

Abstract
The iron-dependent oxidase UndA cleaves one C3–H bond and the C1–C2 bond of dodecanoic acid to produce 1-undecene and CO2. A published x-ray crystal structure showed that UndA has a heme-oxygenase-like fold, thus associating it with a structural superfamily that includes known and postulated nonheme diiron proteins, but revealed only a single iron ion in the active site. Mechanisms proposed for initiation of decarboxylation by cleavage of the C3–H bond using a mono-iron cofactor to activate O2 necessarily invoked unusual or potentially unfeasible steps. Here we present spectroscopic, crystallographic, and biochemical evidence that the cofactor of Pseudomonas fluorescens Pf-5 UndA is actually a diiron cluster and show that binding of the substrate triggers rapid addition of O2 to the Fe2(II/II) cofactor to produce a transient peroxo-Fe2(III/III) intermediate. The observations of a diiron cofactor and substrate-triggered formation of a peroxo-Fe2(III/III) intermediate suggest a small set of possible mechanisms for O2, C3–H and C1–C2 activation by UndA; these routes obviate the problematic steps of the earlier hypotheses that invoked a single iron.
Graphical Abstract
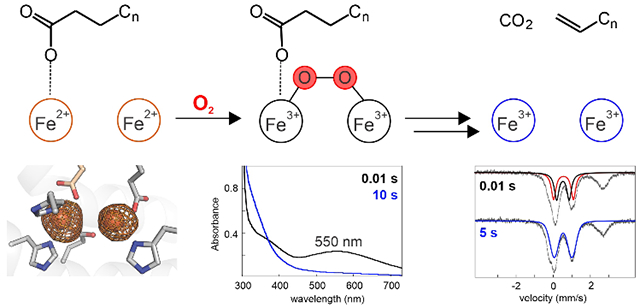
The Pseudomonas enzyme UndA oxidatively decarboxylates medium-chain Cn (n = 8, 10, 12) free fatty acids to Cn-1 terminal olefins.1 This reaction generates hydrocarbons that could be directly employed as liquid fuel or industrial feedstocks.2–4 An initial report established that UndA requires Fe(II), dioxygen and a reducing system for multiple turnovers in vitro.1 A mechanism to account for these requirements and for the observations of a single iron ion in the UndA x-ray crystal structure and retention of both C2 deuteria in the 1-undecene product from 2-[2H2]-dodecanoic acid (lauric acid, LA) was proposed.1 Addition of O2 to the Fe(II) cofactor was envisaged to generate a Fe(III)-superoxide intermediate that would abstract a hydrogen (H•) from C3 (Cβ) of the LA substrate to initiate decarboxylation. However, the C3–H bond has a homolytic dissociation energy (BDE > 95 kcal/mol)5 significantly exceeding that of any C–H bond currently known to be cleaved by a mid-valent metal-superoxo complex. In these known cases, a heteroatom with a non-bonded electron pair or an olefin is invariably α to the scissile C–H bond and is thought to make the initiating hydrogen atom transfer (HAT) thermodynamically feasible.6–7 Moreover, the mid-valent-metal-superoxo reaction manifold often enables four-electron oxidation reactions,6–7 whereas the UndA reaction is a two-electron oxidation. These inconsistencies suggested that a different mechanism could be operant.
UndA has a heme-oxygenase-like fold, similar to the Chlamydia protein associating with death domains (CADD), a protein of unknown function.8–9 The CADD x-ray crystal structure revealed a diiron cluster reminiscent of the nonheme diiron cofactors in proteins of the topologically distinct ferritin-like structural superfamily.10 Many ferritin-like enzymes use this cofactor to activate O2 for cleavage of strong C–H bonds,10 as must also occur in the UndA reaction. Ligands to the dinuclear cluster in CADD are conserved in UndA (Figure S1), and a recent study presented evidence, obtained by Mössbauer spectroscopy and measurement of reaction stoichiometry, that Pseudomonas syringae pv. tomato DC3000 UndA uses a diiron cofactor.11 Here we show that Pseudomonas fluorescens Pf-5 UndA (78 % sequence identity to the P. syringae UndA) also harbors a diiron cluster and that binding of the LA substrate promotes very rapid O2 activation through an intermediate that is identified by its absorption and Mössbauer spectra as a peroxo-Fe2(III/III) complex.
We first obtained evidence by Mössbauer, extended x-ray absorption fine structure (EXAFS), and EPR spectroscopies for a coupled Fe2(III/III) cluster in P. fluorescens UndA. Its Mössbauer features (Figure S2) are similar to those recently reported for the P. syringae UndA.11 EXAFS measurements yielded an Fe-Fe separation of 3.21 Å (Figures S3 and S4). EPR spectra of a partially reduced sample exhibited an axial g < 2 signal characteristic of a mixed-valent Fe2(II/III) cluster (Figure S5). We next sought a high-resolution x-ray crystal structure of the protein with both iron sites occupied. Anomalous difference maps from Fe(II)-soaked UndA crystals (Figure S6) revealed clear evidence for a diiron cofactor, but despite the improved Fe2 occupancy, these datasets failed to yield a complete view of the first coordination sphere in the UndA reactant complex. Specifically, persistent disorder of residues 195-200 prevented modeling of D198 (Figure S6), one of three proposed Fe2 ligands (along with E159 and H201). However, substitution with alanine of any of these three residues abolished 1-undecene production (Figure S1), underscoring the importance of site 2 coordination in catalysis.
Mixing of an anoxic solution containing UndA and Fe(II) with O2-containing buffer resulted in slow (kobs = 0.5 s−1) development of stable absorption at ~350 nm, signifying oxidation of Fe(II) to Fe(III) (Figure 1A).12–15 By contrast, with LA also present in the protein solution, a transient feature centered at λmax ~ 550 nm (Figure 1B) developed to maximum intensity in < 0.01 s (black and red spectra) and decayed by ~1 s (orange spectrum). The substrate-triggered accumulation of the associated complex and its transient nature suggest that it could be an intermediate in the decarboxylation reaction.
Figure 1.
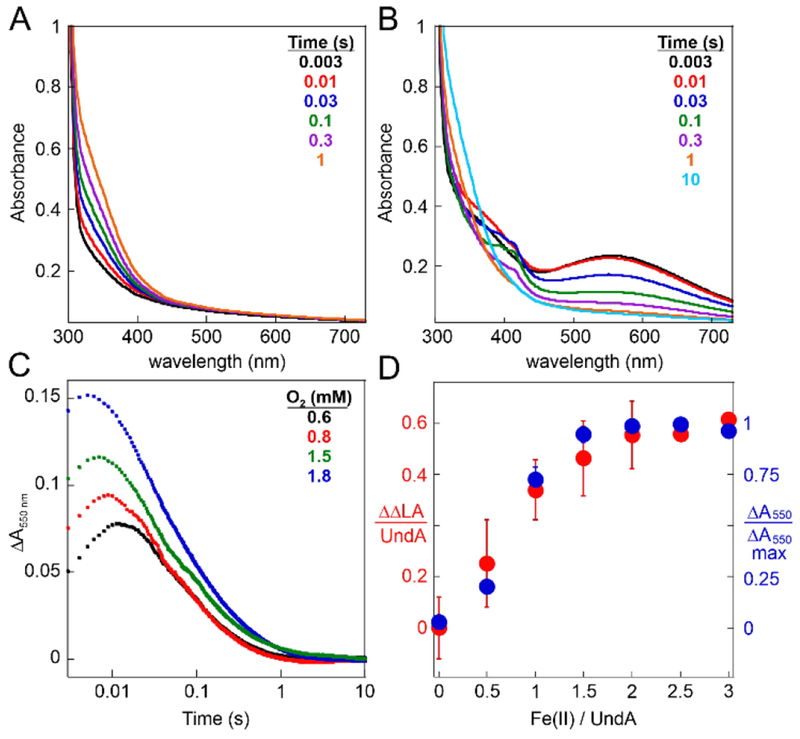
Substrate-triggered, [O2]-dependent, rapid formation of an absorbing, on-pathway intermediate in the reaction of UndA-Fe2(II/II) with O2. (A and B) Absorption spectra acquired after rapid mixing at 5 °C of a solution containing 0.30 mM UndA, 0.54 mM Fe(II) (1.8 equiv), and either 0 mM (A) or 1.0 mM (B) LA with an equal volume of O2-saturated buffer (~1.8 mM O2). (C) Change in absorbance at 550 nm (ΔA550nm) versus time after mixing of the same protein reactant solution as in B with an equal volume of buffer containing O2 at the concentrations indicated in the legend. (D) Comparison of the ΔA550nm amplitudes (differences between maximum and minimum values) and quantities of LA consumed [relative to a reaction with no added Fe(II)] in analogous reactions with varying equivalencies of Fe(II):UndA in the protein solution. Details of the reactions and the procedure for LA quantification are provided in the Supporting Information.
Early adducts between O2 and Fe2(II/II) cofactors have been characterized in a number of ferritin-like proteins, and many of these have visible absorption features with maxima between 420 and 700 nm.10 Most are μ-peroxo-Fe2(III/III) complexes resulting from oxidative addition of O2 to the cluster in a bridging mode.16–22 The rate of formation of the 550 nm-absorbing complex in UndA depends on the concentration of O2 (Figure 1C), confirming that no prior protein-O2 adduct accumulates to a high level in the reaction. We obtained further evidence that the absorbing species is a diiron complex and an on-pathway intermediate by variation of the Fe(II):UndA stoichiometry (Figure 1D). The dependencies of the quantity of LA consumed (red points, left axis) and the amplitude of the 550-nm absorbance transient (blue points, right axis) are nearly identical, and both quantities approach their maximum values at an Fe(II):UndA ratio of ~ 2. The slope of the plot for LA consumed implies a stoichiometry of 0.6 LA consumed per diiron cluster, only 60 % of the theoretical value of unity. This diminished stoichiometry reflects the oxidation of Fe(II) via at least one unproductive pathway. The accumulation of the intermediate complex to a maximum of 60 % of the total added iron and its decay via (a) radical species in a fraction of events (vide infra) are consistent with this explanation.
To further characterize the 550nm-absorbing intermediate, we prepared freeze-quench samples for Mössbauer spectroscopy (Figure 2 and S7). The 4.2-K/53-mT spectrum of the 0.01-s sample exhibits diminished intensity of the high energy line (~ 2.4 mm/s) from the Fe(II) reactant complex and a new asymmetric feature at ~ 1 mm/s. The developing features could be accounted for as a pair of overlapping quadrupole doublets, each contributing 30% of the total 57Fe absorption. The parameters of the doublets [δ1 = 0.59 mm/s, |ΔEQ|1 = 1.07 mm/s (red line) and δ2 = 0.56 mm/s, |ΔEQ|2 = 0.67 mm/s (blue line, see Figure S8 for analysis)] are characteristic of N/O-coordinated high-spin Fe(III), but the isomer shifts (δ) are toward the higher end of the typical range,23 as has been seen for other μ-peroxo-Fe2(III/III) complexes (δ ~ 0.48-0.68 mm/s).10 The 4.2-K/8-T spectrum of the intermediate can be adequately simulated using the values of δ and |ΔEQ| determined from the 53-mT spectrum and assuming a diamagnetic ground state (Figure S9). The rapid two-electron oxidation of the Fe2(II/II) reactant complex upon exposure to O2 is consistent with assignment of the 550-nm absorbing intermediate as a peroxo-Fe2(III/III) species.
Figure 2.
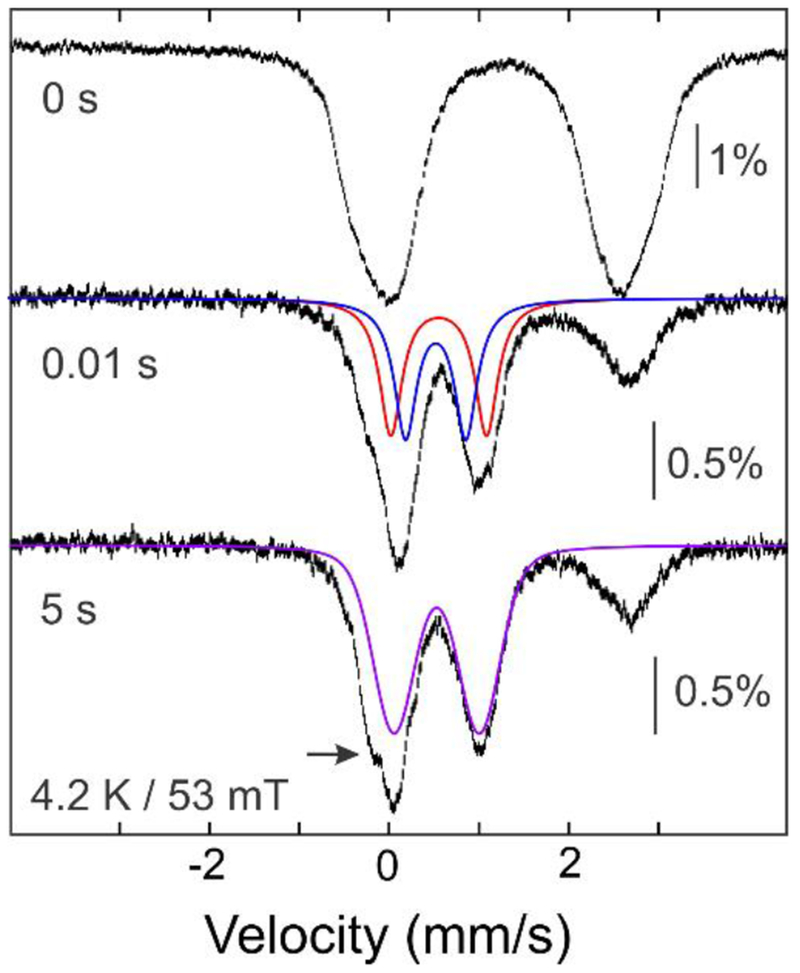
The UndA reaction monitored by freeze-quench Mössbauer spectroscopy. The vertical bars show 4.2-K/53-mT spectra of a sample (top) of an anoxic solution of 1.2 mM reduced, reconstituted 57Fe-UndA and 10 mM LA (see Supporting Information for details) and of samples obtained by mixing the reactant complex with an equal volume of O2-saturated buffer (~1.8 mM O2 at 5°C) and freeze-quenching after 0.01 s (middle) or 5 s (bottom) at 5°C. The solid lines are simulations of the spectra of the peroxo-Fe2(III/III) intermediate (red and blue) and Fe2(III/III) product (purple) with parameters quoted in the text.
Samples quenched at longer reaction times have 4.2-K/53-mT Mössbauer spectra similar to that of the 0.01-s sample (Figure S7). However, the development of a spectral feature at ~ −0.2 mm/s (arrow in Figure 2) indicates decay of the peroxo-Fe2(III/III) intermediate to a new species. Analysis of the (0.01s–5s) difference spectrum (Figure S10) yields δ ~0.52 mm/s and |ΔEQ| ~0.97 mm/s (Figure 2, purple line) for the developing species, which we assign as the product Fe2(III/III) cluster.
Decay of the peroxo-Fe2(III/III) complex leads to development of absorption- and EPR-spectroscopic features characteristic of a tyrosyl radical (Tyr•) (Figure 1B and S11).24–25 Accumulation of a one-electron-oxidized protein side chain in the two-electron-oxidation reaction converting LA to 1-undecene is unexpected, and, indeed, two lines of evidence suggest that the detected radical species is (are) not on the productive pathway. Substitution of any of the three Tyr residues closest to the active site (Tyr71, Tyr107, and Tyr197) with Phe failed to abolish the transient features (Figure S12), implying that multiple Tyr residues, rather than a single functionally essential Tyr, can be oxidized. Substitution of the cofactor proximal Trp190, posited to mediate oxidation of the Tyr(s) by a relay mechanism, with Phe did abolish the Tyr•(s), and yet this W190F variant retained the ability to consume LA (Figure S13). Intriguingly, a Fe2(III/IV) complex accumulated (Figure S14), albeit only to 0.06 equiv, in the reaction of this variant. It is not clear whether this one-electron-reduced successor to the peroxo-Fe2(III/III) complex is an on-pathway species resulting from substrate oxidation by a Fe2(IV/IV) level complex (Scheme 1) or is also off-pathway.
Scheme 1.

Proposed mechanisms for oxidative decarboxylation of LA by UndA.
The conclusion that UndA uses a Fe2(II/II) cluster to form a peroxo-Fe2(III/III) intermediate reveals its analogy to ferritin-like nonheme diiron enzymes10 such as soluble methane monooxygenase (sMMO).16 In the sMMO reaction, the μ-peroxo-Fe2(III/III) complex is thought to convert to an H•-abstracting Fe2(IV/IV) complex (Q).26–28 As recently suggested by Manley, et al.,11 a similar pathway could be operant in the UndA reaction; the observation of substrate-triggered formation of the first of these two key intermediates puts such a hypothesis on firmer ground. Formation of an intermediate analogous to Q from the peroxide-level complex and HAT from C3 to Q would generate a substrate radical that could initiate 1-undecene production by one of three fates (Scheme 1, HAT): (A) oxygen rebound followed by a Grob-type multi-bond fragmentation, (B) radicaloid C1-C2 fragmentation followed by electron transfer from the •CO2− to the Fe2(III/IV) cluster, or (C) electron transfer to the Fe2(III/IV) cluster followed by polar decarboxylation. Alternatively, substrate decarboxylation via a Kolbe-like mechanism could be initiated by one-electron-oxidation of the fatty acid carboxylate (Scheme 1, PCET). Each of these pathways would involve a Fe2(III/IV) intermediate, and so our detection of such a complex would potentially provide experimental support for such mechanistic hypotheses.
These hypotheses draw directly from mechanisms proposed for ferritin-like diiron enzymes, but UndA has the heme-oxygenase-like (HO-like) fold, which features a three-helix metal-binding core that is distinct from the ferritin-like four-helix core (Figure 3). Most known HO-like proteins (HO, Thi4/TenA, PqqC) do not bind metal ions directly,29–31 but two other recently discovered examples, SznF and BesC, share with UndA a dependence on Fe(II) and O2 for their oxidative activities.32–33 Interestingly, prior to the work of Manley, et al.,11 the functionally unassigned CADD was the only HO-like protein shown to harbor a dimetal cluster,8 suggesting that the metallocofactors may be generally unstable in this scaffold. Cluster lability likely results from ligand-contributing core helices with only modestly stable secondary structures, as observed for both UndA and SznF.1,32 Although cofactor instability would seem to be detrimental to catalytic function, it could conceivably serve to obviate in situ cofactor redox recycling by a dedicated reductase protein, thereby enabling a novel modus operandi for this emerging family of nonheme diiron enzymes.
Figure 3.
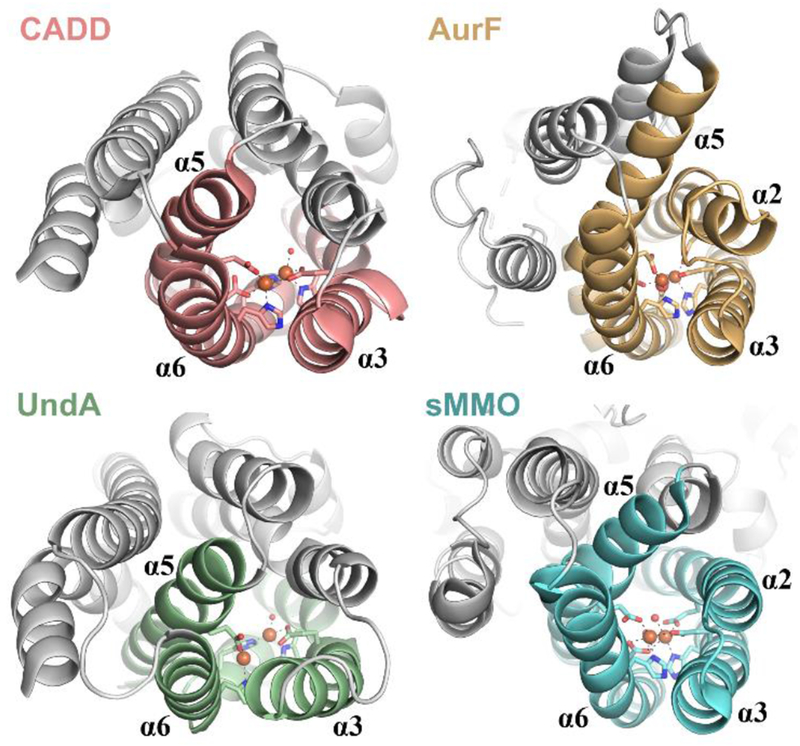
Comparison of the 3-helix HO-like [CADD (1RCW), UndA (6P5Q)] and 4-helix ferritin-like [AurF (3CHH), sMMOH (1FYZ)] architectures.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (CHE-1610676 to C.K., J.M.B., and A.K.B) and the National Institutes of Health (National Research Service Awards GM-116353 to E.J.B. and GM-103220 to B.R.S.). Portions of this work were conducted at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences. Portions of this work were conducted at the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. GM/CA at APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant (085P1000817). We thank Dr. Christopher J. Pollock for helpful discussions regarding EXAFS data collection and analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. This file (PDF) includes the Experimental Methods section, descriptions of the structural and spectroscopic characterizations of the diiron cofactor in P. fluorescens UndA (Figures S1–S6), Figures S7–S15, and Tables S1–S4.
The authors declare no competing financial interests.
REFERENCES
- 1.Rui Z; Li X; Zhu X; Liu J; Domigan B; Barr I; Cate JH; Zhang W, Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc Natl Acad Sci U S A 2014, 111 (51), 18237–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennen RM; Pfleger BF, Microbial production of fatty acid-derived fuels and chemicals. Curr Opin Biotechnol 2013, 24 (6), 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peralta-Yahya PP; Zhang FZ; del Cardayre SB; Keasling JD, Microbial engineering for the production of advanced biofuels. Nature 2012, 488 (7411), 320–328. [DOI] [PubMed] [Google Scholar]
- 4.Ray S; Rao PVC; Choudary NV, Poly-alpha-olefin-based synthetic lubricants: a short review on various synthetic routes. Lubr Sci 2012, 24 (1), 23–44. [Google Scholar]
- 5.Luo YR, Comprehensive Handbook of Chemical Bond Energies. CRC Press: Boca Raton, Fl, 2007. [Google Scholar]
- 6.Bollinger JM Jr.; Krebs C, Enzymatic C-H activation by metal-superoxo intermediates. Curr Opin Chem Biol 2007, 11 (2), 151–158. [DOI] [PubMed] [Google Scholar]
- 7.van der Donk WA; Krebs C; Bollinger JM Jr., Substrate activation by iron superoxo intermediates. Curr Opin Struct Biol 2010, 20 (6), 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarzenbacher R; Stenner-Liewen F; Liewen H; Robinson H; Yuan H; Bossy-Wetzel E; Reed JC; Liddington RC, Structure of the Chlamydia protein CADD reveals a redox enzyme that modulates host cell apoptosis. J Biol Chem 2004, 279 (28), 29320–29324. [DOI] [PubMed] [Google Scholar]
- 9.Stenner-Liewen F; Liewen H; Zapata JM; Pawlowski K; Godzik A; Reed JC, CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem 2002, 277 (12), 9633–9636. [DOI] [PubMed] [Google Scholar]
- 10.Jasniewski AJ; Que L Jr., Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem Rev 2018, 118 (5), 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manley OM; Fan R; Guo Y; Makris TM, Oxidative Decarboxylase UndA Utilizes a Dinuclear Iron Cofactor. J Am Chem Soc 2019, 141 (22), 8684–8688. [DOI] [PubMed] [Google Scholar]
- 12.Kurtz DM Jr., Oxo- and hydroxo-bridged diiron complexes: a chemical perspective on a biological unit. Chem Rev 1990, 90 (4), 585–606. [Google Scholar]
- 13.Fox BG; Shanklin J; Somerville C; Münck E, Stearoyl-acyl carrier protein delta 9 desaturase from Ricinus communis is a diiron-oxo protein. Proc Natl Acad Sci U S A 1993, 90 (6), 2486–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CA; Remar GJ; Musselman RL; Solomon EI, Spectroscopic and Electronic Structure Studies of met-Hemerythrin Model Complexes: A Description of the Ferric-Oxo Dimer Bond. Inorg Chem 1995, 34 (3), 688–717. [Google Scholar]
- 15.Pandelia M-E; Li N; Nørgaard H; Warui DM; Rajakovich LJ; Chang W.-c.; Booker SJ; Krebs C; Bollinger JM Jr., Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J Am Chem Soc 2013, 135 (42), 15801–15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tinberg TE; Lippard SJ, Dioxygen Activation in Soluble Methane Monooxygenase. Acc Chem Res 2010, 44 (4), 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brazeau BJ; Lipscomb JD, Kinetics and Activation Thermodynamics of Methane Monooxygenase Compound Q Formation and Reaction with Substrates. Biochemistry 2000, 39, 13503–13515. [DOI] [PubMed] [Google Scholar]
- 18.Bollinger JM Jr.; Krebs C; Vicol A; Chen SX; Ley BA; Edmondson DE; Huynh BH, Engineering the diiron site of Escherichia coli ribonucleotide reductase protein R2 to accumulate an intermediate similar to H-peroxo, the putative peroxodiiron(III) complex from the methane monooxygenase catalytic cycle. J Am Chem Soc 1998, 120 (5), 1094–1095. [Google Scholar]
- 19.Möenne-Loccoz P; Baldwin J; Ley BA; Loehr TM; Bollinger JM Jr., O2 activation by non-heme diiron proteins: identification of a symmetric mu-1,2-peroxide in a mutant of ribonucleotide reductase. Biochemistry 1998, 37 (42), 14659–14663. [DOI] [PubMed] [Google Scholar]
- 20.Broadwater JA; Achim C; Münck E; Fox BG, Mössbauer studies of the formation and reactivity of a quasi-stable peroxo intermediate of stearoyl-acyl carrier protein Delta 9-desaturase. Biochemistry 1999, 38 (38), 12197–12204. [DOI] [PubMed] [Google Scholar]
- 21.Li N; Korboukh VK; Krebs C; Bollinger JM Jr., Four-electron oxidation of p-hydroxylaminobenzoate to p-nitrobenzoate by a peroxodiferric complex in AurF from Streptomyces thioluteus. Proc Natl Acad Sci U S A 2010, 107 (36), 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makris TM; Vu VV; Meier KK; Komor AJ; Rivard BS; Münck E; Que L Jr.; Lipscomb JD, An unusual peroxo intermediate of the arylamine oxygenase of the chloramphenicol biosynthetic pathway. J Am Chem Soc 2015, 137 (4), 1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Münck E, Aspects of 57Fe Mössbauer spectroscopy In Physical Methods in Bioinorganic Chemistry, Que L Jr., Ed. University Science Books: Sausalito, CA, 2000; pp 287–319. [Google Scholar]
- 24.Bollinger JM Jr.; Edmondson DE; Huynh BH; Filley J; Norton JR; Stubbe J, Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science 1991, 253 (5017), 292–298. [DOI] [PubMed] [Google Scholar]
- 25.Svistunenko DA; Cooper CE, A new method of identifying the site of tyrosyl radicals in proteins. Biophys J 2004, 87 (1), 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu KE; Valentine AM; Wang D; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ, Kinetic and Spectroscopic Characterization of Intermediates and Component Interactions in Reactions of Methane Monooxygenase from Methylococcus capsulatus (Bath). J Am Chem Soc 1995, 177, 10174–10185. [Google Scholar]
- 27.Shu L; Nesheim JC; Kauffmann K; Münck E; Lipscomb JD; Que L Jr., An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science 1997, 275 (5299), 515–518. [DOI] [PubMed] [Google Scholar]
- 28.Nesheim JC; Lipscomb JD, Large kinetic isotope effects in methane oxidation catalyzed by methane monooxygenase: evidence for C-H bond cleavage in a reaction cycle intermediate. Biochemistry 1996, 35 (31), 10240–10247. [DOI] [PubMed] [Google Scholar]
- 29.Toms AV; Haas AL; Park JH; Begley TP; Ealick SE, Structural characterization of the regulatory proteins TenA and TenI from Bacillus subtilis and identification of TenA as a thiaminase II. Biochemistry 2005, 44 (7), 2319–2329. [DOI] [PubMed] [Google Scholar]
- 30.Lad L; Friedman J; Li HY; Bhaskar B; de Montellano PRO; Poulos TL, Crystal structure of human heme oxygenase-1 in a complex with biliverdin. Biochemistry 2004, 43 (13), 3793–3801. [DOI] [PubMed] [Google Scholar]
- 31.Magnusson OT; Toyama H; Saeki M; Rojas A; Reed JC; Liddington RC; Klinman JP; Schwarzenbacher R, Quinone biogenesis: Structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc Natl Acad Sci USA 2004, 101 (21), 7913–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng TL; Rohac R; Mitchell AJ; Boal AK; Balskus EP, An N-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 2019, 566 (7742), 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchand JA; Neugebauer ME; Ing MC; Lin CI; Pelton JG; Chang MCY, Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 2019, 567 (7748), 420–424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.