

Abstract
Uridine insertion deletion editing in kinetoplastid protozoa requires a complex machinery, a primary component of which is the RNA editing substrate binding complex (RESC). RESC contains two modules termed GRBC (guide RNA binding complex) and REMC (RNA editing mediator complex), although how interactions between these modules and their mRNA and gRNA binding partners are controlled is not well understood. Here, we demonstrate that the ARM/HEAT repeat containing RESC protein, MRB10130, controls REMC association with mRNA- and gRNA-loaded GRBC. High-throughput sequencing analyses show that MRB10130 functions in both initiation and 3′ to 5′ progression of editing through gRNA-defined domains. Editing intermediates that accumulate upon MRB10130 depletion significantly intersect those in cells depleted of another RESC organizer, MRB7260, but are distinct from those in cells depleted of specific REMC proteins. We present a model in which MRB10130 coordinates numerous protein–protein and protein–RNA interactions during editing progression.
Keywords: Trypanosoma brucei, kinetoplastid, uridine insertion/deletion RNA editing, mitochondria, guide RNA
INTRODUCTION
A novel and extreme type of RNA editing occurs in the mitochondria of kinetoplastid flagellates, entailing specific insertion and deletion of uridylate (U) residues to correct frame shifts and create translatable open reading frames (ORFs) (Hashimi et al. 2013; Aphasizheva and Aphasizhev 2016; Read et al. 2016; Cruz-Reyes et al. 2018; Zimmer et al. 2018). U insertion/deletion (U-indel) editing is essential for survival of these organisms, which include several deadly human pathogens (Stuart et al. 2008), because fully edited mRNAs encode proteins involved in mitochondrial bioenergetics and translation. The information for this process is specified by small, trans-acting, mitochondrially encoded guide RNAs (gRNAs) that act sequentially to direct U insertions and deletions through base-pairing interactions. In Trypanosoma brucei, the causative agent of human African trypanosomiasis, the majority of edited mRNAs are pan-edited, containing modifications throughout their lengths. A few mRNAs, termed minimally edited, are modified only in smaller domains. Editing proceeds in a general 3′ to 5′ direction along an mRNA, owing to the fact that each gRNA directs the edited mRNA sequence with which the subsequent gRNA will form an anchor duplex. The editing process appears relatively inefficient as the bulk of steady-state mitochondrial mRNAs are partially edited, containing edited sequence at their 3′ ends and pre-edited sequence at their 5′ ends. Adding further complexity to the mitochondrial mRNA pool, partially edited mRNAs typically contain so-called junction regions between 3′ fully edited and 5′ pre-edited sequence (Koslowsky et al. 1991; Simpson et al. 2016, 2017; Carnes et al. 2017; Gerasimov et al. 2018). Junctions are regions of variable lengths that are edited to a sequence other than the final edited sequence. At least some junctions likely represent regions of active editing that will undergo iterative corrections until the final edited sequence is achieved and editing can progress in a 5′ direction (Simpson et al. 2017; Zimmer et al. 2018). Indeed, two components of the editing machinery were recently shown to be essential for both junction formation and 3′ to 5′ editing progression, suggesting a link between the two (Simpson et al. 2017).
The machinery that carries out U-indel editing is broadly composed of two large complexes that undergo repeated transient interactions: the RNA editing core complex (RECC; a.k.a. 20S editosome) and the RNA editing substrate binding complex (RESC; a.k.a. MRB1 complex), each with numerous variants. RECC is a stable complex containing all of the enzymatic components necessary for U insertion and deletion, namely endoribonucleases, a terminal uridylyltransferase, a U specific exoribonuclease, and RNA ligases, as well as several proteins that likely promote protein–protein and protein–RNA interactions (Aphasizheva and Aphasizhev 2016; Read et al. 2016). The three major RECC variants contain 12 common proteins, but differ with respect to their endoribonucleases, partner proteins, and the presence of the U specific exoribonuclease, and these variants have distinct, but overlapping, substrate specificities (Carnes et al. 2008, 2011, 2017). Purified RECC lacks associated RNA and is very poorly active in vitro. RESC is a more recently discovered and less well-understood complex that binds mRNA and gRNA and coordinates interactions between mRNA, gRNA, and RECC (Hashimi et al. 2013; Aphasizheva and Aphasizhev 2016; Read et al. 2016). Early proteomic and yeast two-hybrid studies identified two modules comprising RESC, the guide RNA binding complex (GRBC) and the RNA editing mediator complex (REMC), that appear to associate in a dynamic fashion mediated in part by protein–RNA interactions (Ammerman et al. 2012; Aphasizheva et al. 2014). More recent studies point to the existence of numerous variants of RESC (Kafkova et al. 2012; Madina et al. 2015; Simpson et al. 2017; McAdams et al. 2018), and the organization and functions of RESC are a subject of ongoing research.
A few RESC proteins have been studied in some detail. For example, the founding and defining component of this complex is the GAP1/2 heterotetramer, which is essential for stability of the entire gRNA population (Weng et al. 2008; Hashimi et al. 2009; Kumar et al. 2016). GAP1/2 is canonically referred to as a component of the GRBC module; however, it has now been identified in small complexes apart from fully assembled RESC that contain either REH2, TbRGG3, or MRB7260, indicating a broader function for this protein than first appreciated (Madina et al. 2014; McAdams et al. 2015, 2018). Several components of the REMC module have been studied using biochemical assays, domain interaction and protein–RNA interaction studies, and high-throughput sequencing (HTS), in one case combined with in vivo protein–RNA crosslinking. For example, TbRGG2 contains RNA binding, unwinding, and annealing activities, and is essential for the 3′ to 5′ progression of editing (Ammerman et al. 2010; Foda et al. 2012; Simpson et al. 2017; Travis et al. 2018). HTS studies demonstrated that TbRGG2 modulates gRNA/mRNA interactions and promotes the formation of misedited junctions (Simpson et al. 2017). MRB8180 (named by convention based on its TriTrypDB designation [Ammerman et al. 2012]) is an RNA binding protein that interacts with TbRGG2 and also facilitates junction formation (Simpson et al. 2017). Moreover, sites at which pauses in the 3′ to 5′ progression of editing arise in RPS12 mRNA when either TbRGG2 or MRB8180 are knocked down overlap significantly, indicating a specific function in editing progression for the REMC variant containing these two proteins (Simpson et al. 2017). MRB8170 also binds mRNA and TbRGG2, but appears to have a substantially different function than MRB8180. Dixit et al. (2017), using individual nucleotide cross-linking immunoprecipitation (iCLIP), concluded that MRB8170 marks mRNAs for editing, and their qRT-PCR analysis suggested that MRB8170 is involved in the initiation of editing on at least some mRNAs. Using a very different HTS approach, Simpson et al. (2017) also concluded that MRB8170 promotes editing initiation on some mRNAs and generally confines the region of active editing. Because MRB8180 is 15- to 30-fold less abundant than MRB8170 or TbRGG2, it is likely that REMC variants containing TbRGG2 and MRB8170, but not MRB8180, exist (Simpson et al. 2017).
We recently showed that the RESC component, MRB7260, plays a role in modulating GRBC–REMC interactions within the RESC complex during editing (McAdams et al. 2018). Depletion of MRB7260 caused dissociation of GRBC and REMC, with concomitant disruption of normal mRNA and gRNA trafficking within RESC. Specifically, gRNAs were depleted from GRBC and accumulated on REMC, indicating a role for MRB7260 in modulating proper gRNA–protein interactions. mRNAs associated with REMC also dramatically increased when MRB7260 was knocked down. HTS studies showed that MRB7260 is essential for editing progression, but does not impact editing initiation. Another RESC factor with the potential to coordinate GRBC–REMC interactions is the helical repeat protein, MRB10130 (Tb927.10.10130). In one report, MRB10130 was designated as a component of REMC based on mass spectrometry studies (Aphasizheva et al. 2014). However, yeast two-hybrid analysis revealed direct interactions between MRB10130 and five components of GRBC and one component of REMC, TbRGG2 (Ammerman et al. 2012), making assignation to one or the other module difficult. Here, we analyze the function of MRB10130 using a combined genomic and biochemical approach. Our data show that MRB10130 organizes GRBC–REMC interactions and is necessary for both the initiation and 3′ to 5′ progression of editing.
RESULTS
MRB10130 is an RNA binding protein that is essential for procyclic form growth and RNA editing
In silico analysis predicts that MRB10130 is comprised almost entirely of 29 helical repeats, an organization reminiscent of Armadillo (ARM) or HEAT repeat-containing proteins (Fig. 1A,B). ARM/HEAT proteins often organize protein complexes and some reportedly bind RNA, suggesting multiple mechanisms by which MRB10130 might impact RNA editing (Xu and Kimelman 2007; Cook et al. 2009; Kim et al. 2012; Valenta et al. 2012; Nozawa et al. 2013; Berthon and Stratakis 2014; Cretu et al. 2016; Yoshimura and Hirano 2016; Gul et al. 2017). To begin to examine the role of MRB10130 in RNA editing, we depleted the protein from procyclic form (PF) T. brucei using a tetracycline (tet)-inducible RNAi system (Wickstead et al. 2002), and the level of knockdown was assessed by qRT-PCR and western blot (Fig. 1D and data not shown). MRB10130 knockdown resulted in a rapid growth defect beginning on day two post-induction and a substantial decrease in all edited mRNAs tested, including both pan-edited and minimally edited transcripts, confirming a previous report (Fig. 1C,D; Aphasizheva et al. 2014). Never edited and polycistronic precursor transcripts were essentially unaffected (Fig. 1D). Concomitant with fully edited mRNA decreases, pre-edited versions of most mRNAs increased, particularly for pan-edited mRNAs, where they increased between 1.6- and fourfold. These data indicate that editing initiation is inhibited, such that pre-edited mRNAs cannot enter the editing pathway and thus accumulate. Guanylyltransferase labeling of the gRNA population showed that decreased editing initiation was not caused by an overall decrease in gRNA levels (Fig. 1E). It is noteworthy that the MRB10130 phenotype differs from that of all other RESC factors reported to date in that pan-edited and minimally edited mRNAs exhibit the same level of edited RNA decrease. This includes COII mRNA, whose editing does not depend on trans-acting gRNAs or GAP1/2. Because the effects of editing factor depletion are typically more dramatic on pan-edited than on minimally edited mRNAs (Fisk et al. 2008; Acestor et al. 2009; Ammerman et al. 2011, 2013; Huang et al. 2015; Simpson et al. 2017; McAdams et al. 2018), these data suggest a distinct and fundamental function for MRB10130. To determine whether the MRB10130 impact on editing could involve direct RNA binding, we performed UV cross-linking assays with recombinant MRB10130 and body-labeled gRNA or mRNA. MRB10130 displayed in vitro binding to both RNAs, with a signal even greater than the positive control, TbRGG2 (Fig. 1F). Together, these data demonstrate that MRB10130 is an RNA binding protein that broadly affects the initiation of RNA editing.
FIGURE 1.

MRB10130 predicted protein structure and effect of MRB10130 depletion on T. brucei growth and RNA editing. (A) Amino acid sequence of the MRB10130 ORF. The mitochondrial localization signal was predicted using MitoProt II (Claros and Vincens 1996) and is displayed in bold and underlined. Green ribbons above the sequence represent amino acids predicted for form α-helical structures as modeled using the Phyre2 program (Kelley et al. 2015). (B) Ribbon diagram of the MRB10130 predicted protein structure using Phyre2 intensive protein homology modeling. Each α-helical repeat is displayed using a different color. (C) Repression of MRB10130 by tet-inducible RNAi in PF T. brucei. Cell growth was measured in triplicate for uninduced (tet−) and induced cells (tet+) for 10 d. (D) RNA was isolated from PF T. brucei containing the MRB10130 RNAi construct on day 2 postinduction and quantified by qRT-PCR using primer sets that specifically detect MRB10130 and never-edited, pan-edited, minimally edited, and dicistronic precursor RNAs. Relative RNA abundance represents RNA levels in tet induced cells compared to levels in uninduced cells. RNA levels were normalized to 18S rRNA. Levels and numbers represent the mean and standard error of six determinations. (E) RNA was isolated from PF MRB10130 RNAi cells either grown in the absence (tet−) or presence (tet+) of tet for 2 d, labeled with [α32P]-GTP using guanylyltransferase to identify gRNAs, and resolved on a denaturing gel. Cytoplasmic RNA that was used for normalization and the labeled gRNA are indicated. The level of MRB10130 knockdown was detected for two biological replicates by qRT-PCR (33%–40% of wild-type levels). (F) UV cross-linking assays were performed using radiolabeled gA6[14] gRNA or a 79-nt fragment of pre-edited A6 mRNA with a positive control protein (GST-TbRGG2), a negative control protein (maltose binding protein; MBP), and MBP-MRB10130-His. In vitro transcribed, 32P-labeled RNA was incubated with the indicated purified recombinant protein, and reactions were subjected to UV cross-linking followed by treatment with RNase A. Proteins were resolved by 10% SDS-PAGE. Asterisks indicate the size of full-length proteins (kDa) on both the Coomassie stained gel and the phosphorimage.
Sequence level analysis of RNA editing defects in MRB10130 knockdowns reveals impacts on editing initiation and progression
Having demonstrated by qRT-PCR that MRB10130 impacts editing initiation, we next wanted to confirm these results by another method and determine whether MRB10130 has additional effects on the 3′ to 5′ progression of editing. To this end, we performed HTS analysis of mitochondrial mRNA populations, using our previously described TREAT algorithm to analyze ND7-5′, RPS12, and CYb mRNAs from MRB10130 replete and depleted cells (Simpson et al. 2016, 2017). TREAT identifies the percentage of each mRNA population that comprises pre-edited, partially edited, and fully edited mRNAs. We can then further analyze editing progression by determining the editing stop site in each mRNA, which is defined as the final (5′ most) ES that matches the canonical fully edited sequence correctly (Table 1; Simpson et al. 2016). At a population level, TREAT allows us to define exacerbated pause sites (EPSs; Table 1); EPSs are editing stop sites at which canonical editing pauses to a significant extent in a given knockdown cell line compared to uninduced controls (P < 0.05, q < 0.05) (Simpson et al. 2017; McAdams et al. 2018; Tylec et al. 2019). Additionally, we can analyze the lengths and sequences of misedited junctions 5′ of EPSs to provide insights into specific editing defects that arise when distinct editing factors are depleted (Table 1; Simpson et al. 2017; McAdams et al. 2018; Tylec et al. 2019).
TABLE 1.
Glossary of terms

To confirm our qRT-PCR results indicating an effect of MRB10130 on editing initiation (Fig. 1D), we first analyzed the levels of pre-edited ND7-5′, RPS12, and CYb mRNAs in our high-throughput libraries. For all transcripts, we observed a statistically significant greater than twofold increase in pre-edited mRNAs upon MRB10130 depletion, indicating a defect in editing initiation (Fig. 2A). We next analyzed our libraries to identify EPSs that arise upon MRB10130 knockdown. Identification of EPSs would signify additional MRB10130 effects on the 3′ to 5′ progression of editing, a type of defect that would not be visible in the qRT-PCR assays shown in Figure 1D. No EPSs were identified in ND7-5′ mRNA. However, we observed numerous EPSs arising in both RPS12 and CYb mRNAs when MRB10130 is depleted, and positions of EPSs are indicated in Figure 2B,C. We analyzed EPS positions in RPS12 and CYb mRNAs relative to both edited mRNA sequence and to the positions of reported cognate gRNAs (Koslowsky et al. 2014). In both mRNAs, EPSs were located primarily throughout gRNA-defined regions, rather than at gRNA ends, suggesting an effect of MRB10130 on gRNA–mRNA positioning. Strikingly, the positions of EPSs in RPS12 mRNA when MRB10130 is depleted overlapped significantly (P = 7.66 × 10−06) with those previously reported in cells depleted of RESC factor, MRB7260 (Fig. 2B, black diamonds; McAdams et al. 2018). In contrast, overlap between RPS12 EPSs in the MRB10130 knockdown and the knockdown of TbRGG2, a component of the REMC module, was not statistically significant (P = 0.12) (Simpson et al. 2017; McAdams et al. 2018). To determine if EPSs in CYb mRNA are also similar between MRB10130 and MRB7260 knockdowns, we identified CYb EPSs that arise upon MRB7260 RNAi and again observed a statistically significant overlap with EPSs in MRB10130 depleted cells (P = 0.002) (Fig. 2C; Tylec et al. 2019). EPSs in CYb mRNA upon MRB10130 and TbRGG2 knockdown did not overlap (P = 0.50) (Simpson et al. 2017). Because MRB10130 depletion does not affect total MRB7260 levels (data not shown), these data suggest that MRB10130 and MRB7260 function at similar points in editing progression, or that one factor impacts the function of the other.
FIGURE 2.

High-throughput sequence analysis of mitochondrial transcripts. (A) Average number of normalized pre-edited ND7-5′, RPS12, and CYb transcripts for eight uninduced (Avg Unin) and two MRB10130 RNAi induced samples (MRB10130 KD). P-values (unpaired t-test with Welch's correction): 0.0183 for ND7-5′ (*), <0.0001 for RPS12 (****), and 0.0149 for CYb (*). (B) RPS12 3′ edited mRNA sequence with exacerbated pause sites (EPSs) arising upon MRB10130 RNAi in both biological replicates indicated with red diamonds. Black diamonds represent previously reported EPSs arising upon MRB7260 knockdown (McAdams et al. 2018). No EPSs were detected 5′ of the region shown in either cell line. Gray bars below the sequence indicate gRNAs, numbered 3′ to 5′ (Koslowsky et al. 2014). gRNA anchor regions are depicted with bold lines. Hatched regions at the 5′ and 3′ ends represent variation in gRNA lengths within a gRNA class. RPS12 stop codon is underlined; additional underscores are shown for clarity in stretches of unedited sequence to align numbers with the correct ES. Lower case u indicates a U inserted by editing; asterisks indicate encoded U's deleted by editing. (C) EPSs arising in CYb mRNA upon knockdown of MRB10130 and MRB7260. Shown is the edited domain and flanking sequence. The start codon is underlined, and the underscore indicates ES 580. All symbols as in B. (D) Sequences of the most abundant junctions within the region directed by RPS12 gRNAs 1–3 in MRB10130 replete and depleted cells. These sequences span editing stop sites (ESS) 10–40. Average normalized counts of a given sequence in uninduced samples (AvgUn) and MRB10130 knockdown (KD) are ordered from highest to lowest fold change upon MRB10130 knockdown. ESS refers to the editing stop site for that sequence. Jcn indicates the number of ES spanned by a given junction sequence. Fold indicates the fold change in the number of sequences for each junction in MRB10130 RNAi samples compared to uninduced samples. Pre-edited sequence, black; canonically edited sequence, red; junctions, purple. Gray lines below pre-edited and edited sequences indicate gRNA-directed blocks, with anchor regions depicted as bold lines. ES are indicated with triangles and are numbered from 3′ to 5′. Red diamonds; EPSs that arise following MRB10130 depletion.
To gain further insight into the editing defects that arise when MRB10130 is depleted, we undertook a detailed examination of the most abundant partially edited mRNAs with Editing Stop Sites in the regions directed by the first three gRNAs. Figure 2D shows all sequences with Editing Stop Sites in this region having greater than 100 normalized counts in the MRB10130 knockdown. Sequences are aligned in order of fold change in the MRB10130 knockdown cell line compared to uninduced controls. While the sequences are fairly heterogeneous, those that exhibit the greatest increase upon MRB10130 knockdown either lack junctions or contain very short (≤3 ES) junctions (Fig. 2D). It is striking that the top three sequences in the MRB10130 knockdown are the same as those reported in the MRB7260 knockdown, albeit in a slightly different order and with a greater increase in the MRB10130 knockdown. We also noted the presence of two sequences with longer junctions of similar sequence (ES39, J16 and ES39, J15). These sequences also accumulated in the MRB7260 knockdown (McAdams et al. 2018). The positions of these junctions, beginning at the end of gRNA2, as well as their lengths and sequence similarities, are consistent with utilization of a noncognate gRNA increasing at this site upon MRB10130 knockdown. Collectively, our HTS analysis reveals that MRB10130 impacts both editing initiation and progression through gRNA-defined regions of RPS12 and CYb mRNAs, and that its effects on editing progression overlap those of MRB7260 (McAdams et al. 2018).
MRB10130 affects RESC assembly
The predicted helical repeat structure of MRB10130, coupled with its widespread effect on RNA editing and reported direct binding to components of both the REMC and GRBC modules of RESC, suggest that it might be involved in proper RESC assembly (Figs. 1, 3A; Ammerman et al. 2012). We first confirmed by western blotting with available antibodies that MRB10130 depletion causes no substantial or reproducible changes in the levels of RESC components TbRGG2, MRB8180, MRB8170, MRB3010, GAP1, or MRB7260 (data not shown). With this knowledge in hand, we utilized a cell line harboring in situ His-TEV-myc tagged TbRGG2 (TbRGG2-HTM) (McAdams et al. 2018) and MRB10130 RNAi to ask whether interactions between the REMC and GRBC modules are compromised when MRB10130 is depleted. While intra-REMC interactions between TbRGG2 and MRB8180 and MRB8170 were unaffected, MRB10130 knockdown lead to a modest decrease in TbRGG2 associated GAP1 and a dramatic decrease in the TbRGG2-MRB3010 interaction (Fig. 3B,C). We confirmed these results by knocking down MRB10130 in cell lines containing in situ protein A-TEV-protein C (PTP) tagged versions of the GRBC components, MRB3010 or MRB11870. As expected, GRBC associated TbRGG2 and MRB8180 were decreased upon MRB10130 knockdown (Fig. 4), confirming that MRB10130 promotes interactions between GRBC and at least some variants of REMC. Like the intra-REMC contacts (Fig. 3), intra-GRBC contacts appeared unaffected, as shown by the unchanged MRB11870-PTP association with MRB3010 when MRB10130 is depleted (Fig. 4C,D). Results with MRB8170 and GAP1 were more variable, suggesting that distinct REMC or GRBC variants could be differentially affected by MRB10130 knockdown. These results are consistent with reported functions for MRB8170 distinct from those of TbRGG2 and MRB8180 (Dixit et al. 2017; Simpson et al. 2017), and reported interactions of GAP1 outside of the canonical GRBC module (Ammerman et al. 2012; Madina et al. 2014; McAdams et al. 2015, 2018). The interaction between GRBC and the catalytic RECC complex appears unaffected by MRB10130 depletion, as shown by the unchanged level of KREPA6 associated with MRB11870-PTP (Fig. 4C,D). From these data, we conclude that MRB10130 is essential for normal RESC organization, particularly interactions between REMC and GRBC subcomplexes.
FIGURE 3.

Effect of MRB10130 depletion on TbRGG2 protein–protein interactions. (A) Schematic representation of the RESC complex divided into the GRBC and REMC subcomplexes (Ammerman et al. 2012, 2013; Aphasizheva et al. 2014; Read et al. 2016; McAdams et al. 2018). Solid lines (strong) or dotted lines (weak) indicate the strength of interaction observed in published yeast two-hybrid screens (Ammerman et al. 2012; McAdams et al. 2018). Thick lines represent interactions in both directions, whereas thin lines represent interactions that occurred in one direction of the screen. (B) PF T. brucei cells harboring both the MRB10130 RNAi construct and the TbRGG2-HTM construct were used to assess the proteins associated with the REMC subcomplex upon depletion of MRB10130. Cells were grown for 2 d in the absence or presence of tet, followed by IP of TbRGG2 using the myc tag. Bound protein was eluted from myc beads using TEV protease cleavage, and elutions were then analyzed using western blot with a subset of REMC and GRBC protein antibodies. Biological replicate experiments were performed and qRT-PCR was used to validate the level of MRB10130 knockdown (14%–21% remaining). (C) Quantification of western blots in B using BioRad Image Lab software. Protein levels were normalized to amount of TbRGG2 for that immunoprecipitation (IP). The normalized protein levels from the tet+ IP were then compared to that of the tet− IP (which was set to 100%) to calculate the protein associated with TbRGG2. Bar graphs represent the average and standard deviation of two biological replicates.
FIGURE 4.

Effect of MRB10130 depletion on MRB3010 and MRB11870 protein–protein interactions. PF T. brucei cells harboring both the MRB10130 RNAi construct and the MRB3010-PTP (A,B) or MRB11870-PTP (C,D) construct were used to assess the proteins associated with the GRBC subcomplex in the absence of MRB10130. Cells were grown for 2 d in the absence or presence of tet, followed by IP of PTP-tagged protein using IgG. Bound protein was eluted from IgG beads using TEV protease cleavage, and elutions were analyzed using western blot with a subset of REMC and GRBC protein antibodies. Biological replicate experiments were performed and qRT-PCR was used to validate the level of MRB10130 knockdown (30%–45% remaining). (B,D) Quantification of western blots in (A,C) using BioRad Image Lab software. Protein levels were normalized to amount of PTP-tagged protein for that immunoprecipitation (IP). The normalized protein levels from the tet+ IP were then compared to that of the tet− IP (which was set to 100%) to calculate the protein associated with either MRB3010 or MRB11870. Bar graphs represent the average and standard deviation of two biological replicates.
To better understand the nature of the interactions of MRB10130 with REMC and GRBC modules, we expressed MRB10130 with a PTP tag at its chromosomal locus. We purified PTP tagged MRB10130 by IgG Sepharose affinity column followed by cleavage with TEV protease overnight at 4°C (Fig. 5A; Ammerman et al. 2012). MRB10130-PTP containing complexes were analyzed by glycerol gradient sedimentation followed by western blot (Fig. 5B). When MRB10130-PTP containing complexes purified in this manner were separated on 10%–30% glycerol gradients, the bulk of MRB10130-PTP fractionated in the upper fractions corresponding to values around 5S, as revealed by anti-Protein C western blot (Fig. 5B, fractions 2–5). Faint peaks were also observed at ∼10S (Fig. 5B, fraction 9) and 19S (Fig. 5B, fractions 13–15). We then probed the same gradient fractions for components of REMC and GRBC to characterize MRB10130-PTP complexes. Approximately 50% of TbRGG2 and MRB8180 cosedimented with 5S MRB10130-PTP, although MRB8180 may be present in only a subset of these complexes (Fig. 5B, fractions 2–5). In contrast, the majority of MRB3010 sedimented with the 19S MRB10130 complexes (Fig. 5B, fractions 13–16). A very small fraction of MRB3010 also reproducibly cosedimented with MRB10130 in fraction 9 (Fig. 5B). Thus, MRB10130 is present in assembled RESC (∼19S complexes), and in smaller complexes that appear to contain either REMC components (∼5S complexes with TbRGG2 and MRB8180), or GRBC components (∼10S complexes with MRB3010). However, only a vanishingly small amount of total MRB10130-PTP was present in assembled RESC, despite the presence of 50% of TbRGG2 and almost all MRB3010 being contained in this complex (Fig. 5B, fractions 13–16). We wondered if this distribution might in part reflect MRB10130 disassembly from RESC that occurred during the overnight TEV cleavage. To test this, we separated whole cell lysates from MRB10130-PTP expressing cells directly on 10%–30% glycerol gradients without prior purification of the MRB10130 containing complexes. Figure 5C shows that, under these conditions, MRB10130 sediments predominantly in larger complexes that peak between 11S and 19S, and also in larger complexes at the bottom of the gradient. Although all signals were quite heterodisperse, MRB10130-PTP clearly cosedimented with both TbRGG2 and MRB3010, suggesting organization into various larger RESC complexes, and confirming MRB10130-PTP disassembly due to overnight incubation (Fig. 5B). Given our observations that MRB10130 promotes REMC–GRBC interaction (Figs. 3, 4) and that almost all MRB3010 remains assembled into RESC even when MRB10130-PTP disassembles (Fig. 5B), our data suggest that MRB10130 acts as a scaffold protein that promotes REMC–GRBC interactions but whose continued interaction with the assembled complex is not required.
FIGURE 5.

Visualization of MRB10130-containing complexes using glycerol gradient sedimentation. (A) Stepwise description of the method used to isolate and visualize the MRB10130-containing complexes. (B) MRB10130-PTP was affinity purified from PF T. brucei and then subjected to 10%–30% (v/v) glycerol gradient sedimentation. Fractions collected were analyzed by western blot with antibodies against RESC proteins. Size markers obtained from a parallel gradient are displayed below the blots. Shown are representative western blots of two biological replicate experiments. (C) Total cell lysate from PF T. brucei containing the MRB10130-PTP construct was subject to 10%–30% (v/v) glycerol gradient sedimentation. Fractions were collected and analyzed by western blot. Shown is a representative of two biological replicate experiments.
mRNA and gRNA interactions with RESC are perturbed by MRB10130 knockdown
Having shown that MRB10130 modulates protein–protein interactions within RESC, we next wanted to determine the role of MRB10130 in maintaining normal RESC protein–RNA interactions. We used the MRB3010-PTP/MRB10130 RNAi and TbRGG2-HTM/MRB10130 RNAi lines described above and performed RNA immunoprecipitations (RIPs), measuring both associated mRNA and gRNA. To assess mRNA, we used our previous strategy with primers specific for never-edited and pre-edited mRNA at the extreme 5′ ends of pan-edited transcripts (Fig. 6A; McAdams et al. 2018). These primer pairs detect all but fully edited mRNA, and because the fraction of mRNA that is edited this far 5′ is typically very small, almost all species of mRNA are detected (Simpson et al. 2016; McAdams et al. 2018). Figure 6B shows that the levels of A6, COIII, and RPS12 mRNAs detected by these primer pairs are unchanged upon MRB10130 knockdown. We also used a panel of primers specific to a range of gRNAs (McAdams et al. 2018). We first confirmed that mRNAs and gRNAs are enriched in both MRB3010-PTP and TbRGG2-HTM pulldowns between 20- and 300-fold (data not shown). We then tested the effect of MRB10130 knockdown on RNA association with MRB3010-PTP. We observed no change in levels of MRB3010-PTP associated mRNA upon MRB10130 knockdown, and little to no effect on associated gRNAs, with the exception of a 50% decrease in gCYb[32] (Fig. 6C). In contrast, when we quantified RNAs associated with TbRGG2-HTM, we saw a dramatic decrease in all mRNAs and gRNAs (Fig. 6D). The three tested mRNAs decreased 40%–70%, and gRNAs decreased 50%–95%. From these data, we conclude that when REMC and GRBC dissociate due to MRB10130 knockdown (Figs. 3, 4), both mRNA and gRNA remain with the GRBC module. These data demonstrate that MRB10130, TbRGG2, and MRB8180 are not required for efficient mRNA or gRNA association with GRBC. In sum, our findings are consistent with a model in which MRB10130 facilitates the rearrangement of the RESC RNP to accommodate positioning of the REMC and GRBC modules around the gRNA–mRNA duplex (Fig. 7).
FIGURE 6.

Effect of MRB10130 depletion on RESC protein–RNA interactions. (A) Schematic representation of the qRT-PCR primers used to detect the largest pool of mRNAs for a given transcript in the RNA immunoprecipitations (RIPs). Pre-edited regions are indicated in black, edited RNA is displayed using hashed lines, and never-edited (NE) regions are in white. RIP mRNA primers (indicated with black arrows) hybridize to the 5′ never-edited and 5′-most pre-edited region of the transcripts. RIP primers detect pre-edited and all partially edited RNAs, excluding RNAs that are fully edited. (B) Total cellular RNA from PF cells containing the MRB10130 RNAi construct grown with or without tet for 2 d were analyzed using the RIP qRT-PCR primers. RNA levels were normalized to 18S rRNA levels. The relative RNA abundance represents RNA levels in tet induced cells compared to uninduced. For comparison, primers that detect only fully edited A6 mRNA (as in Fig. 1) are also displayed. qRT-PCR was also used to validate the level of MRB10130 knockdown in the biological replicate experiments (∼40% remaining). (C,D) Comparison of RNA immunoprecipitated with MRB3010-PTP (C) or TbRGG2-HTM (D) grown with or without tet to induce MRB10130 RNAi. Fold change with MRB10130 knockdown represents the RNA levels detected in the RIP from tet-induced cells compared to the RIP from uninduced cells. RNA was detected using RIP mRNA primers described in A and primers designed to detect a subset of gRNAs using qRT-PCR. RNA levels were standardized against 18S rRNA, and numbers represent the mean and standard deviation of two biological replicates with at least eight determinations total.
FIGURE 7.
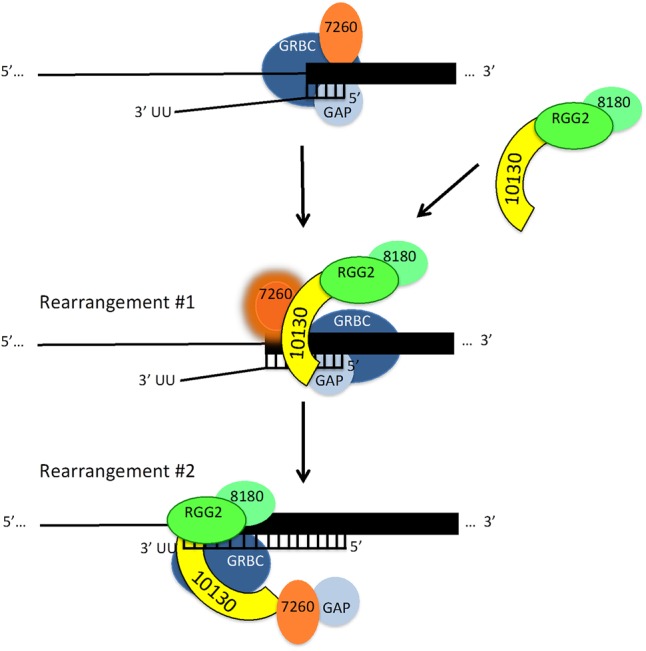
Working model of MRB10130 action during U-indel editing progression. See text for details.
DISCUSSION
U-indel RNA editing involves a complex protein interaction network entailing cooperative and/or competitive binding of numerous proteins, mRNAs, and gRNAs (Hashimi et al. 2013; Aphasizheva and Aphasizhev 2016; Read et al. 2016; Cruz-Reyes et al. 2018). RESC is a dynamic and heterogeneous complex that comprises the primary nonenzymatic component of the U-indel editing machinery in kinetoplastid mitochondria. RESC is believed to coordinate protein–protein and protein–RNA interactions during editing; however, the functions of most RESC proteins and the mechanisms that control rearrangements of the RESC complex are not well understood. Here, we report the role of MRB10130, a helical repeat protein that interacts with components of both the REMC and GRBC modules of RESC. We show that MRB10130 has a fundamental function in editing, affecting both editing initiation and progression and impacting all classes of edited mRNAs. We further show that MRB10130 acts as a scaffold that mediates RESC protein–protein interactions.
qRT-PCR and HTS analyses of edited mRNAs in cells depleted of MRB10130 revealed a role for this protein in editing initiation. That is, although total mRNA abundance does not change (Fig. 6B), fully edited mRNAs are depleted and pre-edited mRNAs accumulate when MRB10130 is knocked down (Figs. 1D, 2A), indicating that pre-edited mRNAs do not enter the editing pathway. Remarkably, the decreases observed for both pan-edited and minimally edited mRNAs were very similar. This phenotype is distinct even from that seen in knockdowns of GRBC components such as MRB3010, MRB11870, or MRB8620, where we typically observe a less dramatic effect on minimally edited mRNAs compared to pan-edited mRNAs (Ammerman et al. 2011, 2013; Huang et al. 2015). The equivalent effect of MRB10130 depletion on both classes of mRNAs, including COII that does not utilize trans-acting gRNAs, implies a fundamental function of this protein in editing initiation. What could account for this effect? One potential mechanism may involve recruitment of MRB8170 to the GRBC component, MRB3010 (Fig. 4); MRB8170 is required for editing initiation on some mRNAs (Dixit et al. 2017; Simpson et al. 2017). However, we did not observe the same defect in recruitment of MRB8170 to another GRBC protein, MRB11870, so the importance of these findings awaits further experimentation and a deeper understanding of RESC heterogeneity. Decreased association of TbRGG2 and MRB8180 with RESC (Figs. 3, 4) cannot account for initiation defects, as these REMC proteins affect only progression and not initiation (Ammerman et al. 2010; Simpson et al. 2017). The across-the-board decrease in editing initiation upon MRB10130 depletion also does not appear to be due to impaired association of RECC and RESC, as we do not observe any change in the association between KREPA6 and MRB11870 (Fig. 4). The role of MRB10130 in editing initiation may involve RESC proteins that were not investigated here due to lack of reagents, such as MRB800 or MRB1860. Alternatively, its effects may be subtler, such as promoting a rearrangement in RNA structure and/or protein–protein interactions that permits editing catalysis.
MRB10130 is also important in the overall 3′ to 5′ progression of editing. HTS analysis showed that partially edited mRNAs in MRB10130 knockdowns exhibit numerous EPSs compared to uninduced cells. This was true in both pan-edited RPS12 mRNA and minimally edited CYb mRNA. These EPSs are abundant throughout gRNA-directed regions, suggesting an effect of MRB10130 on gRNA–mRNA rearrangements rather than gRNA exchange. We unexpectedly found that the EPSs produced by MRB10130 knockdown exhibit significant overlap with those arising in cells depleted of MRB7260, another protein that impacts RESC organization (McAdams et al. 2018). The common EPSs in both MRB10130 and MRB7260 knockdowns indicate a functional association of these two proteins. However, it is unlikely that MRB10130 and MRB7260 act together in a dedicated complex as their phenotypes differ substantially (McAdams et al. 2018). First, MRB7260 affects only editing progression and has no effect on editing initiation. Moreover, while both proteins are required for optimal REMC–GRBC association, MRB7260 knockdown causes a dramatic reduction in GRBC–RNA binding that is not exhibited by MRB10130 knockdowns. We note that these data reveal that MRB7260's role in promoting correct RESC–RNA interactions is an inherent property of MRB7260 and not a secondary effect of REMC–GRBC disruption. Also arguing against a dedicated complex containing MRB7260 and MRB10130 are published data showing that MRB7260 and MRB10130 do not strongly coimmunoprecipitate, except in lysates in which RNA has been removed (McAdams et al. 2018). This does not necessarily mean that the interaction between these proteins occurs entirely apart from mRNA or gRNA. It may simply take place at a point in the editing cycle in which MRB10130, an RNA binding protein (Fig. 1F), is detached from or minimally associated with RNA, even as it is still interacting with RESC (Fig. 7, bottom). It is also possible that the functional similarity between MRB10130 and MRB7260 does not entail direct contact between the two proteins, but is imparted indirectly. For example, MRB10130 may promote critical rearrangements of RESC in a manner that is permissive for MRB7260 action (Fig. 7, middle).
MRB10130 is one of the few RESC proteins with recognizable domains. Structural predictions indicate that it is composed almost entirely of helical repeats resembling ARM or HEAT repeats. ARM and HEAT repeats form similar superhelical structures that are often difficult to distinguish in the absence of structural data (Gul et al. 2017), and proteins containing these repeats frequently act as organizers of multiprotein complexes. For example, the ARM protein, Β-catenin, is the central effector and organizer of the Wnt pathway (Xu and Kimelman 2007; Valenta et al. 2012). Structural and biochemical studies showed that numerous Β-catenin partners have overlapping binding sites within the helix formed by the ARM repeats and cannot bind simultaneously, implying a dynamic cycling of factors during the signaling process. Β-catenin also functions in the nucleus as a transcriptional activator and, together with associated proteins, is thought to modulate chromatin structure, suggesting an analogous function for MRB10130 in modulating RNA structure. HEAT repeat proteins possess structural plasticity and often undergo conformation changes upon binding different partners (Yoshimura and Hirano 2016). Interestingly, they participate in several aspects of RNA biology (Cook et al. 2009; Nozawa et al. 2013; Cretu et al. 2016). tRNA transport entails two HEAT repeat proteins, Xpot and Cex1p (Cook et al. 2009; Nozawa et al. 2013), and cargo-induced conformational changes of HEAT domains likely underlie the functions of both proteins. During pre-mRNA splicing, HEAT repeat protein SF3b155, a component of the U2 snRNP, binds pre-mRNA nucleotides around the branch site (Cretu et al. 2016). It is thought that dynamic hot spots in the Sf3b155 HEAT repeats permit conformational rearrangements of SF3b as it transits different stages of the splicing cycle. The reported mechanisms by which ARM/HEAT proteins modulate multiprotein and ribonucleoprotein complexes, together with biochemical and genomic studies on MRB10130 (Ammerman et al. 2012; Aphasizheva et al. 2014; Simpson et al. 2017; McAdams et al. 2018 and data herein), lead us to propose a working model of MRB10130 action during the progression of U-indel editing (Fig. 7).
The model shown in Figure 7 takes into account data reported herein as well as numerous published studies touching on MRB10130 function and interactions. Our model depicts the GRBC module, MRB7260, and GAP1 associated with an mRNA–gRNA pair that has formed an anchor duplex (Fig. 7, top), based on the following observations. RIP analyses show that mRNA and gRNA associate normally with GRBC in cells depleted of MRB10130 (Fig. 6). MRB10130 RNAi cells exhibit significantly reduced interaction of TbRGG2 and MRB8180 with GRBC (Figs. 3, 4), indicating that these REMC components are also not needed for normal levels of GRBC–RNA binding. We previously reported data suggesting that a complex containing MRB7260 and GAP1 facilitates gRNA–mRNA association during editing progression. Specifically, MRB7260 RNAi cells exhibit a typical 60%–70% reduction in gRNAs associated with GRBC, and RPS12 mRNA editing often pauses at gRNA ends in cells depleted of MRB7260 (McAdams et al. 2018). Thus, we show MRB7260/GAP1 associated with GRBC at this step. We next illustrate that MRB10130 promotes association of TbRGG2 and MRB8180 with GRBC (Fig. 7, middle), as indicated by reciprocal pulldowns with tagged TbRGG2, MRB3010, or MRB11870 (Figs. 3, 4). These data are supported by studies showing that MRB10130 interacts strongly with TbRGG2 following sequential MRB10130 pulldown and glycerol gradient sedimentation (Fig. 5) and in yeast two-hybrid assays (Ammerman et al. 2012). We propose that association of MRB10130–TbRGG2–MRB8180 with RESC and the gRNA–mRNA duplex then promotes a rearrangement of proteins and/or RNA that activates MRB7260 to facilitate editing at anchor duplex-proximal sites (highlighted MRB7260, Fig. 7, middle) in keeping with previously reported HTS studies in MRB7260 RNAi cells (McAdams et al. 2018). Also supporting this rearrangement and activation of MRB7260 are HTS studies reported here showing significant overlap between EPSs in cells depleted of these MRB10130 and MRB7260 (Fig. 2), indicating that these two proteins work together or impact each other's functions. Finally, we envision a second rearrangement that productively positions TbRGG2–MRB8180 in proximity to anchor duplex-distal mRNA regions, where these factors cooperate to promote editing as previously described (Fig. 7, bottom; Simpson et al. 2017). MRB7260–GAP1 may be released from RESC at this point in the editing cycle, as MRB7260 has not always been detected in RESC pulldowns, suggesting it is not stably bound (Hashimi et al. 2008; Panigrahi et al. 2008; Weng et al. 2008; Ammerman et al. 2011). A role for MRB10130 in recruitment and sequential exchange of RESC factors and promotion of protein–protein and protein–RNA rearrangements during editing is consistent with reported functions and the high degree of conformational flexibility exhibited by ARM/HEAT repeat proteins. Future structural and biochemical analyses to determine cooperative and competitive interactions with MRB10130, as well as binding sites for distinct proteins and RNAs will test this model of U-indel editing progression.
MATERIALS AND METHODS
Generation of cell lines and T. brucei culture conditions
To generate MRB10130 RNAi cell lines, a 768 bp fragment of the MRB10130 ORF was amplified using the primers “MRB0150 5′ RNAi” (5′-GGA TGC TCA ACG TTC TCT CGA GCA CAG C-3′) and “MRB0150 3′ HindIII RNAi” (5′-GGA AGC TTA TGC TGC AGG CCA CAC TCC AC-3′). To generate MRB800 cell lines (RNA from which was included in the uninduced controls for analysis of CYb mRNA in MRB7260 knockdowns), a 1021 bp fragment of the MRB800 ORF was amplified using primers “MRB800 5′ HindIII” (5′-GGA AGC TTA TGC GAC GTC GGG TAG TTT TAT G-3′) and “MRB800 3′ BamHI RNAi” (5′-GAG GAT CCG GCT CAA GGT GAA ATG GCG TAA G-3′). In both cases, amplicons were cloned into pJET1.2/Blunt (Thermo Scientific) and plasmids digested with BamHI and HindIII. The resulting inserts were then cloned into p2T7-177 (Wickstead et al. 2002) between opposing tetracycline-regulated T7 RNA polymerase promoters. The resulting plasmids were linearized with NotI and transfected into PF T. brucei strain 29–13. Transformants were selected with 2.5 µg/mL phleomycin, and clones were obtained using limiting dilution.
To generate the cell lines harboring both MRB10130 RNAi and either carboxy-terminally PTP-tagged MRB3010 or MRB11870, or His-TEV-Myc(HTM)-tagged TbRGG2, the p2T7-177 plasmid harboring nucleotides 180–768 of the MRB10130 ORF (see above) was linearized with NotI and transfected into previously described MRB3010-PTP (Ammerman et al. 2011), MRB11870-PTP (Ammerman et al. 2012), or TbRGG2-HTM (McAdams et al. 2018) PF cell lines. Cells were selected with 2.5 µg/mL phleomycin and 1 µg/mL puromycin and cloned by limiting dilution. Repression of MRB10130 was induced in all cell lines by adding 2.5–4 µg/mL tetracycline to cell cultures. The MRB10130-PTP cell line was created as described previously (Ammerman et al. 2012).
All PF T. brucei strains were grown under conditions described previously (Wirtz et al. 1999; Pelletier and Read 2003). For monitoring cell growth, cells harboring the MRB10130 RNAi construct were grown in the presence or absence of 2.5 µg/mL tetracycline and monitored in triplicate for 10 d. MRB10130 repression on day 2 was validated by qRT-PCR (n = 6) (see below).
Quantitative RT-PCR analysis
RNA was isolated from MRB10130 RNAi cells grown in the presence or absence of 2.5 µg/mL tetracycline on day 2 post-induction using TRIzol reagent (Ambion). RNA was then DNase treated using the DNA-free DNase kit (Ambion) and reverse transcribed to cDNA with the Taq-Man reverse transcription kit (Applied Biosciences) using random hexamer primers. Quantitative reverse transcription PCR (qRT-PCR) was then performed in a final volume of 25 µL with designated primers, and cDNA was amplified using a CFX Connect Real-Time System (Bio-Rad). Established primers specific to the pre-edited, edited, and precursor mitochondrial transcripts for T. brucei were used (Carnes et al. 2005; Fisk et al. 2008). In addition, the level of MRB10130 repression was analyzed with primers “MRB10130 3′UTR qPCR Fwd” (5′-ATG CTG TCA CTT CGT TAC AA-3′) and “MRB10130 3′UTR qPCR Rev” (5′-ACC CGC ACA CAT AAA ATA TC-3′). Results were analyzed using the Bio-Rad CFX Manager 3.1 software and the RNA levels were normalized to 18S rRNA using the standard curve method. Guanylyltransferase labeling was performed as described previously (Hayman and Read 1999; Ammerman et al. 2010) to assess the global level of gRNAs in the MRB10130 RNAi cells.
Recombinant protein expression and in vitro RNA binding
Recombinant glutathione S-transferase (GST)-tagged TbRGG2 was purified as described previously (Fisk et al. 2008; Foda et al. 2012). To generate the amino-terminal MBP and carboxy-terminal histidine tagged MRB10130, the MRB10130 ORF without the predicted mitochondrial localization signal (Fig. 1, underlined amino acids 1–21) was optimized for expression in Escherichia coli (GenScript), and a TEV cleavage site followed by 10 histidine residues were encoded at the carboxyl terminus. This optimized sequence was then used as a template for PCR using the primers “For XbaI MBP-MRB10130-No MLS” (5′-GTA TAA TCT AGA CGT CCG TCG GCA CTG-3′) and “Rev HindIII MBP-MRB10130-No MLS” (5′-TGA ATA AAG CTT TCA GTG ATG ATG GTG ATG ATG ATG ATG-3′). The resulting PCR product was digested with XbaI and HindIII and ligated into pMAL-C2 (NEB) to create plasmid for the expression of MBP-MRB10130-His in E. coli. Recombinant protein was first purified using a standard MBP purification method using amylose resin (NEB), followed by purification over ProBond Resin (Novex) as described previously (McAdams et al. 2018).
For in vitro RNA binding studies, guide RNA gA6[14] (Read et al. 1994) and mRNA A6U5 pre-mRNA (79 nt) (Fisk et al. 2008) were synthesized from plasmids using the T7 Maxiscript Kit (Ambion). Internally [α 32P]-UTP (800 Ci/mmol) labeled RNA was then purified on 8% (w/v) acrylamide/7M urea gel. UV cross-linking reactions containing 5 fmol of radiolabeled RNA and 2 µg of protein were performed as described previously (McAdams et al. 2018).
Immunoprecipitation experiments and western blots
Approximately 8 × 109–1.5 × 1010 PF cells containing either MRB3010-PTP, MRB11870-PTP, or TbRGG2-HTM and tetracycline-inducible MRB10130 RNAi were grown either in the presence or absence of tetracycline for 2 d. MRB10130 knockdown was validated by qRT-PCR (see above). Endogenously tagged proteins were affinity purified as described previously (Schimanski et al. 2005; Günzl and Schimanski 2009; Ammerman et al. 2012) using either IgG Sepharose 6 Fast Flow (GE Healthcare) or Agarose Immobilized Rabbit anti-cMYC (ICL) chromatography followed by cleavage using AcTEV protease (Invitrogen). TEV elutions were subsequently analyzed by western blot.
Proteins were electrophoresed on a 10% (w/v) SDS-PAGE gel and transferred to either a nitrocellulose or Immobilon FL (low-fluorescent) PVDF membrane. Membranes were probed with polyclonal antibodies against MRB3010 (Ammerman et al. 2012), GAP1 (Ammerman et al. 2011), TbRGG2 (Fisk et al. 2008), MRB8170 (Kafkova et al. 2012), MRB8180 (Simpson et al. 2017), MRB10130 (Ammerman et al. 2013), KREP-A6 (Tarun et al. 2008), and the Protein C epitope (ICL). Monoclonal antibodies against Histidine (Clonetech) and REAP-1 (gift from S. Madison-Antenucci) were also used. Western blots were subsequently probed with secondary antibodies Goat anti-rabbit IRdye 680RD, Goat anti-mouse IRdye 680RD, Goat anti-rabbit HRP, or Goat anti-mouse HRP. Fluorescent western blots were visualized using the Odyssey CLX Imaging System (LiCor) and chemiluminescent western blots by ChemiDoc MP imaging system (BioRad).
Glycerol gradient sedimentation
Approximately 5 × 1010 cells harboring a carboxy-terminal PTP tagged MRB10130 were used for immunoprecipitation experiments as described previously with minor alterations which include the use of 2 mM DTT instead of 1 mM DTT while being devoid of 20 mM potassium l-glutamate, and 10 µg/mL aprotinin (Schimanski et al. 2005; Günzl and Schimanski 2009). Eluates were normalized to the Protein C epitope retained on the carboxy-terminal end of MRB10130 after TEV cleavage using western blot. Normalized eluates were then loaded on a 10%–30% glycerol gradient and the gradient centrifuged as done previously (Ammerman et al. 2012). Twenty-four 0.5 mL fractions were then collected form the top of the gradient and sedimentation of RESC proteins was analyzed by western blot. This glycerol gradient sedimentation was also performed using MRB10130-PTP cell lysates prior to immunoprecipitation.
RNA immunoprecipitation (RIP)
PF T. brucei harboring the MR10130 RNAi construct along with either the MRB3010-PTP or TbRGG2-HTM construct were grown with or without 4 µg/mL tetracycline for 2 d, and mitochondria were enriched as described previously (Hashimi et al. 2008). Enriched mitochondria were lysed, and tagged proteins were immunoprecipitated as described previously (McAdams et al. 2018) using anti-C-myc agarose (ICL) for TbRGG2-HTM or IgG Sepharose 6 Fast Flow beads (GE Healthcare) for MRB3010-PTP. Prior to treatment with proteinase, 2% of beads were subjected to western blot using antibodies against the myc tag for theTbRGG2-HTM RIPs or Protein-C antibodies for the MRB3010-PTP RIPs to normalize the amount of protein immunoprecipitated. RNA was then extracted from the bound protein using proteinase K treatment followed by phenol/chloroform extraction. RNA was DNase-treated (Ambion DNA-free DNase kit) and converted to cDNA with gene specific primers (McAdams et al. 2018) using iScript Select cDNA synthesis kit (Bio-Rad). Prior to addition to qRT-PCR, cDNA was preamplified using SsoAdvances PreAmp Supermix. qRT-PCR was then performed as described above using 18S rRNA as a normalization gene.
Fold enrichment compared to the mock RIP was calculated for each gene of interest (GOI) using the ΔΔCt method comparing the tet uninduced sample to that of the mock IP. Fold change to mock IP = 2(ΔΔCt), where ΔΔCt = (CtGOI-Mock − Ct18S-Mock) − (CtGOI-Uninduced − Ct18S-Uninduced). To determine the fold change in the RNA associated when MRB10130 was depleted, the RNA detected in the tet-induced sample was compared to the uninduced IP using the ΔΔCt method. Fold change with MRB10130 knockdown = 2(ΔΔCt), where ΔΔCt = (CtGOI-Uninduced − Ct18S-Uninduced) − (CtGOI-Induced − Ct18S-Induced).
High-throughput sequencing and bioinformatic analysis
PF T. brucei RNAi cells were grown in the presence or absence of 4 µg/mL tetracycline for 2 d (MRB10130) or 3 d (MRB7260 [McAdams et al. 2018]) followed by isolation of RNA using TRIzol per manufacturer's instructions. Two biological replicate experiments were performed, and qRT-PCR was used to validate the level of knockdown (MRB10130; average 36.5% remaining and MRB7260; average 33.0% remaining). cDNA was generated from DNase treated RNA using gene specific primers described previously (Simpson et al. 2016). These cDNA samples were PCR amplified within the linear range of PCR to maintain the relative abundance of unique fragments. Amplicons were then sequenced using paired-end Illumina MiSeq and paired as described previously (Simpson et al. 2016). The number of total (de-collapsed) and unique (collapsed) reads for each sample are listed in Supplemental Tables 1 and 2. To normalize the number of reads in each sample, the total (de-collapsed) number of fragments that have no non-T mismatches (standard alignments in Supplemental Tables 1 and 2) are normalized to 100,000 reads. This normalization scales each sample so that their relative abundance can be compared via their normalized counts (Simpson et al. 2016). Two biological replicates of the MRB10130 RNAi samples were compared to 10 uninduced samples, two from this study and eight from a previous study (Simpson et al. 2017). For analysis of CYb mRNA in MRB7260 knockdowns, the biological replicate induced RNAi samples were compared to eight uninduced samples (two replicates of MRB10130 RNAi cells, and two replicates each of MRB7260 RNAi cells [McAdams et al. 2018]); MRB3010 RNAi cells (Ammerman et al. 2011), and MRB800 RNAi cells. TREAT (Trypanosome RNA Editing Alignment Tool) was used as a multiple sequence alignment and visualization tool (Simpson et al. 2016). TREAT consists of a command-line alignment tool along with a built-in web server providing a web-based interface for searching, viewing and analyzing the alignment results. TREAT v0.03 (Simpson et al. 2017) was used in this study. Analysis of pre-edited transcripts and determination of EPSs was performed as described previously (Simpson et al. 2017).
DATA DEPOSITION
TREAT is written in Go and freely available under the GPLv3 license at http://github.com/ubccr/treat. Code for all R analysis used in this paper can be found through the TREAT github repository (ubccr/treat). The sequencing data used in this study have been deposited in the Sequence Read Archive, accession number SRP131478 for the MRB10130 RPS12 and ND7-5′ samples (two uninduced and two induced) and accession number SRP097727 for the additional eight uninduced samples from a previous study used for RPS12 and ND7-5′ controls (Simpson et al. 2017), and SRP185791 for all CYb samples.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University at Buffalo Genomics and Bioinformatics Core, especially Sujith Valiyaparambil and Jonathan Bard. We are also grateful to Andrew Bruno for assistance with TREAT analysis and Susan Madison-Antenucci for providing anti-REAP-1 antibodies. We thank members of the Read laboratory for critical reading of the manuscript. This work was supported by National Institutes of Health grants R01 AI061580 and R01 GM129041 to L.K.R., R01 AI125982 to Y.S. and R.C., and R01 DE024523 to Y.S.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071902.119.
REFERENCES
- Acestor N, Panigrahi AK, Carnes J, Zikova A, Stuart KD. 2009. The MRB1 complex functions in kinetoplastid RNA processing. RNA 15: 277–286. 10.1261/rna.1353209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Presnyak V, Fisk JC, Foda BM, Read LK. 2010. TbRGG2 facilitates kinetoplastid RNA editing initiation and progression past intrinsic pause sites. RNA 16: 2239–2251. 10.1261/rna.2285510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Hashimi H, Novotna L, Cicova Z, McEvoy SM, Lukes J, Read LK. 2011. MRB3010 is a core component of the MRB1 complex that facilitates an early step of the kinetoplastid RNA editing process. RNA 17: 865–877. 10.1261/rna.2446311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Downey KM, Hashimi H, Fisk JC, Tomasello DL, Faktorová D, Kafková L, King T, Lukeš J, Read LK. 2012. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res 40: 5637–5650. 10.1093/nar/gks211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Tomasello DL, Faktorová D, Kafková L, Hashimi H, Lukeš J, Read LK. 2013. A core MRB1 complex component is indispensable for RNA editing in insect and human infective stages of Trypanosoma brucei. PLoS ONE 8: e78015 10.1371/journal.pone.0078015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Aphasizhev R. 2016. U-insertion/deletion mRNA-editing holoenzyme: definition in sight. Trends Parasitol 32: 144–156. 10.1016/j.pt.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Zhang L, Wang X, Kaake RM, Huang L, Monti S, Aphasizhev R. 2014. RNA binding and core complexes constitute the U-insertion/deletion editosome. Mol Cell Biol 34: 4329–4342. 10.1128/MCB.01075-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthon A, Stratakis CA. 2014. From β-catenin to ARM-repeat proteins in adrenocortical disorders. Hormone Metab Res 46: 889–896. 10.1055/s-0034-1389993 [DOI] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619. 10.1073/pnas.0506133102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol 28: 122–130. 10.1128/MCB.01374-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Zelaya Soares C, Wickham C, Stuart K. 2011. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem 286: 19320–19330. 10.1074/jbc.M111.228965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, McDermott S, Anupama A, Oliver BG, Sather DN, Stuart K. 2017. In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases. Nucleic Acids Res 45: 4667–4686. 10.1093/nar/gkx116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros MG, Vincens P. 1996. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241: 779–786. 10.1111/j.1432-1033.1996.00779.x [DOI] [PubMed] [Google Scholar]
- Cook AG, Fukuhara N, Jinek M, Conti E. 2009. Structures of the tRNA export factor in the nuclear and cytosolic states. Nature 461: 60–65. 10.1038/nature08394 [DOI] [PubMed] [Google Scholar]
- Cretu C, Schmitzová J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Lührmann R, Pena V. 2016. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell 64: 307–319. 10.1016/j.molcel.2016.08.036 [DOI] [PubMed] [Google Scholar]
- Cruz-Reyes J, Mooers BHM, Doharey PK, Meehan J, Gulati S. 2018. Dynamic RNA holo-editosomes with subcomplex variants: insights into the control of trypanosome editing. Wiley Interdiscip Rev RNA 9: e1502 10.1002/wrna.1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit S, Müller-McNicoll M, David V, Zarnack K, Ule J, Hashimi H, Lukeš J. 2017. Differential binding of mitochondrial transcripts by MRB8170 and MRB4160 regulates distinct editing fates of mitochondrial mRNA in trypanosomes. MBio 8: e02288-16 10.1128/mBio.02288-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk JC, Ammerman ML, Presnyak V, Read LK. 2008. TbRGG2, an essential RNA editing accessory factor in two Trypanosoma brucei life cycle stages. J Biol Chem 283: 23016–23025. 10.1074/jbc.M801021200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda BM, Downey KM, Fisk JC, Read LK. 2012. Multifunctional G-rich and RRM-containing domains of TbRGG2 perform separate yet essential functions in trypanosome RNA editing. Eukaryot Cell 11: 1119–1131. 10.1128/EC.00175-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimov ES, Gasparyan AA, Kaurov I, Tichý B, Logacheva MD, Kolesnikov AA, Lukeš J, Yurchenko V, Zimmer SL, Flegontov P. 2018. Trypanosomatid mitochondrial RNA editing: dramatically complex transcript repertoires revealed with a dedicated mapping tool. Nucleic Acids Res 46: 765–781. 10.1093/nar/gkx1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul IS, Hulpiau P, Saeys Y, van Roy F. 2017. Metazoan evolution of the armadillo repeat superfamily. Cell Mol Life Sci 74: 525–541. 10.1007/s00018-016-2319-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzl A, Schimanski B. 2009. Tandem affinity purification of proteins. Curr Protoc Protein Sci Chapter 19: Unit 19.19 10.1002/0471140864.ps1919s55 [DOI] [PubMed] [Google Scholar]
- Hashimi H, Zikova A, Panigrahi AK, Stuart KD, Lukes J. 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14: 970–980. 10.1261/rna.888808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Cicova Z, Novotna L, Wen YZ, Lukes J. 2009. Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA 15: 588–599. 10.1261/rna.1411809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Zimmer SL, Ammerman ML, Read LK, Lukeš J. 2013. Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol 29: 91–99. 10.1016/j.pt.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman ML, Read LK. 1999. Trypanosoma brucei RBP16 is a mitochondrial Y-box family protein with guide RNA binding activity. J Biol Chem 274: 12067–12074. 10.1074/jbc.274.17.12067 [DOI] [PubMed] [Google Scholar]
- Huang Z, Faktorová D, Křížová A, Kafková L, Read LK, Lukeš J, Hashimi H. 2015. Integrity of the core mitochondrial RNA-binding complex 1 is vital for trypanosome RNA editing. RNA 21: 2088–2102. 10.1261/rna.052340.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafkova L, Ammerman ML, Faktorova D, Fisk JC, Zimmer SL, Sobotka R, Read LK, Lukes J, Hashimi H. 2012. Functional characterization of two paralogs that are novel RNA binding proteins influencing mitochondrial transcripts of Trypanosoma brucei. RNA 18: 1846–1861. 10.1261/rna.033852.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Kwak H, Lee HK, Hyun S, Jeong S. 2012. β-Catenin recognizes a specific RNA motif in the cyclooxygenase-2 mRNA 3′-UTR and interacts with HuR in colon cancer cells. Nucleic Acids Res 40: 6863–6872. 10.1093/nar/gks331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky DJ, Bhat GJ, Read LK, Stuart K. 1991. Cycles of progressive realignment of gRNA with mRNA in RNA editing. Cell 67: 537–546. 10.1016/0092-8674(91)90528-7 [DOI] [PubMed] [Google Scholar]
- Koslowsky D, Sun Y, Hindenach J, Theisen T, Lucas J. 2014. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res 42: 1873–1886. 10.1093/nar/gkt973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Madina BR, Gulati S, Vashisht AA, Kanyumbu C, Pieters B, Shakir A, Wohlschlegel JA, Read LK, Mooers BH, et al. 2016. REH2C helicase and GRBC subcomplexes may base pair through mRNA and small guide RNA in kinetoplastid editosomes. J Biol Chem 291: 5753–5764. 10.1074/jbc.M115.708164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madina BR, Kumar V, Metz R, Mooers BH, Bundschuh R, Cruz-Reyes J. 2014. Native mitochondrial RNA-binding complexes in kinetoplastid RNA editing differ in guide RNA composition. RNA 20: 1142–1152. 10.1261/rna.044495.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madina BR, Kumar V, Mooers BH, Cruz-Reyes J. 2015. Native variants of the MRB1 complex exhibit specialized functions in kinetoplastid RNA editing. PLoS ONE 10: e0123441 10.1371/journal.pone.0123441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams NM, Ammerman ML, Nanduri J, Lott K, Fisk JC, Read LK. 2015. An arginine-glycine-rich RNA binding protein impacts the abundance of specific mRNAs in the mitochondria of Trypanosoma brucei. Eukaryot Cell 14: 149–157. 10.1128/EC.00232-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams NM, Simpson RM, Chen R, Sun Y, Read L. 2018. MRB7260 is essential for productive protein-RNA interactions within the RNA editing substrate binding complex during trypanosome RNA editing. RNA 24: 540–556. 10.1261/rna.065169.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa K, Ishitani R, Yoshihisa T, Sato M, Arisaka F, Kanamaru S, Dohmae N, Mangroo D, Senger B, Becker HD, et al. 2013. Crystal structure of Cex1p reveals the mechanism of tRNA trafficking between nucleus and cytoplasm. Nucleic Acids Res 41: 3901–3914. 10.1093/nar/gkt010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ziková A, Dalley RA, Acestor N, Ogata Y, Anupama A, Myler PJ, Stuart KD. 2008. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol Cell Proteomics 7: 534–545. 10.1074/mcp.M700430-MCP200 [DOI] [PubMed] [Google Scholar]
- Pelletier M, Read LK. 2003. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA 9: 457–468. 10.1261/rna.2160803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read LK, Göringer HU, Stuart K. 1994. Assembly of mitochondrial ribonucleoprotein complexes involves specific guide RNA (gRNA)-binding proteins and gRNA domains but does not require preedited mRNA. Mol Cell Biol 14: 2629–2639. 10.1128/MCB.14.4.2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read LK, Lukeš J, Hashimi H. 2016. Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley Interdiscip Rev RNA 7: 33–51. 10.1002/wrna.1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Nguyen TN, Günzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell 4: 1942–1950. 10.1128/EC.4.11.1942-1950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RM, Bruno AE, Bard JE, Buck MJ, Read LK. 2016. High-throughput sequencing of partially edited trypanosome mRNAs reveals barriers to editing progression and evidence for alternative editing. RNA 22: 677–695. 10.1261/rna.055160.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RM, Bruno AE, Chen R, Lott K, Tylec BL, Bard JE, Sun Y, Buck MJ, Read LK. 2017. Trypanosome RNA editing mediator complex proteins have distinct functions in gRNA utilization. Nucleic Acids Res 45: 7965–7983. 10.1093/nar/gkx458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. 2008. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest 118: 1301–1310. 10.1172/JCI33945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun SZ J., Schnaufer A, Ernst NL, Proff R, Deng J, Hol W, Stuart K. 2008. KREPA6 is an RNA-binding protein essential for editosome integrity and survival of Trypanosoma brucei. RNA 14: 347–358. 10.1261/rna.763308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis B, Shaw PLR, Liu B, Ravindra K, Iliff H, Al-Hashimi HM, Schumacher MA. 2018. The RRM of the kRNA-editing protein TbRGG2 uses multiple surfaces to bind and remodel RNA. Nucleic Acids Res 47: 2130–2142. 10.1093/nar/gky1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylec BL, Simpson RM, Kirby LE, Chen R, Sun Y, Koslowsky DJ, Read LK. 2019. Intrinsic and regulated properties of minimally edited trypanosome mRNAs. Nucleic Acids Res 47: 3640–3657. 10.1093/nar/gkz012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Hausmann G, Basler K. 2012. The many faces and functions of β-catenin. EMBO J 31: 2714–2736. 10.1038/emboj.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R. 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol Cell 32: 198–209. 10.1016/j.molcel.2008.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K. 2002. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol 125: 211–216. 10.1016/S0166-6851(02)00238-4 [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99: 89–101. 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- Xu W, Kimelman D. 2007. Mechanistic insights from structural studies of β-catenin and its binding partners. J Cell Sci 120: 3337–3344. 10.1242/jcs.013771 [DOI] [PubMed] [Google Scholar]
- Yoshimura SH, Hirano T. 2016. HEAT repeats - versatile arrays of amphiphilic helices working in crowded environments? J Cell Sci 129: 3963–3970. 10.1242/jcs.185710 [DOI] [PubMed] [Google Scholar]
- Zimmer SL, Simpson RM, Read LK. 2018. High throughput sequencing revolution reveals conserved fundamentals of U-indel editing. Wiley Interdiscip Rev RNA 9: e1487 10.1002/wrna.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.