM
Outer mitochondrial membrane
- PAMP
Pathogen associated molecular patterns
- PRR
Pathogen recognition receptors
- PTM
Post-translational modifications
- RAGE
Receptor for advanced glycation end products
- RLR
RIG-I like receptors
- ROS
Reactive oxygen species
- RR
Ruthenium red
- TLR
Toll like receptors
- UCP
Uncoupling protein
- WAT
White adipose tissue
Footnotes
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.abb.2018.12.002.
References
- [1].Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF, Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers, J. Hematol. Oncol 6 (2013) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ryan MT, Hoogenraad NJ, Mitochondrial-nuclear communications, Annu. Rev. Biochem 76 (2007) 701–722. [DOI] [PubMed] [Google Scholar]
- [3].Zeng H, Nanayakkara GK, Shao Y, Fu H, Sun Y, Cueto R, Yang WY, Yang Q, Sheng H, Wu N, Wang L, Yang W, Chen H, Shao L, Sun J, Qin X, Park JY, Drosatos K, Choi ET, Zhu Q, Wang H, Yang X, DNA checkpoint and repair factors are nuclear sensors for intracellular organelle stresses-inflammations and cancers can have high genomic risks, Front. Physiol 9 (2018) 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S, Mitochondrial dynamics, mitophagy and cardiovascular disease, J. Physiol 594 (2016) 509–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Filadi R, Theurey P, Pizzo P, The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance, Cell Calcium 62 (2017) 1–15. [DOI] [PubMed] [Google Scholar]
- [6].Liu Y, Zhu X, Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases, Transl. Neurodegener 6 (2017) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL, Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies, Annu. Rev. Pharmacol. Toxicol 57 (2017) 535–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Soubannier V, McBride HM, Positioning mitochondrial plasticity within cellular signaling cascades, Biochim. Biophys. Acta 1793 (2009) 154–170. [DOI] [PubMed] [Google Scholar]
- [9].Ribas V, Garcia-Ruiz C, Fernandez-Checa JC, Mitochondria, cholesterol and cancer cell metabolism, Clin. Transl. Med 5 (2016) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang XF, Yin Y, Wang H, Vascular inflammation and atherogenesis are activated via receptors for pamps and suppressed by regulatory T cells, Drug Discov. Today Ther. Strat 5 (2008) 125–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, Yang XF, Inflammasomes are differentially expressed in cardiovascular and other tissues, Int. J. Immunopathol. Pharmacol 22 (2009) 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Venereau E, Ceriotti C, Bianchi ME, DAMPs from cell death to new life, Front. Immunol 6 (2015) 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I, Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis, Am. J. Respir. Crit. Care Med 182 (2010) 774–783. [DOI] [PubMed] [Google Scholar]
- [14].Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG, ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors, Science 314 (2006) 1792–1795. [DOI] [PubMed] [Google Scholar]
- [15].Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S, IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction, Nat. Immunol 6 (2005) 981–988. [DOI] [PubMed] [Google Scholar]
- [16].Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J, Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus, Nature 437 (2005) 1167–1172. [DOI] [PubMed] [Google Scholar]
- [17].Seth RB, Sun L, Ea CK, Chen ZJ, Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3, Cell 122 (2005) 669–682. [DOI] [PubMed] [Google Scholar]
- [18].West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S, TLR signalling augments macrophage bactericidal activity through mitochondrial ROS, Nature 472 (2011) 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation, Nature 469 (2011) 221–225. [DOI] [PubMed] [Google Scholar]
- [20].Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM, Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome, Nat. Immunol 12 (2011) 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li X, Fang P, Li Y, Kuo YM, Andrews AJ, Nanayakkara G, Johnson C, Fu H, Shan H, Du F, Hoffman NE, Yu D, Eguchi S, Madesh M, Koch WJ, Sun J, Jiang X, Wang H, Yang X, Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation, Arterioscler. Thromb. Vasc. Biol 36 (2016) 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee YK, Lee DH, Kim JK, Park MJ, Yan JJ, Song DK, Vaziri ND, Noh JW, Lysophosphatidylcholine, oxidized low-density lipoprotein and cardiovascular disease in Korean hemodialysis patients: analysis at 5 years of follow-up, J. Kor. Med. Sci 28 (2013) 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Matsumoto T, Kobayashi T, Kamata K, Role of lysophosphatidylcholine (LPC) in atherosclerosis, Curr. Med. Chem 14 (2007) 3209–3220. [DOI] [PubMed] [Google Scholar]
- [24].Wang X, Li YF, Nanayakkara G, Shao Y, Liang B, Cole L, Yang WY, Li X, Cueto R, Yu J, Wang H, Yang XF, Lysophospholipid receptors, as novel conditional danger receptors and homeostatic receptors modulate inflammation-novel paradigm and therapeutic potential, J Cardiovasc Transl Res 9 (2016) 343–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shao Y, Nanayakkara G, Cheng J, Cueto R, Yang WY, Park JY, Wang H, Yang X, Lysophospholipids and their receptors serve as conditional DAMPs and DAMP receptors in tissue oxidative and inflammatory injury, Antioxidants Redox Signal (2017). [DOI] [PMC free article] [PubMed]
- [26].Nakahira K, Hisata S, Choi AM, The roles of mitochondrial damage-associated molecular patterns in diseases, Antioxidants Redox Signal 23 (2015) 1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature 464 (2010) 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA, Cellular death, reactive oxygen species (ROS) and diabetic complications, Cell Death Dis 9 (2018) 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D, Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production, Nat. Genet 26 (2000) 435–439. [DOI] [PubMed] [Google Scholar]
- [30].Lambeth JD, NOX enzymes and the biology of reactive oxygen, Nat. Rev. Immunol 4 (2004) 181–189. [DOI] [PubMed] [Google Scholar]
- [31].Hamanaka RB, Chandel NS, Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes, Trends Biochem. Sci 35 (2010) 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reczek CR, Chandel NS, ROS-dependent signal transduction, Curr. Opin. Cell Biol 33 (2015) 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Venditti P, Di Stefano L, Di Meo S, Mitochondrial metabolism of reactive oxygen species, Mitochondrion 13 (2013) 71–82. [DOI] [PubMed] [Google Scholar]
- [34].Cueto R, Zhang L, Shan HM, Huang X, Li X, Li YF, Lopez J, Yang WY, Lavallee M, Yu C, Ji Y, Yang X, Wang H, Identification of homocysteine-suppressive mitochondrial ETC complex genes and tissue expression profile - novel hypothesis establishment, Redox Biol 17 (2018) 70–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rich PR, Marechal A, The mitochondrial respiratory chain, Essays Biochem 47 (2010) 1–23. [DOI] [PubMed] [Google Scholar]
- [36].Noji H, Yasuda R, Yoshida M, Kinosita K Jr., Direct observation of the rotation of F1-ATPase, Nature 386 (1997) 299–302. [DOI] [PubMed] [Google Scholar]
- [37].Mitchell P, Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism, Nature 191 (1961) 144–148. [DOI] [PubMed] [Google Scholar]
- [38].Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF, Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species, J. Neurosci. Offic. J. Soc. Neurosci 24 (2004) 7779–7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chance B, Williams GR, Respiratory enzymes in oxidative phosphorylation. III. The steady state, J. Biol. Chem 217 (1955) 409–427. [PubMed] [Google Scholar]
- [40].Kadenbach B, Intrinsic and extrinsic uncoupling of oxidative phosphorylation, Biochim. Biophys. Acta 1604 (2003) 77–94. [DOI] [PubMed] [Google Scholar]
- [41].Murphy MP, Slip and leak in mitochondrial oxidative phosphorylation, Biochim. Biophys. Acta 977 (1989) 123–141. [DOI] [PubMed] [Google Scholar]
- [42].Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, Tian Y, Wang H, Yang X, Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases, Adv. Exp. Med. Biol 982 (2017) 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK, The causes and functions of mitochondrial proton leak, Biochim. Biophys. Acta 1187 (1994) 132–139. [DOI] [PubMed] [Google Scholar]
- [44].Rolfe DF, Newman JM, Buckingham JA, Clark MG, Brand MD, Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR, Am. J. Physiol 276 (1999) C692–C699. [DOI] [PubMed] [Google Scholar]
- [45].Brookes PS, Buckingham JA, Tenreiro AM, Hulbert AJ, Brand MD, The proton permeability of the inner membrane of liver mitochondria from ectothermic and endothermic vertebrates and from obese rats: correlations with standard metabolic rate and phospholipid fatty acid composition, Comp. Biochem. Physiol. B Biochem. Mol. Biol 119 (1998) 325–334. [DOI] [PubMed] [Google Scholar]
- [46].Fontaine EM, Moussa M, Devin A, Garcia J, Ghisolfi J, Rigoulet M, Leverve XM, Effect of polyunsaturated fatty acids deficiency on oxidative phosphorylation in rat liver mitochondria, Biochim. Biophys. Acta 1276 (1996) 181–187. [DOI] [PubMed] [Google Scholar]
- [47].Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ, The basal proton conductance of mitochondria depends on adenine nucleotide translocase content, Biochem. J 392 (2005) 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Parker N, Vidal-Puig A, Brand MD, Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential, Biosci. Rep 28 (2008) 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, Giacobino JP, De Matteis R, Cinti S, The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation, Am. J. Physiol. Endocrinol. Metab 298 (2010) E1244–E1253. [DOI] [PubMed] [Google Scholar]
- [50].Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, During MJ, White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis, Cell Metabol 14 (2011) 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kiefer FW, Browning and thermogenic programing of adipose tissue, Best Pract. Res. Clin. Endocrinol. Metabol 30 (2016) 479–485. [DOI] [PubMed] [Google Scholar]
- [52].Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J, Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes, J. Biol. Chem 285 (2010) 7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Marlatt KL, Ravussin E, Brown adipose tissue: an update on recent findings, Curr Obes Rep 6 (2017) 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerback S, Schrauwen P, Spiegelman BM, Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human, Cell 150 (2012) 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vidal-Puig AJ, Grujic D, Zhang CY, Hagen T, Boss O, Ido Y, Szczepanik A, Wade J, Mootha V, Cortright R, Muoio DM, Lowell BB, Energy metabolism in uncoupling protein 3 gene knockout mice, J. Biol. Chem 275 (2000) 16258–16266. [DOI] [PubMed] [Google Scholar]
- [56].Azzu V, Brand MD, The on-off switches of the mitochondrial uncoupling proteins, Trends Biochem. Sci 35 (2010) 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Brookes PS, Mitochondrial H(+) leak and ROS generation: an odd couple, Free Radical Biol. Med 38 (2005) 12–23. [DOI] [PubMed] [Google Scholar]
- [58].Turrens JF, Boveris A, Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria, Biochem. J 191 (1980) 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Herrero A, Barja G, ADP-regulation of mitochondrial free radical production is different with complex I- or complex II-linked substrates: implications for the exercise paradox and brain hypermetabolism, J. Bioenerg. Biomembr 29 (1997) 241–249. [DOI] [PubMed] [Google Scholar]
- [60].Boveris A, Chance B, The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen, Biochem. J 134 (1973) 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Starkov AA, Fiskum G, Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state, J. Neurochem 86 (2003) 1101–1107. [DOI] [PubMed] [Google Scholar]
- [62].Ganote CE, Armstrong SC, Effects of CCCP-induced mitochondrial uncoupling and cyclosporin A on cell volume, cell injury and preconditioning protection of isolated rabbit cardiomyocytes, J. Mol. Cell. Cardiol 35 (2003) 749–759. [DOI] [PubMed] [Google Scholar]
- [63].Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS, Brand MD, Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer, Aging Cell 3 (2004) 87–95. [DOI] [PubMed] [Google Scholar]
- [64].Green K, Brand MD, Murphy MP, Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes, Diabetes 53 (Suppl 1) (2004) S110–S118. [DOI] [PubMed] [Google Scholar]
- [65].Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z, Protective role of uncoupling protein 2 in atherosclerosis, Circulation 107 (2003) 388–390. [DOI] [PubMed] [Google Scholar]
- [66].Koziel A, Sobieraj I, Jarmuszkiewicz W, Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels, Am. J. Physiol. Heart Circ. Physiol 309 (2015) H147–H156. [DOI] [PubMed] [Google Scholar]
- [67].Brookes PS, Land JM, Clark JB, Heales SJ, Peroxynitrite and brain mitochondria: evidence for increased proton leak, J. Neurochem 70 (1998) 2195–2202. [DOI] [PubMed] [Google Scholar]
- [68].Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD, Superoxide activates mitochondrial uncoupling proteins, Nature 415 (2002) 96–99. [DOI] [PubMed] [Google Scholar]
- [69].Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, Robinson AJ, Gygi SP, Spiegelman BM, Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1, Nature 532 (2016) 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ukropec J, Anunciado RP, Ravussin Y, Hulver MW, Kozak LP, UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1−/− mice, J. Biol. Chem 281 (2006) 31894–31908. [DOI] [PubMed] [Google Scholar]
- [71].Chouchani ET, Kazak L, Spiegelman BM, Mitochondrial reactive oxygen species and adipose tissue thermogenesis: bridging physiology and mechanisms, J. Biol. Chem 292 (2017) 16810–16816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nicholls DG, The physiological regulation of uncoupling proteins, Biochim. Biophys. Acta 1757 (2006) 459–466. [DOI] [PubMed] [Google Scholar]
- [73].Fedorenko A, Lishko PV, Kirichok Y, Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria, Cell 151 (2012) 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, Gao T, Wang H, Yang X, Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells, Can. J. Physiol. Pharmacol (2016) 1–6. [DOI] [PMC free article] [PubMed]
- [75].Li X, Wang L, Fang P, Sun Y, Jiang X, Wang H, Yang XF, Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation, J. Biol. Chem 293 (2018) 11033–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Li X, Shao Y, Sha X, Fang P, Kuo YM, Andrews AJ, Li Y, Yang WY, Maddaloni M, Pascual DW, Luo JJ, Jiang X, Wang H, Yang X, IL-35 (Interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (histone 3 lysine 14), Arterioscler. Thromb. Vasc. Biol 38 (2018) 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Clapham DE, Calcium signaling, Cell 131 (2007) 1047–1058. [DOI] [PubMed] [Google Scholar]
- [78].Deluca HF, Engstrom GW, Calcium uptake by rat kidney mitochondria, Proc. Natl. Acad. Sci. U. S. A 47 (1961) 1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lehninger AL, Rossi CS, Greenawalt JW, Respiration-dependent accumulation of inorganic phosphate and Ca ions by rat liver mitochondria, Biochem. Biophys. Res. Commun 10 (1963) 444–448. [DOI] [PubMed] [Google Scholar]
- [80].Demaurex N, Poburko D, Frieden M, Regulation of plasma membrane calcium fluxes by mitochondria, Biochim. Biophys. Acta 1787 (2009) 1383–1394. [DOI] [PubMed] [Google Scholar]
- [81].Bernardi P, Mitochondrial transport of cations: channels, exchangers, and permeability transition, Physiol. Rev 79 (1999) 1127–1155. [DOI] [PubMed] [Google Scholar]
- [82].Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I, NCLX is an essential component of mitochondrial Na+/Ca2+ exchange, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED, Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission, Circ. Res 122 (2018) 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Harper ME, Dent R, Monemdjou S, Bezaire V, Van Wyck L, Wells G, Kavaslar GN, Gauthier A, Tesson F, McPherson R, Decreased mitochondrial proton leak and reduced expression of uncoupling protein 3 in skeletal muscle of obese diet-resistant women, Diabetes 51 (2002) 2459–2466. [DOI] [PubMed] [Google Scholar]
- [85].Thrush AB, Zhang R, Chen W, Seifert EL, Quizi JK, McPherson R, Dent R, Harper ME, Lower mitochondrial proton leak and decreased glutathione redox in primary muscle cells of obese diet-resistant versus diet-sensitive humans, J. Clin. Endocrinol. Metab 99 (2014) 4223–4230. [DOI] [PubMed] [Google Scholar]
- [86].Tian XY, Wong WT, Xu A, Lu Y, Zhang Y, Wang L, Cheang WS, Wang Y, Yao X, Huang Y, Uncoupling protein-2 protects endothelial function in diet-induced obese mice, Circ. Res 110 (2012) 1211–1216. [DOI] [PubMed] [Google Scholar]
- [87].McLeod CJ, Aziz A, Hoyt RF Jr., McCoy JP Jr., Sack MN, Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia, J. Biol. Chem 280 (2005) 33470–33476. [DOI] [PubMed] [Google Scholar]
- [88].Nadtochiy SM, Tompkins AJ, Brookes PS, Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection, Biochem. J 395 (2006) 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Perrino C, Schiattarella GG, Sannino A, Pironti G, Petretta MP, Cannavo A, Gargiulo G, Ilardi F, Magliulo F, Franzone A, Carotenuto G, Serino F, Altobelli GG, Cimini V, Cuocolo A, Lombardi A, Goglia F, Indolfi C, Trimarco B, Esposito G, Genetic deletion of uncoupling protein 3 exaggerates apoptotic cell death in the ischemic heart leading to heart failure, J. Am. Heart Assoc 2 (2013) e000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen Y, Liu J, Zheng Y, Wang J, Wang Z, Gu S, Tan J, Jing Q, Yang H, Uncoupling protein 3 mediates H(2)O(2) preconditioning-afforded cardioprotection through the inhibition of MPTP opening, Cardiovasc. Res 105 (2015) 192–202. [DOI] [PubMed] [Google Scholar]
- [91].Jiang P, Wang M, Xue L, Xiao Y, Yu J, Wang H, Yao J, Liu H, Peng Y, Li H, Chen Y, Guan MX, A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function, Mol. Cell Biol 36 (2016) 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, Lee PJ, Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia, Arterioscler. Thromb. Vasc. Biol 35 (2015) 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gomez-Hernandez A, Perdomo L, de las Heras N, Beneit N, Escribano O, Otero YF, Guillen C, Diaz-Castroverde S, Gozalbo-Lopez B, Cachofeiro V, Lahera V, Benito M, Antagonistic effect of TNF-alpha and insulin on uncoupling protein 2 (UCP-2) expression and vascular damage, Cardiovasc. Diabetol 13 (2014) 108. [DOI] [PMC free article] [PubMed] [Google Scholar]