

SUMMARY
Antisense piRNAs guide silencing of established transposons during germline development, and sense piRNAs drive ping-pong amplification of the antisense pool, but how the germline responds to genome invasion is not understood. The KoRV-A gammaretrovirus infects the soma and germline and is sweeping through wild koalas by a combination of horizontal and vertical transfer, allowing direct analysis of retroviral invasion of the germline genome. Gammaretroviruses produce spliced Env mRNAs and unspliced transcripts encoding Gag, Pol and the viral genome, but KoRV-A piRNAs are almost exclusively derived from unspliced genomic transcripts and are strongly sense strand biased. Significantly, selective piRNA processing of unspliced proviral transcripts is conserved from insects to placental mammals. We speculate that bypassed splicing generates a conserved molecular pattern that directs proviral genomic transcripts to the piRNA biogenesis machinery, and that this “innate” piRNA response suppresses transposition until antisense piRNAs are produced, establishing sequence-specific adaptive immunity.
Graphical Abstract
INTRODUCTION
Transposons are ubiquitous mobile elements with the potential to trigger genome instability and mutations linked to disease (Belancio et al., 2008; Iskow et al., 2010). Antisense piRNAs guide an adaptive genome immune system that silences established transposons during germline development (Aravin et al., 2007a; Parhad and Theurkauf, 2018). Studies on P-M and I-R hybrid dysgenesis in Drosophila, which are sterility syndromes caused by crossing naïve strains with strains that have adapted to a new transposon, have revealed the importance of maternally deposited piRNAs in silencing transposons during embryogenesis (Brennecke et al., 2008; Chambeyron et al., 2008; Grentzinger et al., 2012; Khurana et al., 2011; Rio, 1991), but how the piRNA pathway responds to initial genome invasion is not understood.
KoRV-A is a gammaretrovirus associated with leukemia, immunodeficiency, and opportunistic chlamydia infection in koalas, and characterization of proviral insertion sites demonstrated that the virus infects the germline and is spreading by an unusual combination of vertical and horizontal transfer mechanisms (Chappell et al., 2017; Denner and Young, 2013; Ishida et al., 2015; Tarlinton et al., 2006). KoRV-A entered the wild population at the northern end of Australia and is sweeping south, where a few naïve populations persist (Tarlinton et al., 2006). Koala infection by KoRV-A thus provides a unique opportunity to directly examine the germline response to retroviral invasion of a mammalian genome. To characterize this response, we analyzed genome organization and long RNA and short RNA transcriptomes in testis, liver, and brain from two wild koalas infected with KoRV-A, while integrating our results with published genomic data from two additional animals. We show that koalas are typical of other mammals and produce piRNAs from genic and intergenic loci called piRNA clusters (referred to as clusters), and that established transposon subfamilies are targeted by roughly equal levels of antisense piRNAs, which are the effectors of trans-silencing, and sense piRNAs, which drive ping-pong amplification of the antisense pool (Brennecke et al., 2007; Ernst et al., 2017; Fu and Wang, 2014; Weick and Miska, 2014). By contrast, KoRV-A piRNAs are strongly sense biased, and appear to be derived from isolated proviral insertions, and not clusters. Gammaretroviruses, including KoRV-A, produce spliced Env mRNAs and unspliced transcripts encoding Gag, Pol, and the viral genome (Hanger et al., 2000). Although KoRV-A Env mRNAs are 5-fold more abundant than the unspliced genomic transcripts, KoRV-A piRNAs are almost exclusively derived from the unspliced transcripts, and we show that unspliced proviral transcripts are preferentially processed into piRNAs in systems ranging from flies to mice. At the organismal level, the innate immune system recognizes conserved features of bacteria and viruses, keeping these invaders in check until pathogen-specific adaptive immunity and immune memory are established (Hansson Göran K. et al., 2002; Paul, 2011). Gammaretroviruses are ancient genome invaders (Hayward, 2017), and bypassed splicing is a conserved and essential step in the viral replication cycle. We propose that bypassed splicing generates a conserved molecular “pattern” that is recognized in an innate genome immune response, which processes genomic proviral transcripts into sense strand piRNAs. We speculate that this system suppresses replication until antisense piRNAs are produced, guiding sequence-specific adaptive immunity and generating a memory of the genome invader.
RESULTS
KoRV-A and three endogenous retrotransposons are active in the koala germline
To define the genomic context of KoRV-A invasion, we characterized endogenous transposons in the koala reference genome (see Methods). This analysis identified 402 transposon subfamilies, which occupy 44% of the genome (Supplementary Table 1). Most of the transposon subfamilies are represented by short, degenerate copies, which appear to be inactive (Fig. S1A, gray points). However, the reference genome carries seven full-length copies of KoRV-A with less than 0.5% sequence divergence (Fig. S1A), confirming that it has been active recently (Ávila-Arcos et al., 2012; Johnson et al., 2018; Tarlinton et al., 2006). We also identified three endogenous retroviruses, designated ERVL.1, ERV.1, and ERVK.14, represented by multiple copies with less than 1% divergence, suggesting that these elements were also recently active (Fig. S1A, as indicated). To determine if these elements are expressed, we performed RNA-sequencing (RNA-seq) on tissues from two previously uncharacterized animals (designated K63464 and K63855), and reanalyzed published RNA-seq data from two additional individuals—Birke and PC (Hobbs et al., 2014). KoRV-A and the handful of endogenous transposons with full-length, low-divergence copies were expressed in multiple tissues, including testis, in all four animals. KoRV-A showed the highest expression levels, followed by ERVL.1, ERV.1, and ERVK.14 (Fig. 1A), suggesting that all four elements are currently active.
Figure 1. KoRV-A is highly expressed and actively transposing in the koala genome.

A. A heatmap depicting the expression levels of transposon subfamilies in tissues from four koalas: K63464 and K63855 in this work and Birke and PC (Hobbs et al., 2014; Johnson et al., 2018). Only the transposons with expression levels higher than 1 RPKM are shown.
B. Numbers of new germline insertions (defined as having reads mapped to both ends of the integration site and penetrance greater than 0.4 in at least one tissue from a koala individual). The nine most highly expressed transposon subfamilies from Figure 1A are graphed individually, and the remaining transposon subfamilies are pooled and labeled as others.
C. Venn diagrams showing the overlap of KoRV-A insertions among tissue samples. (Left) KoRV-A insertions in the testis, liver, and brain genomes of K63464. (Right) KoRV-A germline insertions in K63464, K63855, Birke, and PC.
Ongoing germline transposition would generate individual-to-individual insertion site variation, while recent activity in a common ancestor would produce low-divergence insertions that are shared between individuals. Previous analyses indicate that KoRV-A is currently active and generating substantial individual-to-individual variation (Cui et al., 2016; Tarlinton et al., 2006). To extend these earlier studies and assay for endogenous retrotransposon activity, we directly identified transposon insertion sites in four wild koalas, by sequencing genomic DNA from K63464 and K63855 and analyzing published genome sequences from Birke and PC (Johnson et al., 2018). We mapped new transposon insertions—insertions not in the reference koala genome (Johnson et al., 2018)—and quantified their insertion frequencies as described in Methods. To avoid low-frequency chimeric reads that result from sequencing artifacts during library construction (Treiber and Waddell, 2017), we required both ends of each insertion to be supported by “discordant” read pairs, with one read mapping to the reference genome and the other read of the pair mapping to the inserted element (see Methods). Consistent with earlier studies, we identified new insertions for KoRV-A, but also identified numerous new insertions of the three endogenous retrotransposons (Fig. 1B).
To characterize new germline transposon copies, we identified insertions present at 40% or greater frequency, consistent with heterozygosity, which were also confirmed by reads mapping to both ends of the transposon. For K63464, we sequenced genomic DNA from testis, liver, and brain, and for K63855, we sequenced genomic DNA from liver and brain. For these two animals, germline insertions also had to be present in all of the tissues analyzed (Fig. 1C, Fig. S1B). By these criteria, each of the four animals carried ~60 germline KoRV-A insertions that were not present in the annotated genome. Remarkably, none of these insertions was shared by all four individuals: Birke and PC shared a single KoRV-A insertion, while K63464 and K63855 shared four sites (Fig. 1C). These four animals, which were all collected from the center of the habitat range, thus are likely to represent at least two independent KoRV-A genome invasion events, followed by significant KoRV-A expansion in the germline. By contrast, we identified 22 ERVL.1, 15 ERV.1, and two ERVK.14 germline insertions that were shared by all four animals (Fig. S1C), confirming that these endogenous retrotransposon subfamilies were active in a common ancestor. However, each animal also carried numerous unique insertions of each of these elements (Fig. S1C). For example, K63855 had eight germline ERVK.14 insertions that were not present in any other animals, and PC had 94 unique germline ERVL.1 insertions (Fig. S1C). KoRV-A and the three endogenous retrotransposons are therefore currently active and generating remarkable germline variations in wild koalas.
To determine if KoRV-A and endogenous retrotransposons are active during later development or in adult tissues, we sought to estimate the rate of random transposition. This process is expected to generate insertions that are present in single cells or small groups of related cells in each tissue. The koala genome is approximately the same size as the human genome, with a single genome mass of roughly 3.5 X10−12 grams. Typical genomic library construction starts with 9 x 10−9 g of DNA, or 3000 genome equivalents. We sequenced to 35-fold depth, thus only sampling the genomes in the starting pool. As a result, random insertions will be supported by single pairs of discordant reads, which can arise from bona fide random insertions, or low-frequency sequencing artifacts that randomly join genomic DNA fragments (Treiber and Waddell, 2017). The frequency of artifactual read pairs should be constant across different tissues from the same animal, which share a common genome, and the transposon-mapping ends of these reads should be randomly distributed over the element. By contrast, the frequency of insertion reads should parallel tissue-specific transposon expression, and map only to terminal regions of the inserted element. We therefore analyzed genomic DNA sequences from the testis, brain, and liver of a single animal, K63464, and filtered for reads mapping to terminal regions of mobile elements. This analysis revealed different apparent transposition rates in the three tissues, with the lowest rate in testis, followed by brain and liver (Fig. S1D). KoRV-A showed strikingly elevated numbers of discordant reads in the liver, and RNA-seq indicates that KoRV-A is highly expressed in this tissue (Fig. 1A). While the sample size is small and the nature of the insertions precludes independent verification by PCR, these data suggest that KoRV-A and the endogenous retrotransposons are active in the germline and soma, and that their activities are lower in the testis, where the piRNA pathway operates.
Genomic sources of koala piRNAs
To characterize the piRNA response to KoRV-A and endogenous retrotransposons, we sequenced 18–34 nt long small RNAs from testis, liver, and brain, for both K63464 and K63855 (piRNAs are typically 24–32 nt long). Because piRNAs are 2’-O-methylated at their 3’ termini, which renders them resistant to oxidation (Horwich et al., 2007; Kirino and Mourelatos, 2007), we sequenced both unoxidized and oxidized libraries for each tissue sample. These studies revealed 24–32 nt, oxidation-resistant RNAs only in the testis (Fig. S2A, B), and these RNAs showed a strong 5’-U (1U) bias and significant 10-nt overlaps between sense and antisense species (ping-pong signature, Fig. S2)—hallmarks of piRNAs observed in previously characterized animals, including flies and mice (Brennecke et al., 2007; Gunawardane et al., 2007; Horwich et al., 2007; Saito et al., 2007). Accordingly, most of the 19 genes annotated to function in the piRNA pathway are more highly expressed in the testis than in somatic tissues (Fig. S3).
In the mammals analyzed to date, piRNAs are produced from a combination of long non-coding intergenic clusters, genic clusters that encode proteins in the soma but produce piRNAs in the germline, and isolated transposon insertions (Aravin et al., 2007b; Li et al., 2013). Intergenic and genic clusters can be single transcription units (uni-directional) or divergently transcribed pairs of transcription units (bidirectional), and can produce piRNAs from one genomic strand (uni-strand) or both genomic strands (dual-strand). Mapping piRNAs and long RNA reads from the K63464 testis to the reference genome (see Methods) revealed the same spectrum of piRNA clusters (Fig. 2A–D), with 202 unidirectional genic clusters, 132 unidirectional intergenic clusters, and 40 bidirectional clusters in intergenic loci (Fig. 2E, Supplementary Table 2). The genic clusters are expressed in testis, liver, and brain, while most of the intergenic clusters are expressed only in testis (Fig. S4A). A breakdown of koala cluster families is summarized in the mosaic plot in Figure 2E and is nearly identical to the breakdown of mouse clusters (Fig. 2E). Most clusters produce piRNAs and long RNAs predominantly from one genomic strand (Fig. 2F). These piRNAs exhibit a strong 1U bias, which is typical of piRNAs, and a significant ping-pong signature, indicating that they engage complementary transcripts in piRNA production (Fig. 2G–H). The promoters of the intergenic unidirectional and bidirectional piRNA clusters are also enriched for the binding motif for A-MYB (Fig. S4B), which is a master regulator of pachytene piRNA production in mice (Li et al., 2013). Cumulatively, 376 combined genic and intergenic clusters occupy only 0.17% of the koala genome, but account for up to 68.53% of all piRNAs, and the intergenic clusters account for up to 81% of transposon mapping piRNAs. Isolated transposon insertions, by contrast, occupy 43.99% of the genome, and account for up to 32.09% of all piRNAs. The sum of transposon mapping piRNAs from clusters and isolated elements exceeds 100%, due to reads that map to both classes of source loci. A similar pattern is observed in adult mouse testis (Li et al., 2013). Koalas are therefore similar to other mammals and produce piRNAs from a combination of clusters and isolated transposon insertions.
Figure 2. Characterization of koala piRNA clusters.

A. An intergenic and unidirectional piRNA cluster. The top track shows the location of the piRNA cluster and transposon insertions, where appropriate. Plus-strand piRNA clusters or transposon copies are shown in blue, while minus-strand copies are in red. The middle track in purple shows long-RNA abundance (in RPKM) and the bottom track in green shows piRNA abundance (in PPM; all mappers). These track notations are also used in B-D. B. A pair of intergenic, bidirectional piRNA clusters. C. A genic and unidirectional piRNA cluster. D. An intergenic and unidirectional piRNA cluster in the plus strand with a Ko.ERV.1 inserted in the minus strand. E. A mosaic plot for 376 koala piRNA clusters and 214 mouse piRNA clusters classified by: (1) genomic location (genic clusters in blue, which overlap protein-coding genes, and intergenic in red); and (2) directionality (bidirectional, divergently transcribed piRNA clusters are shown with a green outline, and unidirectional clusters with a yellow outline). Black numbers denote the number of piRNA clusters in each class. F. Size distribution of piRNAs derived from clusters in the oxidized K63855 testis sample. G. A sequence logo showing the nucleotide composition of piRNAs derived from clusters in oxidized K63855 testis. H. Distribution of overlapping nucleotides between plus-strand and minus-strand piRNAs derived from piRNA clusters in oxidized K63855 testis. The prominent peak at the 10-nt overlap is characteristic of ping-pong amplification.
Does transposition into clusters enhance antisense piRNA production?
Transposon insertion into a cluster is proposed to enhance ping-pong amplification and production of trans-silencing antisense piRNAs (Zanni et al., 2013). In koala, transposon-mapping piRNAs are roughly equally sense and antisense oriented (Fig. 3A) and show typical 1U and ping-pong signatures (Fig. 3B, C). High cluster representation is associated with increased sense and antisense piRNA expression (Fig. 3D), but the subfamilies that are highly represented in clusters are also more abundant in the rest of the genome (data not shown) and are not enriched in clusters. We performed the same analysis on adult mouse testis, which showed the same pattern (data not shown). As observed in mice, koala piRNA clusters are also slightly, but significantly, depleted of transposons relative to the rest of the genome (Fig. S4C, Wilcoxon signed-rank test p-value = 3.1 x 10−7 and < 2.2 x 10−16 for intergenic and genic piRNA clusters, respectively). These observations suggest that there is no selective advantage for transposon insertions into clusters.
Figure 3. KoRV-A piRNAs are sense biased.

A. Size distribution for small RNAs targeting all transposons. B. A sequence logo for transposon-targeting piRNAs. C. Distribution of overlapping nucleotides between sense and antisense transposon-targeting piRNAs. D. A scatterplot comparing the abundance (in PPM) of sense piRNAs and antisense piRNAs targeting all transposon subfamilies. Each dot represents a transposon subfamily: subfamilies not present in piRNA clusters are in yellow, while subfamilies present in piRNA clusters are in green. Half and 2-fold are marked with dashed lines. E. Size distributions for piRNAs targeting KoRV-A, Ko.ERV.1, Ko.ERVL.1, and Ko.ERVK.14. Small RNA sequencing was performed on oxidized testis RNA, from K63855.
However, a small number of koala and mouse clusters are enriched for antisense transposon insertions and produce transposon mapping piRNAs with an antisense bias (e.g., Fig. 2D; (Wasik et al., 2015)). These features are shared with the Drosophila flam cluster, which has an established function in transposon silencing in the somatic follicles cells of the ovary (Brennecke et al., 2007; Prud’homme et al., 1995). The three active endogenous retrotransposons in koala all produce antisense-biased piRNAs with ping-pong signatures (Fig. 3E, Fig. S5), but ERV.1 produces more antisense piRNAs than any other transposon subfamily. Intriguingly, a nearly full-length antisense ERV.1 insertion is present in the pi-phaCin-IG-174 intergenic cluster, which is enriched for fragments of other transposons that are also inserted in the antisense orientation and produces piRNAs that are predominantly antisense to the inserted elements (Fig. 2D). ERV.1 is also expressed at very low levels in testis, and shows little random transposition (Fig. 1A, S1D), suggesting that the antisense piRNAs suppress this transposable element. These findings raise the possibility that transposition into a subset of intergenic clusters does enhance piRNA biogenesis, but it is unclear how these loci differ from the majority of clusters, which are depleted of transposon insertions.
Unspliced KoRV-A transcripts are processed into sense strand piRNAs
piRNAs recognize targets through base pairing, with antisense piRNAs targeting established transposon families, leading to silencing and ping-pong amplification, and total transposon mapping piRNAs in koala are targeted by roughly equal numbers of sense and antisense piRNAs (Fig. 3A). However, in both animals analyzed, KoRV-A piRNAs were strongly sense-biased (Fig. 3D and compare histograms in Fig. 3E, S5A). To gain insight into the genomic source of these piRNAs, we characterized KoRV-A proviral insertion sites. KoRV-A is present in a piRNA cluster in the reference genome, but this insertion is not present in the two animals we analyzed (Fig. S4D). These animals each carry over 60 new germline KoRV-A insertions, but none of these insertions map to a piRNA cluster. In Drosophila, a subset of single euchromatic transposon insertions are bound by the cluster-specific HP1 homolog Rhino and produce piRNAs from both genomic strands (Mohn et al., 2014). Cluster chromatin spreads, and these “mini clusters” can be identified by piRNAs mapping to unique sequences flanking the insertion sites (Mohn et al., 2014), but we did not detect piRNAs flanking any of the KoRV-A insertions in the two koalas we analyzed (Fig. S4E). With the caveat there are gaps in the koala genome assembly that might include additional piRNA clusters, these observations strongly suggest that transcripts from dispersed KoRV-A proviral insertions are directly processed into sense piRNAs.
Gammaretroviruses produce spliced mRNAs that encode the Env protein, and unspliced transcripts that encode Gag, Pol and the retroviral genome (Johnson, 2015). In the testis from the animals we analyzed, spliced KoRV-A Env mRNAs are 5-fold more abundant than unspliced genomic transcripts, which is reflected in a striking difference in RNA-seq signal over Env exons relative to the intron (Fig. 4A). By contrast, KoRV-A piRNAs are uniformly distributed over the viral genome, suggesting that they are primarily produced from the lower abundance unspliced genomic transcripts (Fig. 4A). As an estimate of piRNA processing efficiency, we calculated the ratio of piRNAs to long RNAs across full-length KoRV-A (Fig. 4A, bottom track). This processing efficiency index is elevated over the intron relative to the Env exon, supporting preferential processing of the unspliced genomic transcript. To more directly test for preferential processing of the unspliced transcript, we quantified long and short RNA reads mapping to the exon-exon junction, which are specific to mature Env mRNAs, and to splice sites, which are only present in unspliced genomic transcripts. For long RNAs, the exon-exon junction to splice site ratio was 5.0, reflecting the abundance of mature Env mRNAs. In striking contrast, the junction to splice site ratio for piRNAs was 0.08 (Fig. 4A). KoRV-A piRNAs are therefore produced almost exclusively from unspliced proviral transcripts.
Figure 4. Transposon piRNAs are produced primarily from unspliced transcripts.

A. Normalized long RNA and piRNA reads across KoRV-A in koala testis, with sense reads in blue and antisense reads in red. The junction to splice-site ratios for the main exon-exon junction are indicated, defined by long RNA or piRNA reads. The bottom track plots the ratio of piRNAs to long RNAs, and is elevated over the intron, consistent with preferential processing of the unspliced genomic transcript. B. Normalized long RNA and piRNA reads across the fourth to eighth exons of the genic pi-phaCin-PC-ARHGAP20 cluster. Most long RNAs and piRNAs are spliced at the exon-exon junctions, piRNAs are enriched over the exons, and the piRNA to long RNA ratio is higher over exons than introns, indicating preferential processing of the spliced transcripts. C. Scatterplots comparing splicing indices defined by long RNA reads and piRNA reads, for all transposon exon-exon junctions in adult testis from koala, rat, cow, opossum, chicken and fruit fly, and pachytene spermatocytes (pSC) and round spermatids (rST) from mice. Exon-exon junctions with significantly higher splicing indices defined by long RNA reads than by piRNA reads are colored red, otherwise in grey, and the size of the dots depict statistical significance (chi-square test p-values).
These findings suggest that piRNA processing is strongly biased toward unspliced KoRV-A transcripts, but testis is composed of somatic and germline cells, and our data could be explained by an alternative model: KoRV-A proviral transcripts could be inefficiently spliced in the germline, where the piRNA machinery is expressed, and efficiently spliced in somatic cells. However, we find that KoRV-A is very inefficiently spliced in brain and liver, which are exclusively composed of somatic cells (Fig. S6A). While indirect, these observations suggest that KoRV-A is spliced in the germline, and that the unspliced transcripts are preferentially processed into piRNAs.
Does selective piRNA processing destabilize and “silence” unspliced proviral transcripts? Selective destabilization would reduce the steady-state level of unspliced genomic RNAs relative to the spliced Env mRNA, increasing the ratio of spliced to unspliced transcripts. The ratio of spliced to unspliced transcripts in testis, where the piRNA pathway operates, is 5. By contrast, this ratio is between 1.47 to 0.02 in liver and brain, which lack the piRNA biogenesis machinery. We find that protein-coding gene splicing efficiency is essentially identical in all three tissues (Fig. S6B), and proviral transcripts are spliced by the host machinery. Together, these findings suggest that KoRV-A splicing efficiency is similar in all three tissues, and that the unspliced transcripts are less stable in testes, potentially due to selective processing into piRNAs. Our very limited data on spliced to unspliced transcript levels place the range of destabilization between 3.5 (5/1.47) to 250 (5/0.02) fold (Fig. S6). Consistent with this rough range, Mov10L mutations in mice, which disrupt piRNA biogenesis, result in 5 to 30-fold over-expression of piRNA precursors from pachytene and pre-pachytene clusters (FigureS3E in (Vourekas et al., 2015). However, these observations are indirect, and a silencing role for KoRV-A genomic transcript processing into sense piRNAs remains to be rigorously tested.
piRNA processing of established transposon families
Antisense piRNA-guided transcript cleavage initiates processive, phased piRNA production (Han et al., 2015; Mohn et al., 2015). To determine if antisense piRNAs mapping to exons in spliced proviral transcripts initiate piRNA processing across exon-exon junctions, we analyzed long RNAs and piRNAs mapping to exon-exon junctions and splice sites for all endogenous koala retrotransposons, which are targeted by antisense piRNAs. Figure S6C shows long RNA and piRNA profiles for ERV.1, which has two splice donor sites that utilize a common splice acceptor. The piRNA junction to splice site ratio is only 0.09 for both combinations, while the corresponding long-RNA ratios are 3.63 and 2.23, indicating that the spliced transcript is a very poor substrate for phased piRNA production (Fig. S6C). To extend this analysis to all transposon families, we calculated a long RNA and piRNA “splicing index” for each intron, which we defined by the ratio of exon-exon junction reads to the sum of exon-exon junction and splice site reads, expressed as a percentage. For this analysis, introns were defined by long RNA reads mapping to exon-exon junctions, which correspond to consensus splice donor and acceptor sites. Only transposon families producing spliced transcripts by this method were included in our analysis. For this splicing index, 0% indicates that no reads map to the junction, while 100% indicates all of the reads map to the junction. Across all koala transposon families producing verified spliced transcripts, the piRNA splicing index was 0% or near 0%, while the long RNA splicing index ranged from 10% to over 90% (Fig. 4C). In striking contrast, the median splicing index for piRNAs from genic clusters, which are very efficiently spliced, was higher than the long RNA splicing index, indicating that the spliced transcripts are preferentially processed (Fig. S6D, S6E). This is illustrated in the piRNA/long RNA ratio plot for the pi-phaCin-PC-ARHGAP20 genic cluster in Fig. 4B. The exons for the pi-phaCin-PC-ARHGAP20 cluster and the KoRV-A intron show similar piRNA/long RNA ratios, suggesting that unspliced KoRV-A transcripts are as efficiently processed as the spliced genic cluster transcripts. By contrast, the vast majority of spliced gene transcripts are not efficiently processed into piRNAs. It is unclear if unspliced KoRV-A and spliced genic cluster transcripts are processed by the same machinery, or how these transcripts are differentiated from gene transcripts. However, the germline piRNAs in Drosophila are produced primarily from unspliced cluster transcripts (Zhang et al., 2014), while somatic piRNAs in flies are produced from spliced and polyadenylated transcripts, through a genetically separate process (Mohn et al., 2014). These observations raise the possibility that KoRV-A genomic RNAs and genic piRNA cluster transcripts in koala are also processed by distinct mechanisms, which may be related to the germline and somatic piRNA biogenesis pathways in Drosophila.
piRNA processing of unspliced proviral transcripts is deeply conserved
To determine if unspliced proviral transcripts are preferentially processed into piRNAs in other species, we analyzed published long and short RNA data from mouse, rat, cow, opossum, chicken, and fly (Li et al., 2013; Merkin et al., 2012; Meunier et al., 2013; Roovers et al., 2015; Soumillon et al., 2013; Wasik et al., 2015; Zhang et al., 2018). As for koala, only transposon families producing experimentally verified spliced transcripts were included in this analysis. In each species analyzed, piRNAs are almost exclusively produced from unspliced sense-strand transcripts (Fig. 4C, Fig. S6F). An example is illustrated in Fig. S6G—the ERV1_MD_I transposon in opossum testis—which shows a long RNA junction to splice site ratio of 0.5, but no detectable piRNAs from spliced transcripts. The piRNA to long RNA ratio for this element is also elevated over the intron, supporting more efficient processing of unspliced transcripts (Fig. S6G, bottom track). Preferential piRNA processing on unspliced transcripts extends to purified mouse round spermatids (rST) and pachytene spermatocytes, which do not contain somatic cells that could complicate analysis of splicing ratios. In these purified germline cells, we did not detect a single piRNA read mapping to an exon-exon junction for any of the transposons analyzed (Fig. 4C; Fig. S6F). For example, 10 long RNA reads map to the exon-exon junction in the mouse transposon, IAPEz, and no long RNA reads map to its splice sites (Supplementary Table 3). By contrast, no piRNA reads map to the junction, and 1778 piRNA reads map to the splice sites.
These data indicate that preferential piRNA production from the unspliced proviral transcripts of established retrotransposons is deeply conserved. To extend this analysis to an additional recent gammaretroviral genome invader, we analyzed the piRNAs in AKR strain mice. These mice are predisposed to leukemia, which is linked to two closely spaced insertions of AKV murine leukemia virus (Rowe et al., 1972), which appears to be active in the germline (Rowe and Kozak, 1980). RNA-seq shows that AKV is expressed at high levels in the AKR strain testis but is not expressed in several common laboratory strains (Fig. 5A). Together, these data suggest that AKV recently entered the AKR germline. Small RNA sequencing of AKR strain testis revealed piRNAs mapping to established transposons with the expected antisense bias, and a very significant ping-pong amplification signature (Fig. 5B, C). However, AKV piRNAs are strongly sense-biased (Fig. 5D) and do not show a significant 10 nt overlap between sense and antisense species (Fig. 5E), indicating that they do not engage complementary transcripts in ping-pong amplification. The AKV piRNAs also appear to be preferentially produced from unspliced proviral transcripts: We detected 13 piRNA reads mapping to splice sites but did not detect any piRNA reads mapping to the AKV exon-exon junction. By contrast, we detected 56 long RNA reads mapping to the junction, and 222 long RNA reads mapping to splice sites. These data support preferential processing of unspliced proviral transcripts, albeit the number of reads mapping specifically to splice sites and junctions is low. We therefore calculated the average piRNA/long RNA ratio over AKV intron and exons (Fig. 5F, bottom track). The mean ratio over the intron is 0.37, and the mean ratio over the Env exons is 0.16. In koala and mouse, unspliced transcripts from recent genome invaders thus appear to be preferentially processed into piRNAs.
Figure 5. piRNA production from AKV murine leukemia virus.

A. Heatmap depicting expression of transposon subfamilies in testis from five mouse strains: AKR, C3H, C57BL/6J, C57BL/6NJ and LP. Only transposon families with expression levels higher than 1 RPKM are shown. AKV, which is marked in purple, is only over-expressed in AKR mice. B. Histogram summarizing size and strand abundance for small RNAs targeting all transposons in AKR testis. C. Frequency of overlap between sense and antisense transposon-targeting piRNAs in the AKR mouse testis. D. Histogram showing size and strand bias for piRNAs targeting AKV.E. Frequency of sense-antisense overlap for AKV piRNAs, which does not support ping-pong. F. Normalized long RNA and piRNA reads across AKV, with sense reads in blue and antisense reads in red. The ratios of junction to splice site reads for the main intron are indicated: 25% of long RNAs are spliced, but no piRNA reads mapped to the exon-exon junction. The bottom track plots the ratio of piRNAs to long RNAs and is elevated over the intron.
Discussion
Does genome invasion activate resident transposons?
KoRV-A infection of wild koalas offers a unique opportunity to directly analyze the germline response to retroviral invasion of the genome. Surprisingly, genomic sequencing of KoRV-A infected animals revealed ongoing germline transposition of the new invader and three resident retrotransposon subfamilies. P element invasion of the Drosophila genome is associated with activation of endogenous transposon families (Khurana et al., 2011), and HIV infection has recently been linked to LINE-1 activation in somatic cells (Yurkovetskiy et al., 2018), raising the possibility that endogenous koala retrotransposons are activated by KoRV-A invasion. The mechanism of endogenous transposon activation on P element invasion is not understood, but mobilization of this DNA transposon leads to double-stranded DNA breaks, and genotoxic stress disrupts transposon silencing in a wide range of systems (Bradshaw and McEntee, 1989; Farkash et al., 2006; McClintock, 1984). Genome instability caused by KoRV-A transposition could activate a damage response that suppresses silencing of established transposon families. Alternatively, viruses frequently express inhibitors of host defense systems (reviewed by Finlay and McFadden, 2006; Yurkovetskiy et al., 2018), and KoRV-A could encode an RNA or protein that interferes with transposon silencing. Finally, the endogenous elements could have been active prior to KoRV-A infection. A role for KoRV-A in endogenous transposon mobilization can be tested through analysis of naïve animals, which persist at the southern end of the habitat.
A conserved link between transposon silencing and splicing?
Preferential processing of unspliced proviral transcripts into piRNAs dovetails with earlier studies in flies and yeast. A link between splicing and small RNA mediated transposon silencing was first demonstrated in pathogenic yeast, where stalled splicing intermediates are processed into transposon-silencing siRNAs (Dumesic et al., 2013). In Drosophila, transposon-silencing piRNAs are derived from heterochromatic clusters composed of nested transposon fragments (Bergman et al., 2006; Brennecke et al., 2007; Siomi et al., 2011). These genomic domains are bound by the Rhino-Deadlock-Cutoff complex, which suppresses cluster transcript splicing and is required for processing of these unspliced transcripts into germline piRNAs (Klattenhoff et al., 2009; Mohn et al., 2014; Parhad et al., 2017; Thomas et al., 2014; Zhang et al., 2014). Small silencing RNA and splicing pathways also appear to be co-evolving (Tabach et al., 2013), which may reflect a direct link between splicing and small RNA pathways in an ancient host genome-pathogen conflict.
Innate and adaptive genome immunity
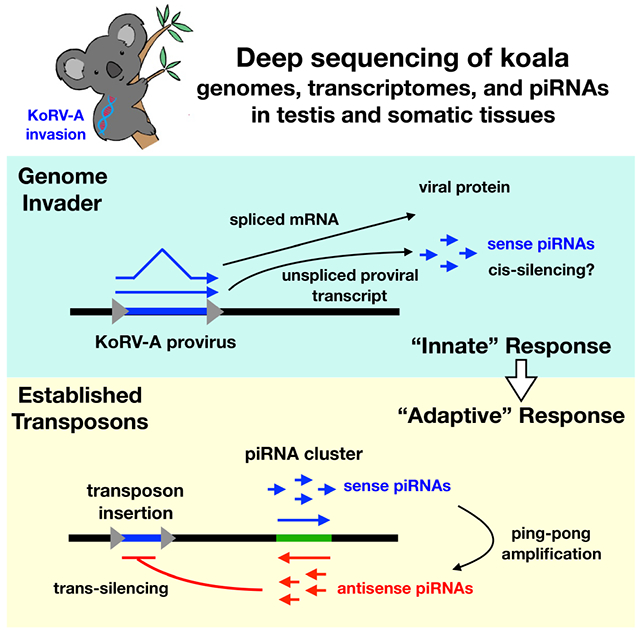
Immune responses to viral or bacterial infection comprise distinct innate and adaptive phases (Paul, 2011). During the initial innate response, the host immune system recognizes conserved molecular “patterns” that are common to pathogens, and suppress invaders until a more effective adaptive response is mounted (Brubaker et al., 2015). Adaptive immunity is produced by differentiation and amplification of lymphocytes, which produce antigen-specific antibodies and T cells, and carry a memory of the infectious agent (Bonilla and Oettgen, 2010). The data presented here, with earlier studies, lead us to propose that the piRNA response is also composed of distinct innate and adaptive phases (Fig. 6). During the initial phase of genome invasion, proviral insertions are transcribed to produce spliced and unspliced transcripts. We propose that the unspliced proviral transcripts carry a “pattern” that differentiates them from spliced viral and genic mRNAs, which is recognized by the innate genome immune system. This system processes target transcripts into sense strand piRNAs that cannot be translated or support viral replication. However, these piRNAs can direct antisense RNA cleavage, which generates precursors of trans-silencing antisense piRNAs. Initial sense strand piRNA production thus “primes” the adaptive genome immune response, which appears to be triggered by proviral insertion into a cluster or epigenetic conversion of isolated insertions into mini-clusters. Both processes produce antisense RNAs that are processed into trans-silencing piRNAs, and generate genetic and epigenetic memory of the genome invader (Fig. 6).
Figure 6. Model for innate and adaptive piRNA responses to genome invasion.

A. The “innate” response to retroviral invasion. Retroviral infection leads to proviral genome insertion. Transcription produces spliced Env mRNAs, which are structurally identical to genic mRNAs, and evade piRNA processing. By contrast, we propose that failed splicing generates a molecular “pattern”, which triggers cis-silencing through processing into sense strand piRNAs. B. “Adaptive” immunity through epigenetic conversion of isolated transposon insertions. Through a process that is not understood, a subset of isolated transposon insertions initiate transcription and piRNA production from both strands, generating antisense piRNAs that guide sequence-specific trans-silencing, and epigenetic memory of the genome invader. C. “Adaptive” immunity through cluster insertions. Partial silencing during the innate response still allows continued transposition. Antisense insertion into an existing piRNA cluster leads to production of trans-silencing antisense piRNAs that guide sequence-specific adaptive immunity, and a hardwired genetic memory of the genome invader.
The “pattern” that leads to piRNA processing during genome invasion remains to be determined, but potential conserved triggers include, but are not limited to, persistent splice donor and acceptor sites in cytoplasmic transcripts, and stop codons and frame shifts associated with the Gag-Pol protein junction. Striking parallels between germline piRNA biogenesis in flies and retroviral genomic transcript export in mammals support a link to failed splicing. In Drosophila, germline piRNAs are produced from unspliced cluster transcripts, which are bound by the DEAD box protein UAP56 and the THO complex, and mutations in uap56 and the THO complex genes thoc5 and thoc7 disrupt piRNA biogenesis, but do not disrupt gene expression or piRNA production from spliced somatic cluster transcripts (Hur et al., 2016; Mohn et al., 2014; Zhang et al., 2018, 2014). Recent studies indicate that these nuclear precursors are transferred to a complex containing Nxt1 and Nxf3, which facilitates Crm1 dependent export and delivery to the perinuclear piRNA processing machinery (ElMaghraby et al., 2019). In mouse somatic cells, export of unspliced Murine Leukemia Virus genomic transcripts also requires UAP56, Thoc5 and Thoc7, CRM1 and Nxt1-Nxf1 dimers (reviewed by Pessel-Vivares et al., 2015). piRNA biogenesis in the fly germline and unspliced proviral transcript export in mouse somatic cells thus coopt the same host factors to bypass nuclear quality control mechanisms and export unspliced transcripts, suggesting a common origin. Selective piRNA processing of unspliced KoRV-A and AKV transcripts suggests that, in the germline, utilization of this export pathway triggers sense strand piRNA biogenesis.
Following export to the cytoplasm, persistent introns could be sensed and trigger piRNA biogenesis. Alternatively, any poorly translated transcript that is exported from the nucleus may be processed into piRNAs. In the latter model, the piRNA pathway functions as an alternative cytoplasmic quality control system. Supporting this possibility, the highly conserved piRNA biogenesis factor Mov10l/Armi is a structural homolog of UPF1, a core component of the nonsense mediated decay (NMD) pathway, which degrades protein coding gene transcripts with retained introns or premature stop codons in somatic cells (He and Jacobson, 2015). The available data cannot distinguish between these alternatives, but the models are testable, and provide a framework for further analysis of the piRNA response to genome invasion.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for further information should be directed to William Theurkauf, (William.theurkauf@umassmed.edu). No unique reagents were generated during the course of these studies.
Data availability
All data have been deposited to GEO with the accession number GSE128122.
EXPERIMENTAL MODEL AND SUBJECT DETIAL
Koala (Phascolarctos cinereus). Brain, liver and testes tissues were isolated from wild male koalas (age unknown) that had been admitted to Currumbin Wildlife Hospital for treatment and had to be euthanized for humanitarian reasons. Sample collection was performed under University of Queenland Animal Ethics Approval Certificate #ANRFA/SVS/335/17. Tissue was imported to the USA under US Fish and Wildlife Service permit #MA65311C-0.
Mice (Mus musculus). Mouse studies were conducted under an approved Institutional Animal Care and Use Committee (IACUC) protocol at University of Massachusetts Medical School (UMMS). Male AKR/J (The Jackson Laboratory Stock No. 000648), C57BL/6J mice (The Jackson Laboratory Stock No. 000664), C57BL/6NJ mice (The Jackson Laboratory Stock No. 005304), C3H/HeJ (The Jackson Laboratory Stock No. 000659), and LP/J (The Jackson Laboratory Stock No. 000676) mice between 7 and 22 weeks of age were used for the mouse experiments in this study. Animals were housed and maintained within the AALAC-certified vivarium at UMMS, and in accordance with institutional guidelines.
Mapping statistics
All mapping statistics for DNA-seq, RNA-seq and small RNA-seq data are available in Supplementary Table 4.
Experimental details
Total DNA/RNA isolation
Total DNA was isolated from koala (K63464 and K63855) tissue samples using the DNeasy® Blood and Tissue Kit (Qiagen). Total RNA was isolated from brain, liver, and testis from two koalas (K63464 and K63855) and testis from the AKR/J (JAX stock #000648), C3H/HeJ (JAX stock #000659), C57BL/6J (JAX stock #000664), C57BL/6NJ (JAX stock #005304), and LP/J (JAX stock #000676) mouse strains using the mirVana™ miRNA Isolation Kit (ThermoFisher Scientific). Total RNA samples were treated with TURBO DNase (ThermoFisher Scientific) and RNA cleanup was done following the manufacturer’s instructions using the RNeasy® Mini Kit (Qiagen).
Library preparation
Small RNA sequencing libraries were prepared as previously described (Li et al., 2009a). Briefly, total RNA was isolated from flash frozen koala or mouse tissue using the mirVana miRNA Isolation Kit (ThermoFisher Scientific). Small RNAs were sized selected and purified from a 15% denaturing polyacrylamide-urea gel using the ZR small-RNA™ PAGE Recovery Kit (Zymo Research). For oxidized RNA library preparations, purified RNA was oxidized with 25 mM NaIO4 in 30 mM borax, 30 mM boric acid, pH 8.6, for 30 min at room temperature followed by ethanol precipitation. The 3’ pre-adenylated adapter was ligated to oxidized or un-oxidized small RNAs. The 3’ ligated product was purified from a 15% denaturing polyacrylamide-urea gel. 5’ RNA adapter ligation was performed and the ligated product was purified from a 10% denaturing polyacrylamide-urea gel and used to synthesize cDNA. The resulting cDNA was PCR amplified and run on a 2% Certified Low Range Ultra Agarose (Bio-Rad) gel with subsequent extraction using the QIAquick® Gel Extraction Kit (Qiagen). Purified small RNA libraries were single-end sequenced using the Illumina NextSeq 500 system.
Strand-specific RNA sequencing libraries for koala K63464 testis, two replicates of koala K63855 testis, and AKR/J, C3H/HeJ, C57BL/6J, C57BL/6NJ, LP/J mouse testes were prepared as previously described (Zhang et al., 2012). In brief, total RNA was isolated from frozen koala or mouse tissue samples via the mirVana miRNA Isolation kit. Koala total RNA was rRNA depleted using the RiboZero™ Gold rRNA Removal Kit for human, mouse, and rat (Illumina). Mouse total RNA samples were rRNA depleted as previously described (Adiconis et al., 2013; Morlan et al., 2012). RNAs longer than 200 nt were selectively recovered using the RNA Clean & Concentrator-5 kit (Zymo Research). RNA samples were fragmented and reverse transcribed. dUTP was incorporated during second strand synthesis for strand specificity. End repair and A-tailing was performed, followed by adapter ligation and uracil-DNA glycosylase (UDG) treatment. Finally, the library was PCR amplified. RNA libraries were paired-end sequenced using the Illumina NextSeq 500 system.
Strand-specific RNA-seq libraries for one replicate of the K63464 testis, K63464 liver, K63464 brain, K63855 liver, and K63855 brain were constructed at BGI. These RNA-seq libraries were sequenced using the HiSeq 101PE platform at BGI.
Short fragments of DNA libraries for K63464 testis, K63464 liver, K63464 brain, K63855 liver, and K63855 brain were constructed at BGI. These DNA-seq libraries were sequenced using the NovaSeq 151PE platform at BGI.
Bioinformatics analysis
Transposon annotation
We annotated transposon consensus sequences and individual copies in the koala reference genome using several algorithms: repeatModeler, repeatMasker, LTRharvest, LTRdigest, TransposonPSI, and ucluster (Edgar, 2010; Ellinghaus et al., 2008; Steinbiss et al., 2009), described in detail as follows.
First, we used three separate algorithms to identify transposons de novo. We ran RepeatModeler on the koala reference genome with default parameters to build a transposon library. We also used LTRharvest with parameters “-index phaCin.fsa -seed 100 -minlenltr 100 -maxlenltr 1000 - mindistltr 1000 -maxdistltr 15000 -xdrop 5 -mat 2 -mis -2 -ins -3 -del -3 -similar 90.0 -overlaps best -mintsd 5 -maxtsd 20 -motif tgca -motifmis 0 -vic 60 -longoutput” to predict LTR retrotransposons. LTRdigest was then used to filter out false positives from LTR transposons predicted by LTRharvest using the hidden Markov models (HMMs) of transposon proteins from Pfam and GyDB. LTR elements without HMM hits were discarded. Finally, TransposonPSI was used to construct all potential transposon sequences based on high homology to existing transposon annotation.
Second, we merged the three transposon libraries by RepeatModeler, LTRharvest/LTRdigest, and TransposonPSI via usearch (version 11.0.667). Transposon clusters with fewer than 3 transposon sequences were discarded. The remaining transposon clusters were deemed de novo discovered transposon families with their centroid sequences regarded as the consensus sequences. We further combined the de novo transposon library with Repbase’s transposon library of koala via usearch and provided the centroid sequences to RepeatClassifier to determine their transposon families, and only those transposons classified as LINE/SINE/LTR/DNA were retained in our final consensus sequence library.
Finally, we provided our final consensus sequences to RepeatMasker with parameters “-s -pa 48 - e ncbi -div 40 -nolow -norna -no_is” to identify individual copies in the genome. To further remove false positives, we discarded those transposon families without at least two copies or 1000 base pairs in the koala reference genome.
Except for KoRV-A and PhER that had been named already, we assigned transposons names that began with Ko, which stood for koala, followed by the family they belonged to. A number was also added for different subfamilies in the same family. For example, ERV family transposons were named Ko.ERV.1, Ko.ERV.2, Ko.ERV.3, etc.
Calculation of average sequence divergence for transposon subfamilies
For each transposon subfamily, all copies annotated in the reference genome were extracted, and copies longer than half of the length of the consensus sequence were aligned to the consensus sequence, and the alignments were used to calculate the average divergence for each transposon subfamily, defined as the number of substituted nucleotides in the alignments divided by the total number of nucleotides of these copies in the reference genome.
Quantification of the expression levels of transposons and piRNA pathway genes
We first removed all RNA-seq reads that mapped to ribosomal RNAs (rRNAs) in the reference koala genome. We annotated the rRNAs using RNAmmer (Lagesen et al., 2007) (version 1.2) with default parameters and mapped RNA-seq reads to rRNAs using Bowtie2 (Langmead and Salzberg, 2012) (version 2.2.5) with default parameters. The remaining RNA-seq reads were mapped to the reference koala genome using STAR (Dobin et al., 2013) (version 020201). The mapping results in the SAM format were transformed into sorted and duplication-removed BAM format using SAMtools (Li et al., 2009b) (version 1.8). The final mapped reads were assigned to protein-coding genes, non-coding RNAs, and piRNA genes using HTSeq (Anders et al., 2015) (version 0.9.1), and the expression levels of these genes, in reads per million unique mapped reads in per thousand nucleotides (RPKM), were calculated using custom bash scripts.
After rRNA removal, RNA-seq reads were also mapped to transposon consensus sequences (defined as described in the previous sub-section) directly using Hisat2 (Kim et al., 2015) (version 2.1.0) with default parameters. Then transposon expression levels were calculated using Bedtools (Quinlan and Hall, 2010) (version 2.27.1). The similarity between some transposon elements may prevent the reads from these high-similarity regions to be counted accurately when these reads were mapped directly to the transposon consensus sequences. Thus, we applied the expectation-maximization (EM) algorithm, which assigns these multiple mappers according to mapping potential in different transposons (Fu et al., 2018). First, multiple mappers were assigned to different transposons according to the number of unique mappers in each transposon. Then the number of assigned reads for each transposon was used as the weight for the second round of multiple mapper assignments. The iterations continued until the process converged, defined as the Manhattan distance between two iterations being less than 0.05.
Identification of new transposon insertions and transposon absences using genomic DNA-seq data
DNA-seq raw reads were mapped to the reference koala genome using the BWA MEM algorithm (Li and Durbin, 2009) (version 0.7.12-r1044) allowing soft clipping. Mapping results in the SAM format were transformed into sorted BAM format via SAMtools (version 1.8). We then used the TEMP algorithm (Zhuang et al., 2014) to detect new transposon insertions. TEMP defines discordant read pairs as those with one end mapping to the reference genome and the other mapping to an inserted transposon element and uses these discordant reads for identifying new transposon insertions. Such discordant reads may also come from low-frequency chimeric reads that result from sequencing library construction (Treiber and Waddell, 2017), but in such case, the transposon mapping ends would be randomly distributed and randomly oriented over the transposon element. Thus, we only retained reads mapping to the ends of the element, with the correct orientation for a bona fide insertion. We made two more modifications to TEMP for detecting transposons that are in the reference genome but not in the new sample. First, we used reads flanking −5 to +5 bp of breakpoints as supporting reads instead of the default 0 to +5 bp in TEMP. Second, the default TEMP uses all reads overlapped with the region from upstream 20 bp of the 5’-end to downstream 20 bp of the 3’-end of a transposon insertion to detect breakpoints. Instead, we only used reads that overlapped with the two ends of a transposon insertion, ±20 bp centered on the 5’-end or ±20 bp centered on the 3’-end, to detect breakpoints, which is more stringent and guards against low-frequency chimeric reads that result from sequencing library construction (Treiber and Waddell, 2017).
Analysis of small RNA-seq data
We first removed the adapter sequence (TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACCGATGTATCTCGT) from all small RNA-seq reads using cutadapt (Martin, 2011) (version 1.15). We then removed all reads that mapped to rRNAs, miRNAs, snoRNAs, snRNAs, and tRNAs. As mentioned above, we defined rRNAs in the koala genome using RNAmmer (version 1.2) with default parameters. We annotated miRNAs in the koala genome using miRDeep2 (Friedländer et al., 2012) (version 2.0.0.8) with default parameters and the unoxidized small RNA-seq datasets in the testis of K63464 and K63855 testis samples as the input. The remaining small RNA-seq reads were mapped to reference koala genome and transposon consensus sequences independently using Bowtie (version 1.1.0). We calculated piRNA abundance at piRNA clusters and individual transposon insertions in the genome using unoxidized and oxidized small RNA-seq samples separately. We normalized the abundance by sequencing depth, i.e., the total number of genome mapping reads after removing rRNAs, miRNAs, snoRNAs, snRNAs, and tRNAs. Nucleotide content and ping-pong amplification were analyzed for all reads that mapped to the genome, reads that mapped to transposons, and reads that mapped to piRNA clusters, respectively. For ping-pong analysis on cluster-mapping reads, 5’ to 5’ overlaps between all pairs of piRNAs that mapped to the opposite genomic strands were calculated, and then the Z-score for the 10-nt overlap was calculated using the 1-9 nt and 11-30 nt overlaps as the background (Li et al., 2009a). For ping-pong analysis on transposon-mapping reads, we only used the reads that mapped to transposon consensus sequences with zero or one mismatch and the 10-nt overlap was computed using the coordinates of reads on the consensus sequences. Reads that mapped to the Ko.RTEBovB.98 transposon were excluded due to one highly abundant sequence.
piRNA cluster annotation
We annotated piRNA clusters using RNA-seq and small RNA-seq data in the K63464 testis. We considered 24–32 nt small RNA reads that could map to the koala genome, after rRNA, miRNA, tRNA, snRNA, and snoRNA removal, as piRNAs. piRNAs were then assigned to 20 kb sliding windows (with a 1 kb step), and windows with more than 100 piRNAs per million uniquely mapped piRNAs were further considered as potential piRNA clusters. To remove false positives due to unannotated miRNAs, rRNAs, tRNAs, snRNAs, and snoRNAs, which mostly produce reads with the same sequences, we also filtered out those 20-kb genomic windows with fewer than 200 distinct reads (called species). We then calculated the first-nucleotide content for each 20-kb window, and those windows with 1U/10A percentage less than 50% were also discarded (15 windows were discarded in total). The remaining contiguous 20-kb windows were deemed putative piRNA clusters. We used the RNA-seq reads after rRNA removal for annotating exon-exon junctions in piRNA clusters. Finally, we performed manual curation for putative piRNA clusters using piRNA profile, long RNA profile, and detected exon-exon junctions. The direction of a piRNA cluster was indicated by the direction of the main long-RNA transcript.
The final piRNA clusters were classified according to their genomic location, directionality, and strandedness. First, piRNA clusters with more than 50% base pairs overlapped with protein-coding genes were defined as genic piRNA clusters and others intergenic piRNA clusters. Second, piRNA cluster pairs that shared their promoters (distance between their TSSs were less than 500 bp) but transcribe in divergent directions were annotated as bidirectional piRNA clusters while others were unidirectional piRNA clusters. Third, piRNA clusters which produced similar or high levels of piRNAs from both genomic strands (< 4 folds difference or > 1000 ppm) were defined as dualstrand piRNA clusters while others were uni-strand clusters.
Exon-exon junction discovery and splicing index calculation in transposons
RNA-seq data (after rRNA removal) were used to identify high-confident exon-exon junctions in transposons. First, RNA-seq reads that could map fully to the reference koala genome (using Bowtie2 in the end-to-end mode and soft clipping disabled) were considered primary transcripts and discarded. The remaining RNA-seq reads were then mapped to the transposon consensus sequences using Hisat2 with default parameters for exon-exon junction detection.
To calculate the splicing index for each detected junction, rRNA-removed long RNA reads and rRNA/miRNA/tRNA/snRNA/snoRNA removed small RNA reads were used as input, and the calculation was performed in the following steps. First, reads were mapped to transposon consensus sequences using Hisat2 given the detected junctions. The output of Hisat2 contained two types of reads, those that mapped to splice sites (unspliced reads) and the remaining that mapped to exon-exon junctions. We further mapped the junction-mapping reads back to the reference genome (Bowtie2 in the end-to-end mode with soft clipping disabled) and discarded the reads that could map. The remaining long RNA reads that spanned at least 7 bps of the exon-exon junctions and piRNA reads that spanned at least 3 bps of the exon-exon junctions were considered spliced reads. After this, splicing index for each exon-exon junction detected by long RNA and piRNA respectively was defined as the ratio of spliced reads and the total reads (splice reads plus unspliced reads).
piRNA processing efficiency calculation
We defined piRNA processing efficiency as the ratio of piRNA to long RNA abundance for exons and introns of transposons and piRNA clusters. Specifically, piRNA reads per million mapped reads divided by long RNA reads per million mapped reads is considered as processing efficiency. Long RNAs cannot be mapped to the edges of transposons by Hisat2, while piRNAs are mappable due to their short length, leading to artificially high processing efficiency. Hence we removed edge sequences (100 nt) of transposons for piRNA processing efficiency analysis.
Supplementary Material
Figure S1, related to Figure 1. KoRV-A and three other ERVs are potentially active in the koala genome.
A. A scatterplot depicting the number of full-length copies of a transposon subfamily (defined as longer than 50% of the consensus sequence length) vs. divergence from the consensus sequence. Four ERVs are labeled, including KoRV-A, which have more than five full-length copies at divergence lower than 1% in the koala genome.
B. Venn diagrams showing the overlap of transposon insertions among the three tissues of K63464. They correspond to the left panel of Figure 1C but for transposon subfamilies other than KoRV-A.
C. Venn diagrams showing the overlap of transposon insertions among the various koalas. They correspond to the right panel of Figure 1C but for transposon subfamilies other than KoRV-A.
D. Numbers of uninherited insertions and germline insertions for all transposon subfamilies in the testis, brain, and liver of K63464. Uninherited insertions are defined as insertions supported by only one discordant read and only present in one tissue. Germline insertions are defined as in Figure 1B. KoRV-A, Ko.ERV.1, Ko.ERVL.1, Ko.ERVK.14 and PhER are colored in red, blue, green, yellow, and purple respectively, while other transposon subfamilies are in black.
Supplementary Table3, linked to Figures 4 and 5. Number of reads mapping to junctions and splice sites of transposons mentioned in this study.
Supplementary Table4, linked to STAR Methods, Experimental details. Mapping statistics of DNA-seq, RNA-seq and small RNA-seq data.
Figure S2, related to Figure 2. Koala piRNAs are produced in the testis but not in the brain or liver.
A-F. Length distribution of piRNAs, nucleotide composition of piRNAs, and distribution of overlapping nucleotides between sense and antisense piRNAs.
A and B. All genome-mapping oxidized small RNA reads in K63464 and K63855 testis samples, after removal of rRNAs, miRNAs, snRNAs, snoRNAs, and tRNAs.
C-H. Unoxidized small RNA reads in testis, liver and brain from K63464 and K63855
Figure S3, related to Figure 2. Most piRNA pathway genes are specifically expressed in the koala testis.
A heatmap showing the expression levels of 19 piRNA pathway genes in various koala tissue samples. The expression level (in RPKM) is written in each cell, and highly expressed genes are shown in shades of red.
Figure S4, related to Figure 2. Koala piRNA clusters have similar features to mouse piRNA clusters.
A. Long RNA expression levels at intergenic piRNA clusters (red) and genic piRNA clusters (blue) in the testis, brain and liver samples of the two koalas assayed by us.
B. Boxplots show the maximal A-MYB motif scores in the ±100 bp window centered on the TSSs of 19,945 mRNAs or three classes of piRNA clusters: 202 genic unidirectional, 132 intergenic unidirectional, and 42 bidirectional piRNA clusters.
C. piRNA clusters are significantly depleted of transposons. The observed number of transposon overlapping nucleotides in each of 376 piRNA clusters is plotted against the expected number of transposon-overlapping nucleotides. Wilcoxon rank-sum test p-value = 3.1 x 10−7 and < 2.2 x 10−16 for intergenic (red) and genic (blue) piRNA clusters, respectively.
D. A genic, unidirectional and uni-strand piRNA cluster with a KoRV-A insertion in the reference koala genome but the insertion is absent in our animals. The top track indicates this piRNA cluster and the KoRV-A insertion annotated in the reference genome. The middle and bottom tracks illustrate DNA-seq reads mapping around the KoRV-A insertion in K63464 and K63855 brain samples. Note the absence of reads that map to the KoRV-A despite plentiful reads in flanking regions. Only the two ends of the KoRV-A insertion are shown and the internal segment is illustrated with dashed lines (not to scale).
E. Profiles showing average piRNA abundance in the ±5kb window around KoRV-A insertions in K63464 and K63855. piRNA abundance in KoRV-A is shown in the center. piRNAs in the same strand as KoRV-A insertions are in blue while piRNAs in the opposite strand in red.
Figure S5, related to Figure 3. Length distribution and ping-pong signature for endogenous retrovirus piRNAs.
A. Size distribution of piRNAs mapping to KoRV-A, Ko.ERV.1, Ko.ERVL.1, or Ko.ERVK.14, corresponding to Figure 3D but for oxidized piRNAs in the K63464 testis and unoxidized piRNAs in the K63464 and K63855 testis tissues, respectively.
B. Distributions of overlapping nucleotides between sense and antisense piRNAs from KoRV-A, Ko.ERV.1, Ko.ERVL.1, and Ko.ERVK.14, for oxidized piRNAs in the K63464 and K63855 testis and unoxidized piRNAs in the K63464 and K63855 testis tissues, respectively.
Figure S6, related to Figure 4. piRNAs derived from genic piRNA clusters are preferentially spliced.
A. Long RNA reads across KoRV-A in liver and brain, with the junction to splice-site ratios for the main exon-exon junction indicated. These figures correspond to Figure 4A but for long RNA reads in liver and brain samples of two Koalas.
B. Barplot at the left showing splicing index of KoRV-A transcripts in K63464 and K63855 testis, brain, and liver tissues. Boxplot at the right showing splicing index of shared expressed transcripts (RPKM ≥ 1) in K63464 and K63855 testis, brain, and liver tissues
C. Normalized long RNA and piRNA reads and piRNA to long RNA ratio across Ko.ERV.1 in koala testis, corresponding to Figure 4A but for Ko.ERV.1 in koala.
D. Boxplots indicating splicing index of 2,460 exon-exon junctions (EEJ) in 204 genic piRNA clusters. Splicing index defined by long RNA reads is marked as green while splicing index defined by piRNA reads are marked as purple. We could only detect junction- or splice site-mapping piRNA reads and long RNA reads at 1,404 junctions.
E. Boxplot indicating piRNA to long RNA ratio for exons and introns of KoRV-A, genic piRNA clusters and intergenic piRNA clusters.
F. A scatterplot comparing the splicing index defined by long RNA reads and piRNA reads for all transposon exon-exon junctions in the mouse adult testis tissue. This panel corresponds to Figure 4C, which were for sorted mouse pachytene spermatocyte and round spermatid cells.
G. Normalized long RNA and piRNA reads across the ERV1_MD_I transposon in opossum testis, with the ratio of junction reads over splice-site reads for the annotated exon-exon junction indicated. This figure corresponds to Figure 4A but for ERV1_MD_I in opossum.
Supplementary Table1, linked to Figure 1. Overall summary of koala transposons.
Supplementary Table2, linked to Figure 2. Detailed information for 379 annotated koala piRNA clusters.
Highlights:
KoRV-A and three endogenous retroviruses are active in the koala genome.
Unspliced KoRV-A proviral transcripts are preferentially processed into piRNAs.
piRNA production from unspliced transposon transcripts is deeply conserved.
ACKNOWLEDGEMENTS
We would like to thank the members of Theurkauf and Weng laboratories for their critical comments and discussion on the manuscript. Special thanks to the Currumbin Wildlife Hospital for supporting koala tissue acquisition, to Lenny Schultz (Jackson Labs) and Jaime Rivera (UMass Med) for providing mouse strains, and to Dr. Paul Young, for introducing WET to KC. We also thank Zoe Huang for drawing the koala clipart in the graphical abstract. This work was supported in part by NIH grants HD078253 and HD049116 and Chinese National Natural Science Foundation grants 31571362, 31871296, and 91640201. T.Y. was partly funded by a Ph.D. student study-abroad fellowship from the China Scholarship Council.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. (2013). Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Hannon GJ, and Brennecke J (2007a). The Piwi-piRNA Pathway Provides an Adaptive Defense in the Transposon Arms Race. Science 318, 761–764. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, and Hannon GJ (2007b). Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316, 744–747. [DOI] [PubMed] [Google Scholar]
- Ávila-Arcos MC, Ho SY, Ishida Y, Nikolaidis N, Tsangaras K, Hönig K, Medina R, Rasmussen M, Fordyce SL, and Calvignac-Spencer S (2012). One hundred twenty years of koala retrovirus evolution determined from museum skins. Mol. Biol. Evol 30, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belancio VP, Hedges DJ, and Deininger P (2008). Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health. Genome Res. 18, 343–358. [DOI] [PubMed] [Google Scholar]
- Bergman CM, Quesneville H, Anxolabéhère D, and Ashburner M (2006). Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 7, R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla FA, and Oettgen HC (2010). Adaptive immunity. J. Allergy Clin. Immunol. 125, S33–S40. [DOI] [PubMed] [Google Scholar]
- Bradshaw VA, and McEntee K (1989). DNA damage activates transcription and transposition of yeast Ty retrotransposons. Mol. Gen. Genet. MGG 218, 465–474. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, and Hannon GJ (2007). Discrete Small RNA-Generating Loci as Master Regulators of Transposon Activity in Drosophila. Cell 128, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, and Hannon GJ (2008). An Epigenetic Role for Maternally Inherited piRNAs in Transposon Silencing. Science 322, 1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol 33, 257–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambeyron S, Popkova A, Payen-Groschêne G, Brun C, Laouini D, Pelisson A, and Bucheton A (2008). piRNA-mediated nuclear accumulation of retrotransposon transcripts in the Drosophila female germline. Proc. Natl. Acad. Sci. U. S. A 105, 14964–14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell KJ, Brealey JC, Amarilla AA, Watterson D, Hulse L, Palmieri C, Johnston SD, Holmes EC, Meers J, and Young PR (2017). Phylogenetic Diversity of Koala Retrovirus within a Wild Koala Population. J. Virol 91, e01820–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P, Löber U, Alquezar-Planas DE, Ishida Y, Courtiol A, Timms P, Johnson RN, Lenz D, Helgen KM, Roca AL, et al. (2016). Comprehensive profiling of retroviral integration sites using target enrichment methods from historical koala samples without an assembled reference genome. PeerJ 4, e1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denner J, and Young PR (2013). Koala retroviruses: characterization and impact on the life of koalas. Retrovirology 10, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR, Bartel DP, and Madhani HD (2013). Stalled Spliceosomes Are a Signal for RNAi-Mediated Genome Defense. Cell 152, 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Ellinghaus D, Kurtz S, and Willhoeft U (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElMaghraby MF, Andersen PR, Pühringer F, Hohmann U, Meixner K, Lendl T, Tirian L, and Brennecke J (2019). A Heterochromatin-Specific RNA Export Pathway Facilitates piRNA Production. Cell 178, 964–979.e20. [DOI] [PubMed] [Google Scholar]
- Ernst C, Odom DT, and Kutter C (2017). The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat. Commun 8, 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkash EA, Kao GD, Horman SR, and Prak ETL (2006). Gamma radiation increases endonuclease-dependent L1 retrotransposition in a cultured cell assay. Nucleic Acids Res. 34, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BB, and McFadden G (2006). Anti-Immunology: Evasion of the Host Immune System by Bacterial and Viral Pathogens. Cell 124, 767–782. [DOI] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, and Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, and Wang PJ (2014). Mammalian piRNAs. Spermatogenesis 4, e27889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Yang Y, Zhang H, Farley G, Wang J, Quarles KA, Weng Z, and Zamore PD (2018). The genome of the Hi5 germ cell line from Trichoplusia ni, an agricultural pest and novel model for small RNA biology. ELife 7, e31628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grentzinger T, Armenise C, Brun C, Mugat B, Serrano V, Pelisson A, and Chambeyron S (2012). piRNA-mediated transgenerational inheritance of an acquired trait. Genome Res. 22, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, and Siomi MC (2007). A Slicer-Mediated Mechanism for Repeat-Associated siRNA 5’ End Formation in Drosophila. Science 315, 1587–1590. [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, and Zamore PD (2015). piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger JJ, Bromham LD, McKee JJ, O’Brien TM, and Robinson WF (2000). The Nucleotide Sequence of Koala (Phascolarctos cinereus) Retrovirus: a Novel Type C Endogenous Virus Related to Gibbon Ape Leukemia Virus. J. Virol 74, 4264–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson Göran K, Libby Peter, Schönbeck Uwe, and Yan Zhong-Qun (2002). Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis. Circ. Res 91, 281–291. [DOI] [PubMed] [Google Scholar]
- Hayward A (2017). Origin of the retroviruses: when, where, and how? Curr. Opin. Virol 25, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, and Jacobson A (2015). Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu. Rev. Genet 49, 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs M, Pavasovic A, King AG, Prentis PJ, Eldridge MD, Chen Z, Colgan DJ, Polkinghorne A, Wilkins MR, Flanagan C, et al. (2014). A transcriptome resource for the koala (Phascolarctos cinereus): insights into koala retrovirus transcription and sequence diversity. BMC Genomics 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich MD, Li C, Matranga C, Vagin V, Farley G, Wang P, and Zamore PD (2007). The Drosophila RNA Methyltransferase, DmHen1, Modifies Germline piRNAs and Single-Stranded siRNAs in RISC. Curr. Biol 17, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Hur JK, Luo Y, Moon S, Ninova M, Marinov GK, Chung YD, and Aravin AA (2016). Splicing-independent loading of TREX on nascent RNA is required for efficient expression of dual-strand piRNA clusters in Drosophila. Genes Dev. 30, 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Zhao K, Greenwood AD, and Roca AL (2015). Proliferation of Endogenous Retroviruses in the Early Stages of a Host Germ Line Invasion. Mol. Biol. Evol 32, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, and Devine SE (2010). Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell 141, 1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE (2015). Endogenous Retroviruses in the Genomics Era. Annu. Rev. Virol 2, 135–159. [DOI] [PubMed] [Google Scholar]
- Johnson RN, O’Meally D, Chen Z, Etherington GJ, Ho SYW, Nash WJ, Grueber CE, Cheng Y, Whittington CM, Dennison S, et al. (2018). Adaptation and conservation insights from the koala genome. Nat. Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Wang J, Xu J, Koppetsch BS, Thomson TC, Nowosielska A, Li C, Zamore PD, Weng Z, and Theurkauf WE (2011). Adaptation to P element transposon invasion in Drosophila melanogaster. Cell 147, 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino Y, and Mourelatos Z (2007). Mouse Piwi-interacting RNAs are 2’-O-methylated at their 3’ termini. Nat. Struct. Mol. Biol 14, 347–348. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, et al. (2009). The Drosophila HP1 Homolog Rhino Is Required for Transposon Silencing and piRNA Production by Dual-Strand Clusters. Cell 138, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, and Ussery DW. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, et al. (2009a). Collapse of Germline piRNAs in the Absence of Argonaute3 Reveals Somatic piRNAs in Flies. Cell 137, 509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009b). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, et al. (2013). An Ancient Transcription Factor Initiates the Burst of piRNA Production during Early Meiosis in Mouse Testes. Mol. Cell 50, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 10–12. [Google Scholar]
- McClintock B (1984). The significance of responses of the genome to challenge. Science 226, 792–801. [DOI] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, and Burge CB (2012). Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, and Kaessmann H (2013). Birth and expression evolution of mammalian microRNA genes. Genome Res. 23, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, and Brennecke J (2014). The Rhino-Deadlock-Cutoff Complex Licenses Noncanonical Transcription of Dual-Strand piRNA Clusters in Drosophila. Cell 157, 1364–1379. [DOI] [PubMed] [Google Scholar]
- Mohn F, Handler D, and Brennecke J (2015). piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348, 812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlan JD, Qu K, and Sinicropi DV (2012). Selective Depletion of rRNA Enables Whole Transcriptome Profiling of Archival Fixed Tissue. PLOS ONE 7, e42882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhad SS, and Theurkauf WE (2018). Rapid evolution and conserved function of the piRNA pathway. Open Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhad SS, Tu S, Weng Z, and Theurkauf WE (2017). Adaptive Evolution Leads to Cross-Species Incompatibility in the piRNA Transposon Silencing Machinery. Dev. Cell 43, 60–70.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul WE (2011). Bridging Innate and Adaptive Immunity. Cell 147, 1212–1215. [DOI] [PubMed] [Google Scholar]
- Pessel-Vivares L, Houzet L, Laine S, and Mougel M (2015). Insights into the nuclear export of murine leukemia virus intron-containing RNA. RNA Biol. 12, 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud’homme N, Gans M, Masson M, Terzian C, and Bucheton A (1995). Flamenco, a gene controlling the gypsy retrovirus of Drosophila melanogaster. Genetics 139, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC (1991). Regulation of Drosophila P element transposition. Trends Genet. 7, 282–287. [DOI] [PubMed] [Google Scholar]
- Roovers EF, Rosenkranz D, Mahdipour M, Han C-T, He N, Chuva de Sousa Lopes SM, van der Westerlaken LAJ, Zischler H, Butter F, Roelen BAJ, et al. (2015). Piwi Proteins and piRNAs in Mammalian Oocytes and Early Embryos. Cell Rep. 10, 2069–2082. [DOI] [PubMed] [Google Scholar]
- Rowe WP, and Kozak CA (1980). Germ-line reinsertions of AKR murine leukemia virus genomes in Akv-1 congenic mice. Proc. Natl. Acad. Sci 77, 4871–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe WP, Hartley JW, and Bremner T (1972). Genetic Mapping of a Murine Leukemia Virus-Inducing Locus of AKR Mice. Science 178, 860–862. [DOI] [PubMed] [Google Scholar]
- Saito K, Sakaguchi Y, Suzuki T, Suzuki T, Siomi H, and Siomi MC (2007). Pimet, the Drosophila homolog of HEN1, mediates 2’-O-methylation of Piwi-interacting RNAs at their 3’ ends. Genes Dev. 21, 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, and Aravin AA (2011). PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 12, 246–258. [DOI] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthès P, Kokkinaki M, Nef S, Gnirke A, et al. (2013). Cellular Source and Mechanisms of High Transcriptome Complexity in the Mammalian Testis. Cell Rep. 3, 2179–2190. [DOI] [PubMed] [Google Scholar]
- Steinbiss S, Willhoeft U, Gremme G, and Kurtz S (2009). Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, et al. (2013). Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature 493, 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton RE, Meers J, and Young PR (2006). Retroviral invasion of the koala genome. Nature 442, 79. [DOI] [PubMed] [Google Scholar]
- Thomas AL, Stuwe E, Li S, Du J, Marinov G, Rozhkov N, Chen Y-CA, Luo Y, Sachidanandam R, Toth KF, et al. (2014). Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev. 28, 1667–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber CD, and Waddell S (2017). Resolving the prevalence of somatic transposition in Drosophila. ELife 6, e28297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Zheng K, Fu Q, Maragkakis M, Alexiou P, Ma J, Pillai RS, Mourelatos Z, and Wang PJ (2015). The RNA helicase MOV10L1 binds piRNA precursors to initiate piRNA processing. Genes Dev. 29, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik KA, Tam OH, Knott SR, Falciatori I, Hammell M, Vagin VV, and Hannon GJ (2015). RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev. 29, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick E-M, and Miska EA (2014). piRNAs: from biogenesis to function. Development 141, 3458–3471. [DOI] [PubMed] [Google Scholar]
- Yurkovetskiy L, Guney MH, Kim K, Goh SL, McCauley S, Dauphin A, Diehl WE, and Luban J (2018). Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat. Microbiol 3, 1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanni V, Eymery A, Coiffet M, Zytnicki M, Luyten I, Quesneville H, Vaury C, and Jensen S (2013). Distribution, evolution, and diversity of retrotransposons at the flamenco locus reflect the regulatory properties of piRNA clusters. Proc. Natl. Acad. Sci 110, 19842–19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Tu S, Yu T, Zhang X-O, Parhad SS, Weng Z, and Theurkauf WE (2018). Co-dependent Assembly of Drosophila piRNA Precursor Complexes and piRNA Cluster Heterochromatin. Cell Rep. 24, 3413–3422.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, and Zamore PD (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang J, Schultz N, Zhang F, Parhad SS, Tu S, Vreven T, Zamore PD, Weng Z, and Theurkauf WE (2014). The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Wang J, Theurkauf W, and Weng Z (2014). TEMP: a computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 42, 6826–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 1. KoRV-A and three other ERVs are potentially active in the koala genome.
A. A scatterplot depicting the number of full-length copies of a transposon subfamily (defined as longer than 50% of the consensus sequence length) vs. divergence from the consensus sequence. Four ERVs are labeled, including KoRV-A, which have more than five full-length copies at divergence lower than 1% in the koala genome.
B. Venn diagrams showing the overlap of transposon insertions among the three tissues of K63464. They correspond to the left panel of Figure 1C but for transposon subfamilies other than KoRV-A.
C. Venn diagrams showing the overlap of transposon insertions among the various koalas. They correspond to the right panel of Figure 1C but for transposon subfamilies other than KoRV-A.
D. Numbers of uninherited insertions and germline insertions for all transposon subfamilies in the testis, brain, and liver of K63464. Uninherited insertions are defined as insertions supported by only one discordant read and only present in one tissue. Germline insertions are defined as in Figure 1B. KoRV-A, Ko.ERV.1, Ko.ERVL.1, Ko.ERVK.14 and PhER are colored in red, blue, green, yellow, and purple respectively, while other transposon subfamilies are in black.
Supplementary Table3, linked to Figures 4 and 5. Number of reads mapping to junctions and splice sites of transposons mentioned in this study.
Supplementary Table4, linked to STAR Methods, Experimental details. Mapping statistics of DNA-seq, RNA-seq and small RNA-seq data.
Figure S2, related to Figure 2. Koala piRNAs are produced in the testis but not in the brain or liver.
A-F. Length distribution of piRNAs, nucleotide composition of piRNAs, and distribution of overlapping nucleotides between sense and antisense piRNAs.
A and B. All genome-mapping oxidized small RNA reads in K63464 and K63855 testis samples, after removal of rRNAs, miRNAs, snRNAs, snoRNAs, and tRNAs.
C-H. Unoxidized small RNA reads in testis, liver and brain from K63464 and K63855
Figure S3, related to Figure 2. Most piRNA pathway genes are specifically expressed in the koala testis.
A heatmap showing the expression levels of 19 piRNA pathway genes in various koala tissue samples. The expression level (in RPKM) is written in each cell, and highly expressed genes are shown in shades of red.
Figure S4, related to Figure 2. Koala piRNA clusters have similar features to mouse piRNA clusters.
A. Long RNA expression levels at intergenic piRNA clusters (red) and genic piRNA clusters (blue) in the testis, brain and liver samples of the two koalas assayed by us.
B. Boxplots show the maximal A-MYB motif scores in the ±100 bp window centered on the TSSs of 19,945 mRNAs or three classes of piRNA clusters: 202 genic unidirectional, 132 intergenic unidirectional, and 42 bidirectional piRNA clusters.
C. piRNA clusters are significantly depleted of transposons. The observed number of transposon overlapping nucleotides in each of 376 piRNA clusters is plotted against the expected number of transposon-overlapping nucleotides. Wilcoxon rank-sum test p-value = 3.1 x 10−7 and < 2.2 x 10−16 for intergenic (red) and genic (blue) piRNA clusters, respectively.
D. A genic, unidirectional and uni-strand piRNA cluster with a KoRV-A insertion in the reference koala genome but the insertion is absent in our animals. The top track indicates this piRNA cluster and the KoRV-A insertion annotated in the reference genome. The middle and bottom tracks illustrate DNA-seq reads mapping around the KoRV-A insertion in K63464 and K63855 brain samples. Note the absence of reads that map to the KoRV-A despite plentiful reads in flanking regions. Only the two ends of the KoRV-A insertion are shown and the internal segment is illustrated with dashed lines (not to scale).
E. Profiles showing average piRNA abundance in the ±5kb window around KoRV-A insertions in K63464 and K63855. piRNA abundance in KoRV-A is shown in the center. piRNAs in the same strand as KoRV-A insertions are in blue while piRNAs in the opposite strand in red.
Figure S5, related to Figure 3. Length distribution and ping-pong signature for endogenous retrovirus piRNAs.
A. Size distribution of piRNAs mapping to KoRV-A, Ko.ERV.1, Ko.ERVL.1, or Ko.ERVK.14, corresponding to Figure 3D but for oxidized piRNAs in the K63464 testis and unoxidized piRNAs in the K63464 and K63855 testis tissues, respectively.
B. Distributions of overlapping nucleotides between sense and antisense piRNAs from KoRV-A, Ko.ERV.1, Ko.ERVL.1, and Ko.ERVK.14, for oxidized piRNAs in the K63464 and K63855 testis and unoxidized piRNAs in the K63464 and K63855 testis tissues, respectively.
Figure S6, related to Figure 4. piRNAs derived from genic piRNA clusters are preferentially spliced.
A. Long RNA reads across KoRV-A in liver and brain, with the junction to splice-site ratios for the main exon-exon junction indicated. These figures correspond to Figure 4A but for long RNA reads in liver and brain samples of two Koalas.
B. Barplot at the left showing splicing index of KoRV-A transcripts in K63464 and K63855 testis, brain, and liver tissues. Boxplot at the right showing splicing index of shared expressed transcripts (RPKM ≥ 1) in K63464 and K63855 testis, brain, and liver tissues
C. Normalized long RNA and piRNA reads and piRNA to long RNA ratio across Ko.ERV.1 in koala testis, corresponding to Figure 4A but for Ko.ERV.1 in koala.
D. Boxplots indicating splicing index of 2,460 exon-exon junctions (EEJ) in 204 genic piRNA clusters. Splicing index defined by long RNA reads is marked as green while splicing index defined by piRNA reads are marked as purple. We could only detect junction- or splice site-mapping piRNA reads and long RNA reads at 1,404 junctions.
E. Boxplot indicating piRNA to long RNA ratio for exons and introns of KoRV-A, genic piRNA clusters and intergenic piRNA clusters.
F. A scatterplot comparing the splicing index defined by long RNA reads and piRNA reads for all transposon exon-exon junctions in the mouse adult testis tissue. This panel corresponds to Figure 4C, which were for sorted mouse pachytene spermatocyte and round spermatid cells.
G. Normalized long RNA and piRNA reads across the ERV1_MD_I transposon in opossum testis, with the ratio of junction reads over splice-site reads for the annotated exon-exon junction indicated. This figure corresponds to Figure 4A but for ERV1_MD_I in opossum.
Supplementary Table1, linked to Figure 1. Overall summary of koala transposons.
Supplementary Table2, linked to Figure 2. Detailed information for 379 annotated koala piRNA clusters.
Data Availability Statement
All data have been deposited to GEO with the accession number GSE128122.