

Abstract
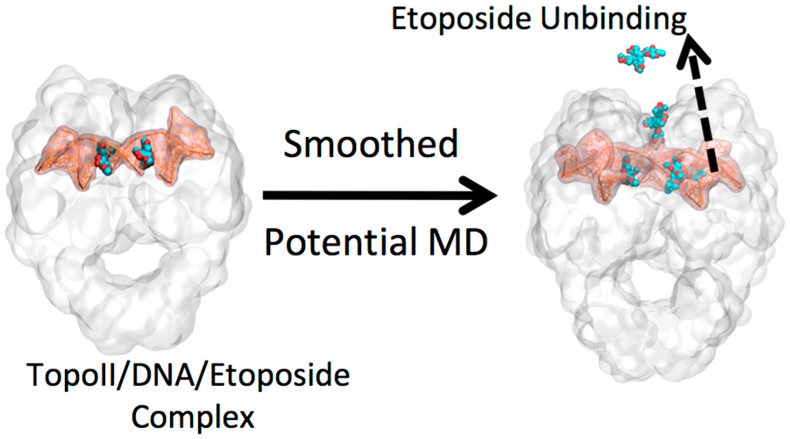
Human type II topoisomerases (TopoII) are essential for controlling DNA topology within the cell. For this reason, there are a number of TopoII-targeted anticancer drugs that act by inducing DNA cleavage mediated by both TopoII isoforms (TopoIIα and TopoIIβ) in cells. However, recent studies suggest that specific poisoning of TopoIIα may be a safer strategy for treating cancer. This is because poisoning of TopoIIβ appears to be linked to the generation of secondary leukemia in patients. We recently reported that enzyme-mediated DNA cleavage complexes (in which TopoII is covalently linked to the cleaved DNA during catalysis) formed in the presence of the anticancer drug etoposide persisted approximately 3-fold longer with TopoIIα than TopoIIβ. Notably, enhanced drug-target residence time may reduce the adverse effects of specific TopoIIα poisons. However, it is still not clear how to design drugs that are specific for the α isoform. In this study, we report the results of classical molecular dynamics (MD) simulations to comparatively analyze the molecular interactions formed within the TopoII/DNA/etoposide complex with both isoforms. We also used smoothed potential MD to estimate etoposide dissociation kinetics from the two isoform complexes. These extensive classical and enhanced sampling simulations revealed stabilizing interactions of etoposide with two serine residues (Ser763 and Ser800) in TopoIIα. These interactions are missing in TopoIIβ, where both amino acids are alanine residues. This may explain the greater persistence of etoposide-stabilized cleavage complexes formed with Topo TopoIIα. These findings could be useful for the rational design of specific TopoIIα poisons.
Introduction
Human type II topoisomerases (TopoII) undo DNA tangles and remove knots from the double helix during vital processes such as DNA repair and replication.1−5 To do so, TopoII generates double-stranded (ds) breaks in the genetic material in a metal ion-dependent reaction,6−9 forming a transient TopoII/DNA cleavage complex.4,8,10,11 Importantly, this complex is targeted and trapped by clinical anticancer drugs (i.e., TopoII poisons).12−23 For example, etoposide is a chemotherapy agent that acts by targeting the cleavage complex (Figure 1) and inhibiting DNA religation. This leads to the accumulation of DNA breaks, which ultimately ends with the death of (cancerous) cells.1,21,24−27 Etoposide is effective against a wide spectrum of cancers, including lymphoma, lung tumors, and ovarian tumors.28−30
Figure 1.
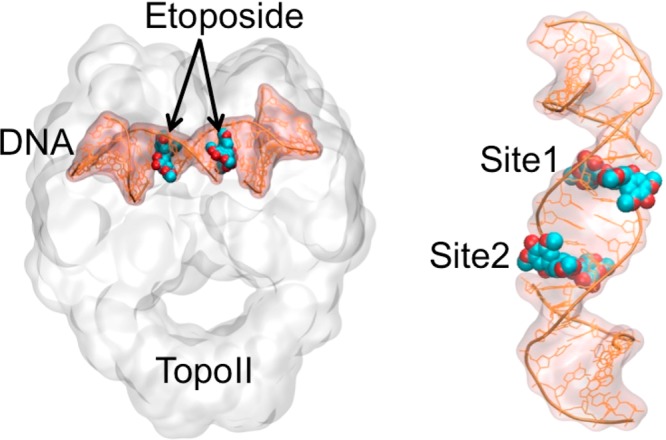
Ternary TopoII/DNA/etoposide complex (right). Close view of the two binding sites of etoposide inserted into the DNA strand.
Two isoforms of TopoII exist in humans: TopoIIα and TopoIIβ. The two isoforms have distinct cellular roles and expression patterns.31 TopoIIα is essential for the survival of proliferating cells and is required for chromosome segregation.4,32−35 It is needed in growth-related cellular processes and has a proliferation-dependent expression.33−35 In contrast, TopoIIβ plays a role in transcription, and its cellular levels are independent of proliferation status.32,33,36−39 Indeed, TopoIIβ is expendable at the cellular level and cannot compensate for the loss of TopoIIα in human cells.4,32,33
To date, all clinical TopoII poisons are nonspecific with regard to TopoIIα and TopoIIβ and affect the DNA cleavage activity of both isoforms.1,34,40−45 However, the contribution of each isoform to the therapeutic effects of drugs is not well understood.40,46−48 In this regard, both cellular and in vivo studies suggest that TopoIIβ is primarily responsible for generating the breaks in the mixed lineage leukemia (MLL) gene that initiate the acute myelogenous leukemias that are associated with etoposide treatment.21,35,49 Strong support for this hypothesis comes from studies with a skin carcinogenesis model, where the incidence of secondary malignancies was curtailed in a TopoIIβ-knockout mouse.50 Further, TopoIIβ was also related to etoposide-induced DNA sequence rearrangements and double-strand breaks in a murine cell model.50 In another experiment, TopoIIβ was shown to stimulate the majority of MLL breaks generated by etoposide, as well as the genotoxic effects of the drug.49,51
Taken together, these results suggested that TopoIIα-specific poisoning might help mitigate the side effects observed with nonspecific TopoII drugs.51−55 However, it is difficult to rationally design selective TopoIIα inhibitors because the two isoforms are 68% identical to each other.56−58 Furthermore, the catalytic sites of the enzymes share approximately 78% identity, differing only in two amino acids (i.e., Met762 and Ser763 in TopoIIα, respectively, changed to Gln778 and Ala779 in TopoIIβ; Figure 2). Of these residues, Gln778 is a good interaction site for targeting TopoIIβ via H-bonds formed with basic amines of polyamine-containing etoposide derivatives.56,59−61 However, etoposide has a sugar moiety instead of a polyamine tail.62 Hence, it cannot engage Gln778(β) for binding. Consequently, it exhibits a slight preference for TopoIIα. In this regard, we recently used DNA cleavage assays to demonstrate that etoposide generates more cleavage complex in TopoIIα than in TopIIβ (approximately 4-fold difference: TopoIIα vs TopIIβ).59 We also measured the persistence of cleavage complexes (half-life) by assessing the loss of double-strand breaks following dilution of cleavage complexes. We found that the cleavage complex formed by etoposide persisted much longer in TopoIIα (>150 min; 100 μM etoposide) than in TopoIIβ (56.5 min; 50 μM etoposide). This finding suggests that there are additional stabilizing interactions of etoposide in the DNA cleavage site of TopoIIα.59 In earlier experiments, Bandele and Osheroff measured the persistence of TopoII(α/β) cleavage complexes following the removal of etoposide from cultured human T lymphoblastic leukemia cells. The half-life of the cleavage complex formed by etoposide was approximately 120 and 25 min for TopoIIα and TopoIIβ, respectively.63 Importantly, the prolonged persistence of the TopoIIα/DNA/etoposide cleavage complex correlated with greater cell kill.
Figure 2.

Etoposide binding site in TopoII. The active site residues are depicted in gray (ball and stick with TopoIIα residues in red).
The crystal structures of cleavage complexes formed by both TopoII isoforms in the presence of etoposide were solved recently.64,65 These new data represent the optimal starting point for computational simulations to elucidate the structural difference between the two isoforms complexed with etoposide and provide a rational approach of how to specifically target TopoIIα.9 Here, we present extended classical molecular dynamics (MD) simulations to comparatively examine drug-target interactions in the ternary TopoII(α/β)/DNA/etoposide cleavage complex. We also used smoothed potential MD to investigate the dissociation kinetics of etoposide from the two isoform metal-aided complexes.66,67 Results have identified specific interaction points for drug binding and unbinding in TopoIIα vs TopoIIβ, including Ser800 in TopoIIα (Alanine 816 in TopoIIβ), which sits approximately 20 Å from the DNA cleavage active site.68
Methods
Structural Models
The structures of the α and β isoforms of the human TopoII(α/β)/DNA/etoposide ternary complex were taken from PDB entries 5GWK and 3QX3, respectively.64,65 Missing loops in the homodimeric protein structures were reconstructed with the Schrödinger2017 suite.69 The protein component of each complex was assigned parameters from the AMBER force field ff99SB70 with ff99Bildn71 modifications. The Parmbsc172 force field was adopted for DNA. Ligand (etoposide) parameters were generated using GAFF2 with restrained electrostatic potential (RESP) atomic charges.73 Each complex was centered in a rhombic dodecahedral simulation cell with a minimum box-solute distance of 1.0 nm. The unit cell was then filled with TIP3P water74 and Na+ counterions sufficient to neutralize the net charge on each complex. All ionizable amino acids were assigned their protonation state at pH 7.4 according to pKa predictions by the H++ server,75 except the aspartic acid residues that coordinate Mg2+ ions in each active site. For these residues, after calculating the charge on the Mg ions via quantum mechanical calculations, the residual charge was distributed over the oxygen atoms of the anionic amino acid residues.76
Classical MD Simulations
The structural models prepared for the two TopoII isoforms were used for MD simulations with GROMACS version 5.1.77 All bonds were constrained using the P-LINCS algorithm, with an integration time step of 2 fs. The Verlet cutoff scheme was used with a minimum cutoff of 1.0 nm for short-range Lennard-Jones interactions and the real-space contribution to the smooth particle mesh Ewald algorithm, which was used to compute long-range electrostatic interactions. Dispersion correction was applied to energy and pressure terms. Periodic boundary conditions were applied in all three dimensions. Each system was equilibrated in two phases, during which restraints were placed on protein and DNA heavy atoms. The first equilibration was done under an NVT ensemble for 100 ps, using a weak coupling algorithm with stochastic rescaling, to maintain the temperature at 310 K. The NVT equilibration was followed by NPT equilibration for 100 ps using the same thermostat and the Parrinello–Rahman barostat to maintain pressure at 1 bar. Production simulations were carried out under an NPT ensemble in the absence of any restraints. Three independent unbiased simulations of approximately 500 ns for each model were accumulated for a total of 3 μs of sampling. The analysis was carried out using programs within the GROMACS package.
Smoothed Potential MD Simulations for Dissociation Kinetics
Smoothed potential MD is a multiple-replica scaled molecular dynamics protocol developed to cost effectively rank congeneric drug (or drug-like) compounds based on their computed residence times.66 This enhanced sampling method is used here to simulate and accelerate, using scaled potentials, ligand unbinding events from protein–ligand systems. The main stabilizing interactions of the ligand within a protein are broken under scaled potential energy, which facilitates ligand unbinding. In this way, smoothed potential MD can help to decipher mechanistic details for ligand unbinding, especially in the vicinity of the target–ligand bound state.66
We employed this enhanced sampling technique to uncover the differences between the unbinding of etoposide from TopoIIα and TopoIIβ. The equilibrated structures for each complex (TopoII(α/β)/DNA/etoposide) were used to perform a series of 32 partially unrestrained smoothed potential MD66 production runs for each ligand. As each isoform TopoII structure contains two symmetric binding sites (thus, containing two etoposide molecules, Figure 1), a total of 64 simulations were performed for each isoform (hence, a total number of 128 simulations were performed). For each replica of smoothed potential MD, we considered the unbinding of one single etoposide molecule at a time (i.e., the second bound etoposide was restrained at the catalytic site). The smoothed potential MD simulations were stopped when one etoposide molecule was fully unbound. This was defined as the instant when the etoposide molecule ceased to interact with the binding site (i.e., no contacts with the target protein, with etoposide fully immersed in the bulk solvent approximately 30 Å from the catalytic site). Harmonic restraints were used to accelerate the unbinding event while preserving the native TopoII structure.66 That is, a set of weak restraints (50 kJ mol–1 nm–1) was applied on the protein backbone heavy atoms, with the exception of residues with at least one atom within 8 Å of the ligand (heavy atoms).66 Initial simulations were performed with the scaling factor varying from 0.6 to 0.3, as recommended by Mollica et al.66 and performed using BiKi Life Sciences.78 We found that a value of λ = 0.4 was the best compromise between a reasonable CPU time and computed ligand-unbinding times.
Results and Discussion
First, we investigated the stability and dynamics of the ternary TopoII(α/β)/DNA/etoposide via classical MD simulations.79 Three approximately 500 ns-long simulations were performed for each of the TopoII/DNA/etoposide ternary complexes (TopoIIα and TopoIIβ), resulting in a total of approximately 3 μs of dynamics. The convergence of all trajectories and the stability of the system were assessed by monitoring the root-mean-square deviations (RMSD) over time (Figures S1 and S2).
More Compact Cleavage Site in TopoIIα/DNA/Etoposide Complex
From our MD simulations, we observed that etoposide makes, on average, closer contacts with the surrounding residues in TopoIIα, compared to TopoIIβ.65 This is reported in Figure 3, which shows the frequency plots of the interaction distance between etoposide and key surrounding residues (i.e., Asp463α/Asp479β, Leu486α/Leu502β, and Met762α/Gln778β; plots for Gly462α/ Gly478β, Arg487α/Arg503β, Ser763α/Ala779β, and Met766α/Met782β in the SI) in the DNA cleavage active site.80,81 Interestingly, a previous analysis by Griffith and co-workers demonstrated that Thr468α/Ser483β, Met762α/Gln778β, Ser763α/Ala779β, Ile769α/Val785β, and Ser800α/Ala816β are located in the extended vicinity of the binding pocket, which are not conserved between the two isoforms.82 Of these residues, Met762α/Gln778β, Ser763α/Ala779β, and Ser800α/Ala816β changes may maximize differences in drug binding, given the nature of the amino acids in the two isoforms. In fact, the Thr468α/Ser483β change conserves the ability to form the hydrogen bond, while the Ile769α/Val785β maintains the apolar character of the amino acid in both isoforms. In contrast, the Met762α/Gln778β mutation alters the chemical nature of the amino acid residue from a hydrophobic methionine to a polar glutamine. Also, in the other two changes (serine to alanine), the potential for hydrogen bond interactions is lost in TopoIIβ. For these reasons, the amino acid alterations in the two isoforms form a different interaction network, which may be relevant to attaining TopoIIα specificity.
Figure 3.

Plots of the distance between the center of mass (COM) of the etoposide E-ring and COM of Asp463α and Asp479β, Leu486α and Leu502β, and COM of the etoposide sugar moiety and COM of Ser800α and Ala816β, and Met762α and Gln778β, which reflect an enhanced compactness of the active site of the TopoIIα isoform (Figure 2). In the frequency plots, the x-axis indicates the distance of the residues to the ligand. Dashed lines show the corresponding distance in the crystal structure (red, TopoIIα; black, TopoIIβ). For clarity, the plots are shown for four of the ten analyzed residues. These four residues are methionine/glutamine (M762α/Q778β), serine/alanine (S800α/A816β), aspartate (D463α/D479β), and leucine (L486α/L502β). These are chosen in such a way that two of the amino acids are selected from the three known mutations. The residue selection also ensures that interactions with different fragments of the bound ligand are considered. In fact, Ser800α/Ala816β and Met762α/Gln778β are near the sugar moiety of etoposide, whereas Asp463α/Asp479 and Leu486α/Leu502β interact with the E-ring of the drug molecules. The plots of Gly462α/Gly478β, Thr467α/Ser483β, Met766α/Met782β, Ser763α/Ala779β, Ile769α/Val785β, and Arg487α/Arg503β are reported in the Supporting Information.
First, we noted that in the TopoIIα crystal structure (PDB 5GWK) the distance between the center of mass of Asp463, Leu486, and the E-ring of etoposide (Figure 2) is 5.1 and 6.6 Å, respectively. The corresponding distances in TopoIIβ (PDB 3QX3) are slightly longer, at 5.9 and 7.1 Å (Asp479 and Leu502), respectively (Figure 3). In TopoIIα, the distance between residues Met762 and Ser800 and the sugar moiety of etoposide is 5.8 and 11.9 Å, respectively. This distance becomes 6.8 and 12.4 Å for the corresponding residues in TopoIIβ (i.e., Gln778 and Ala816, respectively). In our MD simulations (Figure 3), the most frequent interaction distance between the drug and these specific amino acids is maintained close to these experimental values. We further noted that the TopoIIα residues consistently maintained the general trend of making slightly shorter interactions with etoposide, in comparison to the same interactions in TopoIIβ. This difference between pairs of residues in the two isoforms is minimal for the conserved residues, namely, Asp463α (4.7 ± 0.4 Å)/Asp479β (5.6 ± 0.4 Å) and Arg487α (3.8 ± 0.2 Å)/Arg503β (4.2 ± 0.3 Å). In contrast, the difference increases for the nonconserved residues, namely, Ser800α (11.2 ± 0.7 Å)/Ala816 (13.9 ± 0.6 Å) and Met762α (5.0 ± 0.6 Å)/Gln778β (7.6 ± 0.7 Å). Conserved residues thus seem to form an interaction framework that has been preserved in the two isoforms.
The H-bond formed between the carboxyl group of the aspartate residues (Asp463α/Asp479β) and the E-ring hydroxyl group in etoposide is known to be important for drug binding.65 In this respect, in our MD simulations, the OH (E-ring of etoposide)–COO–(aspartate) H-bond is maintained for about 71% and 73% of the simulation time for Asp463α and Asp479β, respectively, restraining the motion of the residue side chain. This is further reflected by the narrow distribution of the distance values in the frequency plot for the pair Asp463α/Asp479β, which are residues that are stabilized by this specific H-bond interaction (Figure 3). For the two isoforms, we also compared the distance between the center of mass of these aspartates and that of the E-ring of etoposide. In TopoIIα, this distance varies between approximately 4.0 and approximately 5.0 Å. In TopoIIβ, this distance varies between 5.0 and 6.6 Å. This further demonstrates the relevance of this drug-target interaction in locking etoposide at the cleavage site.
Similarly, the key arginines (Arg487α/Arg503β) known to form favorable interactions with etoposide65 remain in closer contact with the drug molecule in both TopoIIα (i.e., Arg487 at 3.8 ± 0.2 Å) and TopoIIβ (i.e., Arg503 at 4.2 ± 0.3 Å) (Figure S3). Interestingly, this key arginine residue forms a number of interactions with several fragments of etoposide (A-, B-, E-rings).65 Gly462α/Gly478β and Leu486α/Leu502β are two other conserved amino acids that interact with the E-ring of etoposide (Figure 2). However, these residues form slightly shorter interactions in TopoIIα (at approximately 6.0 ± 0.3 Å) than in TopoIIβ (at 7.6 ± 0.4 Å). Notably, the H-bonding and van der Waals interactions between the glycosidic group of etoposide and the TopoIIβ binding site (Gln778β and Met782β) are reported to be less extensive than those with the E-ring.65,83 In our MD simulations, this seems to be reflected by the wider distribution for the etoposide glycosidic group interactions with both Met762α/Gln778β and Met766α/Met782β. Still, M762α is approximately 2.1 Å closer to the etoposide sugar moiety, relative to the corresponding Gln778 in TopoIIβ. In addition, Met766α is at 6.8 ± 0.3 Å, which is at a closer distance than Met782β (at 7.9 ± 0.3 Å in TopoIIβ). Notably, from our MD trajectories, this analysis was performed on both drug binding sites within the two isoforms, indicating that amino acid residues in the surroundings of the cleavage site are always closer to etoposide in TopoIIα than in TopoIIβ.
Hence, despite the similarities between the active sites of the two TopoII isoforms, etoposide seems to elicit a slightly different structural response from the cleavage site upon binding and complexation. The more compact active site in TopoIIα might cause a greater barrier for drug dissociation. In this respect, we have previously shown that the DNA cleavage complex in TopoIIα has enhanced persistence compared to TopoIIβ.59 The half-life of the TopoIIα cleavage complex is at least 3 times longer than that formed with the β isoform. Taken together, both the experimental evidence and our classical MD simulations point to a difference in the drug dissociation kinetics in the two TopoII isoforms. To elucidate this difference, we continued our study by performing smoothed potential MD simulations for both isoforms.66
Pathways and Relative Kinetics for Etoposide Dissociation from TopoII Isoforms
To evaluate the dissociation kinetics of etoposide from the α and β isoforms of TopoII, we used a multiple-replica smoothed potential MD protocol.66 The crystal structure exhibits minor differences between the two cleavage sites located in each TopoII complex (Figure S4, Table S1) However, for completeness, smoothed potential MD simulations were performed independently for both cleavage sites in both TopoII isoforms. Please note that the computed residence time relates to single dissociation events. We calculated the average dissociation time over both sites (including all single unbinding events) for a qualitative comparison of residence times and possible mechanisms that might occur during drug unbinding (Table 1).
Table 1. Summary of Computed Unbinding Times for Etoposide from Two Topoisomerase Isoformsa.
| TopoIIα |
TopoIIβ |
|||
|---|---|---|---|---|
| Site1 | Site2 | Site1 | Site2 | |
| Avg. unbinding time [tr± σe (ns)] | 75.4 ± 7.6 | 98.8 ± 8.6 | 34.0 ± 2.7 | 64.6 ± 7.0 |
| Avg. unbinding time over both sites [tr± σe (ns)] | 87.1 ± 8.1 | 87.1 ± 8.1 | 49.3 ± 4.8 | 49.3 ± 4.8 |
| Total smulation time (μs) | 2.6 | 3.3 | 1.25 | 2.1 |
Computational unbinding (dissociation) times averaged over replicas are reported in nanoseconds. The unbinding times are shown together with the standard error of mean over a sample size of 32 simulations (of several tens of ns) for both Site1 and Site2 in both TopoII isoforms.
The total simulation time collected for a set of 64 runs each of TopoIIα and TopoIIβ is approximately 5.9 μs and approximately 3.4 μs, respectively (Table 1). The longer simulation time accumulated for TopoIIα reflects the delayed unbinding of etoposide from this isoform compared to TopoIIβ. In fact, the average computed dissociation times over both sites are 87.1 ± 8.1 and 49.3 ± 4.8 ns for TopoIIα and TopoIIβ, respectively. These values are in qualitative agreement with the experimental difference in the overall persistence of the etoposide-stabilized cleavage complex, which lasts 3-fold longer in TopoIIα than in TopoIIβ.59 Notably, these values are averaged over two etoposide dissociation events, each from one of the two cleavage sites in TopoII. The difference in residence time between the two sites within the same isoform (Site1 vs Site 2) may be due to minor structural variance. However, the difference in residence time suggests a dissimilar stabilization of DNA cleavage at the two scissile bonds in each TopoII isoform, which should be further investigated. It would be interesting to connect the observed variability in residence time among isoforms (and between each site of a given isoform) with the experimental evidence that compared to TopoIIα, TopoIIβ has a higher ratio of double-strand (ds) over single-strand (ss) breaks in DNA formed by etoposide.59 This mechanistic aspect, however, deserves further investigations.
To better elucidate the origin of the difference in the dissociation times of etoposide from TopoIIα and TopoIIβ, we examined each unbinding event. We determined three distinct unbinding pathways of etoposide from TopoII, which were consistently present in both isoforms, as shown in Figure 4. From the unbinding trajectories (see for an example Movie S1), we noted that the drug makes transient interactions with several amino acids while escaping from the cleavage site. In order to identify the key residues influencing the unbinding times between the two TopoII isoforms, the interactions of all these residues (Asp463α/Asp479β, Leu486α/Leu502β, Met762α/Gln778β, Gly462α/Gly478β, Arg487α/Arg503β, Ser763α/Ala779β, Met766α/Met782β, Thr468α/Ser483β, Met762α/Gln778β, Ser763α/Ala779β, Ile769α/Val785β, and Ser800α/Ala816β) with both isoforms are plotted in Figure 5.84 Of these, we identified those that seem to most strongly affect the etoposide dissociation kinetics in the two isoforms, as discussed in detail in the following section.
Figure 4.
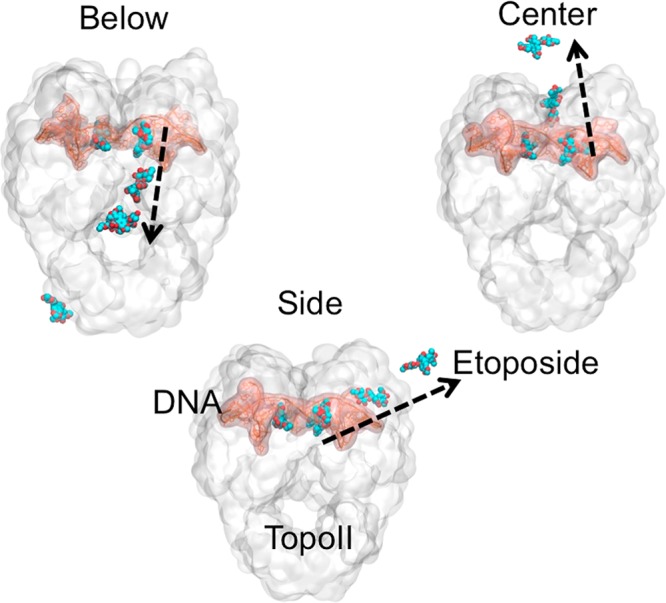
Possible unbinding pathways of etoposide from human Topoisomerase II. Below mode: Etoposide unbinds via the C-gate of TopoII. Center mode: Etoposide unbinds through the dimer intersection. Side mode: Unbinding occurs from the side of the monomer to which the drug molecule is bound. The modes are defined based on the direction of unbinding, relative to the enzyme structure. Color Code: DNA (pink), TopoII (white), Etoposide (blue); black arrows show the direction of unbinding.
Figure 5.

Barcode graph of the interaction of key residues with etoposide, while etoposide unbinds from TopoIIα and TopoIIβ.
Ser763 and Ser800 Residues Hinder Etoposide Dissociation from the TopoIIα Isoform
The objective of this work is to discern how the structural differences between TopoIIα and TopoIIβ can be harnessed to develop isoform-specific drugs. Hence, we first inspected the interactions of etoposide with the three amino acids that differ between TopoIIα (Met762, Ser763, and Ser800) and TopoIIβ (Gln778, Ala779, and Ala816). Figure 6 shows the fluctuations in distances between these residues and etoposide during unbinding. For example, H-bond interactions with these residues are formed more frequently in TopoIIα than in TopoIIβ. However, none of these amino acid changes (Met762, Ser763, and Ser800) in the TopoIIα active site exhibit short electrostatic interactions with the drug in the crystal structure.65,80,81 In contrast, our simulations indicate that as the ligand strives to leave, each of these residues transiently forms H-bond interactions with the drug molecule. For example, Figure 7 shows representative snapshots that reveal how during unbinding, short H-bonds (1.5–2.5 Å) are formed by etoposide with the side chain of these residues. In particular, the hydroxyl group of Ser763 and sulfur in Met762 tightly interacts with the etoposide sugar moiety in TopoIIα. These interactions likely contribute to the stabilization of the drug-target complex, thus prolonging the residence time.
Figure 6.

Interaction (H-bond) of amino acid residues in TopoIIα/β with etoposide during unbinding from each of the two isoforms.
Figure 7.

Stabilizing interactions formed during the etoposide unbinding with (A) Met762α, (B) Ser763α, and (C) and (D) Ser800α in the TopoIIα isoform and with (E) Gln778β in the TopoIIβ isoform, mutated to Met762 in TopoIIα.
The corresponding residue of Ser763 in human TopoIIα is Ser83 in Escherichia coli gyrA, which is located along the α4 helix region. Interestingly, this residue has been associated with quinolone resistance (structural comparison and sequence alignment in Figure 8). Indeed, Ser83 is one of the most frequently mutated residues in strains with high levels of quinolone resistance.85 Moreover, the mutation of Ser83Trp in gyrA considerably reduces ciprofloxacin binding in comparison to the wild-type protein.86,87 Changing the corresponding residue in Saccharomyces cerevisiae (S. cerevisiae, Ser740) leads to resistance to the inhibitor CP-115,953 and hypersensitivity to etoposide.87−89 However, Pommier and co-workers have further shown that Ser740 in S. cerevisiae TopoII is required not only for forming favorable drug interactions but also for DNA binding (needed for TopoII function).90 It remains to be seen if this residue can act as an effective anchor point for developing specific drugs to target TopoIIα.
Figure 8.
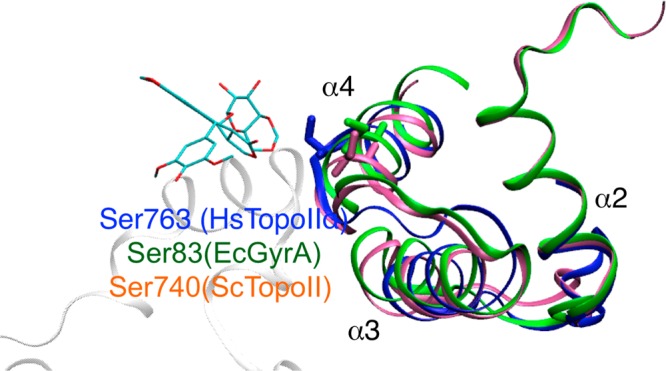
α2−α4 helix region indicates the breakage/cleavage domain of human TopoII (blue) and the corresponding domain in E. coli (green) and S. cerevisiae (pink). The residue Ser763α human TopoII (corresponding to Ser83 in E. coli and Ser740 in S. cerevisiae) is represented in licorice. Quinoline resistance in EcGyrA has been attributed to Ser83 mutations. S740W in ScTopoII has been shown to be hypersensitive to etoposide and resistant to CP-115,953.93
Beyond the alterations in the cleavage site, we also noted the transient interactions with S800 during drug unbinding in TopoIIα. This serine residue (Ser800α/Ala816β) sits quite far from etoposide at the DNA cleavage active site. Still, we found short and transient interactions between etoposide, on its way out of the cleavage site, and Ser800 (Figure 6). Indeed, as the ligand attempts to leave the enzyme pocket at the cleavage site, the hydroxyl group of Ser800 anchors etoposide to TopoII via multiple H-bonds with its glycosidic moiety (2.2–3.0 Å, Figure 7, lower panel). Ser800 also interacts with the D- and E-rings of etoposide, forming H-bonds of 1.6–2.5 and 1.9–2.5 Å, respectively (Figure 7). At times, the drug molecule flips in order to break the interactions formed by the sugar moiety. However, even in these events, etoposide is trapped by the multiple possible interactions formed with Ser800. Taken together, our simulations and analyses show that the three main points (residues) of difference in TopoIIα (i.e., Met762, Ser763, and Ser800) play a role in keeping the drug molecule in contact with the enzyme, although some of these residues are not located close to the cleavage site.
Notably, as shown in Figure 9, E. coli gyrase has a serine (Ser116) in the same position of Ser800 in human TopoIIα, which might also be relevant for the drug binding/unbinding in the bacterial enzyme. To the best of our knowledge, mutations of this residue have not been reported to generate resistance to the drug (as, for example, Arg487 and Glu495 are reported to give rise to etoposide resistance in small-cell lung cancer patients).91,92 We thus hypothesize Ser800 as a possible anchor point for favorable drug-target interactions.
Figure 9.

Region of human TopoII (Hs TopoIIα, blue) containing Ser800 and the corresponding domain in E. coli gyrase (EcGyrA, green) (corresponding to Ser800 in human TopoIIα and Ser116 in E. coli) is represented in licorice. This serine residue, located along these structure motifs, forms transient H-bond interactions during our smoothed potential MD unbinding trajectories. A short sequence alignment of human TopoIIα and EcGyrA is also reported to indicate the conservation of this serine residue.
Of the three amino acid changes in TopoIIβ, Gln778, Ala779, and Ala816, only Gln778 contributes to securing the drug molecule within the active site (Figure 7E). The amide group of Gln778 interacts with the etoposide D-ring, strengthening drug binding to the enzyme. In fact, in our simulations, the distance between the center of mass of etoposide and Gln778 fluctuates around 5 Å (Figure 6) during the first approximately 10 ns of simulations. Here, the Gln778 side chain forms H-bonds (1.6–2.5 Å) with the D-ring oxygen atom of etoposide. However, after the first approximately 10 ns, no further H-bonds are formed by Gln778 with etoposide. In accordance with this, the interaction distance between Gln778 and etoposide increases continuously, reaching approximately 20 Å within the next approximately 10 ns (Figure 6). We also note that Gln778 in TopoIIβ interacts only with the D-ring. This is in contrast to Ser800 in TopoIIα, which interacts with the D-ring, E-ring, and the glycosidic group of etoposide. Hence, as soon as etoposide flips and finds an opportunity to break the D-ring/Gln778 H-bond, it smoothly leaves the enzyme. Also, unlike Ser763 and Ser800 in TopoIIα, Ala779 and Ala816 in TopoIIβ have a side chain (methyl group) that is unable to form H-bonds. Hence, in TopoIIα, Ser763 and Ser800 could contribute to anchoring the drug molecule to the protein in our simulations, while these drug–target interactions are missing in TopoIIβ. Based on these results, we propose these residues as potential new interaction points for targeting and developing TopoIIα-specific drugs.
Conclusions
Poisoning of human TopoII has been harnessed for decades as an effective strategy against a wide variety of cancers. However, TopoII drugs are plagued with the challenge of patients developing drug resistance or secondary malignancies upon drug treatment. One factor is likely the lack of specificity of the current drugs, which unselectively affect both TopoIIα and TopoIIβ. Indeed, recent studies have suggested that selective inhibition of TopoIIα would generate beneficial pharmacological effects, possibly decreasing the side effects caused by the inhibition of TopoIIβ. In this context, we performed classical molecular dynamics (MD) simulations to comparatively examine the molecular interactions of the anticancer drug etoposide in the TopoII/DNA cleavage complex, considering both TopoII isoforms. We also used smoothed potential MD simulations to investigate etoposide dissociation kinetics from the two isoform complexes. We found that etoposide is slower in leaving TopoIIα, which may explain the prolonged persistence of the TopoIIα/DNA cleavage complex formed in the presence of the drug.59 We also found stabilizing interactions of etoposide with two serine residues (Ser763 and Ser800) in TopoIIα, which appear to be responsible for the delayed departure of the drug from the enzyme. Notably, these interactions are not present in TopoIIβ, where both of these serine residues are changed to an alanine. Taken together, these results provide a structural and kinetic rationale for the design of novel TopoIIα-specific drugs able to stably engage these serine residues.
Acknowledgments
This research was supported by the European Union’s Horizon 2020 Research and Innovation Program under Grant Agreement No. 746309 to J.A.K. M.D.V. thanks the Italian Association for Cancer Research (AIRC) for financial support (IG 18883). N.O. thanks the National Institutes of Health (Grant GM126363) and the U.S. Veterans Administration (Merit Review Award I01 Bx002198) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.9b00605.
The authors declare no competing financial interest.
Supplementary Material
References
- Pommier Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum D.; Joshi N.; Tao W.; Karp J. M.; Peer D. Progress and Challenges Towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410. 10.1038/s41467-018-03705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Sun Y.; Huang S. N.; Nitiss J. L. Roles of Eukaryotic Topoisomerases in Transcription, Replication and Genomic Stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. DNA Topoisomerase II and Its Growing Repertoire of Biological Functions. Nat. Rev. Cancer 2009, 9, 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey R. H. Jr.; Pendleton M.; Ashley R. E.; Mercer S. L.; Deweese J. E.; Osheroff N. Catalytic Core of Human Topoisomerase IIα: Insights into Enzyme-DNA Interactions and Drug Mechanism. Biochemistry 2014, 53, 6595–6602. 10.1021/bi5010816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Burgin A. B.; Osheroff N. Human Topoisomerase IIα Uses a Two-Metal-Ion Mechanism for DNA Cleavage. Nucleic Acids Res. 2008, 36, 4883–4893. 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. The Use of Divalent Metal Ions by Type II Topoisomerases. Metallomics 2010, 2, 450–459. 10.1039/c003759a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Cavalli A.; Klein M. L.; Alfonso-Prieto M.; Dal Peraro M.; De Vivo M. Catalytic Metal Ions and Enzymatic Processing of DNA and Rna. Acc. Chem. Res. 2015, 48, 220–228. 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- De Vivo M. Bridging Quantum Mechanics and Structure-Based Drug Design. Front. Biosci., Landmark Ed. 2011, 16, 1619–1633. 10.2741/3809. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Stenta M.; Cavalli A.; Dal Peraro M.; De Vivo M. Molecular Simulations Highlight the Role of Metals in Catalysis and Inhibition of Type II Topoisomerase. J. Chem. Theory Comput. 2013, 9, 857–862. 10.1021/ct300691u. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Singh A.; Kaur N.; Singh G.; Sharma P.; Bedi P.; Sharma S.; Nepali K. Topoisomerase I and II Inhibitors: A Patent Review. Recent Pat. Anti-Cancer Drug Discovery 2016, 11, 401–423. 10.2174/0929866523666160720095940. [DOI] [PubMed] [Google Scholar]
- Ortega J. A.; Riccardi L.; Minniti E.; Borgogno M.; Arencibia J. M.; Greco M. L.; Minarini A.; Sissi C.; De Vivo M. Pharmacophore Hybridization to Discover Novel Topoisomerase II Poisons with Promising Antiproliferative Activity. J. Med. Chem. 2018, 61, 1375–1379. 10.1021/acs.jmedchem.7b01388. [DOI] [PubMed] [Google Scholar]
- Nitiss J. L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor N.; Dominguez I.; Orta M. L.; Campanella C.; Mateos S.; Cortes F. The DNA Topoisomerase II Catalytic Inhibitor Merbarone Is Genotoxic and Induces Endoreduplication. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2012, 738–739, 45–51. 10.1016/j.mrfmmm.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Murphy M. B.; Mercer S. L.; Deweese J. E. Inhibitors and Poisons of Mammalian Type II Topoisomerases. Adv. Mol. Toxic. 2017, 11, 203–240. 10.1016/B978-0-12-812522-9.00005-1. [DOI] [Google Scholar]
- Pommier Y.; Leo E.; Zhang H.; Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly C. Contemporary Challenges in the Design of Topoisomerase II Inhibitors for Cancer Chemotherapy. Chem. Rev. 2012, 112, 3611–3640. 10.1021/cr200325f. [DOI] [PubMed] [Google Scholar]
- Dong G.; Wu Y.; Sun Y.; Liu N.; Wu S.; Zhang W.; Sheng C. Identification of Potent Catalytic Inhibitors of Human DNA Topoisomerase II by Structure-Based Virtual Screening. MedChemComm 2018, 9, 1142–1146. 10.1039/C8MD00219C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap M.; Kandekar S.; Baviskar A. T.; Das D.; Preet R.; Mohapatra P.; Satapathy S. R.; Siddharth S.; Guchhait S. K.; Kundu C. N.; Banerjee U. C. Indenoindolone Derivatives as Topoisomerase II-Inhibiting Anticancer Agents. Bioorg. Med. Chem. Lett. 2013, 23, 934–938. 10.1016/j.bmcl.2012.12.063. [DOI] [PubMed] [Google Scholar]
- Delgado J. L.; Hsieh C. M.; Chan N. L.; Hiasa H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. 10.1042/BCJ20160583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankanala J.; Ribeiro C. J. A.; Kiselev E.; Ravji A.; Williams J.; Xie J.; Aihara H.; Pommier Y.; Wang Z. Novel Deazaflavin Analogues Potently Inhibited Tyrosyl DNA Phosphodiesterase 2 (Tdp2) and Strongly Sensitized Cancer Cells toward Treatment with Topoisomerase II (Top2) Poison Etoposide. J. Med. Chem. 2019, 62, 4669–4682. 10.1021/acs.jmedchem.9b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Huang X. S.; Wu J. F.; Yang L.; Zheng Y. T.; Shen Y. M.; Li Z. Y.; Li X. Discovery of Novel Topoisomerase II Inhibitors by Medicinal Chemistry Approaches. J. Med. Chem. 2018, 61, 8947–8980. 10.1021/acs.jmedchem.7b01202. [DOI] [PubMed] [Google Scholar]
- Froelich-Ammon S. J.; Osheroff N. Topoisomerase Poisons: Harnessing the Dark Side of Enzyme Mechanism. J. Biol. Chem. 1995, 270, 21429–21432. 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- Pommier Y.; Marchand C. Interfacial Inhibitors: Targeting Macromolecular Complexes. Nat. Rev. Drug Discovery 2012, 11, 25–36. 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann K. R.; Ergun Y.; Zencir S.; Oncuoglu S.; Osheroff N.; Topcu Z. Inhibition of Human DNA Topoisomerase IIα by Two Novel Ellipticine Derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 1809–1812. 10.1016/j.bmcl.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson E. G.; King M. M.; Mercer S. L.; Deweese J. E. Two-Mechanism Model for the Interaction of Etoposide Quinone with Topoisomerase IIα. Chem. Res. Toxicol. 2016, 29, 1541–1548. 10.1021/acs.chemrestox.6b00209. [DOI] [PubMed] [Google Scholar]
- Hande K. R. Topoisomerase II Inhibitors. Cancer Chemother. Biol. Response Modif. 2003, 21, 103–125. 10.1016/S0921-4410(03)21005-X. [DOI] [PubMed] [Google Scholar]
- Pogorelcnik B.; Perdih A.; Solmajer T. Recent Developments of DNA Poisons--Human DNA Topoisomerase IIα Inhibitors--as Anticancer Agents. Curr. Pharm. Des. 2013, 19, 2474–2488. 10.2174/1381612811319130016. [DOI] [PubMed] [Google Scholar]
- Baldwin E. L.; Osheroff N. Etoposide, Topoisomerase II and Cancer. Curr. Med. Chem.: Anti-Cancer Agents 2005, 5, 363–372. 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- Harkin L. F.; Gerrelli D.; Gold Diaz D. C.; Santos C.; Alzu’bi A.; Austin C. A.; Clowry G. J. Distinct Expression Patterns for Type II Topoisomerases IIα and IIβ in the Early Foetal Human Telencephalon. J. Anat. 2016, 228, 452–463. 10.1111/joa.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessner R. D.; Mattern M. R.; Mirabelli C. K.; Johnson R. K.; Drake F. H. Proliferation- and Cell Cycle-Dependent Differences in Expression of the 170 Kilodalton and 180 Kilodalton Forms of Topoisomerase II in Nih-3t3 Cells. Cell Growth Differ. 1991, 2, 209–214. [PubMed] [Google Scholar]
- Christensen M. O.; Larsen M. K.; Barthelmes H. U.; Hock R.; Andersen C. L.; Kjeldsen E.; Knudsen B. R.; Westergaard O.; Boege F.; Mielke C. Dynamics of Human DNA Topoisomerases IIα and IIβ in Living Cells. J. Cell Biol. 2002, 157, 31–44. 10.1083/jcb.200112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. The DNA Cleavage Reaction of Topoisomerase II: Wolf in Sheep’s Clothing. Nucleic Acids Res. 2009, 37, 738–748. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M.; Lindsey R. H. Jr.; Felix C. A.; Grimwade D.; Osheroff N. Topoisomerase II and Leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. 10.1111/nyas.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin C. A.; Marsh K. L. Eukaryotic DNA Topoisomerase II Beta. BioEssays 1998, 20, 215–226. . [DOI] [PubMed] [Google Scholar]
- Yang X.; Li W.; Prescott E. D.; Burden S. J.; Wang J. C. DNA Topoisomerase IIbeta and Neural Development. Science 2000, 287, 131–134. 10.1126/science.287.5450.131. [DOI] [PubMed] [Google Scholar]
- Ju B. G.; Lunyak V. V.; Perissi V.; Garcia-Bassets I.; Rose D. W.; Glass C. K.; Rosenfeld M. G. A Topoisomerase IIβ-Mediated dsDNA Break Required for Regulated Transcription. Science 2006, 312, 1798–1802. 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Heng X.; Le W. D. The Function of DNA Topoisomerase IIbeta in Neuronal Development. Neurosci. Bull. 2010, 26, 411–416. 10.1007/s12264-010-0625-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. A.; Byl J. A.; Mercer S. L.; Deweese J. E.; Osheroff N. Etoposide Quinone Is a Covalent Poison of Human Topoisomerase IIβ. Biochemistry 2014, 53, 3229–3236. 10.1021/bi500421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon A. K.; Osheroff N. DNA Topoisomerase II, Genotoxicity, and Cancer. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2007, 623, 83–97. 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmore E.; Frank A. J.; Padget K.; Tilby M. J.; Austin C. A. Etoposide Targets Topoisomerase IIα and IIβ in Leukemic Cells: Isoform-Specific Cleavable Complexes Visualized and Quantified in Situ by a Novel Immunofluorescence Technique. Mol. Pharmacol. 1998, 54, 78–85. 10.1124/mol.54.1.78. [DOI] [PubMed] [Google Scholar]
- Turnbull R. M.; Meczes E. L.; Perenna Rogers M.; Lock R. B.; Sullivan D. M.; Finlay G. J.; Baguley B. C.; Austin C. A. Carbamate Analogues of Amsacrine Active against Non-Cycling Cells: Relative Activity against Topoisomerases IIα and IIβ. Cancer Chemother. Pharmacol. 1999, 44, 275–282. 10.1007/s002800050978. [DOI] [PubMed] [Google Scholar]
- Fief C. A.; Hoang K. G.; Phipps S. D.; Wallace J. L.; Deweese J. E. Examining the Impact of Antimicrobial Fluoroquinolones on Human DNA Topoisomerase IIα and IIβ. ACS Omega 2019, 4, 4049–4055. 10.1021/acsomega.8b03428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell I. A.; Agama K.; Park G. Y.; Pommier Y.; Lippard S. J. Phenanthriplatin Acts as a Covalent Poison of Topoisomerase II Cleavage Complexes. ACS Chem. Biol. 2016, 11, 2996–3001. 10.1021/acschembio.6b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Sun Y.; Ji P.; Kopetz S.; Zhang W. Topoisomerase IIα in Chromosome Instability and Personalized Cancer Therapy. Oncogene 2015, 34, 4019–4031. 10.1038/onc.2014.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris W. H.; Ngo L.; Wilson J. T.; Medawala W.; Brown A. R.; Conner J. D.; Fabunmi F.; Cashman D. J.; Lisic E. C.; Yu T.; Deweese J. E.; Jiang X. Structural and Metal Ion Effects on Human Topoisomerase IIα Inhibition by Alpha-(N)-Heterocyclic Thiosemicarbazones. Chem. Res. Toxicol. 2019, 32, 90–99. 10.1021/acs.chemrestox.8b00204. [DOI] [PubMed] [Google Scholar]
- Vann K. R.; Ekiz G.; Zencir S.; Bedir E.; Topcu Z.; Osheroff N. Effects of Secondary Metabolites from the Fungus Septofusidium Berolinense on DNA Cleavage Mediated by Human Topoisomerase IIα. Chem. Res. Toxicol. 2016, 29, 415–420. 10.1021/acs.chemrestox.6b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell I. G.; Sondka Z.; Smith K.; Lee K. C.; Manville C. M.; Sidorczuk-Lesthuruge M.; Rance H. A.; Padget K.; Jackson G. H.; Adachi N.; Austin C. A. Model for Mll Translocations in Therapy-Related Leukemia Involving Topoisomerase IIbeta-Mediated DNA Strand Breaks and Gene Proximity. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8989–8994. 10.1073/pnas.1204406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarova A. M.; Lyu Y. L.; Lin C. P.; Tsai Y. C.; Lau J. Y.; Wang J. C.; Liu L. F. Roles of DNA Topoisomerase II Isozymes in Chemotherapy and Secondary Malignancies. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 11014–11019. 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell I. G.; Austin C. A. Mechanism of Generation of Therapy Related Leukemia in Response to Anti-Topoisomerase II Agents. Int. J. Environ. Res. Public Health 2012, 9, 2075–2091. 10.3390/ijerph9062075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baviskar A. T.; Madaan C.; Preet R.; Mohapatra P.; Jain V.; Agarwal A.; Guchhait S. K.; Kundu C. N.; Banerjee U. C.; Bharatam P. V. N-Fused Imidazoles as Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. J. Med. Chem. 2011, 54, 5013–5030. 10.1021/jm200235u. [DOI] [PubMed] [Google Scholar]
- Baviskar A. T.; Amrutkar S. M.; Trivedi N.; Chaudhary V.; Nayak A.; Guchhait S. K.; Banerjee U. C.; Bharatam P. V.; Kundu C. N. Switch in Site of Inhibition: A Strategy for Structure-Based Discovery of Human Topoisomerase IIα Catalytic Inhibitors. ACS Med. Chem. Lett. 2015, 6, 481–485. 10.1021/acsmedchemlett.5b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minniti E.; Byl J. A. W.; Riccardi L.; Sissi C.; Rosini M.; De Vivo M.; Minarini A.; Osheroff N. Novel Xanthone-Polyamine Conjugates as Catalytic Inhibitors of Human Topoisomerase IIα. Bioorg. Med. Chem. Lett. 2017, 27, 4687–4693. 10.1016/j.bmcl.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Shen X.; Wang X.; Li A.; Wang P.; Jiang P.; Zhou J.; Feng Q. Egcg Regulates the Cross-Talk between Jwa and Topoisomerase IIα in Non-Small-Cell Lung Cancer (Nsclc) Cells. Sci. Rep. 2015, 5, 11009. 10.1038/srep11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani A.; Bartoli A.; Atwal M.; Lee K. C.; Austin C. A.; Rodriguez R. Differential Targeting of Human Topoisomerase II Isoforms with Small Molecules. J. Med. Chem. 2015, 58, 4851–4856. 10.1021/acs.jmedchem.5b00473. [DOI] [PubMed] [Google Scholar]
- Austin C. A.; Lee K. C.; Swan R. L.; Khazeem M. M.; Manville C. M.; Cridland P.; Treumann A.; Porter A.; Morris N. J.; Cowell I. G. Top2B: The First Thirty Years. Int. J. Mol. Sci. 2018, 19, 2765. 10.3390/ijms19092765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Huang K. C.; Yamasaki E. F.; Chan K. K.; Chohan L.; Snapka R. M. Xk469, a Selective Topoisomerase IIβ Poison. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 12168–12173. 10.1073/pnas.96.21.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviatt A. A.; Kuriappan J. A.; Minniti E.; Vann K. R.; Onuorah P.; Minarini A.; De Vivo M.; Osheroff N. Polyamine-Containing Etoposide Derivatives as Poisons of Human Type II Topoisomerases: Differential Effects on Topoisomerase IIα and IIβ. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. 10.1016/j.bmcl.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Minniti E.; Greco M. L.; Riccardi L.; Simoni E.; Convertino M.; Marchetti C.; Rosini M.; Sissi C.; Minarini A.; De Vivo M. An Optimized Polyamine Moiety Boosts the Potency of Human Type II Topoisomerase Poisons as Quantified by Comparative Analysis Centered on the Clinical Candidate F14512. Chem. Commun. 2015, 51, 14310–14313. 10.1039/C5CC05065K. [DOI] [PubMed] [Google Scholar]
- Infante Lara L.; Fenner S.; Ratcliffe S.; Isidro-Llobet A.; Hann M.; Bax B.; Osheroff N. Coupling the Core of the Anticancer Drug Etoposide to an Oligonucleotide Induces Topoisomerase II-Mediated Cleavage at Specific DNA Sequences. Nucleic Acids Res. 2018, 46, 2218–2233. 10.1093/nar/gky072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry A. C.; Pitts S. L.; Jablonsky M. J.; Bailly C.; Graves D. E.; Osheroff N. Interactions between the Etoposide Derivative F14512 and Human Type II Topoisomerases: Implications for the C4 Spermine Moiety in Promoting Enzyme-Mediated DNA Cleavage. Biochemistry 2011, 50, 3240–3249. 10.1021/bi200094z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandele O. J.; Osheroff N. The Efficacy of Topoisomerase II-Targeted Anticancer Agents Reflects the Persistence of Drug-Induced Cleavage Complexes in Cells. Biochemistry 2008, 47, 11900–11908. 10.1021/bi800981j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. R.; Chen S. F.; Wu C. C.; Liao Y. W.; Lin T. S.; Liu K. T.; Chen Y. S.; Li T. K.; Chien T. C.; Chan N. L. Producing Irreversible Topoisomerase II-Mediated DNA Breaks by Site-Specific Pt(II)-Methionine Coordination Chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. 10.1093/nar/gkx742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C.; Li T. K.; Farh L.; Lin L. Y.; Lin T. S.; Yu Y. J.; Yen T. J.; Chiang C. W.; Chan N. L. Structural Basis of Type II Topoisomerase Inhibition by the Anticancer Drug Etoposide. Science 2011, 333, 459–462. 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- Mollica L.; Decherchi S.; Zia S. R.; Gaspari R.; Cavalli A.; Rocchia W. Kinetics of Protein-Ligand Unbinding Via Smoothed Potential Molecular Dynamics Simulations. Sci. Rep. 2015, 5, 11539. 10.1038/srep11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi L.; Genna V.; De Vivo M. Metal-Ligand Interactions in Drug Design. Nat. Rev. Chem. 2018, 2, 100–112. 10.1038/s41570-018-0018-6. [DOI] [Google Scholar]
- De Vivo M.; Cavalli A. Recent Advances in Dynamic Docking for Drug Discovery. WIREs Comput. Mol. Sci. 2017, 7, e1320 10.1002/wcms.1320. [DOI] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J.; Honig B.; Shaw D. E.; Friesner R. A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct., Funct., Genet. 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Genet. 2006, 65, 712–725. 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved Side-Chain Torsion Potentials for the Amber Ff99sb Protein Force Field. Proteins: Struct., Funct., Genet. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivani I.; Dans P. D.; Noy A.; Perez A.; Faustino I.; Hospital A.; Walther J.; Andrio P.; Goni R.; Balaceanu A.; Portella G.; Battistini F.; Gelpi J. L.; Gonzalez C.; Vendruscolo M.; Laughton C. A.; Harris S. A.; Case D. A.; Orozco M. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. 10.1038/nmeth.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanquelef E.; Simon S.; Marquant G.; Garcia E.; Klimerak G.; Delepine J. C.; Cieplak P.; Dupradeau F. Y. R.E.D. Server: A Web Service for Deriving Resp and Esp Charges and Building Force Field Libraries for New Molecules and Molecular Fragments. Nucleic Acids Res. 2011, 39, W511–517. 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926. 10.1063/1.445869. [DOI] [Google Scholar]
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. H++: A Server for Estimating Pkas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33, W368–371. 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraro M. D.; Spiegel K.; Lamoureux G.; De Vivo M.; DeGrado W. F.; Klein M. L. Modeling the Charge Distribution at Metal Sites in Proteins for Molecular Dynamics Simulations. J. Struct. Biol. 2007, 157, 444–453. 10.1016/j.jsb.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Decherchi S.; Bottegoni G.; Spitaleri A.; Rocchia W.; Cavalli A. Biki Life Sciences: A New Suite for Molecular Dynamics and Related Methods in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 219–224. 10.1021/acs.jcim.7b00680. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Bender R. P.; Jablonksy M. J.; Shadid M.; Romaine I.; Dunlap N.; Anklin C.; Graves D. E.; Osheroff N. Substituents on Etoposide That Interact with Human Topoisomerase IIα in the Binary Enzyme-Drug Complex: Contributions to Etoposide Binding and Activity. Biochemistry 2008, 47, 4501–4509. 10.1021/bi702019z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilstermann A. M.; Bender R. P.; Godfrey M.; Choi S.; Anklin C.; Berkowitz D. B.; Osheroff N.; Graves D. E. Topoisomerase II - Drug Interaction Domains: Identification of Substituents on Etoposide That Interact with the Enzyme. Biochemistry 2007, 46, 8217–8225. 10.1021/bi700272u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drwal M. N.; Marinello J.; Manzo S. G.; Wakelin L. P.; Capranico G.; Griffith R. Novel DNA Topoisomerase IIα Inhibitors from Combined Ligand- and Structure-Based Virtual Screening. PLoS One 2014, 9, e114904 10.1371/journal.pone.0114904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsea S. H.; Westergaard M.; Burden D. A.; Lomenick J. P.; Osheroff N. Quinolones Share a Common Interaction Domain on Topoisomerase II with Other DNA Cleavage-Enhancing Antineoplastic Drugs. Biochemistry 1997, 36, 2919–2924. 10.1021/bi962488f. [DOI] [PubMed] [Google Scholar]
- Thomas T.; Fang Y.; Yuriev E.; Chalmers D. K. Ligand Binding Pathways of Clozapine and Haloperidol in the Dopamine D2 and D3 Receptors. J. Chem. Inf. Model. 2016, 56, 308–321. 10.1021/acs.jcim.5b00457. [DOI] [PubMed] [Google Scholar]
- Reece R. J.; Maxwell A. DNA Gyrase: Structure and Function. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 335–375. 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- Willmott C. J.; Maxwell A. A Single Point Mutation in the DNA Gyrase a Protein Greatly Reduces Binding of Fluoroquinolones to the Gyrase-DNA Complex. Antimicrob. Agents Chemother. 1993, 37, 126–127. 10.1128/AAC.37.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung Y.; Elsea S. H.; Osheroff N.; Nitiss J. L. A Mutation in Yeast Top2 Homologous to a Quinolone-Resistant Mutation in Bacteria. Mutation of the Amino Acid Homologous to Ser83 of Escherichia Coli GyrA Alters Sensitivity to Eukaryotic Topoisomerase Inhibitors. J. Biol. Chem. 1995, 270, 20359–20364. 10.1074/jbc.270.35.20359. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Wang J. C. Identification of Active Site Residues in the “GyrA” Half of Yeast DNA Topoisomerase II. J. Biol. Chem. 1998, 273, 20252–20260. 10.1074/jbc.273.32.20252. [DOI] [PubMed] [Google Scholar]
- Strumberg D.; Nitiss J. L.; Dong J.; Walker J.; Nicklaus M. C.; Kohn K. W.; Heddle J. G.; Maxwell A.; Seeber S.; Pommier Y. Importance of the Fourth Alpha-Helix within the Cap Homology Domain of Type II Topoisomerase for DNA Cleavage Site Recognition and Quinolone Action. Antimicrob. Agents Chemother. 2002, 46, 2735–2746. 10.1128/AAC.46.9.2735-2746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D.; Nitiss J. L.; Rose A.; Nicklaus M. C.; Pommier Y. Mutation of a Conserved Serine Residue in a Quinolone-Resistant Type II Topoisomerase Alters the Enzyme-DNA and Drug Interactions. J. Biol. Chem. 1999, 274, 7292–7301. 10.1074/jbc.274.11.7292. [DOI] [PubMed] [Google Scholar]
- Kubo A.; Yoshikawa A.; Hirashima T.; Masuda N.; Takada M.; Takahara J.; Fukuoka M.; Nakagawa K. Point Mutations of the Topoisomerase IIα Gene in Patients with Small Cell Lung Cancer Treated with Etoposide. Cancer Res. 1996, 56, 1232–1236. [PubMed] [Google Scholar]
- Ganapathi R. N.; Ganapathi M. K. Mechanisms Regulating Resistance to Inhibitors of Topoisomerase II. Front. Pharmacol. 2013, 4, 89. 10.3389/fphar.2013.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda N.; Ito Y.; Imai T.; Kikumori T.; Kikuchi A.; Nishiyama Y.; Yoshida S.; Suzuki M. The Alpha4 Residues of Human DNA Topoisomerase IIα Function in Enzymatic Activity and Anticancer Drug Sensitivity. Nucleic Acids Res. 2004, 32, 1767–1773. 10.1093/nar/gkh339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.