

Abstract
Iron(II)- and 2-(oxo)-glutarate-dependent (Fe/2OG) oxygenases catalyze a diverse array of oxidation reactions via a common iron(IV)-oxo (ferryl) intermediate. Although the intermediate has been characterized spectroscopically, its short lifetime has precluded crystallograhic characterization. In solution, the ferryl was first observed directly in the archetypal Fe/2OG hydroxylase, taurine:2OG dioxygenase (TauD). Here, we substitute the iron cofactor of TauD with the stable vanadium(IV)-oxo (vanadyl) ion to obtain crystal structures mimicking the key ferryl complex. Intriguingly, whereas the structure of the TauD·(VIV-oxo)·succinate·taurine complex exhibits the expected orientation of the V≡O bond—trans to the His255 ligand and toward the C−H bond to be cleaved, in what has been termed the in-line configuration—the TauD·(VIV-oxo) binary complex is best modeled with its oxo ligand trans to Asp101. This off-line-like configuration is similar to one recently posited as a means to avoid hydroxylation in Fe/2OG enzymes that direct other outcomes, though neither has been visualized in an Fe/2OG structure to date. Whereas an off-line (trans to the proximal His) or off-line-like (trans to the carboxylate ligand) ferryl is unlikely to be important in the hydroxylation reaction of TauD, the observation that the ferryl may deviate from an in-line orientation in the absence of the primary substrate may explain the enzyme’s mysterious self-hydroxylation behavior, should the oxo ligand lie trans to His99. This finding reinforces the potential for analogous functional off-line oxo configurations in halogenases, desaturases, and/or cyclases.
Graphical Abstract
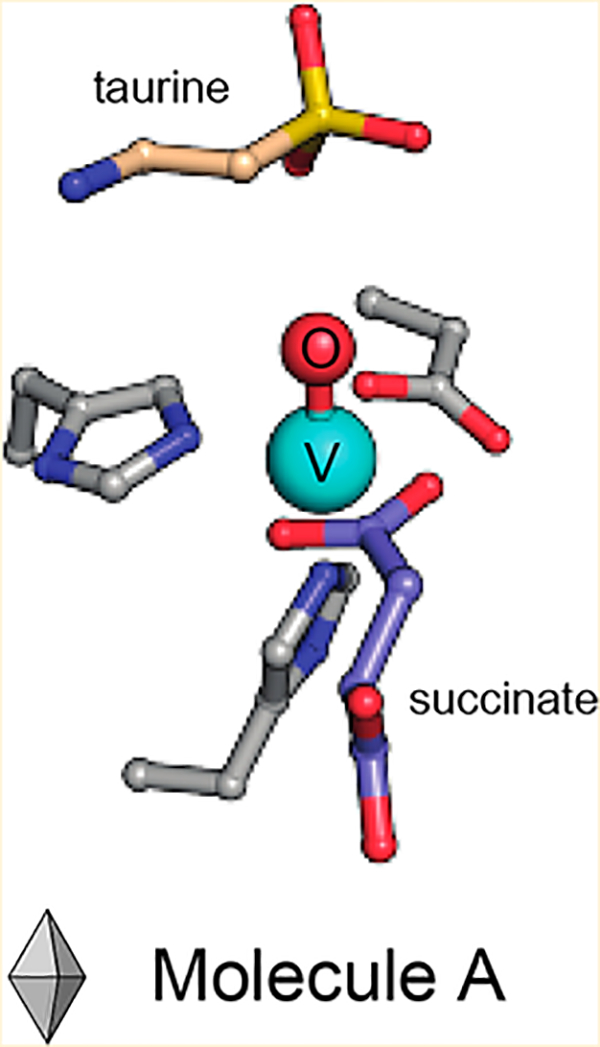
Functionalization of chemically inert carbon centers is a key step in the biosynthesis of many natural products.1,2 To catalyze these challenging transformations, nature has evolved a variety of enzymatic strategies for activating very strong C−H bonds. Iron(II)- and 2-(oxo)-glutarate-dependent (Fe/2OG) oxygenases harness the power of molecular oxygen for this purpose. Members of the ubiquitous Fe/2OG superfamily share a common mechanistic pathway, in which reaction of a mononuclear FeII cofactor with O2 yields an FeIV -oxo (ferryl) intermediate upon decarboxylation of 2OG to succinate (Figure 1).3,4 Abstraction of a substrate hydrogen atom by the ferryl complex most commonly results in hydroxylation via oxygen rebound; however, desaturation, cyclization, stereo-inversion, and halogenation reactions have also been documented.5 Recent work with Fe/2OG-dependent halogenases suggests that the location of the ferryl oxo ligand, and its orientation relative to the C−H bond to be activated, may play a key role in suppressing hydroxylation for those enzymes that effect such alternative outcomes.6,7
Figure 1.

Proposed TauD mechanism. States of the catalytic cycle that are highlighted in gray have been characterized or mimicked in crystallo.3,8–10 The structure at the bottom right is presented herein with vanadyl substituted for the functional ferryl complex. We have chosen to depict succinate in a bidentate coordination mode throughout the cycle, on the basis of its observed binding mode in the vanadyl complex. It should be noted, however, that ligation of the coproduct could be dynamic in solution.
Although visualization of the ferryl intermediate is limited by its short-lived nature,11 metal ion substitution with the vanadyl ion (VIV-oxo) was recently shown to yield a complex that structurally resembles the reactive species in Fe/2OG enzymes.12 Martinie et al. demonstrated that incorporation of vanadyl into taurine:2OG dioxygenase (TauD) closely mimics the active site geometry associated with its ferryl complex. Moreover, vanadyl substitution of the Fe/2OG-dependent halogenase, SyrB2, induces global conformational effects analogous to those detected in the native enzyme-substrate complex upon formation of the ferryl intermediate. Determination of the structure of the Fe/2OG-dependent hydroxylase, VioC, in complex with vanadyl, succinate, and its native substrate, l-Arg, provided further evidence of the utility of this approach.13 A computationally derived model of the ferryl state obtained by geometry optimization following replacement of vanadium by iron accurately predicted the experimentally observed Mössbauer parameters of the ferryl intermediate, suggesting that vanadyl faithfully mimics its structure. Additional structures of Fe/2OG enzymes containing this stable metal oxycation could provide insight into the active site geometries that tailor reactivity and facilitate catalysis. However, its application as a ferryl mimic has not been demonstrated in the structural characterization of substrate-bound systems other than VioC. A structure of the herbicide-degrading Fe/2OG-dependent enzyme arylalkanoate dioxygenase, AAD-1, was recently determined with vanadyl and succinate bound, but the complex lacks the hydroxylation substrate, limiting insight into its mechanism.14 Herein, we report the first crystallographic model for the metal-oxo complex of TauD, historically the most well-characterized member of the Fe/2OG superfamily, via cocrystallization with vanadyl, succinate, and its substrate taurine (2-aminoethanesulfonate). TauD is responsible for the microbial catabolism of taurine initiated by hydroxylation of C1.15 The structures reported here are consistent with the metal-oxo orientation observed crystallographically in VioC and also provide new insights into enigmatic self-hydroxylating behavior detected biochemically in TauD and related enzymes.
RESULTS AND DISCUSSION
Crystals of Escherichia coli TauD grown in the presence of vanadyl, taurine, and the coproduct succinate yielded a 1.73 Å resolution structure containing two molecules in the asymmetric unit. As in previous structures, the enzyme forms a homotetramer.9,10,15 Here, each molecule is a member of an independent oligomer, in which the constituent monomers are related by crystallographic symmetry. Whereas density consistent with vanadyl is clearly visible in both molecules, taurine and succinate are present only in chain B (Figure 2 and Figure S1A,B). The active site of chain A is instead populated by seven ordered water molecules and an adventitious acetate ion, originating from the precipitant solution. This acetate ion forms a salt bridge with the conserved active site side chain, Arg266, analogous to that formed by the C5 carboxylate of 2OG in the reactant complex. The water molecules, by contrast, are found both directly coordinated to vanadium and occupying the taurine binding pocket. Intriguingly, this water-stabilized H-bonding network is not only unique among determined structures of TauD, but also remarkably consistent with calorimetric studies that suggest the ordering of approximately 6.3 ± 2.3 water molecules upon FeII binding.16 We identify eight possible solvent binding locations, including the position held by the vanadyl-oxo in this model (Figure 2A).
Figure 2.

Influence of substrate binding on the metal-oxo orientation in TauD. Active site structures are shown for chains (A) A and (B) B with corresponding 2F0 − Fc electron density maps contoured at 1.0σ around key residues and substrates. Density surrounding spheres (e.g., vanadyl) is contoured at 0.7σ for the sake of clarity. See Figure S1A,B for ligand omit maps. Minimal changes are observed between the vanadyl complex with taurine and succinate bound (gray) and (C) the reactant complex10 (turquoise, PDB entry 1OS7, chain A). However, ordering of Arg270 upon taurine binding appears to promote in-line oxo formation via H-bonding interactions similar to those observed in (D) the VioC·(VIVO)·l-Arg·succinate structure13 (turquoise, PDB entry 6ALR). Sequence-independent alignments were performed over (C) 237 and (D) 116 Cα atoms and resulted in root-mean-square deviations of 0.26 and 3.21 Å, respectively.
Chains A and B overlay well with previously determined structures of the substrate-free [chain D of Protein Data Bank (PDB) entries 1OTJ and 1OS7] and substrate-bound (PDB entries 1GY9 and 1GQW, as well as chains A−C of entries 1OTJ and 1OS7) enzyme, respectively, having root-mean-square deviations (rmsds) of the peptide backbone of <0.33 Å over 224 Cα atoms.8,10 Conformational heterogeneity observed upon alignment of the monomers is confined to the N-terminal lid-loop region and reflects changes previously associated with taurine binding (Figure S1C).10 Moreover, the network of H-bonding interactions with the taurine amine and sulfonate moieties is the same as in the reactant complex (PDB entry 1OS7, chains A−C).10 The succinate binding mode, however, is of particular interest as no structure of TauD in complex with its coproduct has been reported, and computationally derived models have been unable to reach a consensus regarding its binding mode.17 In the vanadyl complex, succinate is best modeled as a bidentate ligand, resulting in a distorted six-coordinate geometry with vanadium−ligand distances ranging from 2.06 to 2.17 Å. This observation contrasts with the more asymmetric succinate coordination environment described for VioC, in which the two V−O interactions are best modeled at 2.15 and 2.34 Å. The latter value is outside the range of typical metal−O ligand bond distances.13 Comparison of the vanadyl-substituted structures of the two enzymes reveals surprising heterogeneity in the coordination geometry given their similar reactivity. Nevertheless, the observed asymmetric bidentate binding mode in the TauD vanadyl structure is consistent with Mössbauer and density functional theory studies of the ferryl intermediate in this system, as well as recent X-ray absorption spectroscopic studies (Figure S2A).12,17 All observations are consistent with a six-coordinate distorted octahedral complex.
A bidentate succinate ligand further obligates the oxo ligand of vanadyl to the in-line site, trans to the distal His of the conserved HXD/E…H facial-triad metal-binding motif (Figure 2B).18 Such an orientation situates the oxo ligand proximal to the target carbon, in preparation for the hydroxylation reaction. Although the stereoselectivity of TauD has yet to be conclusively determined, the pro-R hydrogen of C1 is directed downward in the vanadyl complex, poised for abstraction by the nearby oxo species, in agreement with other substrate-bound TauD reactant complex structures.8,10 The associated C···O distance in TauD is approximately 2.8 Å, noticeably shorter than that determined for VioC (~3.1 Å) (Figure S2B),13 perhaps enabled by downward rotation of the acidic metal-coordinating residue (Asp101) and second-sphere electrostatic interaction between Arg270 and the negatively charged substrate (Figure 2B). Despite elongation of the V−oxo bond due to photoreduction of the metal center, the C1−H···VIV−oxo distances and angles in crystallo show good agreement with hyperfine sublevel correlation (HYSCORE) and X-ray absorption fine structure (EXAFS) models of the TauD·(VIV-oxo)·taurine·succinate complex (Figure 3).12 The outer-sphere active site residues deviate minimally from their positions in the reactant complex (Figure 2C). However, Arg270, a dynamic residue implicated in stabilization of the in-line oxo in vanadyl-VioC structures,13,19 becomes ordered upon taurine binding (Figure 2A,B).
Figure 3.

Comparison of crystallographic and spectroscopic models of the ferryl mimic. Key bond angles and distances derived from HYSCORE/EXAFS (black)12 are juxtaposed to those from the chain B active site (turquoise). Depicted hydrogens were added in standard geometries using phenix.reduce.
The Arg270 side chain likely stabilizes the in-line metal-oxo configuration via H-bonding to O3 of succinate. Detailed analysis of its geometry, however, suggests that the conserved second-sphere residue does not directly H-bond with the oxo ligand even though it is located at a suitable distance.13,20 Rather, its interactions with the taurine sulfonate moiety and 2OG/succinate likely indirectly influence the Fe−O geometry through steric crowding and ionic bonding (Figure 2B). This phenomenon was also observed in structures of the hydroxylase VioC with vanadyl bound (Figure 2D)13,19 and contrasts with models of Fe/2OG halogenase ferryl complexes, in which an off-line geometry stabilized by direct H-bonding interactions has been proposed.6,7 Such donation of H-bonds to the oxo ligand in reactive FeIV=O intermediates of model complexes has been shown to increase the activation barrier to H atom transfer reactivity.21 This distinction may partly explain observed differences in the rates of H atom abstraction in the reactions of hydroxylases and halogenases. However, the most significant factor is likely the increased distance between the oxo ligand and substrate HAT target, with H-bonding playing a more indirect role in stabilizing the off-line ferryl configuration that gives rise to this scenario.22 Nevertheless, crystallographic application of the vanadyl probe thus provides new opportunities to directly visualize subtle second-sphere interactions that may influence ferryl reactivity.
Taken together, the structural models of hydroxylases TauD and VioC complexed with vanadyl provide strong support for previous mechanistic proposals suggesting oxygen rebound to the substrate radical after H atom abstraction by an in-line oxo species. In both cases, this active site geometry is stabilized, in part, by interactions with second-sphere residues. Fewer conformational changes are observed in the TauD ferryl mimic relative to the reactant complex than in the corresponding VioC complexes. However, rearrangement of a conserved active site arginine is equally critical for orientation of the target carbon proximal to the reactive intermediate at distances compatible with H atom transfer (Figure 2D and Figure S2B). While attempts to visualize the true ferryl species in VioC resulted in detection of a peroxysuccinate precursor, the limited reorganization of active site residues required to generate the TauD·(VIV-oxo)·taurine·succinate complex suggests a lower barrier to ferryl formation in this archetypal hydroxylase. Perhaps future experiments to trap the ferryl intermediate in TauD by crystallographic methods would be more successful as a consequence.
Quite unexpectedly, chains A and B of this TauD structure do not share the same vanadyl-oxo orientation. The oxygen adduct is instead coplanar with the metal ion and the first two residues in the facial-triad in the substrate/product-free monomer (Figure 2A). The position of the oxo ligand was determined by modeling waters at all relevant sites around the vanadium ion and relaxing their restraints. Upon refinement, one of the equatorial waters settles a very short distance (~1.82 Å) from the metal, inconsistent with a simple aqua ligand. We assign this position as the nominal vanadyl oxo ligand, with an increased V−O distance due to photoreduction. In contrast, the ligands in the other two coordination positions refine to distances more typical of water ligands (2.04−2.12 Å). This model resembles an off-line metal-oxo; however, instead of being trans to the proximal His, the oxo ligand occupies an unorthodox position trans to Asp101. Upon generation of the true ferryl intermediate, the initial 2OG coordination mode necessitates occupancy of this position by succinate. It follows that a true off-line species, should it occur in TauD, can be located only trans to the proximal His99.
An increased level of disorder of the Arg270 guanidino group observed in the absence of taurine would weaken the H-bonding interactions expected to stabilize succinate coordination (Figure 2), perhaps opening the coordination site trans to His99. The related alkylsulfatase, AtsK, provides precedent for such a monodentate succinate binding mode in the absence of primary substrate (Figure S3).23,24 We hypothesize that these features could allow an off-line ferryl to form unproductively in TauD under these circumstances. For example, uncoupled reactions occurring in the absence of a primary substrate have been shown to result in self-hydroxylation of TauD and other members of the superfamily (Figure S4).8,25–27 Modifications of Tyr73, as well as Trp128, Trp240, and Trp248, members of the putative Tyr and Trp electron transfer pathways proposed by Winkler and Gray,28 have all been observed; however, only Tyr modification is readily rationalized by an in-line oxo (Figure 4).8,26,29,30 Off-line orientation of the ferryl intermediate trans to His99, by contrast, would locate the oxo ligand at a suitable distance, approximately 4.0 Å from the benzene ring of Trp248, for self-hydroxylation of sites distal to an in-line oxo (Figure 4B). An off-line oxo/hydroxo formed in the absence of taurine might also be relevant for one-electron chemistry to rescue uncoupled iron oxidation.
Figure 4.

Self-hydroxylation behavior in TauD is rationalized by both in-line and off-line configurations of the ferryl intermediate. (A) The phenolic OH of Tyr73 is positioned proximal to the in-line oxo. (B) Formation of an off-line species trans to His99 would locate the oxo within a suitable distance for modification of the putative Trp radical transfer pathway.28 (C) The Trp pathway is conserved among related Fe/2OG enzymes such as AtsK and TfdA. Residues that have been observed hydroxylated are colored red.8,30
The unusual orientation of the vanadyl oxo ligand observed in chain A raises another interesting point, namely that the oxo ligand of the ferryl intermediate can potentially occupy three other coordination sites in certain Fe/2OG enzymes. In addition to the in-line orientation, there is also evidence suggesting productive use of open equatorial positions. Active site rearrangements precipitated by loss of CO2 from the 2OG cosubstrate, coordinated trans to the distal His, have been proposed to suppress hydroxylation in Fe/2OG-dependent halogenases. It is also possible that additional rearrangements of halide, small molecule, or amino acid ligands could take place at intermediate stages of the reaction to open other positions for ferryl occupancy. These possibilities motivate continued efforts to trap and mimic intermediates in X-ray crystal structures, particularly in noncanonical Fe/2OG enzymes. The observation of an off-line-like structure in TauD forecasts the utility of the vanadyl probe in elucidating the mechanistic behavior of these nonhydroxylating systems.
CONCLUSION
In conclusion, we present further evidence supporting the use of vanadyl as a structural mimic for the Fe(II)/2OG-oxygenase ferryl intermediate, while providing new insight into the formation of the reactive species in TauD. Observation of an off-line-like oxo in the absence of substrate not only helps to rationalize observed self-hydroxylation behavior, but also lends further credence to proposals suggesting that such a configuration could occur in more catalytically relevant contexts in which the outcome is not simple hydroxylation.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02–06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Funding
K.M.D. is grateful for support from the National Institutes of Health (NIH) K99 Pathway to Independence Award (1K99GM129460–01). R.J.M. was supported by the National Science Foundation Graduate Research Fellowship Program under Grant DGE1255832. This work was additionally supported by NIH Grants GM119707 to A.K.B., GM69657 to J.M.B. and C.K., and GM127079 to C.K. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00598.
Materials and methods, including information about the overexpression and purification of TauD, crystallization procedures, as well as data collection and processing; Figures S1−S4; and Table S1 (PDF)
Accession Codes
PDB entry 6EDH, UniProt entry P37610.
The authors declare no competing financial interest.
REFERENCES
- (1).Cochrane RVK, and Vederas JC (2014) Highly selective but multifunctional oxygenases in secondary metabolism. Acc. Chem. Res 47, 3148–3161. [DOI] [PubMed] [Google Scholar]
- (2).Lewis JC, Coelho PS, and Arnold FH (2011) Enzymatic functionalization of carbon-hydrogen bonds. Chem. Soc. Rev 40, 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Price JC, Barr EW, Tirupati B, Bollinger JM Jr., and Krebs C (2003) The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: A high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (4).Krebs C, GalonicĆ Fujimori D, Walsh CT, and Bollinger JM Jr. (2007) Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bollinger JM Jr., Chang W. c., Matthews ML, Martinie RJ, Boal AK, and Krebs C (2015) Mechanisms of 2-oxoglutarate-dependent oxygenases: The hydroxylation paradigm and beyond In 2-Oxoglutarate-Dependent Oxygenases, pp 95–122, The Royal Society of Chemistry, Cambridge, U.K. [Google Scholar]
- (6).Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, and Boal AK (2016) Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat. Chem. Biol 12, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB III, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM Jr., and Solomon EI (2013) Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Elkins JM, Ryle MJ, Clifton IJ, Dunning Hotopp JC, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, and Roach PL (2002) X-ray crystal structure of Escherichia coli taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry 41, 5185–5192. [DOI] [PubMed] [Google Scholar]
- (9).Knauer SH, Hartl-Spiegelhauer O, Schwarzinger S, Hänzelmann P, and Dobbek H (2012) The Fe(II)/α-ketogluta-rate-dependent taurine dioxygenases from Pseudomonas putida and Escherichia coli are tetramers. FEBS J. 279, 816–831. [DOI] [PubMed] [Google Scholar]
- (10).O’Brien JR, Schuller DJ, Yang VS, Dillard BD, and Lanzilotta WN (2003) Substrate-induced conformational changes in Escherichia coli taurine/α-ketoglutarate dioxygenase and insight into the oligomeric structure. Biochemistry 42, 5547–5554. [DOI] [PubMed] [Google Scholar]
- (11).Bollinger JM Jr., and Krebs C (2006) Stalking intermediates in oxygen activation by iron enzymes: Motivation and method. J. Inorg. Biochem 100, 586–605. [DOI] [PubMed] [Google Scholar]
- (12).Martinie RJ, Pollock CJ, Matthews ML, Bollinger JM Jr., Krebs C, and Silakov A (2017) Vanadyl as a stable structural mimic of reactive ferryl intermediates in mononuclear nonheme-iron enzymes. Inorg. Chem 56, 13382–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mitchell AJ, Dunham NP, Martinie RJ, Bergman JA, Pollock CJ, Hu K, Allen BD, Chang W. c., Silakov A, Bollinger JM Jr., Krebs C, and Boal AK (2017) Visualizing the reaction cycle in an iron(II)- and 2-(oxo)-glutarate-dependent hydroxylase. J. Am. Chem. Soc 139, 13830–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chekan JR, Ongpipattanakul C, Wright TR, Zhang B, Bollinger JM Jr., Rajakovich LJ, Krebs C, Cicchillo RM, and Nair SK (2019) Molecular basis for enantioselective herbicide degradation imparted by aryloxyalkanoate dioxygenases in transgenic plants. Proc. Natl. Acad. Sci. U. S. A 116, 13299–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Eichhorn E, van der Ploeg JR, Kertesz MA, and Leisinger T (1997) Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J. Biol. Chem 272, 23031–23036. [DOI] [PubMed] [Google Scholar]
- (16).Henderson KL, Müller TA, Hausinger RP, and Emerson JP (2015) Calorimetric assessment of Fe2+ binding to α-ketoglutarate/taurine dioxygenase: Ironing out the energetics of metal coordination by the 2-His-1-carboxylate facial triad. Inorg. Chem 54, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM Jr., Neese F, and Krebs C (2007) Spectroscopic and computational evaluation of the structure of the high-spin Fe(IV)-oxo intermediates in taurine: A-ketoglutarate dioxygenase from Escherichia coli and its His99Ala ligand variant. J. Am. Chem. Soc 129, 6168–6179. [DOI] [PubMed] [Google Scholar]
- (18).Hegg EL, and Que L Jr. (1997) The 2-His-1-carboxylate facial triad — an emerging structural motif in mononuclear non heme iron(II) enzymes. Eur. J. Biochem 250, 625–629. [DOI] [PubMed] [Google Scholar]
- (19).Dunham NP, Chang W. c., Mitchell AJ, Martinie RJ, Zhang B, Bergman JA, Rajakovich LJ, Wang B, Silakov A, Krebs C, Boal AK, and Bollinger JM Jr. (2018) Two distinct mechanisms for C-C desaturation by iron(II)- and 2-(oxo)glutarate-dependent oxygenases: Importance of α-heteroatom assistance. J. Am. Chem. Soc 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kortemme T, Morozov AV, and Baker D (2003) An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol 326, 1239–1259. [DOI] [PubMed] [Google Scholar]
- (21).Xue G, Geng C, Ye S, Fiedler AT, Neese F, and Que L Jr. (2013) Hydrogen-bonding effects on the reactivity of [X-FeIII-O-FeIV=O] (X = OH, F) complexes toward C-H bond cleavage. Inorg. Chem 52, 3976–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, and Bollinger JM Jr. (2009) Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry 48, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kahnert A, and Kertesz MA (2000) Characterization of a sulfur-regulated oxygenative alkylsulfatase from Pseudomonas putida S-313. J. Biol. Chem 275, 31661–31667. [DOI] [PubMed] [Google Scholar]
- (24).Müller I, Stückl C, Wakeley J, Kertesz M, and Usón I (2005) Succinate complex crystal structures of the α-ketoglutarate-dependent dioxygenase AtsK: Steric aspects of enzyme self-hydroxylation. J. Biol. Chem 280, 5716–5723. [DOI] [PubMed] [Google Scholar]
- (25).Mantri M, Zhang Z, McDonough MA, and Schofield CJ (2012) Autocatalysed oxidative modifications to 2-oxoglutarate dependent oxygenases. FEBS J. 279, 1563–1575. [DOI] [PubMed] [Google Scholar]
- (26).Ryle MJ, Liu A, Muthukumaran RB, Ho RYN, Koehntop KD, McCracken J, Que L Jr., and Hausinger RP (2003) O2- and α-ketoglutarate-dependent tyrosyl radical formation in TauD, an α-keto acid-dependent non-heme iron dioxygenase. Biochemistry 42, 1854–1862. [DOI] [PubMed] [Google Scholar]
- (27).Liu A, Ho RYN, Que L Jr., Ryle MJ, Phinney BS, and Hausinger RP (2001) Alternative reactivity of an α-ketoglutarate-dependent iron(II) oxygenase: Enzyme self-hydroxylation. J. Am. Chem. Soc 123, 5126–5127. [DOI] [PubMed] [Google Scholar]
- (28).Winkler JR, and Gray HB (2015) Electron flow through biological molecules: Does hole hopping protect proteins from oxidative damage? Q. Rev. Biophys 48, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Koehntop KD, Marimanikkuppam S, Ryle MJ, Hausinger RP, and Que L Jr. (2006) Self-hydroxylation of taurine/α-ketoglutarate dioxygenase: Evidence for more than one oxygen activation mechanism. J. Biol. Inorg. Chem 11, 63–72. [DOI] [PubMed] [Google Scholar]
- (30).Ryle MJ, Koehntop KD, Liu A, Que L Jr., and Hausinger RP (2003) Interconversion of two oxidized forms of taurine/α-ketoglutarate dioxygenase, a non-heme iron hydroxylase: Evidence for bicarbonate binding. Proc. Natl. Acad. Sci. U. S. A 100, 3790–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.