

Abstract
Minor intron splicing plays a central role in human embryonic development and survival. Indeed, biallelic mutations in RNU4ATAC, transcribed into the minor spliceosomal U4atac snRNA, are responsible for three rare autosomal recessive multimalformation disorders named Taybi–Linder (TALS/MOPD1), Roifman (RFMN), and Lowry–Wood (LWS) syndromes, which associate numerous overlapping signs of varying severity. Although RNA-seq experiments have been conducted on a few RFMN patient cells, none have been performed in TALS, and more generally no in-depth transcriptomic analysis of the ∼700 human genes containing a minor (U12-type) intron had been published as yet. We thus sequenced RNA from cells derived from five skin, three amniotic fluid, and one blood biosamples obtained from seven unrelated TALS cases and from age- and sex-matched controls. This allowed us to describe for the first time the mRNA expression and splicing profile of genes containing U12-type introns, in the context of a functional minor spliceosome. Concerning RNU4ATAC-mutated patients, we show that as expected, they display distinct U12-type intron splicing profiles compared to controls, but that rather unexpectedly mRNA expression levels are mostly unchanged. Furthermore, although U12-type intron missplicing concerns most of the expressed U12 genes, the level of U12-type intron retention is surprisingly low in fibroblasts and amniocytes, and much more pronounced in blood cells. Interestingly, we found several occurrences of introns that can be spliced using either U2, U12, or a combination of both types of splice site consensus sequences, with a shift towards splicing using preferentially U2 sites in TALS patients’ cells compared to controls.
Keywords: MOPD1, RNU4ATAC, minor splicing, U12-type introns, RNA sequencing, intron retention
INTRODUCTION
Pre-mRNA splicing is a crucial step that needs accurate execution for proper eukaryotic gene expression. Multiexonic pre-mRNA species can be spliced in a variety of ways as one or several exons may be skipped, introns retained or spliced with alternative donor or acceptor sites, either as part of a physiological process named alternative splicing or as the result of anomalies in the splicing process. Splicing misregulation may occur during cell proliferation, differentiation, survival or death, and is well documented in the context of numerous human diseases (Scotti and Swanson 2016).
Two types of introns coexist in the genome of most eukaryotes, major and minor introns (respectively also named U2- and U12-type introns) (Burge et al. 1998; Sheth et al. 2006). U12-type introns were first discovered due to their unusual AT-AC dinucleotide donor and acceptor splice sites and believed to harbor exclusively these sequences (Jackson 1991). They are now computationally identified based on their specific donor splice site and branch point sequence (BPS) consensus sequences, the latter being located within a specific window of 10–13 nt before the acceptor splice site (Dietrich et al. 2001a, 2005). In 2007, these criteria enabled to identify 695 introns of the U12-type in the human genome, thus representing <1% of all human introns (Alioto 2007). It turned out that 70% of these introns had the classical GT-AG termini.
Each type of intron is spliced by a distinct nuclear machinery: the major, or U2-dependent, spliceosome, and the minor, or U12-dependent, spliceosome. Both contain two small nuclear ribonucleoproteins (snRNPs) involved in intron recognition (respectively, U1 and U2, and U11/U12 di-snRNPs) and three snRNPs involved in the catalytic reaction (respectively, U4/U6.U5 and U4atac/U6atac.U5 tri-snRNPs), U5 being the only snRNA shared between the two spliceosomes. While major spliceosome- and minor spliceosome-specific snRNAs have divergent sequences, they share a similar secondary structure (Tarn and Steitz 1996). Spliceosome specificity relies mostly on splice sites recognition by the major U1 and U2 snRNPs or the minor U11/U12 di-snRNP, and protein composition of the two spliceosomes is highly similar apart from seven proteins which are specific to the minor spliceosome (Schneider et al. 2002).
Minor splicing conservation through evolution implies an important role for this cellular process, but a more direct evidence of its central role came with the identification of mutations in a component of the minor spliceosome in patients afflicted with a severe developmental disease. Indeed, an autosomal recessive disorder named microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1, OMIM 210710) or Taybi–Linder syndrome (TALS) was found by our team and others to be due to biallelic mutations in the gene transcribed into U4atac, RNU4ATAC (Edery et al. 2011; He et al. 2011). This very rare syndrome is characterized by multiple malformations including severe microcephaly and cortical brain malformations (neuronal migration defects), corpus callosum agenesis/dysgenesis, cerebellar vermis hypoplasia, intellectual disability, dysmorphic features, sparse or absent hair, dry skin, short stature and bone anomalies. It leads to early unexplained death occurring within the first three years of life in more than 70% of the cases. Interestingly, other very rare congenital disorders, namely Roifman syndrome (RFMN, OMIM 616651) and Lowry–Wood syndrome (LWS, OMIM 226960) have also been recently attributed to biallelic RNU4ATAC mutations (Merico et al. 2015; Farach et al. 2018). Both RFMN and LWS have features overlapping with TALS (i.e., microcephaly, intellectual deficiency, growth retardation, skeletal dysplasia) but these disorders are not associated with early mortality, they do not include visible structural brain anomalies, and they have less pronounced microcephaly and growth retardation. Of note, RFMN cases exhibit a specific antibody deficiency that is the hallmark of this rare immunodeficiency syndrome.
The U4atac/U6atac bi-molecule has a Y-shaped structure which consists of two intermolecular stems, stem I and stem II, separated by a secondary U4atac structure called the 5′ stem–loop. The U4atac terminal region also contains a 3′ stem–loop and a Sm protein-binding site (for review, see Turunen et al. 2013). To date, mutations have been identified at the homozygous or compound heterozygous states in RNU4ATAC in 53 TALS, 14 RFMN and 5 LWS patients or fetuses (from 30 TALS, 10 RFMN, and 4 LWS families, respectively) (Ferrell et al. 2016; Putoux et al. 2016; Bogaert et al. 2017; Dinur Schejter et al. 2017; Farach et al. 2018; Hallermayr et al. 2018; Heremans et al. 2018; Lionel et al. 2018; Shelihan et al. 2018; Wang et al. 2018; Shaheen et al. 2019). Quite clear, although preliminary, phenotype–genotype correlations stand out across the growing number of cases: Early death in TALS patients (usually before 3 yr of age) is associated with homozygosity for the most common pathogenic variant, g.51G > A, located in the 5′ stem–loop which contains most of the TALS mutations; RFMN is always associated with the location of at least one of the two mutations in Stem II, a region never found mutated in TALS patients.
While germline mutations in genes encoding core protein components of the spliceosome had been already involved in genetic diseases (some forms of retinitis pigmentosa and rare craniofacial, skeletal and skin disorders), U4atac was the first spliceosomal snRNA in which mutations were identified (for reviews, see Padgett 2012; Verma et al. 2018). Since then, mutations in RNU12 were associated with early onset cerebellar ataxia in a large consanguineous family (Elsaid et al. 2017). Mutations in spliceosome components are expected to cause global splicing dysregulation that should manifest in most, if not all tissues, an assumption difficult to reconcile with the highly restricted phenotypes observed in spliceosomopathies. Despite recent technological advances allowing in-depth analyses at the transcriptomic level, very few RNA-seq studies have been performed in these pathologies, precluding comprehensive description of the molecular events associated with the identified mutations. There is now a total of three published analyses of RNA-seq data from RFMN patients that revealed massive U12-type intron retention (IR), but each study focused on only two patients and was restricted to a single cell type, either mononuclear blood cells or megakaryocytes (Merico et al. 2015; Dinur Schejter et al. 2017; Heremans et al. 2018). In contrast, the transcriptomic profile of TALS patients has not been described yet.
We present here for the first time the analysis of RNA-seq data sets performed on cells derived from skin biopsies, amniotic fluids and peripheral blood taken from seven unrelated TALS cases carrying various RNU4ATAC mutations and 13 control individuals matched for tissue, age and gender, hence providing the first whole genome splicing pattern and expression data for this disease. The thorough analysis of this unique data set enables us to study how minor splicing is carried out in physiological and pathological conditions, in various cell types, and sheds new light on this cellular process.
RESULTS
Presentation of RNA-seq data generation and analysis
Biological samples
A total of nine biological samples, that is, five skin, three amniotic fluid and one peripheral blood biospecimens, were obtained from seven unrelated previously published TALS cases (Table 1). This represents the largest collection of TALS samples, to the best of our knowledge. Among these seven cases, four (three children, one fetus) are homozygous for the most common RNU4ATAC mutation, g.51G > A, and three (one child, two fetuses) are compound heterozygous for g.50G > C;g.51G > A, g.40C > T;g.124G > A, and g.51G > A;g.124G > A, respectively. All the affected children died before the age of three, regardless of their mutation(s). Importantly, two different biospecimens were obtained for two g.51G > A homozygous patients, skin and blood for one child and maternal amniotic fluid and skin for the other. Biological samples (eight skin, four amniotic fluid, and one peripheral blood samples) were also obtained from 13 age- and sex-matched controls (Table 1).
TABLE 1.
Description of the samples analyzed by RNA-seq

RNA-seq protocol
We extracted total RNA from fibroblasts (derived from the skin biopsies), amniocytes (derived from the amniotic fluids), and lymphoblastoid cell lines (LCL, established by EBV immortalization of B lymphocytes obtained from blood samples). RNA-seq data were then generated in two experimental setups by Illumina sequencing of (1) poly(A)-selected, non strand-specific sequencing libraries (100 nt paired-end reads) on three patient samples in the pilot study; (2) poly(A)-selected, strand-specific sequencing libraries (75 nt paired-end reads) in the extended study. The extended study was technically more comprehensive and comprised all the samples which had been sequenced in the pilot study. Consequently, we will present and discuss the results obtained in this latter study only. However, in the LCL in-depth analysis, we also used the data set of our pilot experiment as a technical replicate, in order to make up for the lack of biological replicates.
Data sets analysis
Our analysis of these 24 transcriptomes (nine patient and 13 control data sets from the extended study; one patient and one control LCL data sets from the pilot study) examined both gene expression and splicing alterations with a special focus on IR. To this aim, we set up three bioinformatic pipelines (see Materials and Methods), that is, a bioinformatic pipeline that uses a recently developed mapping-first approach dedicated to accurate IR detection, IRFinder (Middleton et al. 2017), and two other pipelines that allow us to identify other types of alternative splicing events, one with a mapping-first approach, vast-tools (Tapial et al. 2017), and the other with an assembly-first approach that we previously reported to have the ability to detect the use of unannotated splice sites, KisSplice (Benoit-Pilven et al. 2018a). Statistical significance of the results obtained with these three pipelines was determined using the same analytical tool, kissDE (Benoit-Pilven et al. 2018b), which allows the identification of significant changes in relative intron or exon inclusion across conditions. To quantify the magnitude of the changes, we computed the Percent Spliced In (PSI) metric, which is the ratio of the reads including the intron over the sum of the reads including or excluding it, for each intron and each condition. This metric provides values close to 100% for fully retained introns and to 0% for fully spliced introns. The PSI metric was also used for quantifying other types of alternative splicing events (see Materials and Methods). The difference between conditions, ΔPSI = PSI_Patients − PSI_Controls, is a measure of the magnitude of the splicing alteration; the sign of this metric indicates in which condition the retention is more frequently seen (patients for positive values or controls for negative values), and its absolute value indicates the level of the difference (the closer to 100%, the higher the difference).
All U12-type intron alternative splicing events identified in patients’ cells are reported in Supplemental Table S1 and described in details in Supplemental Table S2. The processed underlying data can be explored in a Shiny Interface at http://lbbe-shiny.univ-lyon1.fr/TALS-RNAseq/.
Expression levels and splicing efficiency of U12 genes in control fibroblasts, amniocytes and LCL
Global mRNA expression levels of U12 genes in control cell types
To date, despite the large number of transcriptomic studies performed in human tissues and cell types, the spatial and temporal pattern of expression of the transcribed genes containing at least one U12-type intron (hereafter called U12 genes, while U2 genes are those not containing any U12-type intron) has never been described and is largely unknown. We therefore first evaluated which U12 genes were expressed in the eight fibroblast, four amniocyte and one LCL samples derived from control children and fetuses to set the frame of reference for the comparison with TALS patients. We based our analysis on the set of 699 genes containing at least one U12-type intron that we identified in the human genome through a computational scan of the latest annotation of the GRCh37 assembly (Ensembl Release 75) with a U12-type intron annotation tool (Alioto 2007) (846 minor introns annotated in total, Supplemental Table S3), and fixed a threshold for expression at a mean of 5 Transcripts Per Million (TPM) both for U2 and U12 genes. Among these 699 genes, 528 (76%) are expressed in at least one cell type in our control data sets and 427 (61%) are expressed in the three of them, suggesting that the majority of the U12 genes are expressed in various cell types (Supplemental Fig. S1A). The distribution of the expression levels of U12 genes is highly similar between the three cell types and shows a peak at around 30 TPM (Supplemental Fig. S1B). However, we found that the mean number of transcripts per U12 gene was higher in the LCL than in amniocytes and fibroblasts (56, 51 and 48 TPM, respectively). When considering U2 genes, an extra peak of genes expressed at a level <1 TPM is seen (Supplemental Fig. S1B); this bimodal distribution has already been reported and most likely corresponds to noise from the transcriptional machinery (Hebenstreit et al. 2011). Principal component analysis (PCA) of the expression levels of U12 and U2 genes demonstrated that the control transcriptome data sets partitioned depending on the cell type (Supplemental Fig. S1C), indicating that the U12 genes expression level pattern is specific to each cell type. Gender or prenatal vs. post-natal origin of the skin biopsies from which fibroblasts were derived did not strongly influence U2 and U12 genes expression level patterns (Supplemental Fig. S1C).
Global U12- and U2-type intron retentions in U12 genes
After examining mRNA expression levels, we focused on introns and their splicing efficiency in fibroblasts, amniocytes and LCL by analyzing the extent of IR using PSI value calculations. To alleviate potential biases due to the large difference in the number of U2 and U12 genes, we chose to restrict the analysis comparing U12- and U2-type intron splicing efficiency to introns located in U12 genes. In order to obtain robust PSI estimations, we focused on intronic regions with sufficient read coverage (i.e., number of exon-intron + exon–exon junction reads ≥10 in at least 4/8 fibroblast, 2/4 amniocyte, and the LCL control samples). The few annotated introns that were never found spliced out in our data sets were also removed (see Materials and Methods). The analysis was performed on a set of 366 U12-type introns and 1887 U2-type introns scattered in 337 U12 genes with a mean expression of at least 5 TPM in each cell type. We found that the mean PSI for the U12-type introns is 2.2% (median = 0.7%) in fibroblasts, 2.7% (median = 0.9%) in amniocytes and 4.4% (median = 2.0%) in LCL, whereas the mean PSI for the U2-type introns are respectively 3.9% (median = 1.1%), 4.7% (median = 1.5%), and 4.8% (median = 1.5%) in these cells. In contrast with a previous result obtained with HEp-2 cells (Niemela et al. 2014), we did not observe in our data sets that U12-type introns were spliced less efficiently than their neighboring U2-type counterparts. We further observed that splicing was most efficient in fibroblasts, and that U12-type intron splicing was less efficient in LCL (Supplemental Fig. S2A). PCA of the PSI values for U12- and U2-type introns also separated cell types, although less clearly than expression values as one of the four amniocyte data sets segregated with fibroblasts consistently in both U12- and U2-type introns analyses (Supplemental Fig. S2B). We noticed that the LCL data set singled out in PCA of U12-type IR, as it does in PCA of U12 gene expression levels, a finding confirmed when incorporating the pilot study data set in the analyses.
U12-type intron alternative splicing
Besides IR, more complex patterns of U12-type intron alternative splicing have on some occasions also been observed, although less frequently than for U2-type introns (Levine and Durbin 2001; Chang et al. 2007). To identify these events in our data sets we used both a mapping-first approach (vast-tools) and an assembly-first approach (KisSplice), as we previously showed that these approaches were complementary (Benoit-Pilven et al. 2018a). We focused on events with sufficient read coverage (same filter as that used for IR) and with exon–exon junctions covered by an average of at least five reads. We found 9 U12 genes for which a total of 10 complex minor splicing patterns were observed through the use of alternative U12 splice sites in all control data sets. In 9/10 cases, an alternative U12 acceptor site was used, leading to exon skipping in a few instances, while in the remaining one, both alternative U12 donor and acceptor sites were used. The use of the least common donor and/or acceptor splice sites was supported by more than 10% of the reads in all three cell types for six of these events, indicating that they are not marginal. It should be noted that half of the splicing events produced alternative forms considered as noncoding in databases because they contain premature termination codons (PTCs).
U12/U2 splice site switching
Most interestingly, we also found U12-type introns for which nearby U2 splice site(s) were sometimes favored over U12 splice site(s), probably in the context, in most cases, of a switch from the minor to the major spliceosome for splicing the intron. This phenomenon was first described for the D. melanogaster prospero gene (Scamborova et al. 2004); lately, the existence of these introns called U2/U12-type twintrons was extended to several other U12 genes in different species, including humans (for review, see Hafez and Hausner 2015). We identified 21 of such alternative events comprising or not the skipping of an exon in 16 U12 genes. In four of these events, both U2 alternative donor and acceptor splice sites were used. In 10 of them, a U2 alternative donor site was used in combination with the U12 acceptor site, and in the remaining seven, a U12 donor was used with an alternative U2 acceptor site. Such mixed patterns had not yet been observed, to the best of our knowledge. In 13/21 cases, the least abundant form represented more than 10% of all the reads in all three control cell types. A striking example of this situation was observed for the CCDC84 gene (Supplemental Fig. S2C), for which the transcripts derived from the use of U12 splice sites (producing PTC-containing transcripts) or U2 donor and U12 acceptor splice sites (coding the full length protein of unknown function) are found in similar abundance. Hence, the type of splice sites selected to remove this intron from the CCDC84 pre-mRNA can regulate the amount of the full length protein which is produced without changing the transcriptional expression level of the gene.
Overall, beyond the description of these novel mechanisms, this first global analysis of U12-type intron splicing in cells from control children and fetuses provides a reference for studying the consequences of RNU4ATAC biallelic mutations on the transcriptome of cells derived from TALS patients.
Global impact of RNU4ATAC biallelic mutations on transcriptomes of fibroblasts, amniocytes and LCL derived from TALS patients
U12 mRNA expression levels in patients and controls
The PCA performed on either U2 or U12 genes expression levels (TPM measures) in the 22 data sets of the extended RNA-seq study (nine patient and 13 control samples) separated cell types again but failed to separate patients from controls (Supplemental Fig. S3). The fact that we did not see any global impact of RNU4ATAC mutations on U12 gene expression levels using PCA was surprising because one could expect that IR would trigger transcript degradation through quality control pathways, which would in turn lower their amount. These quality control pathways dealing with transcripts with retained introns could be the Nonsense Mediated mRNA Decay (NMD) acting in the cytoplasm (Wong et al. 2013, 2016), or could include both exosome-mediated mRNA turnover following nuclear sequestration, and NMD (Braunschweig et al. 2014). More specifically, U12-type IR have been shown to lead to nuclear retention and nuclear decay by the RNA exosome (Niemela et al. 2014). In order to investigate U12 genes expression levels further, we ran DESeq2 on the fibroblast data sets from controls and patients to identify differentially expressed (DE) genes (i.e., genes for which the number of produced polyA+ mRNAs differs). Using standard cutoffs, that is, False Discovery Rate (FDR) ≤ 5% and |log2(FC)| ≥ 1, we found only 13 DE genes (eight up-, five down-regulated), none of them containing any U12-type intron. The same analysis performed in the patient amniocyte data set collection produced a list of 32 DE genes (11 up-, 21 down-regulated), again all U2 genes, and all different from those identified in fibroblasts except one [RP11-305K5, log2(FC) = 1.6]. To evaluate the biological relevance of these DE genes, we calculated how many were identified in our RNA-seq fibroblast data sets in every possible combination of patients and age- and sex-matched controls and found that this number markedly decreased with the increasing number of patient samples (Supplemental Fig. S4). This pattern is similar to that obtained with false negative results in a study evaluating the number of biological replicates needed to ensure detection of valid significantly differentially expressed genes (Schurch et al. 2016). We therefore conclude that the DE genes we identified here are likely not associated to the pathology itself.
U12-type intron splicing in patients and controls
When performing PCA on U12-type IR levels using PSI values, we observed a clear partitioning of the patients and the controls, as expected, while the same analysis on U2-type IR failed to separate patients from controls (Fig. 1, top and bottom left). Axis 1 of the U12-type IR PCA (PC1: 88% of the variance) was essentially supported by LCL, showing that this cell type has a specific “sensitivity” to defects in U12-type intron splicing. Nevertheless, even when removing LCL data from the analysis, we find that the partition between patients and controls remains clear (Fig. 1, bottom right). We can thus conclude from the PCA analyses that U12-type intron splicing appears indeed globally altered in TALS patients, and that the splicing default appears somehow different in the LCL compared to fibroblasts and amniocytes. We next looked into more details at the global splicing anomalies associated with RNU4ATAC mutations in each cell type.
FIGURE 1.

Patterns of U2- and U12-type IRs in TALS patient and control cells. Principal component analyses of the most variable mean PSI values of U2- and U12-type introns are presented. PCA for U12-type introns was performed with (left) and without the LCL data sets (right). Fibroblasts, amniocytes and LCL were derived from tissues taken from control or TALS fetuses and children. The sex of the donor from which was derived each sample is indicated (M, Male; F, Female), as well as the RNU4ATAC mutation(s) for the patients’ samples. (ns) not significant (the percentage of variance explained by the axis is smaller or equal to the percentage of variance expected by chance, see Materials and Methods).
Splicing efficiency of U12- and U2-type introns in fibroblasts and amniocytes derived from TALS patients
Because the separate analysis of TALS fibroblasts and amniocytes produced similar results, we present them together. The fibroblast data sets (F) were obtained from three homozygous g.51G > A patients, two compound heterozygous g.51G > A;g.50G > C and g.40C > T;g.124G > A patients and eight controls; the amniocyte data sets (A) from two homozygous g.51G > A patients, one compound heterozygous g.51G > A;g.124G > A patient and four controls.
U2-type intron retentions
As expected, the mean PSI values for the U2-type introns passing our filters (see Materials and Methods) were similar in patients and controls [respectively 4.6% vs. 4.3% (F) and 5.1% vs. 5.2% (A)], suggesting that the TALS patients’ cells exhibit unchanged U2-type intron splicing profiles. Indeed, a very small fraction of U2-type introns were found markedly retained (ΔPSI ≥ 10% and FDR ≤ 5%) in patients: 79 out of 54922 (F); 133 out of 59255 (A). Only eight of them were found in both data sets, six of which occurring in U12 genes. As the current annotation is conservative and splice sites that show poor homology with U2- and U12-type intron consensus sequences tend to be considered U2-type, we suspected that some of the retained “U2-type introns” could be misclassified and should be reclassified as U12-type introns. Indeed when examining them, we identified four introns with non consensus splice site sequences located within the RECQL5, DERL2, KIAA0556 and LZTR1 genes (Fig. 2 left, red dots; Supplemental Table S3; Supplemental Fig. S5). For these atypical donor or acceptor splice site sequences, at least one score regarding both U12- and U2- type introns is inferior to −1 (scores are log-likelihood ratios: A sequence with a negative score resembles more the background sequence than the consensus one). Such borderline cases are difficult to classify, and depending on the genome annotation used, their score slightly increases or decreases, causing the classifier to call them U12 or U2. Of note, they were originally classified as U12- or U2/U12-types in U12db (which uses U12 classification scripts, genomic sequences and annotations of 2007), not only in humans but also in macaques and chimpanzees for RECQL5 and in many other species including zebrafish and mouse for DERL2, LZTR1, and KIAA0556. The fact that these introns are markedly retained in TALS samples strongly suggests that they are genuine U12-type introns, consistent with their higher scores for U12 compared to U2 splice site sequences. We thus propose to reclassify them as U12-type introns.
FIGURE 2.

Comparison of U2- and U12-type IR levels in TALS patient and control cells. Analysis of the (A) fibroblast data sets or (B) amniocyte data sets. (Left panels) Plots of the mean U2- and U12-type IR levels expressed with the Percent Spliced In (PSI) metric and obtained for the patients’ versus the controls’ data sets (PSI-plots). Each circle represents an intron: the color indicates its type (U12* means U2-type intron proposed to be reclassified as U12-type in this study), the size indicates the amount of the corresponding transcript, and the filing status indicates the significance of the IR level (filled circle: FDR ≤ 5%; unfilled circle: FDR > 5%). The intron position respective to the line indicates whether the intron is more retained in patients (above the line) or controls (below the line). The further a point is from this line, the greater the intron's ΔPSI. (Right panels) Boxplots of U2- and U12-types intron PSI values of each patient's and control's data set (PSI-boxplots). Mean values are represented as black dots. The numbers of U2- and U12-type introns indicated correspond to those with robust PSI estimation and sufficient coverage in each sample.
U12-type intron retentions
PSI computation was achieved for 482 (F) and 430 (A) U12-type introns, including the four introns that we previously reclassified as U12. As expected, we found that the mean PSI was higher for patients (∼6%) than for controls (∼3%) in both fibroblasts and amniocytes (Table 2; Fig. 2, right), testifying that minor splicing was indeed impaired in patients. Of note, mean PSI were higher in fibroblasts derived from the two RNU4ATAC compound heterozygous patients (Fig. 2A right, last two boxplots; Supplemental Table S4) than in the homozygous patients. However, surprisingly, the magnitude of the effect was limited. As a matter of fact, the vast majority of U12-type introns were statistically significantly misspliced (FDR < 5%), but most of them were only marginally affected (ΔPSI < 10%) (Table 2). The larger fraction of statistically significant U12-type IR observed in fibroblasts compared to amniocytes is most likely due to the larger patient/control sample set in the former (13 vs. 7, respectively), hence increasing the statistical power and enabling us to find more statistically significant small effects.
TABLE 2.
Summary of the U12-type introns results from TALS and RFMN patients’ data sets compared to controls’ data sets

Concomitant U12- and U2-type intron retentions
For some U12-type IR, we noticed that the 5′ or 3′ neighboring U2-type intron was also retained. The example of the DYNC1LI2 gene is given in Figure 3, top. The analysis of the 55 (F) and 33 (A) U12-type marked IR revealed respectively nine and three instances of concomitant U12- and neighboring U2-type IRs suggesting that the missplicing of some U12-type introns could lead to the retention of the 5′ or 3′ adjacent U2-type intron. The scores of the splice sites of these U2-type introns are not different from those of the other U2-type introns, and they have weak U12 splice site scores (Supplemental Fig. S5, black points). Interactions between the minor and the major spliceosomes have already been suggested (Wu and Krainer 1996; Lewandowska et al. 2004; Tapial et al. 2017; Horiuchi et al. 2018), and our study provides additional observations further supporting this hypothesis.
FIGURE 3.
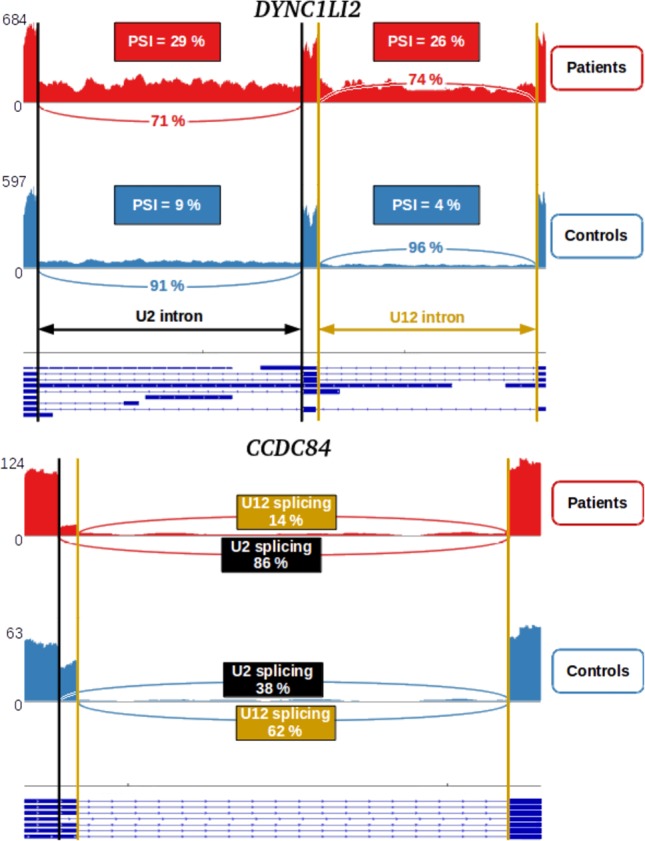
Alternative splicing of U12-type introns in TALS patients’ fibroblasts. Sashimi plots showing a U2-type intron/U12-type intron coupled retention in the DYNC1LI2 gene (top) and a minor/major spliceosome switching event in the CCDC84 gene (bottom). The y-axis corresponds to the mean coverage of each base of the genomic coordinates (x-axis). Reference annotations are given on the lowest part of the figure, with annotated exons and introns shown as thick and thin horizontal lines, respectively. U12 and U2 splice sites are marked with yellow and black vertical bars, respectively. Splice junction reads are drawn as arcs connecting a pair of exons. Mean percentage of reads supporting the splicing of either the U12- or U2-type intron are indicated in yellow and black boxes, respectively.
Fibroblasts and amniocytes obtained from the same patient
We took advantage of the availability of both amniocytes and post-natal fibroblasts for a homozygous g.51G > A patient to assess U12- and U2-type IR in the same genetic background: This analysis revealed that the mean PSI and the PSI distributions of both U2- and U12-type introns were similar in these two cell-types (Supplemental Fig. S6, left).
Other types of U12-type intron alternative splicing
Vast-tools and KisSplice identified respectively four and two U12/U2 splice site switchings in the fibroblast and amniocyte data sets, all of them in favor of the use of U2 splice sites in TALS patients. In particular, both cell types exhibited the same splice site switching event in the CCDC84 gene (shown for the fibroblast data sets in Fig. 3, bottom). The balance of the transcripts derived from the use of either the U12- or the U2-type splice sites observed in controls (∼65%/35%, respectively) was strongly shifted toward the U2 sites-derived coding transcript in TALS patients (∼15%/85%) in both cell types, hence probably increasing the abundance of the functional full-length protein.
Splicing efficiency of U12-type introns in LCL derived from a TALS patient
The PCA analysis of PSI values obtained with our RNA-seq study showed that in TALS patients, the pattern of U12-type IR in the LCL markedly differed from that seen in fibroblasts and amniocytes (Fig. 1, bottom left). Analysis of the data sets from two TALS LCL technical replicates revealed that U2-type intron splicing was globally unaffected (mean PSI: 4.9% vs. 4.8% for the patient and the control, respectively), while U12-type intron splicing was severely affected. Indeed, the mean ΔPSI obtained from TALS patient and control data sets was 19.7% in the LCL, compared to 4.4% and 3.1% in fibroblasts and amniocytes, respectively (Table 2). Looking into further details, we found that 98% of the 480 U12-type introns with a sufficient number of reads for the analysis were more retained in the TALS than in the control LCL sample and that, strikingly, 79% (370/468) of these retentions had a ΔPSI ≥ 10%, as seen when comparing Figure 4A with Figure 2. Other types of U12-type intron alternative splicing were also far more frequent [69 U12/U2 splice site switching vs. 4 (F) and 2 (A)]. On the other hand, a high level of U2-type IR was not observed, ruling out a sequencing or sample preparation problem. The high magnitude of the U12-type intron splicing defects observed in the LCL of the TALS patient was also unlikely to be due to individual particularities because the comparison in this patient of the mean PSI values obtained for U12-type introns in the LCL versus the fibroblast data sets revealed a marked difference (Supplemental Fig. S6, right). We also found more adjacent U12- and U2-type IRs [18 vs. 9 (F) and 3 (A)], among which two, in DYNC1LI2 and DERL2, were common to all cell type data sets.
FIGURE 4.

Comparison of U12-type IR levels in TALS and RFMN patient and control blood cells. Analysis of the (A) TALS LCL (lymphoblastoid cell line) data sets or (B) RFMN MBC (mononuclear blood cells) data sets. The TALS patient's and control's LCL data sets consist of two technical replicates for each. (Left panels) U12-type intron PSI-plots obtained for the patients’ versus the controls’ data sets. (Right panels) U12-type intron PSI-boxplots of each patient's and control's data set. Legend as in Figure 2.
The high U12-type IR observed in the present work in the TALS LCL were reminiscent of the massive deregulation of U12-type intron splicing reported in the transcriptomes of blood cells derived from six RFMN patients belonging to six families (Merico et al. 2015; Dinur Schejter et al. 2017; Heremans et al. 2018). In order to investigate further the extent of similarity of U12-type intron splicing patterns in blood cells derived from patients with these two RNU4ATAC-associated pathologies, we reanalyzed with the pipelines that we set-up for our study the raw data of the transcriptomic sequences of mononuclear blood cells (MBC) taken from two unrelated RFMN patients (Merico et al. 2015), along with that of three of their heterozygous unaffected relatives (brothers or father of the patients). Different expression and splicing profiles were expected as TALS and RFMN are distinct pathologies and, besides the cells’ common tissue's origin (blood), MBC and LCL have marked differences, that is, all types of mononuclear blood cells are present in MBC, while LCL consists of B lymphocytes only, furthermore immortalized by EBV infection, which has been shown to impact gene expression (Lopes-Ramos et al. 2017). Besides, the age at which the blood samples were obtained widely differs between the two studies (babies vs. adults) and the TALS patient and her control are female while the RFMN patients and their controls are male (Table 1). Finally, another important difference was that the TALS data sets were sequenced with higher depth compared to RFMN data sets (125 vs. 47 million of mean aligned reads).
Indeed, not surprisingly given their specificities, PCA showed that U2 and U12 gene expression levels clearly distinguished LCL from MBC; patients and controls from the same collection of data sets grouped together related to the first axis, which explains in both cases more than 65% of the variance (Supplemental Fig. S7A). Concerning U2-type IR, PCA of the mean PSI values did not separate LCL from MBC samples, but separated four of the five MBC samples from the LCL and the fifth MBC samples on the first axis (60% of the variance, Supplemental Fig. S7B, left). These four MBC samples derived from the oldest studied individuals (38, 43, 57, and 67 yr old, compared to 2 mo: LCL sample and 21 yr old: fifth MBC sample), suggesting that age may have an impact on the extent of U2-type IR in blood cells, as previously suggested in the brain (Mazin et al. 2013). Accordingly, we found more than 2000 U2-type IR in the older controls compared to the younger RFMN patients (Fig. 4B, black dots).
Concerning U12-type IR, PCA of the mean PSI values separated TALS and RFMN patients from controls on the first axis (79% of the variance, Supplemental Fig. S7B, right). We did observe separation between TALS LCL and RFMN MBC on the second axis of the PCA, but it explained only 10% of the total variance. When looking at mean U12-type IR values, we observed a strong similarity between the two data sets, as illustrated in Figure 4 (left, yellow dots). Mean PSI were 28.7% in RFMN MBC and 6.0% in control MBC compared to 27.5% and 4.8% in the TALS LCL study, respectively (Table 2), and the mean ΔPSI was 28.9% compared to 27.6%. Because of cell-type specificities and/or different sequencing depths between them, 140 marked U12-type IR found in TALS LCL could not be analyzed in RFMN MBC (13 reciprocally). After filtering them out, we found that 171 marked U12-type IR were common to TALS LCL and RFMN MBC samples (representing 74% and 87% of them, respectively). Of note, only one alternative U12-type intron splicing event, the splice site switching in the uncharacterized CCDC84 gene, had high and similar ΔPSI in all the patient data sets (TALS fibroblasts, amniocytes, LCL and RFMN MBC, mean |ΔPSI| = 54%). Altogether, our results suggest that the magnitude of U12-type intron splicing dysfunction could be, firstly, quite similar in blood cells from TALS and RFMN patients, and secondly, highly tissue-dependent, trends that will need to be investigated further.
qRT-PCR validation of U12-type intron missplicing
To confirm the RNA-seq results, we determined the level of retention of nine U12-type introns with various statistically significant mean ΔPSI values ranging from 0 to 37% using a quantitative RT-PCR (qRT-PCR) approach on RNA extracted from fibroblasts derived from five patients and five age- and sex-matched controls. We found a strong concordance between RNA-seq and qRT-PCR mean ΔPSI values using the same metrics (r² = 0.86, Fig. 5). Of note, even weak effects (mean ΔPSI = 6%) could be confirmed by qRT-PCR.
FIGURE 5.
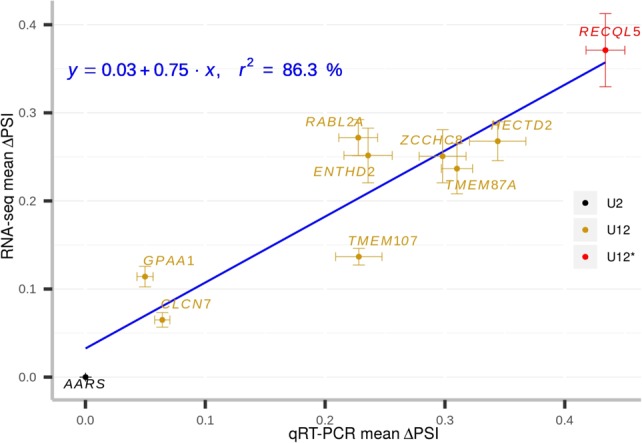
Comparison of U12-type IR levels measured by qRT-PCR and RNA-seq. Correlation between the mean ΔPSI values obtained by qRT-PCR when testing introns from ten genes in fibroblasts derived from five patients and their age- and sex-matched controls and those obtained by RNA-seq. Error bars represent standard errors of the mean in both experiments (vertical: RNA-seq; horizontal: qRT-PCR). The linear regression is shown, together with the squared correlation coefficient. The names of the genes whose intron was tested are indicated. The color of gene names indicates the intron type (U12*: U2-type intron proposed to be reclassified as U12-type in this study).
Gene pathways affected in cells derived from TALS patients
Because 97% of the U12-type introns were retained in the TALS LCL data set, precluding classical enrichment analysis, we focused our attention on identifying genes and pathways impacted by U12-type intron missplicing on TALS fibroblasts and amniocytes. As a preliminary study, we first scrutinized the 26 genes with marked U12-type IR common to both data sets (Supplemental Table S1), and found that a high proportion of them were involved in signal transduction (11/26), notably through Notch (C3orf17) or Sonic Hedgehog (IFT22, TMEM107) signaling pathways; genes involved in protein degradation were also represented in a substantial proportion (6/26). We next wanted to look into more details at U12 genes with misspliced transcripts, potentially leading to reduced level of functional proteins, taking into account all the statistically significant differential U12-type IR and U12/U2 splice sites switching found in the two cell types. Toward this goal, we performed a Gene Ontology (GO) term analysis with TopGO (Alexa and Rahnenfuhrer 2016) and compared misspliced to correctly spliced U12 genes, using either the FDR or ΔPSI values as weights. These two analyses revealed 34 and 12 enriched terms, respectively, and we found, in both of them, instances related to developmental processes, response to stimulus, signaling and interestingly, immune system processes (Supplemental Table S5).
DISCUSSION
Transcriptome analysis by RNA-seq has tremendously enhanced our knowledge on gene expression and intron splicing, shedding light on alternative splicing at a large scale and on its relevance in various cellular contexts. However, this technological revolution has mostly benefited to the understanding of U2-type intron splicing. On the other hand, the U12-type introns and U12 genes, as very small minorities, have been largely neglected, despite their acknowledged importance in embryonic development and survival. The few published analyses focusing on U12-type intron splicing were conducted in plants (Gault et al. 2017), fish (Markmiller et al. 2014), or human cancer cells in order to study gene expression regulation (Younis et al. 2013; Niemela et al. 2014). A few additional studies were conducted in the context of pathologies associated with a minor splicing defect, either due to mutations in snRNA components of the minor spliceosome, mainly RFMN syndrome (Merico et al. 2015; Dinur Schejter et al. 2017; Heremans et al. 2018) and early onset autosomal recessive cerebellar ataxia (Elsaid et al. 2017), or in protein components specific to the minor spliceosome, such as observed in isolated familial growth hormone deficiency (Argente et al. 2014), and myelodysplastic syndrome (Madan et al. 2015). Actually, little is known about global U12 gene expression and U12-type intron splicing in physiological conditions in human cells. Therefore, we started our study by tackling these questions in our control data sets consisting of eight fibroblast, four amniocyte, and one lymphoblastoid cell line (LCL) samples derived from control fetuses and children. In these control cells, we found that (i) ∼60% of the 699 U12 genes are consistently expressed in the three different cell types, and (ii) the distribution of the levels of transcriptional expression of U12 genes is highly homogenous between these cell types and peaks at around 30 TPM, as observed for U2 genes. We also observed several occurrences of U12/U2 splice site switching. Alternative splicing of U12-type introns using U2 cryptic donor and acceptor sites, originally described in insects (for review, see Hafez and Hausner 2015), had already been reported in human cells as the result of U6atac snRNA inactivation (Younis et al. 2013), knockdown of the 48K protein (Turunen et al. 2008), and in the context of isolated familial growth hormone deficiency (Argente et al. 2014), and myelodysplastic syndrome (Madan et al. 2015). However, this is the first time that such alternative splicing events are found to occur physiologically in humans. Because the consensus sequences for the acceptor sites of U2- and U12-type introns are less divergent than that of the donor sites, we suppose that the major spliceosome was used for splicing the U2 donor-U12 acceptor mixed introns and the minor spliceosome for the less abundant U12 donor-U2 acceptor mixed ones.
After having determined the frame of U12 gene expression and U12-type intron splicing in the context of a functional minor spliceosome, we set out to identify the consequences of biallelic RNU4ATAC mutations within these cell types in five fibroblast, three amniocyte and one LCL samples derived from seven unrelated TALS patients. Rather surprisingly, we did not observe any impact of such mutations on U12 or U2 gene expression in fibroblasts or amniocytes derived from TALS patients, although we used the tool (DESeq2) and cutoffs [FDR ≤ 5%; |log2(FC)| ≥ 1] recommended for such a data set size (five patients vs. eight controls) (Schurch et al. 2016). Our previous qRT-PCR study on fibroblasts derived from two homozygous g.51G > A TALS patients and two age- and sex-matched controls (biosamples also included in the present study) had shown that 12 of the 22 tested U12 genes—chosen randomly among those reported as being expressed in the skin—presented a differential expression (Edery et al. 2011). However, we now believe that this previous result most likely stemmed from biological and/or inter-individual variations that could not be correctly modeled due to the small number of samples, and illustrate the necessity to use more stringent criteria when studying a very small number of biological samples. Although we cannot rule out the possibility that a number of U12 genes may be slightly differentially expressed—but identifying them would require more than 20 biological replicates (Schurch et al. 2016)—we conclude that U12 gene expression levels, that is, the number of poly(A)+ transcripts produced, are essentially unchanged in TALS fibroblasts and amniocytes compared to controls.
Then, we studied splicing efficiencies and found that most U12-type introns were significantly retained in the TALS transcriptomes whatever the cell type studied. Hence, even though the number of poly(A)+ transcripts is unchanged for most genes in patients’ compared to controls’ cells, a fraction of them, larger in patients, contains U12-type IR and cannot lead to functional proteins. Although these IR were statistically significant, we found that their magnitudes were small in the fibroblast and amniocyte data sets, with only 14% and 18% of the retained U12-type introns showing a ΔPSI ≥ 10%, respectively. In contrast, these U12-type IR were much more pronounced in the LCL data set, as 79% had a ΔPSI ≥ 10%. Considering that the overall transcript levels are unchanged but splicing is altered, we conclude that the number of transcripts that could be translated into functional proteins is therefore mildly decreased in fibroblasts and amniocytes, and largely decreased in lymphocytes. The extreme rarity of the TALS syndrome and the premature death of the children affected with this disease did not permit us to collect additional blood samples up to now and hence analyze LCL biological replicates. Nevertheless, several lines of evidence support the assumption that peripheral blood cells may exhibit particularly pronounced U12-type IR: (i) This difference in U12-type intron splicing efficiency was clearly visible when comparing cells derived from skin and blood taken the same day on the same TALS child, while no difference was seen for another TALS child between amniotic fluid taken in utero and skin at 10 mo of age (Supplemental Fig. S6); (ii) similar high levels of U12-type IR were observed in the RFMN MBC and TALS LCL data sets, despite the different pathologies, blood cell subtypes analyzed, gender and age of the patients, and RNA-seq settings (Fig. 4); (iii) a comprehensive analysis of IR performed on 52 human samples from different cell and tissue types showed that the highest percentage of retention was found in white blood cells (>30%, compared to <5% in fibroblasts) (Braunschweig et al. 2014).
We observed that the competition between the major and minor spliceosomes for splicing some introns, which we show here for the first time to occur physiologically in humans, is more favorable to the major spliceosome in TALS amniocytes, fibroblasts and LCL as compared to the situation seen in control cells. This was particularly pronounced for the CCDC84 gene, thereby increasing the amount of the full length protein of as yet uncharacterized function.
Unexpectedly, exclusively in the TALS LCL data set (Supplemental Fig. S9), we found reads for all spliceosomal snRNAs at the exception of U6 and U6atac, an observation also made in a previous analysis of RFMN data sets (Dinur Schejter et al. 2017). This was unexpected because snRNAs belong to the nonpolyadenylated class of RNAs, yet we performed RNA-seq experiments on poly(A)+ RNAs. We postulate that the accumulation of polyadenylated snRNA precursors may have resulted from a deficient Integrator complex, which plays a pivotal role in the 3′-end processing of the snRNAs transcribed by RNA Polymerase II, that is, all snRNAs apart from U6 and U6atac (for review, see Guiro and Murphy 2017). Integrator contains at least 14 subunits, of which four are encoded by U12 genes, namely INTS4, INTS7, INTS8, and INTS10, markedly differentially misspliced in TALS LCL (ΔPSIINTS4 = 11.4%; ΔPSIINTS7 = 28.6%; ΔPSIINTS8 = 16.5%; ΔPSIINTS10 = 34.1%). In contrast, the U12-type introns of the three U12 Integrator genes expressed in the TALS fibroblast data sets had a very low ΔPSI value (ΔPSIINTS7 = 2.5%; ΔPSIINTS8 = 1.6%; ΔPSIINTS10 = 6.3%). Interestingly, mutations in INTS1 and INTS8 are associated with impaired RNA splicing in rare recessive neurodevelopmental syndromes with developmental delay and distinctive appearance (Oegema et al. 2017). However, the absence of massive U2-type intron splicing defects in LCL attests that despite this maturation default, the amount of functional snRNAs of the major spliceosome is sufficient for efficient U2-type intron splicing and that U12 Integrator genes missplicing is unlikely to be the primary cause of the high magnitude of U12-type intron missplicing in this LCL sample.
We observed that the level of IR is quite variable among U12-type introns, even in TALS fibroblasts and amniocytes where most introns are retained in only a marginal fraction of transcripts. To try to understand why some U12-type introns are more sensitive to a defective spliceosome than others, we considered a number of intron features previously shown to influence IR in mammals, for example, donor/acceptor splice site scores, GC content, intron length (Braunschweig et al. 2014), and correlated them with the level of U12-type intron missplicing using a linear model (Supplemental Table S6). Among the many features tested, only two were found to significantly correlate with PSI values in patients. The first one is the PSI value in controls (50% of the variance), which means that introns poorly spliced in controls are even more poorly spliced in patients. The second one is the gene expression level (10% of the variance), which means that poorly expressed genes are more subject to missplicing than the more expressed ones, as had been previously reported for U2-type introns (Saudemont et al. 2017). We also searched for enriched motifs such as splicing enhancers that might bind a splicing factor (Dietrich et al. 2001b) for explaining high PSI values but we were unable to identify such sequences, leaving open the question of the remaining features causing U12-type intron “ultra sensitivity” to a defective spliceosome for some of them.
Transcriptome analyses show much promise in elucidating the pathogenesis of genetic diseases, even more in those due to a splicing defect. However, it is well known that expression programs for genes involved in development are highly time- and tissue-specific, and even cell-specific in the early stages of embryogenesis. The understanding of the molecular mechanisms involved in the pathogenesis of TALS will require additional transcriptomic analyses to be performed on different cell types at various developmental or differentiation stages, hence necessitating to generate induced pluripotent stem cells and/or develop animal models. Nevertheless, the present finding that TALS and RFMN blood cells share a similar pattern of U12-type IR and that the GO term analysis performed on the TALS fibroblast and amniocyte data sets showed an enrichment in immunity-linked terms suggest that thorough investigation of TALS immune phenotype should be carried out.
MATERIALS AND METHODS
Identification of U12-type introns in the human genome
U12DB, the U12 Intron Database (http://genome.crg.es/cgi-bin/u12db/u12db.cgi) released in 2006 by T. Alioto with the aim to catalog U12-type introns of completely sequenced eukaryotic genomes (Alioto 2007), has not been updated since its launching. We updated the list of human U12-type introns using T. Alioto's scoring matrices on a more recent genome annotation [Gencode v19 (GRCh37)/Ensembl v75], the latest one at the time of the analysis of the pilot project data sets. Out of 289,023 introns annotated in Gencode v19, the pipeline classifies 846 of them as U12-type introns (Supplemental Table S3). Those are located in 699 genes, of whom 105, 20, 3, and 1, respectively contained 2, 3, 4, and 5 U12-type introns. When more than one U12-type intron is present in a gene, in most cases (85/129), the coordinates of at least two of these U12-type introns overlap, indicating that the same U12-type intron can be spliced out using alternative U12 consensus splice sites.
Biological samples
Biospecimens were obtained from seven unrelated TALS cases, four children (three RNU4ATAC homozygotes and one RNU4ATAC compound heterozygote) and three fetuses (one RNU4ATAC homozygote and two RNU4ATAC compound heterozygotes), and deposited to the Lyon University Hospital Biobank dedicated to genetic diseases for processing, storage and management (CBC Biotec of the Hospices Civils de Lyon, certified with a specific French standard for biobanks, NF S96-900). These biospecimens consisted of skin biopsies and amniotic fluid from which primary fibroblasts and amniocytes were respectively derived, and peripheral blood from which lymphoblastoid cell lines (LCL) were established, following standard procedures. For two children, two different types of samples were obtained: peripheral blood and skin biopsy for one, amniotic fluid during gestation and skin biopsy after birth for the other. Adequate biological samples from age- and sex-matched controls were provided by the CBC Biotec biobank. Informed written consent for the use of these samples in research was obtained from all parents of TALS patients, TALS fetuses and control fetuses and children. The detailed characteristics of the analyzed samples, including the information on whether they derived from post-mortem material, are described in Table 1.
RNA extraction
RNA extractions were performed using the Nucleospin RNA kit (Macherey Nagel) according to the manufacturer's recommendations. A further round of DNase (Promega) treatment was systematically performed to remove any possible residual amount of DNA. Total RNA concentration was then quantified with a NanoDrop spectrophotometer (Nanodrop Technologies) and RNA quality assessed using the Agilent 2100 Bioanalyzer (Agilent). RNA integrity number (RIN) was >7 in all cases.
cDNA library preparation, high-throughput sequencing
One to two micrograms of RNA were sent for RNA-sequencing to IntegraGen Genomics (Evry, France), where a DNA library was generated with the “TruSeq Stranded mRNA Sample Prep” kit (Illumina) that comprises a step of mRNA purification using oligo(dT) beads. A total of 28 RNA-seq experiments have been performed at two different times: (i) A pilot study was performed on a HiSeq 2000 sequencer (Illumina), yielding approximately 716 million of nonstranded two time 100 bp paired-end reads, with librairies obtained with RNA extracted from skin fibroblasts taken on two TALS children homozygous for g.51G > A and from the LCL derived from one of these children, and from their matched controls (six RNA-seq experiments, see Table 1). The reads thus obtained were analyzed as described in the following paragraph: it showed that the extent of IR being low, additional samples needed to be analyzed in order to obtain reliable results. (ii) An extended study was later performed on a HiSeq 4000 sequencer (Illumina), yielding approximately 2670 million of stranded two time 75 bp paired-end reads, with librairies obtained with RNA extracted from all the samples, including those already sequenced in the pilot study in order to have technical replicates for some of them (22 RNA-seq experiments, see Table 1). Sequencing metrics are given for each sample in Supplemental Table S7. Raw RNA-seq data are available upon request.
qRT-PCR
cDNA synthesis was carried out with 1 µg DNA-free RNA (the same batches as those used for RNA-seq) using GoScript Reverse Transcription System and oligodT primers (Promega) according to the manufacturer's protocol. qRT-PCR was performed using the Rotor-Gene SYBR Green PCR kit and Rotor-Gene Q (Qiagen) according to the manufacturer's protocol. All experiments were done in three replicates.
Bioinformatics analysis of RNA-seq data
Splicing analyses
Our three bioinformatics pipelines, shown in Supplemental Figure S10, are composed of multiple steps executed by various tools to achieve three goals: (i) read alignment/assembly; (ii) read quantification; (iii) alternative splicing event quantification and statistical analysis, with a special focus on IR.
IR identification and quantification in RNA-seq data is a difficult bioinformatics task for multiple reasons (discussed in detail in Vanichkina et al. 2018). To date, only four dedicated tools are available: vast-tools (Tapial et al. 2017) (which can also detect other types of alternative splicing events); IRcall/IRclassifier (Bai et al. 2015); intEREst (Oghabian et al. 2018), and IRFinder (Middleton et al. 2017). Their main difference lies in the intronic read quantification method: Vast-tools outputs the number of exon-intron junctions reads, IRcall/IRclassifier the number of reads aligned to the full intron, intEREst and IRFinder the read coverage of specific intronic regions that do not correspond to low complexity regions or alternative exons, hence improving precision. Furthermore, IRFinder reduces the impact of heterogeneous coverage by discarding 60% of the intronic regions’ bases containing the highest and lowest covered bases, and it also outputs the number of exon–intron junctions reads. We thus chose to use IRFinder, being the most precise tool.
IR detection and quantification method (IRFinder v1.2.0, mapping-first)
RNA-seq read quality control was performed using FastQC v0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped with STAR v2.5.0b (Dobin et al. 2013) using IRFinder's custom STAR index of the latest annotation of the GRCh37 assembly (Ensembl v75) and default parameters.
Splicing-supporting reads are reads aligning to an exon-exon junction. An overhang (minimal number of bases around a junction covered by an aligned read) of five was required to consider that any read is aligned to a junction. Hence, the number of unique positions on a junction where a 75 bp read (in the case of our study) can be aligned (referred to as “effective size”) is:
Any read fully covering one of these 66 positions will be counted as a splicing-supporting read.
We then used two strategies to select splicing- and retention-supporting reads depending on the number of an intron's informative bases.
Retention-supporting reads can either be reads aligning to the intron body or reads aligning to one of the two exon-intron junctions. In general, IRFinder will use both intron body reads and exon-intron reads. In the cases where the number of informative bases is too low (≤40 bases or <70% of the total number of bases of the intron, 31% of U2- and U12-type introns), for example, most of the intron length is covered by repeats or annotated features, IRFinder is conservative and reasonably chooses not to compute the intronic read coverage. However, it also does not compute exon–intron junction reads coverage. We argue that this latter quantification could still be of interest, as although it does not indicate the full intronic coverage, it still testifies that this peculiar intron's splice sites were not used for splicing. In other words, exon–intron junctions quantification does not indicate the type of the alternative splicing event (it could either be an IR or a alternative donor + acceptor), but still indicates the amount of unspliced intron. In order to force IRFinder to do the exon–intron quantification for all introns, we rewrote a specific test in its code (intronExclusion.pl, line 83: if ($newlen > 40 && ($newlen/$len) ≥ 0.7) { replaced by if ($newlen > 0) }). For the special case of U12-type introns with no informative base (167 cases), IRFinder 1.2.0 could not be run and we had to develop a custom python script (junctionsCover2IRF.py) to do the quantification. For a given list of introns, a read length and BAM files, this script uses samtools view to quantify the number of aligned reads on the exon–exon junction and the two exon–intron junctions and creates a file formatted in the same way as a conventional IRFinder file, allowing us to merge them together (Supplemental Fig. S10). In the following analyses, the retention-supporting reads will either be reads aligned to the intron body, or reads aligned to the exon–intron junctions if the number of informative bases of the intron was smaller than the effective length of the exon–intron junction (66 nt).
The list of introns we analyze corresponds to constitutively spliced introns, but also to alternatively spliced introns, some of which are spliced out only in specific tissues. Out of all introns analyzed, some are never seen spliced out in our data sets. We chose not to consider them as introns as they would otherwise artificially increase IR rates. In practice, we did not analyze IR for all introns with less than five splicing-supported reads on average in the control samples and for each cell-type. Among the IR sufficiently covered (see Filters for PSI and ΔPSI computation below), this filters out 4687, 5468, 4469, and 3239 introns (among which 12, 13, 18, and 7 were U12-type introns) in the primary fibroblast, amniocyte, LCL, and MBC data sets, respectively.
Alternative splicing events detection with annotation (vast-tools v2.0.0, mapping-first)
In addition to IR, vast-tools (Tapial et al. 2017) can also detect three other types of alternative splicing events: alternative donor, alternative acceptor and exon skipping. Vast-tools results concerning IR are not presented because >99% of the differential U12-type IR detected by vast-tools were also found by IRFinder, but only 35% or less of the differential U12-type IR detected by IRFinder were also found by vast-tools. All results are however available in the supporting shiny interface (http://lbbe-shiny.univ-lyon1.fr/TALS-RNAseq/). Briefly, vast-tools aligns the reads from each sample on different references (genome, exon–exon junction, …) using BOWTIE (v1.1.2.), and then analyzes the alignment file to quantify the number of reads supporting the inclusion or exclusion of an exon for each of its 213,087 possible alternative splicing events annotated in its database.
Some introns from the vast-tools’ splicing event database were not annotated in Ensembl v75. In order to determine their type, we ran T. Alioto's scripts on these introns if both of their splice sites were annotated in Ensembl v75. This resulted in 56 new U12-type introns (of which 55 overlapped with a known U12-type intron but used a different acceptor site, Supplemental Table S3).
Because this method cannot detect alternative splicing events which are absent from its database, we also used an assembly-first and annotation-free method for alternative splicing events detection.
Alternative splicing events detection without annotation (KisSplice v2.4.0-p1, assembly-first)
Briefly, KisSplice (Sacomoto et al. 2012) assembles the reads in a de Bruijn graph and searches for so-called bubbles in this graph, which correspond to alternative splicing events. The two paths of the bubble are then mapped to a reference genome using STARlong v2.5.0b, and the resulting alignments are processed by KisSplice2RefGenome to annotate the event, by assigning it notably a gene name and an AS subtype. We recently showed (Benoit-Pilven et al. 2018a) that this assembly-first approach was particularly adapted to identify novel splice sites. This advantage comes at the expense of poorer performance for long and unfrequent variants, because de novo assembly requires more coverage. This is the reason why we do not use KisSplice to analyze IR.
Counts normalization, PSI/ΔPSI computation, and differential analysis (kissDE)
IRFinder, vast-tools and KisSplice all output the number of reads supporting the inclusion (i.e., a retained intron or exon) or exclusion (i.e., a spliced intron or skipped exon) transcript in each sample and for each IR/alternative splicing event. The bioconductor R package kissDE (https://www.bioconductor.org/packages/devel/bioc/html/kissDE.html) (Benoit-Pilven et al. 2018b) was then used for counts normalization, splicing event strength estimation (PSI and ΔPSI) and differential analysis between two conditions (FDR).
Briefly, kissDE starts by normalizing the read counts for the library sizes using DESeq2, and by normalizing the inclusion-supporting reads by the length of the inclusion (that can be very large for IR events) compared to the exclusion. Then, for each splicing event, kissDE computes a PSI for each sample. In the context of this study, where several replicates are available for the patients and controls, a mean PSI is calculated for each condition, and corresponds to the patients/controls PSI used throughout this article. In the Results section, we also used the mean ΔPSI of all U12- or U2-type introns in a data set. Finally, a differential analysis is run that models counts with either a Poisson (for technical replicates) or a negative binomial (for biological replicates) distribution, and uses the generalized linear model framework to model the expected signal intensity. A likelihood ratio test is used to estimate the probability of an interaction between the splice-forms (inclusion and exclusion) and the condition. The Benjamini–Hochberg procedure is used to account for multiple testing and compute FDR values.
We considered an alternative splicing event statistically significant if its FDR ≤ 5%, and markedly significant if, in addition, its |ΔPSI| ≥ 10%.
Local expression value
The local expression (locExp) value, calculated for each intron, is the number of reads attesting to either the inclusion or exclusion of an intron, and is defined as:
with excReads the number of reads on the exon–exon junction and incReads the number of reads on both exon–intron junctions. A star indicates library-sized normalized counts.
The main advantage of using the local expression value is that there is no need to infer full-length transcripts and their abundance, a notoriously difficult and error-prone task (Steijger et al. 2013), to derive an estimation of transcripts expression. It also has the advantage of directly focusing on transcripts which contain the exons flanking the intron of interest. In contrast, a measure of gene expression based on counting all reads falling within the gene boundaries will also include reads stemming from transcripts which do not overlap the intron of interest, for instance in the case of alternative transcription start/end. It will also be confronted with the difficult task of correctly estimating gene length, in the presence of multiple alternative transcripts.
Filters for PSI and ΔPSI computation
To compute robust metrics, we apply a coverage threshold on the local expression of an alternative splicing event. In a sample, both the local expression and the normalized local expression values must be ≥10 to compute the PSI value of an intron. At least half of the patients and half of the controls must have a computed PSI in order to have a ΔPSI estimation.
Differential gene expression analysis method (DESeq2)
We tested if genes were differentially expressed between our two conditions with the DESeq2 conventional pipeline (Love et al. 2014) HTSeq tool to generate gene expression values (Anders et al. 2015).
Principal component analyses (PCA)
We used the dudi.pca function from the R package ade4 v1.7-11 (https://github.com/sdray/ade4) (Bougeard and Dray 2018) on either a table of TPM or PSI. For each PCA, the most variable values (up to 500) were used (as conventionally done in DESeq2) and the first (PC1) and second (PC2) most explanatory axes were plotted. We compared the percentage of the variance explained by each axis of these PCA (PCAvar) to the mean of the ones obtained after randomizing independently each row of the TPM or PSI table 100 times (randomVar). Axes with explained variance smaller or equal to our randomized data (PCAvar ≤ randomVar) are denoted with ns (not significant) and should not be interpreted.
Intron retention validations
IR validations were carried out with RNA extracted from fibroblast cell lines derived from patients TALS2, TALS4, TALS6 (all g.51G > A homozygous), TALS10 (g.50G > C;g.51G > A) and TALS3 (g.40C > T;g.124G > A) and from five control children or fetuses matched for age and gender. We tested introns with various extent of IR (i.e., mean ΔPSI) from eight U12 genes (CLCN7: 6.5%, GPAA1: 11.4%, TMEM107: 13.7%, TMEM87A: 23.7%, ZCCHC8: 25.1%, ENTHD2: 25.2%, HECTD2: 26.8%, RABL2A: 27.2%), one U2 gene reclassified as U12 in this study (U12*, RECQL5: 37.1%) and one control U2 gene that did not display IR (AARS: 0%). The ACTB gene (encoding β actin) was chosen as the endogenous control. To be able to compare IR measured by qRT-PCR to that measured by RNA-seq, we computed the mean ΔPSI for each gene from qRT-PCR experiments as follows:
with Ct the number of qRT-PCR cycle needed for the fluorescence to cross a given threshold (125), * denoting the mean Ct (from the three technical replicates) of the endogenous control (ACTB), i the technical replicate, t the type of transcript quantified (either r or s for transcript retaining or splicing the intron), C the experimental conditions (either Ctrl or Patient) and Rq the relative quantification of the DNA with respect to the endogenous control. The RNA-seq mean ΔPSI was computed for each gene by subtracting the PSI of each matched control/patient pair, and calculating the mean.
GO terms enrichment analysis
We searched for GO terms enriched in our set of genes with U12-type differentially spliced introns to highlight potential biological processes specifically disrupted in patients and thus, possibly related to the phenotype. We used the topGO (Alexa and Rahnenfuhrer 2016) (v2.30.1) R package using the genes for which a U12-type intron alternative splicing event had been tested and a user provided quantitative score for each gene. We performed two different analyses using distinct scores. The first one defined the score based on the FDR of a gene's U12-type intron (minimum FDR in the special case where a gene harbors multiple U12-type introns or multiple splicing events for the same U12-type intron), which correspond to the classical use of TopGO. Genes gain more weight as their FDR value is close to 0. This should detect GO terms enriched in genes with the most reproducible U12-type intron alternative splicing events compared to unaffected genes. The second analysis defined the score based on the |ΔPSI| of a gene's U12-type intron: The score is either 0 for genes without any significant U12-type intron alternative splicing event or the |ΔPSI| (maximum |ΔPSI| in the special case described above). Genes gain more weight as their |ΔPSI| is close to 1. This should detect GO terms enriched in genes with the highest differences of missplicing between patients and controls compared to unaffected genes. For each analysis, we used the Kolmogorov–Smirnov test to account for the weights and we reported a GO term as enriched if its P-value was ≤5% for either of our two analyses.
In each analysis, the default “weight01” algorithm was used. The following describes the parameters used to create the topGOdata object in R: We searched for biological process (ontology = “BP”) in the Gene Ontology DataBase version from October 2018 (mapping = “org.Hs.eg.db”, annot = annFUN.org) using Ensembl ID (ID = “ensembl”). The enriched GO terms were mapped to a subset of more generic GO terms (GO slim) using the GSEABase R package v1.44.0 and the GO slim AGR subset (go_slim.agr) downloaded on the GeneOntology website (http://geneontology.org/docs/go-subset-guide/).
Features influencing U12-type IR
In order to identify features that could have an impact on the level of U12-type IR, we used a linear model. We worked on all analyzed introns (using filters described in Materials and Methods) in the fibroblasts data set. We wanted to explain the U12-type introns’ mean PSI (mPSI) of the patients with a set of 32 explicative variables (hereafter referred to as predictors), see Supplemental Table S4. We used a log transformation of mPSI (Supplemental Fig. S11A), since diagnostic plots (Supplemental Fig. S11B–E) show that the assumptions of linear regression are much better satisfied with this transformation. Zero values were replaced by the minimum nonzero mPSI divided by two to guarantee that the transformed value for zero is still lower than all other values. For 15/32 predictors, we needed to define a major transcript for each intron. In the case of multiple transcripts, we chose the CCDS, as annotated in APPRIS (Rodriguez et al. 2013). In the case of multiple CCDS, we chose the longest ORF. In case of ties, we chose the longest transcript. We first performed a simple linear regression to test each predictor in an independent way [model = log(mPSI) ∼ predictor] using R version 3.5.1 and anova[lm(model)], for the fitting and the variance analysis. P-values and R-squared (R2) values (indicating the percentage of variance explained) for each predictor are both reported in the Supplemental Table S4. Then, we ordered the significant (P-value ≤ 5%) predictors by decreasing R2 value (predictor1, predictor2, …, predictorN). From the initial model m0 = log(mPSI) ∼ predictor1, we created the multiple linear regression model m∞ = m0 + predictor2. We then compared these two nested models, using a likelihood ratio test [anova(lm(m0), lm(m∞))], to decide whether the additional predictor could be considered as significantly associated to IR. If the P-value was ≤5% and R2 ≥ 1%, we set m0 = m∞, else we kept the same unchanged m0. We did this up to predictorN to build the complete model. Each R2 value is computed by dividing the sum-of-square of each predictor by the sum of the sum-of-square of all predictors. The same analysis was run to explain the U12-type introns’ mean PSI of the controls.
Motif sequences analysis
In order to identify motifs enriched in differentially retained U12-type introns compared to other analyzed U12-type introns, we used the MEME Suite 5.0.1. software (Machanick and Bailey 2011; Bailey et al. 2015). Tested U12-type introns were separated into two groups: candidates (n = 49), for which a strong differential IR was detected in Patients (FDR ≤ 5% and ΔPSI ≥ 10%), and unchanged (n = 45), for which no differential IR was detected in Patients (FDR ≥ 20% and |ΔPSI| < 1%). In the case of overlapping U12-type introns, the largest one was conserved. Sequences of each intron, with 100 bp upstream and downstream the intron, were retrieved with bedtools’ fastaFromBed (v2.25.0). Sequences of introns on the minus strand were reverse complemented. In order to have groups with comparable intron length, we calculated the minimum length ratio of each sequence from the candidates group with each sequence from the unchanged group (minRatioLength) and we selected the sequences with a minRatioLength ≥ 0.95. This step resulted in 34 selected sequences in the candidates group and 39 sequences selected in the unchanged group. With MEME, we searched for ungapped motifs of length 8 to 50. The OOPS (One Occurrence Per Sequence), ZOOPS (Zero or One Occurrence Per Sequence) and ANR (Any Number of Repetitions) mode of MEME were used (-mod oops|zoops|anr) with the “differential enrichment” objective function (-objfun de) to detect motifs significantly enriched either in the candidates or in the unchanged sequences (E-value ≤ 5%, -evt 0.05). All other parameters were set to default values. With DREME, we searched for small (up to 8 nt) ungapped motifs differentially enriched in either the candidates or unchanged sequences. The -norc option was used; other parameters were set to default value.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by CNRS, Inserm, Université Paris 7 and Université Lyon 1 through recurrent funding, the ANR Aster (no. ANR-16-CE23-0001) and U4ATAC-BRAIN (no. ANR-18-CE12-0007-01) grants and an Inserm/Hospices Civils de Lyon grant to P.E. (Contrat d'Interface pour Hospitaliers). A.C. was supported by a grant from Inria (Thése Inria-Inserm “Médecine Numérique” - 2016) and C.B.P. by a grant from the Fondation pour la Recherche Médicale to P.E. (Financement d‘un ingénieur - ING20160435660). We thank the families for their participation in this study, the CBC Biotec biobank for sample management (Emilie Chopin, Isabelle Rouvet), the students for their contribution (Clément Saccaro, Gabriel Sala, Nabil Sersoub), Integragen SA for performing the RNA-seq experiments, Daniele Merico for critical reading of the manuscript and helpful suggestions, Tyler Alioto for providing us with the scripts for predicting U12 introns and Cyril Bourgeois, Vincent Navratil and Marion Delous for stimulating discussions.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071423.119.
Freely available online through the RNA Open Access option.
REFERENCES
- Alexa A, Rahnenfuhrer J. 2016. topGO: enrichment analysis for gene ontology. R package version 2320.
- Alioto TS. 2007. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 35: D110–D115. 10.1093/nar/gkl796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argente J, Flores R, Gutiérrez-Arumí A, Verma B, Martos-Moreno GÁ, Cuscó I, Oghabian A, Chowen JA, Frilander MJ, Pérez-Jurado LA. 2014. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol Med 6: 299–306. 10.1002/emmm.201303573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Ji S, Wang Y. 2015. IRcall and IRclassifier: two methods for flexible detection of intron retention events from RNA-seq data. BMC Genomics 16(Suppl 2): S9 10.1186/1471-2164-16-S2-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS. 2015. The MEME suite. Nucleic Acids Res 43: W39–W49. 10.1093/nar/gkv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Pilven C, Marchet C, Chautard E, Lima L, Lambert M-P, Sacomoto G, Rey A, Cologne A, Terrone S, Dulaurier L, et al. 2018a. Complementarity of assembly-first and mapping-first approaches for alternative splicing annotation and differential analysis from RNAseq data. Sci Rep 8: 4307 10.1038/s41598-018-21770-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Pilven C, Marchet C, Kielbassa J, Brinza L, Cologne A, Siberchicot A, Lacroix V. 2018b. kissDE: retrieves condition-specific variants in RNA-seq data. R package version 113.
- Bogaert DJ, Dullaers M, Kuehn HS, Leroy BP, Niemela JE, De Wilde H, De Schryver S, De Bruyne M, Coppieters F, Lambrecht BN, et al. 2017. Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Sci Rep 7: 3702 10.1038/s41598-017-02434-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougeard S, Dray S. 2018. Supervised multiblock analysis in R with the ade4 package. J Stat Softw 86: 1–17. 10.18637/jss.v086.i01 [DOI] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CB, Padgett RA, Sharp PA. 1998. Evolutionary fates and origins of U12-type introns. Mol Cell 2: 773–785. 10.1016/S1097-2765(00)80292-0 [DOI] [PubMed] [Google Scholar]
- Chang W-C, Chen Y-C, Lee K-M, Tarn W-Y. 2007. Alternative splicing and bioinformatic analysis of human U12-type introns. Nucleic Acids Res 35: 1833–1841. 10.1093/nar/gkm026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich RC, Peris MJ, Seyboldt AS, Padgett RA. 2001a. Role of the 3′ splice site in U12-dependent intron splicing. Mol Cell Biol 21: 1942–1952. 10.1128/MCB.21.6.1942-1952.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich RC, Shukla GC, Fuller JD, Padgett RA. 2001b. Alternative splicing of U12-dependent introns in vivo responds to purine-rich enhancers. RNA 7: 1378–1388. [PMC free article] [PubMed] [Google Scholar]
- Dietrich RC, Fuller JD, Padgett RA. 2005. A mutational analysis of U12-dependent splice site dinucleotides. RNA 11: 1430–1440. 10.1261/rna.7206305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinur Schejter Y, Schejter YD, Ovadia A, Alexandrova R, Thiruvahindrapuram B, Pereira SL, Manson DE, Vincent A, Merico D, Roifman CM. 2017. A homozygous mutation in the stem II domain of RNU4ATAC causes typical Roifman syndrome. NPJ Genom Med 2: 23 10.1038/s41525-017-0024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery P, Marcaillou C, Sahbatou M, Labalme A, Chastang J, Touraine R, Tubacher E, Senni F, Bober MB, Nampoothiri S, et al. 2011. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 332: 240–243. 10.1126/science.1202205 [DOI] [PubMed] [Google Scholar]
- Elsaid MF, Chalhoub N, Ben-Omran T, Kumar P, Kamel H, Ibrahim K, Mohamoud Y, Al-Dous E, Al-Azwani I, Malek JA, et al. 2017. Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Ann Neurol 81: 68–78. 10.1002/ana.24826 [DOI] [PubMed] [Google Scholar]
- Farach LS, Little ME, Duker AL, Logan CV, Jackson A, Hecht JT, Bober M. 2018. The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. Am J Med Genet A 176: 465–469. 10.1002/ajmg.a.38581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell S, Johnson A, Pearson W. 2016. Microcephalic osteodysplastic primordial dwarfism type 1. BMJ Case Rep 2016: Jun 16 10.1136/bcr-2016-215502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault CM, Martin F, Mei W, Bai F, Black JB, Barbazuk WB, Settles AM. 2017. Aberrant splicing in maize rough endosperm3 reveals a conserved role for U12 splicing in eukaryotic multicellular development. Proc Natl Acad Sci 114: E2195–E2204. 10.1073/pnas.1616173114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiro J, Murphy S. 2017. Regulation of expression of human RNA polymerase II-transcribed snRNA genes. Open Biol 7: 170073 10.1098/rsob.170073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafez M, Hausner G. 2015. Convergent evolution of twintron-like configurations: one is never enough. RNA Biol 12: 1275–1288. 10.1080/15476286.2015.1103427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermayr A, Graf J, Koehler U, Laner A, Schönfeld B, Benet-Pagès A, Holinski-Feder E. 2018. Extending the critical regions for mutations in the non-coding gene RNU4ATAC in another patient with Roifman Syndrome. Clin Case Rep 6: 2224–2228. 10.1002/ccr3.1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Liyanarachchi S, Akagi K, Nagy R, Li J, Dietrich RC, Li W, Sebastian N, Wen B, Xin B, et al. 2011. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 332: 238–240. 10.1126/science.1200587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA. 2011. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol 7: 497 10.1038/msb.2011.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heremans J, Garcia-Perez JE, Turro E, Schlenner SM, Casteels I, Collin R, de Zegher F, Greene D, Humblet-Baron S, Lesage S, et al. 2018. Abnormal differentiation of B cells and megakaryocytes in patients with Roifman syndrome. J Allergy Clin Immunol 142: 630–646. 10.1016/j.jaci.2017.11.061 [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Perez-Cerezales S, Papasaikas P, Ramos-Ibeas P, López-Cardona AP, Laguna-Barraza R, Fonseca Balvís N, Pericuesta E, Fernández-González R, Planells B, et al. 2018. Impaired spermatogenesis, muscle, and erythrocyte function in U12 intron splicing-defective Zrsr1 mutant mice. Cell Rep 23: 143–155. 10.1016/j.celrep.2018.03.028 [DOI] [PubMed] [Google Scholar]
- Jackson IJ. 1991. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res 19: 3795–3798. 10.1093/nar/19.14.3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A, Durbin R. 2001. A computational scan for U12-dependent introns in the human genome sequence. Nucleic Acids Res 29: 4006–4013. 10.1093/nar/29.19.4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowska D, Simpson CG, Clark GP, Jennings NS, Barciszewska-Pacak M, Lin C-F, Makalowski W, Brown JWS, Jarmolowski A. 2004. Determinants of plant U12-dependent intron splicing efficiency. Plant Cell 16: 1340–1352. 10.1105/tpc.020743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, Thiruvahindrapuram B, Merico D, Jobling R, Nalpathamkalam T, et al. 2018. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 20: 435–443. 10.1038/gim.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Ramos CM, Paulson JN, Chen C-Y, Kuijjer ML, Fagny M, Platig J, Sonawane AR, DeMeo DL, Quackenbush J, Glass K. 2017. Regulatory network changes between cell lines and their tissues of origin. BMC Genomics 18: 723 10.1186/s12864-017-4111-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042 10.1038/ncomms7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Cloonan N, Lardelli RM, Doggett K, Keightley M-C, Boglev Y, Trotter AJ, Ng AY, Wilkins SJ, Verkade H, et al. 2014. Minor class splicing shapes the zebrafish transcriptome during development. Proc Natl Acad Sci 111: 3062–3067. 10.1073/pnas.1305536111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, He L, Somel M, Yuan Y, Phoebe Chen Y-P, et al. 2013. Widespread splicing changes in human brain development and aging. Mol Syst Biol 9: 633 10.1038/msb.2012.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Roifman M, Braunschweig U, Yuen RKC, Alexandrova R, Bates A, Reid B, Nalpathamkalam T, Wang Z, Thiruvahindrapuram B, et al. 2015. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat Commun 6: 8718 10.1038/ncomms9718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ-L, Bomane A, Cosson B, Eyras E, Rasko JEJ, et al. 2017. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18: 51 10.1186/s13059-017-1184-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela EH, Oghabian A, Staals RHJ, Greco D, Pruijn GJM, Frilander MJ. 2014. Global analysis of the nuclear processing of transcripts with unspliced U12-type introns by the exosome. Nucleic Acids Res 42: 7358–7369. 10.1093/nar/gku391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema R, Baillat D, Schot R, van Unen LM, Brooks A, Kia SK, Hoogeboom AJM, Xia Z, Li W, Cesaroni M, et al. 2017. Correction: human mutations in integrator complex subunits link transcriptome integrity to brain development. PLoS Genet 13: e1006923 10.1371/journal.pgen.1006923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oghabian A, Greco D, Frilander MJ. 2018. IntEREst: intron-exon retention estimator. BMC Bioinformatics 19: 130 10.1186/s12859-018-2122-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA. 2012. New connections between splicing and human disease. Trends Genet 28: 147–154. 10.1016/j.tig.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoux A, Alqahtani A, Pinson L, Paulussen ADC, Michel J, Besson A, Mazoyer S, Borg I, Nampoothiri S, Vasiljevic A, et al. 2016. Refining the phenotypical and mutational spectrum of Taybi-Linder syndrome. Clin Genet 90: 550–555. 10.1111/cge.12781 [DOI] [PubMed] [Google Scholar]
- Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink J-J, Lopez G, Valencia A, Tress ML. 2013. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res 41: D110–D117. 10.1093/nar/gks1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacomoto GAT, Kielbassa J, Chikhi R, Uricaru R, Antoniou P, Sagot M-F, Peterlongo P, Lacroix V. 2012. KISSPLICE: de-novo calling alternative splicing events from RNA-seq data. BMC Bioinformatics 13(Suppl 6): S5 10.1186/1471-2105-13-S6-S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudemont B, Popa A, Parmley JL, Rocher V, Blugeon C, Necsulea A, Meyer E, Duret L. 2017. The fitness cost of mis-splicing is the main determinant of alternative splicing patterns. Genome Biol 18: 208 10.1186/s13059-017-1344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scamborova P, Wong A, Steitz JA. 2004. An intronic enhancer regulates splicing of the twintron of Drosophila melanogaster prospero pre-mRNA by two different spliceosomes. Mol Cell Biol 24: 1855–1869. 10.1128/MCB.24.5.1855-1869.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Will CL, Makarova OV, Makarov EM, Lührmann R. 2002. Human U4/U6.U5 and U4atac/U6atac.U5 tri-snRNPs exhibit similar protein compositions. Mol Cell Biol 22: 3219–3229. 10.1128/MCB.22.10.3219-3229.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch NJ, Schofield P, Gierliński M, Cole C, Sherstnev A, Singh V, Wrobel N, Gharbi K, Simpson GG, Owen-Hughes T, et al. 2016. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 22: 839–851. 10.1261/rna.053959.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, Swanson MS. 2016. RNA mis-splicing in disease. Nat Rev Genet 17: 19–32. 10.1038/nrg.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Maddirevula S, Ewida N, Alsahli S, Abdel-Salam GMH, Zaki MS, Tala SA, Alhashem A, Softah A, Al-Owain M, et al. 2019. Genomic and phenotypic delineation of congenital microcephaly. Genet Med 21: 545–552. 10.1038/s41436-018-0140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelihan I, Ehresmann S, Magnani C, Forzano F, Baldo C, Brunetti-Pierri N, Campeau PM. 2018. Lowry-Wood syndrome: further evidence of association with RNU4ATAC, and correlation between genotype and phenotype. Hum Genet 137: 905–909. 10.1007/s00439-018-1950-8 [DOI] [PubMed] [Google Scholar]
- Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. 2006. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res 34: 3955–3967. 10.1093/nar/gkl556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engström PG, Kokocinski F, RGASP Consortium, Hubbard TJ, Guigó R, Harrow J, Bertone P. 2013. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10: 1177–1184. 10.1038/nmeth.2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapial J, Ha KCH, Sterne-Weiler T, Gohr A, Braunschweig U, Hermoso-Pulido A, Quesnel-Vallières M, Permanyer J, Sodaei R, Marquez Y, et al. 2017. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res 27: 1759–1768. 10.1101/gr.220962.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. 1996. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 273: 1824–1832. 10.1126/science.273.5283.1824 [DOI] [PubMed] [Google Scholar]
- Turunen JJ, Will CL, Grote M, Lührmann R, Frilander MJ. 2008. The U11-48K protein contacts the 5′ splice site of U12-type introns and the U11-59K protein. Mol Cell Biol 28: 3548–3560. 10.1128/MCB.01928-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Niemelä EH, Verma B, Frilander MJ. 2013. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 4: 61–76. 10.1002/wrna.1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanichkina DP, Schmitz U, Wong JJ-L, Rasko JEJ. 2018. Challenges in defining the role of intron retention in normal biology and disease. Semin Cell Dev Biol 75: 40–49. 10.1016/j.semcdb.2017.07.030 [DOI] [PubMed] [Google Scholar]
- Verma B, Akinyi MV, Norppa AJ, Frilander MJ. 2018. Minor spliceosome and disease. Semin Cell Dev Biol 79: 103–112. 10.1016/j.semcdb.2017.09.036 [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu X, Du L, Zheng J, Deng S, Bi X, Chen Q, Xie H, Férec C, Cooper DN, et al. 2018. Identification of compound heterozygous variants in the noncoding RNU4ATAC gene in a Chinese family with two successive foetuses with severe microcephaly. Hum Genomics 12: 3 10.1186/s40246-018-0135-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJ-L, Ritchie W, Ebner OA, Selbach M, Wong JWH, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. 2013. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154: 583–595. 10.1016/j.cell.2013.06.052 [DOI] [PubMed] [Google Scholar]
- Wong JJ-L, Au AYM, Ritchie W, Rasko JEJ. 2016. Intron retention in mRNA: no longer nonsense: known and putative roles of intron retention in normal and disease biology. Bioessays 38: 41–49. 10.1002/bies.201500117 [DOI] [PubMed] [Google Scholar]
- Wu Q, Krainer AR. 1996. U1-mediated exon definition interactions between AT-AC and GT-AG introns. Science 274: 1005–1008. 10.1126/science.274.5289.1005 [DOI] [PubMed] [Google Scholar]
- Younis I, Dittmar K, Wang W, Foley SW, Berg MG, Hu KY, Wei Z, Wan L, Dreyfuss G. 2013. Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA. Elife 2: e00780 10.7554/eLife.00780 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.