

Summary
We report an ancient genome from the Indus Valley Civilization (IVC). The individual we sequenced fits as a mixture of people related to ancient Iranians (the largest component) and Southeast Asian hunter-gatherers, a unique profile that matches ancient DNA from 11 genetic outliers from sites in Iran and Turkmenistan in cultural communication with the IVC. These individuals had little if any Steppe pastoralist-derived ancestry, showing it was not ubiquitous in northwest South Asia during the IVC as it is today. The Iranian-related ancestry in the IVC derives from a lineage leading to early Iranian farmers, herders and hunter-gatherers before their ancestors separated, contradicting the hypothesis that the shared ancestry between early Iranians and South Asians reflects a large-scale spread of western Iranian farmers east. Instead, sampled ancient genomes from the Iranian plateau and IVC descend from different groups of hunter-gatherers who began farming without being connected by movement of people.
Keywords: Population Genetics, Archaeology, Anthropology, South Asia
In Brief
Skeletal DNA from a member of the ancient Indus Valley Civilization shows ancestry from ancient Iranians before their adoption of farming and from Southeast Asian hunter-gatherers, while completely lacking Steppe pastoralist ancestry.
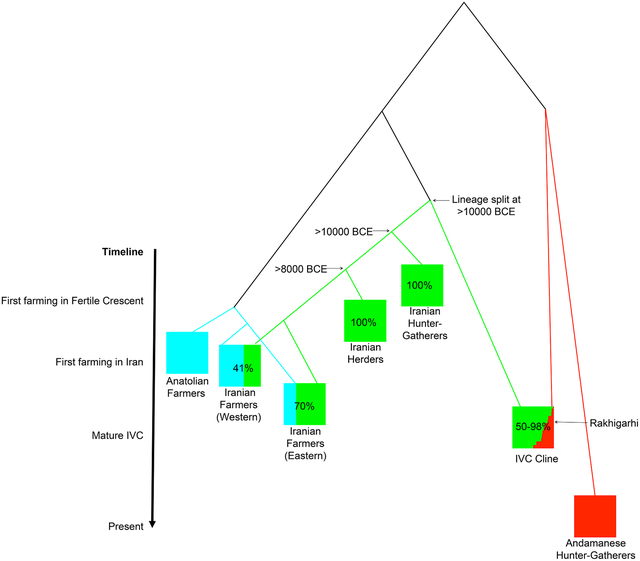
Graphical Abstract
Introduction
The mature Indus Valley Civilization (IVC), also known as the Harappan Civilization, was spread over northwestern South Asia from 2600–1900 BCE and was one of the first large-scale urban societies of the ancient world, characterized by systematic town planning, elaborate drainage systems, granaries, and standardization of weights and measures. The inhabitants of the IVC were cosmopolitan, with multiple cultural groups living together in large regional urban centres like Harappa (Punjab), Mohenjo-daro (Sindh), Rakhigarhi (Haryana), Dholavira (Kutch/Gujarat) and Ganweriwala (Cholistan) (Figure 1A) (Mughal, 1990; Possehl, 1982; Possehl, 1990; Shaffer and Lichtenstein, 1989; Thapar, 1979). Rakhigarhi, the largest known IVC site, was spread over 550 hectares (Figure 1B,C), and seven dates from charcoal at depths of 9–23 meters have point estimates of 2800–2300 BCE, which largely fall within the mature phase of the IVC (Shinde et al., 2018; Vahia et al., 2016). As part of the archaeological effort we attempted to generate ancient DNA data for a subset of the excavated burials.
Figure 1. Archeological context of the individual that yielded ancient DNA.

(A) We label the geographic location of the archaeological site of Rakhigarhi (blue) and other significant Harappan sites (red) to define the geographic range of the IVC. We label in black sites in the north and west with which IVC people were in cultural contact, and specifically highlight in yellow the sites of Gonur and Shahr-i-Sokhta, which are the source of the 11 outlier individuals who genetically form a cline of which the Rakhigarhi individual is a part. (B) Photograph of the I6113 burial (skeletal code RGR7.3, BR-01, HS-02) and associated typical IVC grave goods and illustrating typical North-South orientation of IVC burials. High resolution images of IVC-style ceramics associated with the grave are shown in Figure S1.
Results
In dedicated clean rooms, we obtained powder from 61 skeletal samples from the Rakhigarhi cemetery, which lies ~1 km west of the ancient town (Table S1). We extracted DNA (Dabney et al., 2013; Korlević et al., 2015) and converted the extracts into libraries (Rohland et al., 2015), some of which we treated with uracil-DNA glycosylase (UDG) to greatly reduce the error rates associated with the characteristic cystosine-to-uracil lesions of ancient DNA (Rohland et al., 2015). For a subset of libraries that passed basic laboratory quality controls showing they contained DNA, we enriched for sequences overlapping both the mitochondrial genome and ~3000 targeted nuclear positions (Mathieson et al., 2015), and sequenced the enriched libraries either on Illumina NextSeq500 instrument using paired 2×76 base pair (bp) reads, or on Illumina HiSeq X10 instruments using paired 2×150 bp reads. After trimming adapters and merging sequences overlapping by at least 15 bp (allowing up to one mismatch), we mapped to both the mitochondrial genome rsrs (Behar et al., 2012) and the human genome reference hg19 (Li and Durbin, 2010) (Table S1). After inspecting the screening results we enriched a subset of libraries for ~1.24 million single nucleotide polymorphisms (SNPs), and sequenced the enriched libraries and processed the data computationally (Fu et al., 2015; Haak et al., 2015; Mathieson et al., 2015). For the most promising sample, which had the genetic identification code I6113 and the archaeological skeletal code RGR7.3, BR-01, HS-02, we created, enriched, and sequenced a total of 108 libraries from 5 extractions to maximize the amount of data (Meyer et al., 2012; Rohland et al., 2015) (the initial library was UDG-treated, while all 107 subsequent libraries were not UDG-treated). After removing 40 libraries (from one extraction) that had significantly lower coverage as well as significantly lower damage rates compares to the other libraries, and merging data from the remaining 68 libraries, we had 208,111 SNPs covered at least once. Almost all of these 68 libraries showed cytosine-to-thymine mismatch rates to the human reference genome in the final nucleotide greater than 10%, consistent with the presence of authentic ancient DNA. However, when we stratified the pooled data by sequence length we found lower damage rates particularly for sequences of length >50 bp (Star Methods). Based on this evidence of length-dependent contamination, we restricted the data to molecules that showed cytosine-to-thymine mismatches characteristic of ancient DNA. This resulted in data at 31,760 SNPs. The ratio of sequences mapping to the Y chromosome to sequences mapping to both the Y and X chromosomes was in the range expected for a female, consistent with the morphology of the skeleton. After building a mitochondrial DNA consensus sequence, we determined that its haplogroup was U2b2, which is absent in data from about 400 ancient Central Asians and today is nearly exclusive to South Asia (Narasimhan et al., 2019).
In PCA (Figure 2A), I6113 projects onto a previously defined genetic gradient represented in 11 individuals from two sites in Central Asia in cultural contact with the IVC (3 from Gonur in present-day Turkmenistan and 8 from Shahr-i-Sokhta in far eastern Iran); these individuals were previously identified via a formal statistical procedure as significant outliers relative to the majority of samples at these two sites (they represent only 25% of the total) and were called the Indus Periphery Cline (Narasimhan et al., 2019). Despite having only modest SNP coverage, the error bars for the positioning of I6113 in the PCA are sufficiently small to show that this individual is not only significantly different in ancestry from the primary ancient populations of Bronze Age Gonur and Shahr-i-Sokhta, but also does not fall within the variation of the present-day South Asians. We obtained qualitatively consistent results when analyzing the data using ADMIXTURE (Alexander et al., 2009), with I6113 again similar to the 11 outlier individuals in harboring a mixture of ancestry related to ancient Iranians and tribal southern Indians. None of these individuals had evidence of “Anatolian farmer-related” ancestry, a term we use to refer to the lineage found in ancient genomes from 7th millennium BCE farmers from Anatolia (Mathieson et al., 2015). This Anatolian farmer-related ancestry was absent in all sampled ancient genomes from Iranian herders or hunter-gatherers dating from the 12th through the 8th millennia BCE, which instead carried a very different ancestry profile also present in mixed form in South Asia that we call “Iranian-related” (Broushaki et al., 2016; Lazaridis et al., 2016).
Figure 2. Population genetic analysis.

(A) PCA of ancient DNA from South and Central Asia projected onto a basis of whole genome sequencing data from present-day Eurasians. I6113 and I11042 (a non-outlier individual from the site of Gonur of similar data quality), are shown along with black error bars indicating 1 standard error as estimated using a chromosomal block jackknife. I6113’s position in the PCA is inconsistent with that of present-day South Asians and with Iranian groups, but is consistent with a set of 11 outliers who represent 25% of analyzed individuals at the sites of Gonur and Shahr-i-Sokhta and who with I6113 for the IVC Cline. (B) ADMIXTURE analysis of individuals from South and Central Asia shown with components in Green, Teal, Orange, Blue and Red maximized in Iranian farmers, Anatolian farmers, Eastern European Hunter-Gatherers, Western European Hunter-Gatherers, and Andamanese Hunter-Gatherers, respectively. (C) Estimated proportions of three ancestry profiles in ancient and present-day individuals. The three components are maximized in Middle to Late Bronze Age Steppe Pastoralists (Central_Steppe_MLBA), the reconstructed hunter-gatherer population of South Asia (represented by Andamanese Hunter-Gatherers (AHG) as proxy for with greatest relatedness to southeast Asian hunter-gatherers), and Indus_Periphery_West, an individual on the IVC Cline represented with the lowest proportion of AHG-related ancestry. Individuals that fit a two-way model of mixture between these three sources are shown on the triangle edges, whereas individuals that could only be fit with a three way model are shown in the interior. I6113 is shown on the IVC Cline as a red dot; the other IVC cline individuals are shown as orange dots; later individuals who formed as mixtures between people on the IVC Cline and people with Steppe ancestry are shown as green dots; and diverse modern South Asian groups who formed as a mixture of two later mixed groups are shown as blue dots
We used qpAdm to test highly divergent populations that have been shown to be effective for modeling diverse West and South Eurasian groups as potential sources for I6113 (Narasimhan et al., 2019). If one of these population fits, it does not mean it is the true source; instead, it means that it and the true source population are consistent with descending without mixture from the same homogeneous ancestral population that potentially lived thousands of years before. The only fitting two-way models were mixtures of a group related to herders from the western Zagros mountains of Iran, and also to either Andamanese Hunter-Gatherers (73 ± 6% Iranian-related ancestry; p=0.103 for overall model fit) or East Siberian Hunter-Gatherers (63 ± 6% Iranian-related ancestry; p=0.24 (the fact that the latter two populations both fit reflects that they have the same phylogenetic relationship to the non-West Eurasian-related component of I6113 likely due to shared ancestry deeply in time). This is the same class of models previously shown to fit the 11 outliers that form the Indus Periphery Cline (Narasimhan et al., 2019), and indeed, I6113 fits as a genetic clade with the pool of Indus Periphery Cline individuals in qpAdm (p=0.42). Multiple lines of evidence suggest that the genetic similarity of I6113 to the Indus Periphery Cline individuals is due to gene flow from South Asia rather than in the reverse direction. First, of the 44 individuals with good quality data we have from Gonur and Shahr-i-Sokhta, only 11 (25%) have this ancestry profile; it would be surprising to see this ancestry profile in the one individual we analyzed from Rakhigarhi if it was a migrant from regions where this ancestry profile was rare. Second, of the three individuals at Shahr-i-Sokhta that have material culture linkages to Baluchistan in South Asia, all are IVC Cline outliers, specifically pointing to movement out of South Asia (Narasimhan et al., 2019). Third, both the IVC Cline individuals and the Rakhigarhi individual have admixture from people related to present-day South Asians (ancestry deeply related to Andamanese hunter-gatherers) that is absent in the non-outlier Shahr-i-Sokhta samples and is also absent in Copper Age Turkmenistan and Uzbekistan (Narasimhan et al., 2019), implying gene flow from South Asia into Shahr-i-Sokhta and Gonur, whereas our modeling does not necessitate reverse gene flow. Based on these multiple lines of evidence it is reasonable to conclude that individual I6113’s ancestry profile was widespread among people of the IVC at sites like Rakhigarhi, and supports the conjecture (Narasimhan et al., 2019), that the 11 outlier individuals in the Indus Periphery Cline are migrants from the IVC living in non-IVC towns. We rename the genetic gradient represented in the combined set of 12 individuals the “IVC Cline,” and then use higher coverage individuals from this cline in lieu of I6113 to carry out fine-scale modeling of this ancestry profile.
Modeling the individuals on the IVC Cline using the two-way models previously fit for diverse present-day South Asians (Narasimhan et al., 2019), we find that as expected from the PCA it does not fit the two-way mixture that drives variation in modern South Asians as it is significantly depleted in Steppe pastoralist-related ancestry adjusting for its proportion of Iranian-related ancestry (p=0.018 from a two-sided Z-test). Modeling the IVC Cline using the simpler two-way admixture model without Steppe pastoralist-derived ancestry previously shown to fit the 11 outliers (Narasimhan et al., 2019), I6113 falls on the more Iranian-related end of the gradient, revealing that Iranian-related ancestry extended to the eastern geographic extreme of the IVC, and was not restricted to individuals at its Iranian and Central Asian periphery. The estimated proportion of ancestry related to tribal groups in southern India in I6113 is smaller than in present-day groups, suggesting that since the time of the IVC there has been gene flow into the part of South Asia where Rakhigarhi lies from both the northwest (bringing more Steppe ancestry) and southeast (bringing more ancestry related to tribal groups in southern India). The genetic profile that we document in this individual, with large proportions of Iranian-related ancestry, but no evidence of Steppe pastoralist-related ancestry, is no longer found in modern populations of South Asia or Iran, providing further validation that the data we obtained from this individual reflects authentic ancient DNA.
To obtain insight into the origin of the Iranian-related ancestry in the IVC Cline, we co-modeled the highest-coverage individual from the IVC Cline (who also happens to have the highest proportion of Iranian-related ancestry) with other ancient individuals from across the Iranian plateau representing early hunter-gatherer and food producing groups: a ~10000 BCE individual from Belt Cave in the Alborsz Mountains, a pool of ~8000 BCE early goat herders from Ganj Dareh in the Zagros Mountains, a pool of ~6000 BCE farmers from Hajji Firuz in the Zagros Mountains, and a pool of ~4000 BCE farmers from Tepe Hissar in Central Iran. Using qpGraph (Patterson et al., 2012), we tested all possible simple trees relating the Iranian-related ancestry component of these groups, accounting for known admixtures (Anatolian farmer-related admixture into Hajji Firuz and Tepe Hissar, and Andamanese Hunter-Gatherer-related admixture in the IVC Cline), using an acceptance criterion for the model fitting that the maximum |Z|-scores between observed and expected f-statistics was <3, or that the Akaike Information Criterion (AIC) was within 4 of the best-fit (Burnham and Anderson, 2004). The only consistently fitting models specified the IVC Cline Iranian-related ancestry lineage as splitting before the other Iranian-related lineages separated from each other (Figure 3 represents one such model consistent with our data). We confirmed this result by applying symmetry tests to evaluate the relationships among the Iranian-related lineages, correcting for the effects of Anatolian farmer-related, Andamanese hunter-gatherer-related, and West Siberian hunter-gatherer-related admixture (Star Methods). We find that 94% of the resulting trees supported the Iranian-related lineage in the IVC Cline being the first to separate from the other lineages, consistent with our modeling results.
Figure 3. Best-fitting admixture graph relating populations with Iranian-related ancestry.

The Iranian-related subtree is shown in green, the Anatolian farmer-related subtree in blue, the southeast Asian-related subtree in red, and mixed populations as pie charts (other fitting topologies all share the feature that the IVC Cline descends from the first-splitting lineage in the subtree of Iranian-related ancestry). The dates of the analyzed populations are shown on the vertical axis and provide minima on the population split dates. The observation that the Iranian-related lineage contributing to the IVC Cline split earlier than Belt Cave at ~10000 BCE and Ganj Dareh at ~8000 BCE is incompatible with the hypothesis that the advent of farming in South Asia after ~7000–6000 BCE was associated with a large-scale eastward migration bringing ancestry from people related to western Zagros mountain farmers or herders across the Iranian plateau to South Asia.
Our evidence that the Iranian-related ancestry in the IVC Cline diverged from lineages leading to ancient Iranian hunter-gatherers, herders, and farmers prior to their ancestors’ separation places constraints on the spread of Iranian-related ancestry across the combined region of the Iranian plateau and South Asia, where it is represented in all ancient and modern genomic data sampled to date. The Belt Cave individual dates to ~10000 BCE, before the advent of farming anywhere in the world, which implies that the split leading to the Iranian-related component in the IVC Cline predates the advent of farming as well (Figure 3). Even if we do not consider the results from the low-coverage Belt Cave individual, our analysis shows that the Iranian-related lineage present in the IVC Cline individuals split before the date of the ~8000 BCE Ganj Dareh individuals, who lived in the Zagros mountains of the Iranian plateau before crop farming began there around ~7000–6000 BCE. Thus, the Iranian-related ancestry in the IVC Cline descends from a different group of hunter-gatherers from the ancestors of the earliest known farmers or herders in the western Iranian plateau. We also highlight a second line of evidence against the hypothesis that eastward migrations of descendants of western Iranian farmers or herders were the source of the Iranian-related ancestry in the IVC Cline. An independent study has shown that all ancient genomes from Neolithic and Copper Age crop farmers of the Iranian plateau harbored Anatolian farmer-related ancestry not present in the earlier herders of the western Zagros (Narasimhan et al., 2019). This includes western Zagros farmers (~59% Anatolian farmer-related ancestry at ~6000 BCE at Hajji Firuz) and eastern Alborsz farmers (~30% Anatolian farmer-related ancestry at ~4000 BCE at Tepe Hissar). That the 12 sampled individuals from the IVC Cline harbored negligible Anatolian farmer-related ancestry thus provides an independent line of evidence (in addition to their deep-splitting Iranian-related lineage that has not been found in any sampled ancient Iranian genomes to date) that they did not descend from groups with ancestry profiles characteristic of all sampled Iranian crop-farmers (Narasimhan et al., 2019). While there is a small proportion of Anatolian farmer-related ancestry in South Asians today, it is consistent with being entirely derived from Steppe pastoralists who carried it in mixed form and who spread into South Asia from ~2000–1500 BCE (Narasimhan et al., 2019).
Discussion
These findings suggest that in South Asia as in Europe, the advent of farming was not mediated directly by descendants of the world’s first farmers who lived in the fertile crescent. Instead, populations of hunter-gatherers--in Western Anatolia in the case of Europe (Feldman et al., 2019), and in a yet-unsampled location in the case of South Asia--began farming without large-scale movement of people into these regions. This does not mean that movements of people were unimportant in the introduction of farming economies at a later date: for example, ancient DNA studies have documented that the introduction of farming to Europe after ~6500 BCE was mediated by a large-scale expansion of Western Anatolian farmers who descended largely from early hunter-gatherers of western Anatolia (Feldman et al., 2019). It is possible that in an analogous way, an early farming population expanded dramatically within South Asia causing large-scale population turnovers that helped to spread this economy within the region; whether this occurred is still unverified and could be determined through ancient DNA studies from just before and after the farming transitions in South Asia.
Our results also have linguistic implications. One theory for the origins of the now-widespread Indo-European languages in South Asia is the “Anatolian hypothesis,” which posits that the spread of these languages was propelled by movements of people from Anatolia across the Iranian plateau and into South Asia associated with the spread of farming. However, we have shown that the ancient South Asian farmers represented in the IVC Cline had negligible ancestry related to ancient Anatolian farmers, as well as an Iranian-related ancestry component distinct from sampled ancient farmers and herders in Iran. Since language spreads in pre-state societies are often accompanied by large-scale movements of people (Bellwood, 2013) these results argue against the model (Heggarty, 2019) of a trans-Iranian-plateau route for Indo-European language spread into South Asia. However, a natural route for Indo-European languages to have spread into South Asia is from Eastern Europe via Central Asia in the first half of the 2nd millennium BCE, a chain-of-transmission now documented in detail with ancient DNA. The fact that the Steppe pastoralist ancestry in South Asia matches that in Bronze Age Eastern Europe (but not Western Europe (de Barros Damgaard et al., 2018; Narasimhan et al., 2019)) provides additional evidence for this theory, as it elegantly explains the distinctive shared distinctive features of Balto-Slavic and Indo-Iranian languages (Ringe et al., 2002)
Our analysis of data from one individual from the IVC, in conjunction with 11 previously reported individuals from sites in cultural contact with the IVC, demonstrates the existence of an ancestry gradient that was widespread in farmers in the northwest of peninsular India at the height of the IVC, that had little if any genetic contribution from Steppe pastoralists or western Iranian farmers or herders, and that had a primary impact on the ancestry of later South Asians. While our study is sufficient to demonstrate that this ancestry type was a common feature of the IVC, a single sample--or even the gradient of 12 likely IVC samples we have identified--cannot fully characterize a cosmopolitan ancient civilization. An important direction for future work will be to carry out ancient DNA analysis of additional individuals across the IVC range to obtain a quantitative understanding of how the ancestry of IVC people was distributed, and to characterize other features of its population structure.
STAR Methods
Lead Contact and Materials Availability
This study did not generate new unique reagents. Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, David Reich (reich@genetics.med.harvard.edu).
Experimental Model and Subject Details
We attempted to generate genome-wide data from skeletal remains of 61 ancient individuals from the IVC site of Rakhigarhi. Only a single sample yielded enough authentic ancient DNA for analysis: I6113, Rakhigarhi, Haryana, India (n=1). We report the archeological context dates for this burial in Method Details. The skeletal samples from Rakhigarhi were excavated by the archaeological team led by V.S. at the Deccan College Post-Graduate and Research Institute in Pune India and sampled by the ancient DNA group led by N.Ra. at the Birbal Sahni Institute of Palaeosciences in Lucknow India. Analysis using the methods implemented by the ancient DNA group led by D.R. at Harvard Medical School in Boston USA was approved by a Memorandum of Understanding between Deccan College and Harvard Medical School executed in February 2016.
Method Details
Contextual date for individual I6113
There is insufficient collagen preservation for the human bones at Rakhigarhi cemetery to allow Accelerator Mass Spectrometry radiocarbon dating; multiple attempts showed a carbon-to-nitrogen ratio outside the range appropriate for dating, including five attempts we made specifically on skeletal elements from I6113. However, the cemetery can be securely dated based on archaeological context. First, the only evidence of human occupation of the site is in the Harappan period and hence all the excavated remains are likely to belong to that period. Second, all the characteristic features of the Harappan burial customs and features are present in the cemetery, including a separation from the main habitation area (about 1 kilometer), and typical Harappan artifacts including pots, beads made of semi-precious stones, and bangles of copper, shell or terracotta, all of which are indistinguishable from artifacts found in the main habitation area. As discussed in the text, the main habitation area has 7 radiocarbon dates based on charcoal spanning 2800–2300 BCE, largely falling within the mature IVC (Shinde et al., 2018; Vahia et al., 2016). Third, the pottery associated with the burial (Figure S1), appear to be stylistically similar to others made during the mature Harappan period.
Ancient DNA Data Generation
Excel Table Titles and Legends Table S 1 presents details of genetic results on the 251 libraries we generated on 61 distinct samples. To represent I6113, we generated data from 108 libraries (27 double stranded (Rohland et al., 2018) and 81 single stranded (Meyer et al., 2014; Rohland et al., 2015)), and then filtered out 40 single-stranded libraries (all the libraries from a single extraction) that had extremely low coverage and low levels of the cytosine-to-thymine mismatch to the human reference sequence expected for authentic ancient DNA. For the remaining 68 libraries, we restricted the data to sequences with evidence of characteristic ancient DNA damage in the final nucleotide using PMDtools (Skoglund et al., 2014).
Assessing samples for authenticity of ancient DNA
Based upon the rate of cytosine-to-thymine mismatches to the reference sequence in the final nucleotide of the libraries (Dabney et al., 2013; Korlević et al., 2015), we prioritized the individual with relatively high ancient rates of characteristic damage, I6113 for additional library preparation and sequencing (Excel Table Titles and Legends Table S 1).
For I6113 we generated a total of 27 double stranded libraries (Rohland et al., 2018) (1 UDG-treated and 26 not UDG-treated) using powder from both the otic capsule and semicircular canals of the petrous bone, and also generated an additional 81 single stranded libraries (all non-UDG-treated) using powder from the semicircular canal and one of two different extraction procedures (Meyer et al., 2014; Rohland et al., 2015). Out of these 108 libraries, nearly all of the 40 made from single-stranded libraries prepared using the extract made with Buffer G (Korlević et al., 2015) had low coverage (<100 targeted SNPs covered) and low damage in the final nucleotide (Excel Table Titles and Legends Table S 1), consistent with the extreme sensitivity of extracts made using this buffer to inhibition especially for single-stranded libraries (Korlević et al., 2015). We therefore removed all libraries prepared from this extract from analysis and proceeded with the remaining 68.
The number of DNA sequences obtained from each library of I6113 was insufficient for assessment of contamination on a per-library basis. We therefore examined the datasets obtained by pooling 208,111 sequences across the 68 libraries for I6113. Examining the number of sequences mapping to the Y chromosome as a fraction of that mapping to both the X and Y, we found a ratio of 0.047. On data for many other ancient individuals subject to ~1.24 million SNP enrichment, we have empirically found that this ratio is less than about 0.03 for uncontaminated libraries from females, and above 0.35 for uncontaminated males. Thus, I6113 has evidence of a mixture of human DNA from both males and females, and thus contamination.
To identify subsets of the molecules that are highly likely to be authentic, we analyzed the fraction of sequences that retained typical signatures of ancient DNA damage based on a characteristic cytosine-to-thymine mismatches to a reference sequence at their ends (Meyer et al., 2014; Skoglund et al., 2014), stratified by the lengths of the molecules (Figure S2A,B). We carried out this analysis not only for I6113, but also for a previously published ancient DNA sample from Southeast Asia from a similar time period (I4011) comprised of a merge of data from 21 double-stranded libraries (Lipson et al., 2018). The libraries from I6113 have high rate of damage (up to ~50%) indicative of a high proportion of genuine ancient DNA. The rate of damage for I6113 decreases for lengths greater that 40 bp, suggesting that longer molecules are more likely to be contaminated. We further found that sequences that were damaged on one end of the ancient DNA molecules (showing cytosine-to-thymine (C-to-T) mismatches relative to the reference sequence) also had an enhanced chance of damage on the other, as expected if damage restriction enriches for authentic DNA (Meyer et al., 2014) (Figure S2C).
To maximize the number of SNPs available for analysis while minimizing contamination, we analyzed multiple subsets of sequences for I6113, restricting to ones with characteristic ancient DNA damage in the final nucleotides. The resulting dataset contains sequences covering 31,760 SNPs at least once. Its ratio of Y chromosome sequences to X+Y chromosome sequences is 0.026, consistent with being an uncontaminated female (and the anthropological determination).
Autosomal Contamination Estimates
We estimated contamination using an algorithm based on breakdown of linkage disequilibrium (Posth et al., 2018). This software measures contamination levels by comparing the haplotype distribution of a tested sample to the haplotype distribution of an external reference panel. We used Sri Lankan Tamils sampled from the United Kingdom (STU) from the 1000 Genomes Project (Auton et al., 2015) as the reference panel. The algorithm was run in the usually conservative “uncorrected” mode to attain maximal power.
Quantification and Statistical Analysis
ADMIXTURE clustering analysis
We pruned the data using PLINK2 to retain only sites for which at least 70% of individuals had a non-missing genotype (Chang et al., 2015). We then ran ADMIXTURE (Alexander et al., 2009) with 10 replicates and report the replicate with the highest likelihood. In Figure 2, we show the results for clustering using K=6 components.
f-statistics
We computed f-statistics using the packages in ADMIXTOOLS (Patterson et al., 2012). To test for admixture we ran f3-statistics using the inbreed:YES parameter with an ancient population as a target. To estimate the ancestry proportion for a test population given a set of source populations and a set of outgroups, we used the qpAdm methodology (18) in ADMIXTOOLS.
Hierarchical modeling
To model a given sample as part of an established genetic cline determined by a set of other populations, we used an approach described in (Narasimhan et al., 2019) (the Supplementary Materials of (Narasimhan et al., 2019) gives the full details). We begin by obtaining ancestry proportions for a set of samples on a genetic cline, and jointly model these in a single generative model taking advantage of the fact that the proportions for the three ancestral sources must sum to 1. We estimate the mean and covariance of these sources using a bivariate normal distribution via maximum likelihood. We evaluate if the test population can be fit as deriving from the same original three sources as those we just modeled on the genetic cline using qpAdm, and if there is a fit, evaluate if the observed ratios of the ancestry proportions of the test population fit with the expected values from the generative model established by the cline. We compute a p-value based on the empirically determined covariance matrices.
Determination of the phylogeny of Iranian-related ancestry
We wished to examine the relationship of the Iranian-related ancestry present in the IVC Cline to that of ancient Iranian plateau individual reported in the ancient DNA literature.
We first focused on a set of populations chosen to represent a diverse group of early hunter-gatherers and farmers from across the geographic area of present day Iran. Our approach was to identify a set of allowable phylogenies and then, based on the known dates of the samples, to make inferences on minimum split times between lineages.
The individuals or groups of individuals we examined were:
Belt_Cave_M (BC) (n=1) – A Mesolithic individual from the Alborz mountains of Central Iran. Due to the evidence of contamination in the data from this individual, we used a damage restricted version of the sample resulting in 30,722 SNPs.
Ganj_Dareh_N (GD) (n=8) – Early goat herders from the Zagros Mountains of western Iran. The highest coverage individual has data from 938,523 SNPs.
Hajji_Firuz_C (HF) (n=5) – Late Neolithic and early Copper Age individuals from the Zagros Mountains of Western Iran. The highest coverage individual has data from 916,581 SNPs.
Tepe_Hissar_C (TH) (n=12) – Copper Age individuals from the Central Iranian Plateau. The highest coverage individual has data from 745,066 SNPs.
Indus_Periphery_West (IP) (n=1) – Member of the IVC Cline which includes the Rakhigarhi individual I6113. We represent the Iranian-related ancestry in this cline with I8728, the individual with the highest Iranian-related ancestry and also the highest coverage on this cline with data from 657,401 SNPs.
As documented in ref. (Narasimhan et al., 2019), the Hajji Firuz and Tepe Hissar pools of individuals have evidence of admixture related to Anatolian farmers while the Indus Periphery individuals (of which we show several including the individual with the highest West Eurasian-related ancestry I8728) have significant proportions of ancestry related to southeast Asian hunter-gatherers.
Building scaffolds of all possible topologies of Iranian-related ancestry
We were interested in understanding the relationship of the Iranian-related component of the ancestry of these 5 populations, treating the non-Iranian-related components such as the Anatolian farmer-related ancestry as nuisances that we need to model out assuming a topology in which the lineages lead to Tepe Hissar (PTA) and Hajji Firuz (PHA) formed a separate clade from Anatolian farmers (in the next sub-section we show that our results are robust to the choice of the topology relating the Anatolian farmer-related lineages).
There are 3 distinct topologies according to which these 5 populations could be related, which we call “Serial Founder” (Figure S3A), “Single Outgroup” (Figure S3B), and “Two Clades” (Figure S3C). Within these topologies where are multiple permutations for how the 5 individual populations could relate, depending on how the 5 Iranian-related populations fit into “slots” on the topology.
We used qpWave (Patterson et al., 2012) to evaluate all 120 possible ways for the 5 populations to be grafted onto each of the open “Slots” or positions, taking care to account for the correct admixing source for the populations that were admixed. In some topologies the assignment of populations to the slots did not alter the graph topology (when two populations were a clade with respect to the others. Therefore, there were only 30, 15 and 60 different models that were unique for the Serial Founder, Single Outgroup and Two Clades phylogenies respectively, though in our results we show all 120 possibilities for the assignment of a “Slot” to a population.
Methodology for model selection of admixture graphs
In the previous section we described the assignment of populations to “Slots” and the creation of a large number of admixture graphs.
For each admixture graph produced there are two metrics that we used to evaluate fit. The first is a list of residuals above a particular Z-score and the second is a score for the weighted error for the fitted statistics based on the graph in comparison to the empirically observed statistics, S(G) = −1/2(g − f)′Q−1(g − f). Here f is the vector of observed f-statistics and g is the corresponding vector of statistics fit on the graph with the specified topology, and Q is an estimated covariance matrix determined empirically (Patterson et al., 2012).
As a first screen for a working model, we begin by selecting models that have their largest residual with an absolute Z-score below 3. This is a standard approach in the literature and while this may provide a practical threshold for rejecting models, this alone is not sufficient to adjudicate between two models whose worst fitting residuals are both close to the threshold. We wanted to get an idea of how similar two models were with respect to their statistical likelihood. If our admixture graph models were nested within one another, we could do this using a Likelihood Ratio Test as described in (Lipson and Reich, 2017). In this approach, the log-likelihood of two models, one involving admixture from a certain population and one without were compared and their difference can then be compared using a chi-squared test.
However this approach cannot be applied in the present setting as the models we are testing are not nested within one another and the number of parameters of all of the models is the same. To enable us to examine the level of support one particular model has when compared to another we use Akaike Information Criterion (AIC) as a measure of model fit. Since the number of parameters between two models remains the same, the actual computed score remains the same whether we use AIC or a Bayesian Information Criterion (BIC).
We used a set of guidelines outlined in (Burnham and Anderson, 2004) for performing model selection using AIC. Specifically we computed δi = AICi − AICmin where AICi is the AIC of the i-th model, and AICmin is the lowest AIC obtained amongst all the models we tested across all topologies (SO, SF and TC). The models can then be compared using the following guidelines from (Burnham and Anderson, 2004):
δi ≤ 2, the i-th model is nearly as plausible as the best fitting model;
2 < δi ≤ 4, the i-th model is consistent with the data but considerably less probable than the best fitting model;
4 < δi ≤ 7, the i-th model is much less likely than the best fitting model;
7 < δi, the i-th model has essentially no support.
Based on this published set of criteria we chose to accept all models for which the difference in AIC was <4.
Among the samples we analyzed was a Mesolithic individual from Central Iran, Belt_Cave_M, which after restricting to damaged sequences (to address the evidence of contamination in this individual) reflects data from just 30,722 autosomal SNPs, and thus co-modeling with this individual restricts the number of SNPs available for admixture graph fitting. To address this, we repeated the admixture graph fitting removing this particular individual which improved our SNP coverage by more than 10 fold, allowing us to remove models that worked simply because of a lack of data and ensuring that the fit of a particular admixture graph was not due to our inability to reject it at lower coverage. As a further criterion for model selection, we restricted to the intersection between the fitting models analyzed with and without the Belt Cave individual.
Results from the model selection of tested admixture graphs
We report the successful graph topology results of our admixture graphs in Table S3.
In Table S3A, we observe that all working models exclude the “Two Clades” topology, regardless of how populations get assigned to “slots”. We also observe that all fitting populations have Indus_Periphery_West or Hajji_Firuz as an outgroup with respect to all other groups at the AIC < 4 threshold, with only models with Indus_Periphery_West if we use AIC<2.
Robustness to altering the topology of the non-Iranian-related admixing sources
We explored if modifying the ordering of both the South Asian hunter-gatherer-related components (Table S 3B) and Anatolian farmer-related admixture events (Table S 3C) change our inferences and found that they did not except in one notable way. Previously our model selection criterion had not been successful at distinguishing the earliest diverging of the Iranian-related populations. i.e. either Indus_Periphery_West or Hajji_Firuz_C, but under a different topology of the Anatolian farmer-related component in Hajji_Firuz_C we see that models with Indus_Periphery_West as the earliest diverging split are strongly supported over the other working models.
The inference is robust to treating all of the test populations as admixed
As another way of perturbing the resulting graphs, we chose to allow all the five populations to be admixed with all source populations thereby allowing a much freer model. We find that our results showing that the Indus_Periphery_West being the first to split are robust to this perturbation (Table S 3D).
Taken together, this analysis shows a clear branching structure which involves the Indus_Periphery_West ancestry as the first to split, followed by the others which are not distinguishable. There are two marginally fitting models in which Hajji_Firuz is the first to split (Table S 3A), but even if these models are correct they do not change the inference that the Iranian-related ancestry in Indus_Periphery_West split from the lineages leading to those in Belt Cave, Tepe Hissar, and Ganj Dareh before they separated from each other, which is the only inference we need for our main conclusions.
Alternative approaches to determining phylogeny
The major confounder when inferring trees and examining their topology as determined by shared drift amongsdifferent populations is admixture. In the analyses above based on testing of admixture graphs, we dealt with this by modeling known admixtures into populations, and showed that the changing of the topology of the admixing sources does not affect the inference we obtain about the internal phylogeny of the Iranian related component of the ancestry of our test populations. As an alternative approach to exploring these issues, we obtained unbiased estimates of the allele frequencies for the Iranian-related component in each of the samples by subtracting the expectation from the admixing sources, and then performed symmetry tests to reconstruct the phylogenetic relationships.
Prior to implementing this procedure on real data, we began by confirming that if the relevant admixing source populations or populations related to those source populations were available and the proportion of their admixture known, then it was possible to recover the internal phylogeny of the populations even though there is significant admixture present in the data.
To do this we simulated the phylogeny described in Figure S4 using the msprime coalescent simulator (Kelleher et al., 2016). We used standard mutation and recombination rates and sampled 1 million positions in 10 individuals from each population. We converted these to haploid genotypes by random sampling. We were interested in whether we could recover the internal phylogeny of the pp5 node. The choice of this particular topology and set of admixing populations mirrors the structure of the admixture graph that we think may be a reasonable match to our real data.
In the first step of the process, we computed the allele frequencies per SNP for the populations for which we were interested in obtaining a phylogeny, namely 3, 4, 5 and 6. We then subtracted the relevant allele frequencies of the admixing populations which were known in this setting. For example, we subtracted the allele frequency of population 8 from population 6, weighted by the admixture proportion 50%. We then computed all statistics of the form f4(0,A,B,Test), where A, B and Test could be any of the populations 3, 4, 5 and 6. In Table S 3E, we show that for samples without admixture correction there are no simple trees that are compatible with the data and it is not possible to uncover the internal phylogeny of these populations (at the |Z|>3 level).
However, after subtracting the allele frequencies (Table S 3F), it is possible to infer the internal phylogeny of the graph under our threshold. This suggests that if we account for the correct admixing population as well as the proportion of admixture, it is possible to recover the phylogeny of a set of populations even though they might be admixed even to levels of 50% as was the case in simulations. In the next section, we apply this procedure to the admixture graph that we constructed using the real data.
Accounting for admixture at the genotype level on real data
We began by examining if our inference procedure that we used on the simulated data could be applied as an additional validation of our best fitting admixture graph. To do this, we computed the allele frequencies of the Iranian-related populations, Ganj_Dareh_N, Hajji_Firuz_C, Tepe_Hissar_C and Indus_Periphery_West. We dropped Belt_Cave_M as it was too low in coverage to produce meaningful results. We then examined the inferred admixture proportions of the non-Iranian-related ancestry in these populations. To do this we utilized the qpAdm methodology for distal populations that was developed in (Narasimhan et al., 2019), and tested the populations that were required for admixture graph fitting, AHG, and Anatolia_N as sources for all of the populations. This produced point estimates that were in line with our admixture graph fits for the relevant admixture proportions as well as a covariance matrix measuring uncertainty.
To account for uncertainty in this procedure, we carried out this procedure sampled 1000 times from the point estimates and covariance matrix of admixture proportions and produced 1000 samples. For each of these samples we subtracted the allele frequencies of the AHG- and Anatolia_N-related ancestries and computed all possible triplets of f4-statistics as we had done for the simulated data. We computed f4-statistics using a |Z|>3 threshold to determine whether there continued to remain significant evidence of admixture relating the populations. Unlike with the simulated data where we knew a priori the exact mixing proportions and admixing sources, the uncertainty in the admixture proportion resulted in reduced power. We observed 465 cases where the inferred tree was (IP,(GD,(HF,TH))), 29 cases where the inferred tree was (IP,(HF,(GD,TH))), and 506 cases where a single tree determination could not be made due to the presence of additional significant admixture events between the populations. These results suggest that using this procedure we produce only two viable tree topologies, both of which involve Indus_Periphery_West as the population splitting first, mirroring the topology produced using the admixture graph methodology.
Data and Code Availability
All newly reported sequencing data are available from the European Nucleotide Archive, accession number PRJEB34154.
Additional Resources
Supplemental Data include an Excel spreadsheet detailing all ancient samples for which attempts were made to extract ancient DNA data and excel spreadsheets with other statistics detailed in the paper.
Supplementary Material
Figure S1. Ceramics in the Grave of Individual I6113/RGR7.3, BR-01, HS-02, Related to Figure 1 and STAR Methods. (A) Broken Goblet from Harappan level placed at the top of the head. (B) Complete specimen of a Red Slipped ware medium-sized globular pot from the Harappan level placed at the top of the head. (C) Central Column of a Stand from the Harappan level that appears to have been burned, placed at the top of the head. (D) Broken specimen of Red Slipped ware medium size globular pot from the Harappan level placed at the top of the head. There are lines as well as indentations on the upper right side, just below the rim. (E) Broken specimen of a Red Slipped ware medium-sized globular pot from Harappan level placed at the top of the head. There are a series of linear marks suggesting that these pots were wheel-thrown. Additional indication of this is seen in the interior of the vessel. (F) Complete specimen of a Red Slipped ware beaker from the Harappan level placed at the top of the head. (G) Complete specimen of Red Slipped ware large vessel from the Harappan level placed at the top of the head. The distinct rim suggests that this was made separately and then attached to the pot. (H) Broken stand of a dish on stand from the Harappan level placed at the top of the head. (I) Complete beaker from the Harappan level placed near the head. (J) Complete Goblet from the Harappan level placed at the top of the head. (K) Broken black painted Goblet from the Harappan level placed at the top of the head. (L) Complete beaker from the Harappan level placed near the right leg.
Figure S2. Quality control to identify a subset of authentic sequences, Related to STAR Methods. (A) The distribution of the number of sequences of a given length for two different samples - I6113 from Rakhigarhi, and I4011 which is previously reported data of a similar chronological age from Myanmar. For I6113, sequence length distributions are shown separately for pools of the double and single stranded libraries. (B) The fraction of sequences showing characteristic ancient DNA damage as a function of sequence length for I6113 and I4011. For I6113, sequence length distributions are shown separately for double and single stranded libraries. (C) Frequency of C-to-T substitutions for both ends of the sequences as a function of distance from the end for I6113, after merging all libraries. The profile shows damage patterns characteristic of authentic ancient DNA. Restricting to sequences with C-to-T substitutions on one end of a fragment result in an increase in the damage rate on the other end.
Figure S3. Phylogenies of Iranian-related populations tested for fits to the data, Related to STAR Methods. (A) “Serial Founder” model: A first population splits, then a second, then a third, then a fourth and fifth. (B) “Single Outgroup” model: A single population splits. Thereafter two pairs of populations diverge from a common source. (C) “Two Clades” model: The 5 populations split into two groups, one with 3 populations in a clade and the second with 2 populations in a clade.
Figure S4. Simulated phylogeny, Related to STAR Methods. This was the phylogeny used to test our inference procedure.
Table S 1, Related to STAR Methods. Detailed information for the 251 libraries generated for this study
Table S 2, Related to STAR Methods. Summary information for each of the 61 individuals screened for this study
Table S 3a-d, Related to STAR Methods. Z-scores, likelihoods and AIC of difference from best performing model for the admixture graph topologies shown in Figure S3 along with results from simulations of admixture graphs and all possible f-statistics before and after correcting for admixture.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||
|---|---|---|---|---|---|
| Biological Samples | |||||
| Ancient skeletal element | This study | Sample ID: I6113 | |||
| Chemicals, Peptides, and Recombinant Proteins | |||||
| Pfu Turbo Cx Hotstart DNA Polymerase | Agilent Technologies | Cat# 600412 | |||
| Herculase II Fusion DNA Polymerase | Agilent Technologies | Cat# 600679 | |||
| 2x HI-RPM hybridization buffer | Agilent Technologies | Cat# 5190–0403 | |||
| 0.5 M EDTA pH 8.0 | BioExpress | Cat# E177 | |||
| Sera-Mag Magnetic Speed-beads Carboxylate-Modified (1 μm, 3EDAC/PA5) | GE LifeScience | Cat# 65152105050250 | |||
| USER enzyme | New England Biolabs | Cat# M5505 | |||
| UGI | New England Biolabs | Cat# M0281 | |||
| Bst DNA Polymerase2.0, large frag. | New England Biolabs | Cat# M0537 | |||
| PE buffer concentrate | QIAGEN | Cat# 19065 | |||
| Proteinase K | Sigma Aldrich | Cat# P6556 | |||
| Guanidine hydrochloride | Sigma Aldrich | Cat# G3272 | |||
| 3M Sodium Acetate (pH 5.2) | Sigma Aldrich | Cat# S7899 | |||
| Water | Sigma Aldrich | Cat# W4502 | |||
| Tween-20 | Sigma Aldrich | Cat# P9416 | |||
| Isopropanol | Sigma Aldrich | Cat# 650447 | |||
| Ethanol | Sigma Aldrich | Cat# E7023 | |||
| 5M NaCl | Sigma Aldrich | Cat# S5150 | |||
| 1M NaOH | Sigma Aldrich | Cat# 71463 | |||
| 20% SDS | Sigma Aldrich | Cat# 5030 | |||
| PEG-8000 | Sigma Aldrich | Cat# 89510 | |||
| 1 M Tris-HCl pH 8.0 | Sigma Aldrich | Cat# AM9856 | |||
| dNTP Mix | Thermo Fisher Scientific | Cat# R1121 | |||
| ATP | Thermo Fisher Scientific | Cat# R0441 | |||
| 10x Buffer Tango | Thermo Fisher Scientific | Cat# BY5 | |||
| T4 Polynucleotide Kinase | Thermo Fisher Scientific | Cat# EK0032 | |||
| T4 DNA Polymerase | Thermo Fisher Scientific | Cat# EP0062 | |||
| T4 DNA Ligase | Thermo Fisher Scientific | Cat# EL0011 | |||
| Maxima SYBR Green kit | Thermo Fisher Scientific | Cat# K0251 | |||
| 50x Denhardt’s solution | Thermo Fisher Scientific | Cat# 750018 | |||
| SSC Buffer (20x) | Thermo Fisher Scientific | Cat# AM9770 | |||
| GeneAmp 10x PCR Gold Buffer | Thermo Fisher Scientific | Cat# 4379874 | |||
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher Scientific | Cat# 65602 | |||
| Salmon sperm DNA | Thermo Fisher Scientific | Cat# 15632–011 | |||
| Human Cot-I DNA | Thermo Fisher Scientific | Cat# 15279011 | |||
| DyNAmo HS SYBR Green qPCR Kit | Thermo Fisher Scientific | Cat# F410L | |||
| Methanol, certified ACS | VWR | Cat# EM-MX0485–3 | |||
| Acetone, certified ACS | VWR | Cat# BDH1101–4LP | |||
| Dichloromethane, certified ACS | VWR | Cat# EMD-DX0835–3 | |||
| Hydrochloric acid, 0.6N, 0.5N & 0.01N | VWR | Cat# EMD-HX0603–3 | |||
| Critical Commercial Assays | |||||
| High Pure Extender from Viral Nucleic Acid Large Volume Kit | Roche | Cat# 5114403001 | |||
| MinElute PCR Purification Kit | QIAGEN | Cat# 28006 | |||
| NextSeq 500/550 High Output Kit v2 (150 cycles) | Illumina | Cat# FC-404-2002 | |||
| Deposited Data | |||||
| Raw and analyzed data | This paper | ENA: PRJEB34154 | |||
| Software and Algorithms | |||||
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ | |||
| BWA | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ | |||
| ADMIXTOOLS | Patterson et al., 2012 | https://github.com/DReichLab/AdmixTools | |||
| R | https://www.r-project.org/ | https://www.r-project.org/ | |||
| EAGER | Peltzer et al., 2016 | https://github.com/apeltzer/EAGER-GUI | |||
| Schmutzi | Renaud et al., 2015 | https://github.com/grenaud/schmutzi | |||
| SeqPrep | https://github.com/jstjohn/SeqPrep | https://github.com/jstjohn/SeqPrep | |||
| smartpca | Patterson et al., 2006 | https://www.hsph.harvard.edu/alkes-price/software/ | |||
| ADMIXTURE | Alexander et al., 2009 | https://www.genetics.ucla.edu/software/admixture/download.html | |||
| PMDtools | Skoglund et al., 2014a | https://github.com/pontussk/PMDtools | |||
| Haplogrep 2 | Weissensteiner et al., 2016 | http://haplogrep.uibk.ac.at/ | |||
Highlights.
The sample was from a population that is the largest source of ancestry for S Asians
Iranian-related ancestry in S Asia split from Iranian plateau lineages >12000 yr ago
First farmers of the Fertile Crescent contributed little to no ancestry to S Asians
Acknowledgements
We thank Richard Meadow and several anonymous reviewers for critical comments on the manuscript and suggestions for improvements, and Anna Koutoulas and the Genomics Platform at the Broad Institute for sequencing support. We thank Elena Essel and Sarah Nagel for help in the laboratory. K.T. was supported by NCP fund (MLP0117) of the Council of Scientific and Industrial Research (CSIR), Government of India. D.R. is an Investigator of the Howard Hughes Medical Institute and his ancient DNA laboratory work was supported by National Science Foundation HOMINID grant BCS-1032255, by National Institutes of Health grant GM100233, by an Allen Discovery Center grant, and by grant 61220 from the John Templeton Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Alexander DH, Novembre J, and Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome research 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, van Oven M, Rosset S, Metspalu M, Loogväli E-L, Silva NM, Kivisild T, Torroni A, and Villems R (2012). A Copernican reassessment of the human mitochondrial DNA tree from its root. American journal of human genetics 90, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellwood PS (2013). 11 Human migrations and the histories of major language families In The Encyclopedia of Global Human Migration, Ness I, ed. (Chichester, UK: Wiley-Blackwell; ), pp. 87–95. [Google Scholar]
- Broushaki F, Thomas MG, Link V, López S, van Dorp L, Kirsanow K, Hofmanová Z, Diekmann Y, Cassidy LM, Díez-Del-Molino D, et al. (2016). Early Neolithic genomes from the eastern Fertile Crescent. Science (New York, NY) 353, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham KP, and Anderson DR (2004). Multimodel Inference: Understanding AIC and BIC in Model Selection. Sociological Methods & Research 33, 261–304. [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney J, Knapp M, Glocke I, Gansauge M-T, Weihmann A, Nickel B, Valdiosera C, García N, Pääbo S, Arsuaga J-L, et al. (2013). Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proceedings of the National Academy of Sciences of the United States of America 110, 15758–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Barros Damgaard P, Martiniano R, Kamm J, Moreno-Mayar JV, Kroonen G, Peyrot M, Barjamovic G, Rasmussen S, Zacho C, Baimukhanov N, et al. (2018). The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science (New York, NY) 360, eaar7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M, Fernández-Domínguez E, Reynolds L, Baird D, Pearson J, Hershkovitz I, May H, Goring-Morris N, Benz M, Gresky J, et al. (2019). Late Pleistocene human genome suggests a local origin for the first farmers of central Anatolia. Nature Communications 10, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Hajdinjak M, Moldovan OT, Constantin S, Mallick S, Skoglund P, Patterson N, Rohland N, Lazaridis I, Nickel B, et al. (2015). An early modern human from Romania with a recent Neanderthal ancestor. Nature 524, 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K, et al. (2015). Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heggarty P (2019). Prehistory through language and archaeology. In (Routledge; ), pp. 616–644. [Google Scholar]
- Kelleher J, Etheridge AM, and McVean G (2016). Efficient Coalescent Simulation and Genealogical Analysis for Large Sample Sizes. PLOS Computational Biology 12, e1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlević P, Gerber T, Gansauge M-T, Hajdinjak M, Nagel S, Aximu-Petri A, and Meyer M (2015). Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. BioTechniques 59, 87–93. [DOI] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, et al. (2016). Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson M, Cheronet O, Mallick S, Rohland N, Oxenham M, Pietrusewsky M, Pryce TO, Willis A, Matsumura H, Buckley H, et al. (2018). Ancient genomes document multiple waves of migration in Southeast Asian prehistory. Science 361, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson M, and Reich D (2017). A Working Model of the Deep Relationships of Diverse Modern Human Genetic Lineages Outside of Africa. Molecular biology and evolution 34, 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, et al. (2015). Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Fu Q, Aximu-Petri A, Glocke I, Nickel B, Arsuaga J-L, Martínez I, Gracia A, de Castro JMB, Carbonell E, et al. (2014). A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505, 403–406. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, et al. (2012). A high-coverage genome sequence from an archaic Denisovan individual. Science (New York, NY) 338, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal MR (1990). Further evidence of the Early Harappan Culture in the Greater Indus Valley. South Asian Studies 6, 175–2000. [Google Scholar]
- Narasimhan VM, Patterson N, Moorjani P, Rohland N, Bernardos R, Mallick S, Lazaridis I, Nakatsuka N, Olalde I, Lipson M, et al. (2019). The formation of human populations in South and Central Asia. Science 365, eaat7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, and Reich D (2012). Ancient admixture in human history. Genetics 192, 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possehl GL (1982). The Harappan Civilization: A contemporary perspective In Harappan Civilization (Oxford University Press; ), pp. 16–28. [Google Scholar]
- Possehl GL (1990). Revolution in the Urban Revolution: The Emergence of Indus Urbanization. In Annual Review of Anthropology (Annual Reviews), pp. 261–282. [Google Scholar]
- Posth C, Nakatsuka N, Lazaridis I, Skoglund P, Mallick S, Lamnidis TC, Rohland N, Nägele K, Adamski N, Bertolini E, et al. (2018). Reconstructing the Deep Population History of Central and South America. Cell 175, 1185–1197.e1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringe D, Warnow T, and Taylor A (2002). Indo-European and Computational Cladistics. Transactions of the Philological Society 100, 59–129. [Google Scholar]
- Rohland N, Glocke I, Aximu-Petri A, and Meyer M (2018). Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nature Protocols 13, 2447–2461. [DOI] [PubMed] [Google Scholar]
- Rohland N, Harney E, Mallick S, Nordenfelt S, and Reich D (2015). Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philosophical Transactions of the Royal Society B: Biological Sciences 370, 20130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer JG, and Lichtenstein DA (1989). Ethnicity and change in the Indus Valley cultural tradition In Wisconsin Archeological Reports, Kenoyer JM, ed., pp. 117–126. [Google Scholar]
- Shinde VS, Kim YJ, Woo EJ, Jadhav N, Waghmare P, Yadav Y, Munshi A, Chatterjee M, Panyam A, Hong JH, et al. (2018). Archaeological and anthropological studies on the Harappan cemetery of Rakhigarhi, India. PLOS ONE 13, e0192299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Pääbo S, Krause J, and Jakobsson M (2014). Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences 111, 2229–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapar R (1979). The Mosaic of the Indus Civilization Beyond the Indus Valley. In International Conference on Mohenjo-daro, Karachi. [Google Scholar]
- Vahia MN, Kumar P, Bhogale A, Kothari DC, Chopra S, Shinde VS, Jadhav N, and Shastri R (2016). Radiocarbon dating of charcoal samples from Rakhigarhi using AMS. Current Science 111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ceramics in the Grave of Individual I6113/RGR7.3, BR-01, HS-02, Related to Figure 1 and STAR Methods. (A) Broken Goblet from Harappan level placed at the top of the head. (B) Complete specimen of a Red Slipped ware medium-sized globular pot from the Harappan level placed at the top of the head. (C) Central Column of a Stand from the Harappan level that appears to have been burned, placed at the top of the head. (D) Broken specimen of Red Slipped ware medium size globular pot from the Harappan level placed at the top of the head. There are lines as well as indentations on the upper right side, just below the rim. (E) Broken specimen of a Red Slipped ware medium-sized globular pot from Harappan level placed at the top of the head. There are a series of linear marks suggesting that these pots were wheel-thrown. Additional indication of this is seen in the interior of the vessel. (F) Complete specimen of a Red Slipped ware beaker from the Harappan level placed at the top of the head. (G) Complete specimen of Red Slipped ware large vessel from the Harappan level placed at the top of the head. The distinct rim suggests that this was made separately and then attached to the pot. (H) Broken stand of a dish on stand from the Harappan level placed at the top of the head. (I) Complete beaker from the Harappan level placed near the head. (J) Complete Goblet from the Harappan level placed at the top of the head. (K) Broken black painted Goblet from the Harappan level placed at the top of the head. (L) Complete beaker from the Harappan level placed near the right leg.
Figure S2. Quality control to identify a subset of authentic sequences, Related to STAR Methods. (A) The distribution of the number of sequences of a given length for two different samples - I6113 from Rakhigarhi, and I4011 which is previously reported data of a similar chronological age from Myanmar. For I6113, sequence length distributions are shown separately for pools of the double and single stranded libraries. (B) The fraction of sequences showing characteristic ancient DNA damage as a function of sequence length for I6113 and I4011. For I6113, sequence length distributions are shown separately for double and single stranded libraries. (C) Frequency of C-to-T substitutions for both ends of the sequences as a function of distance from the end for I6113, after merging all libraries. The profile shows damage patterns characteristic of authentic ancient DNA. Restricting to sequences with C-to-T substitutions on one end of a fragment result in an increase in the damage rate on the other end.
Figure S3. Phylogenies of Iranian-related populations tested for fits to the data, Related to STAR Methods. (A) “Serial Founder” model: A first population splits, then a second, then a third, then a fourth and fifth. (B) “Single Outgroup” model: A single population splits. Thereafter two pairs of populations diverge from a common source. (C) “Two Clades” model: The 5 populations split into two groups, one with 3 populations in a clade and the second with 2 populations in a clade.
Figure S4. Simulated phylogeny, Related to STAR Methods. This was the phylogeny used to test our inference procedure.
Table S 1, Related to STAR Methods. Detailed information for the 251 libraries generated for this study
Table S 2, Related to STAR Methods. Summary information for each of the 61 individuals screened for this study
Table S 3a-d, Related to STAR Methods. Z-scores, likelihoods and AIC of difference from best performing model for the admixture graph topologies shown in Figure S3 along with results from simulations of admixture graphs and all possible f-statistics before and after correcting for admixture.
Data Availability Statement
All newly reported sequencing data are available from the European Nucleotide Archive, accession number PRJEB34154.