

Abstract
The cardinal motor symptoms that define Parkinson’s disease (PD) clinically have been recognized for over two hundred years. That these symptoms arise following the loss of dopamine neurons in the substantia nigra has been known for the last fifty. These long-established facts have fueled a broadly held expectation that degenerating dopaminergic neurons alone hold the key to understanding and curing PD. This prevalent expectation is at odds with the observation that many nonmotor symptoms, including depression and cognitive inflexibility among others, can appear years earlier than the overt dopaminergic neuron degeneration that drives motor abnormalities and are not improved by levodopa treatment. Thus, preserving or rescuing dopamine neuron health and function is of paramount importance, but this alone fails to capture the underlying neurobiology of earlier-appearing nonmotor symptoms. Insight into the complete landscape of disease-related abnormalities and the context in which they arise can be gleaned from a more comprehensive consideration of the PARK genes that are known to cause PD. Here, we make the case that a full incorporation of research showing when and where PARK genes are expressed as well as the impact of gene mutation on function throughout life, in tandem with research studying how dopaminergic neuron degeneration begins, is essential for a full understanding of the multi-dimensional etiology of PD. A broad view may also reveal something about long-term adjustments cells and systems make in response to gene mutation and help to identify mechanisms conferring the resilience or susceptibility of some cells and systems over others.
Keywords: Parkinson’s Disease, PARK Genes, nonmotor symptoms, development, critical periods, protein trafficking, protein degradation
Graphical Abstract
Why study genetic forms of Parkinson’s Disease?
The idea that PD can have a genetic component is relatively new in the context of a 200-year history. In 1997, point mutations in the gene encoding alpha-synuclein, SNCA, were found to cause an autosomal dominant form of PD (Polymeropoulos et al., 1997). Since then, a handful of PARK genes have been identified as monogenic causes of PD (Table 1). PARK genes account for only about 10% of total PD (de Lau and Breteler, 2006), but they are the subject of intensive research efforts with the hope that the PD arising from gene mutation will share features with sporadic PD and lead the way to cures. Laboratory efforts have been focused on (1) producing animal models of the disease that could be used to interrogate disease progression and for testing therapies; (2) gaining an understanding of the functional consequences of particular mutations in order to discover triggers responsible for initiating the disease; and (3) identifying mechanisms shared by PARK proteins that point toward common pathways.
TABLE 1.
| GENE | Inheritance | Mutation | Typical age of onset (years) | Progression | Prominent symptoms outside of the motor symptoms defining PD | Response to L-DOPA | Lewy Pathology | Citations | |
|---|---|---|---|---|---|---|---|---|---|
| Group I | PARKIN/PARK2 | AR | probable loss of function | 24-40 | slow | dystonia; depression; anxiety; autonomic | Yes | Rare | Kitada et al, 1998; Kahn et al, 2003 |
| PINK1/PARK6 | AR | probable loss of function | 18-56 | slow | dystonia; depression; anxiety; autonomic | Yes | Yes (n=1) | Hatano et al, 2004; Valente et al, 2004; Steinlechner et al, 2007; Samaranch et al, 2010 | |
| DJ-1/PARK7 | AR | probable loss of function | 24-40 | slow | dystonia; anxiety | Yes | Yes (n=1) | Bonifati et al, 2003; Taipa et al, 2016 | |
| ATP13A2/PARK9 | AR | probable loss of function | 10-18 | rapid | Kufor Rakeb Syndrome. supranuclear gaze palsy,spasticity,cognitive decline,pyramid atrophy; psychotic episodes. | Yes | Yes | Ramirez et al, 2006 | |
| FBXO7/PARK15 | AR | probable loss of function | 35-50 | slow | dystonia; spasticity; pyramidal symptoms | Yes (non pyramidal) | ? | Di Fonzo et al, 2009 | |
| VPS13C/PARK23 | AR | probable loss of function | 25-46 | rapid | dysautonomia; cognitive decline; incontinence | Yes | Yes (n=1) | Lesage et al, 2016 | |
| Group II | SNCA/PARK1 and PARK4 | AD | mutation; duplication; change and/or gain of function triplication; gain of function |
35-65 30’s |
variable rapid | cognitive decline; depression; sleep disorders dementia; paranoia; hallucinations |
Yes Yes | Yes Yes |
Polymeropoulos et al, 1997; Ibanez et al, 2004; Chartier-Harlin et al, 2004 Singleton et al, 2003 |
| LRRK2/PARK8 | AD | change or gain of function | 50-65 | slow | depression; sleep disorders; cognitive decline | Yes | not always | Paisan-Ruiz et al, 2004; Zimprich et al, 2004 | |
| VPS35/PARK17 | AD | partial loss of function | 50-52 | slow | Yes | No (n=1) | Vilarino-Guell et al, 2011; Zimprich et al, 2011 | ||
| Group III | DNAJC6/auxillin/PARK19 | AR | probable loss of function | 10-42 | rapid | seizures; pryamidal symptoms;intellectual disability | Mixed | ? | Edvardson et al, 2012 |
| SJ1/synaptojanin/PARK20 | AR | partial loss of function | 20-30 | rapid | generalized seizures; cognitive decline; supranuclear gaze palsy | Yes but develop dyskinesia | ? | Krebs et al, 2013 |
Despite substantial progress towards each of these goals (e.g. Chesselet et al., 2012; Vingill et al., 2016; Cao et al., 2017), to date, no single PD gene mutation has produced a bona fide PD model in laboratory animals having the defining motor deficiencies arising as a consequence of the progressive degeneration of dopaminergic neurons in the SN (Blesa and Przedborski, 2014). This suggests either that PD is a uniquely human disease or that the appropriate conditions (environment, age, etc.) have not yet been met in the animal models (usually rats and mice). The difficulty of identifying suitable rodent models for neurodegenerative diseases is a roadblock that extends well beyond PD, as several lines of evidence indicate that rodents are more resistant to many forms of neurodegeneration than humans and cellular mechanisms underlying neurodegeneration may display species-specific differences (Seok et al., 2013; Kreiner, 2015; Burbulla et al., 2017). Human inducible pluripotent stem cell (hiPSC)-based models are particularly valuable for elucidating human-specific cell biological functions, but they are currently limited as a model for understanding the impact of such cell biology on synaptic circuit function and the behaviors they support. This limitation is partly a reflection of the relative state of immaturity of the neurons, as their gene profiles best match human neurons in their first trimester of life (Brennand et al., 2015) and partly because they do not generate complex circuits, although this may change with continued progress in the generation of 3-d organoid stem cell cultures (Lancaster et al., 2013).
Challenges associated with producing bona fide disease models should not obscure the enormous gains that have been made in understanding the biology of PD-causing genes and the impact of PD-causing gene mutations on the activity and function of neural circuits generally and in systems relevant to PD. In contrast to neural degeneration, basic principles of cell biology and circuit development in rodents have proven to be good predictors for the function of cells and circuits in humans.
Three PARK Gene Groups in Space and Time
For the purposes of this review, we have placed the PARK genes into three groups (I-III) based on pattern of inheritance, age of motor symptom onset, and cellular actions (Table 1). Binning genes into groups inevitably obscures some important nuances and to counter this we highlight some areas of crosstalk between the groups, but the strategy provides a useful construct for discussion.
Group I comprises six genes, PARKIN, PINK1, DJ-1, FBXO7, VPS13C and ATP13A2. Mutations identified in these genes produce autosomal recessive forms of PD that appear to be loss of function. Symptoms typically emerge much earlier than sporadic PD and as early as the first decade of life, consistent with a developmental disorder. All of the encoded proteins in this group act in cellular pathways that are relevant for marking and clearing defective mitochondria or for maintaining mitochondrial health, and this has been the subject of multiple reviews (Abou-Sleiman et al., 2006; Farrer, 2006; Corti and Brice, 2013; Itoh et al., 2013; Scarffe et al., 2014; Menzies et al., 2015; Ryan et al., 2015). Briefly summarized, PARKIN, PINK1 and FBXO7 work coordinately to promote mitophagy (Clark et al., 2006; Springer et al., 2006; Yang et al., 2006; Narendra et al., 2008; Narendra et al., 2010; Pompilio and Kacelnik, 2010; Burchell et al., 2013; Bingol and Sheng, 2016). In parallel, Parkin can also regulate mitochondrial biogenesis indirectly by controlling levels of the transcriptional repressor, PARIS (Shin et al., 2011). DJ-1, VPS13C and ATP13A2 appear to act in pathways that lie upstream (DJ-1 and ATP13A2) or in parallel (VPS13C) to the others indirectly regulating mitochondrial health (Canet-Aviles et al., 2004; Park et al., 2014; Moscovitz et al., 2015; Lesage et al., 2016; Burbulla et al., 2017) or degradation (Dehay et al., 2012; Kong et al., 2014; Tsunemi and Krainc, 2014). Whether direct or indirect, the actions of this cohort of genes on mitochondria dovetail with data showing that neurotoxins known or suspected to cause PD-like motor dysfunction including MPTP, rotenone and paraquat interfere with mitochondrial metabolism (Martinez and Greenamyre, 2012). Early disease onset suggests the impact of these genes is direct and cumulative. In this light it is interesting that PARKIN mutations rarely produce Lewy Bodies, the neuronal cytoplasmic inclusions that are the pathological hallmark of sporadic Parkinson’s Disease (Postuma et al., 2015), suggesting that some mechanisms may lie downstream of or can bypass production of Lewy pathology.
In situ hybridization data show that Group I genes are expressed broadly and almost homogeneously throughout the brain (Fig. 1) and body (www.proteinatlas.org; Uhlen et al., 2015; Allen Institute; Lein et al., 2007) beginning prior to birth and continuing through life (Fig. 2). On its surface, this pattern contradicts the apparent selectivity of PD symptoms. However, with respect to dopaminergic neurons, specificity may arise from intrinsic properties since strong data support that their tonic pacemaking activity is uniquely dependent on calcium currents that render them vulnerable to oxidative stress (Puopolo et al., 2007; Guzman et al., 2010; Burbulla et al., 2017). Selective vulnerability of dopamine neurons could also reflect the huge bioenergetic demands of maintaining extensive and uniquely complex axonal arbors (Bolam and Pissadaki, 2012). Conversely, the broad expression pattern of Group I genes suggests that most cells may be able to tolerate or can functionally compensate for loss of gene function. Additional populations may not die, but may suffer consequences mediated by the mutations. A recent study by Vingill and colleagues (Vingill et al., 2016) pinpoints the relevance of restricted populations of cells to particular disease symptoms while also highlighting the presence of pathology that sits outside the usual suspects. In mice, conditional deletion of Fbxo7 from forebrain glutamatergic neurons produces pyramidal tract–like motor impairments such as hindlimb clasping beginning at two months of age (Vingill et al., 2016) and consistent with symptoms seen in humans lacking FBXO7 (Di Fonzo et al., 2009). In contrast, conditional deletion in dopaminergic and noradrenergic neurons produces PD-like deficits in motor coordination and mobility beginning at six months. Germline Fbxo7 knockout mice display motor deficits consistent with combining the impairments observed in the two conditional mutants, but the phenotypes are more severe, the age of onset much earlier and death ensues by P28 (Vingill et al., 2016). Thus, while gene mutations in restricted cell populations are sufficient to produce particular symptoms, germline knockout reveals additional effects that extend well beyond the conditional phenotypes and which are likely contributed by other cell populations that also normally express FBX07.
Fig. 1. Where PARK genes are expressed.
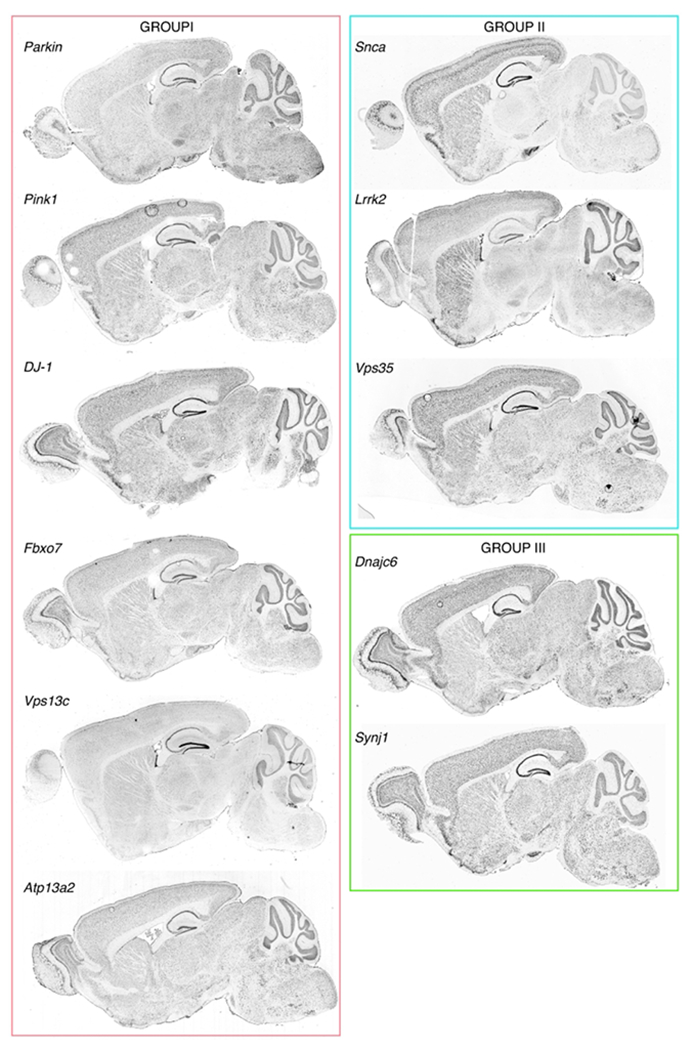
In situ hybridization showing regional distribution patterns of the PARK gene transcripts indicated in adult mice. All data shown are from the Allen Mouse Brain Atlas (Lein et al., 2007). Groups are defined and colored as indicated in Table 1 and text.
Fig. 2. When PARK genes are expressed.
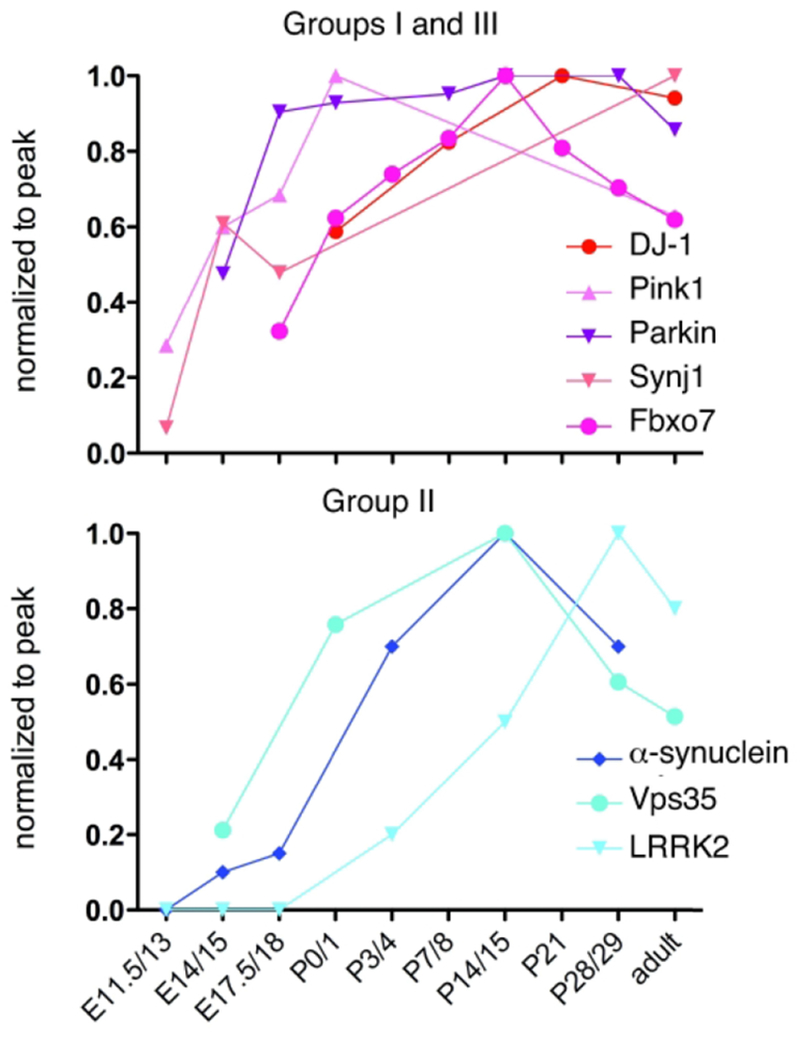
Data from published papers was normalized to peak expression and then plotted in order to provide a gross comparison of the timing of expression of PARK genes(Petersen et al., 1999; Kuhn et al., 2004; Westerlund et al., 2008; Wang et al., 2012; Giesert et al., 2013; Beccano-Kelly et al., 2014; Bjorkblom et al., 2014; Choi et al., 2016). Colors are assigned by Groups defined in Table 1.
In addition to their impact on mitochondria, most Group I genes play additional roles that would also be expected to affect cell health. For example, FBX07, also regulates proteasome assembly (Vingill et al., 2016), Parkin-mediated ubiquitination targets several non-mitochondrial proteins, including Kainate receptors, to the proteasome (Shimura et al., 2000; Maraschi et al., 2014), DJ-1 has chaperone activity (Shendelman et al., 2004; Zhou and Freed, 2005), and ATP13A2 plays an important role in the normal regulation of lysosome function and α-synuclein clearance (Dehay et al., 2012; Kong et al., 2014; Tsunemi and Krainc, 2014). These data suggest that mechanisms supporting protein homeostasis are also negatively impacted by the loss of Group I proteins in a variety of cells, and would be expected to be deleterious, or at the very least, to exacerbate the impact of cellular stressors.
Group II contains SNCA, LRRK2 and VPS35 (Table 1). The genes DNAJC13 or TMEM230 may also be part of this group, but they were identified in the same extended family dataset and have yet to be confirmed, so we have excluded them here (Vilarino-Guell et al., 2014; Deng et al., 2016). Group II genes cause autosomal dominant, late-onset forms of PD. Disease symptoms and progression closely resemble idiopathic PD, making this group particularly relevant for understanding PD in general. The members of this group show interesting anatomical and cellular expression patterns that are more restricted than Group I genes and help to define cell populations most relevant to PD. Group II gene expression is most enriched in cerebral cortex and neostriatum, the major postsynaptic targets for SN dopaminergic neurons, underscoring the idea that retrograde signaling from postsynaptic targets may be relevant to the degeneration of dopamine-expressing neurons (for review, see Bjorklund et al., 1997; Volta et al., 2015; Tagliaferro and Burke, 2016). Group II genes also show a striking increase in expression during synaptogenesis (Fig. 2) in a pattern that is consistent with them playing roles in shaping neural circuits. This developmental^ late onset of expression is also consistent with research showing that Group II proteins are largely dispensable for earlier events in brain development such as regionalization and neuronal cell migration (Abeliovich et al., 2000; Herzig et al., 2011; Wang et al., 2012). All of the encoded Group II proteins regulate intracellular membrane trafficking.
SNCA encodes α-synuclein (Maroteaux and Scheller, 1991), which is the main component of Lewy bodies (Spillantini et al., 1997). SNCA mutation, duplication or triplication in humans is associated with the generation of Lewy Bodies (Singleton et al., 2003; Singleton and Gwinn-Hardy, 2004) and data from animal models support that α-synuclein mutation, and in particular, increased levels, produce Lewy pathology (Fernagut and Chesselet, 2004). Lewy pathology can also develop in the absence of endogenous SNCA mutation and can originate outside the central nervous system from exogenously introduced α-synuclein fibrils and propagate extracellularly like a prion-mediated disease (Kordower et al., 2008; Volpicelli-Daley et al., 2011; Luk et al., 2012; Brundin and Melki, 2017). Aggregated exogenous α-synuclein oligomers can also interfere with lysosomal (Decressac et al., 2013) and mitochondrial function (Luth et al., 2014).
α-Synuclein is enriched in brain and erythroid cells (Nakai et al., 2007). Within the brain, it is expressed almost exclusively in neurons (Gokce et al., 2016), and highest levels are observed in olfactory bulb, cerebral cortex, dorsal and ventral striatum, hippocampus, and SN (Fig. 1). α-Synuclein and its closely related family members, β- and γ-synucleins, concentrate in presynaptic terminals, loosely associated with vesicle membranes (Iwai et al., 1995; Fortin et al., 2005) where they appear to negatively regulate vesicle tethering to one another and to the presynaptic active zone (Vargas et al., 2017). It is likely that it is through these interactions and effects on membrane curvature (Lautenschlager et al., 2017) that synucleins modulate vesicle endo- and exocytosis. Mice lacking all three isoforms show slower synaptic vesicle endocytosis (Vargas et al., 2014) and increased exocytosis (Greten-Harrison et al., 2010; Anwar et al., 2011), while overexpression attenuates release of both synaptic and large dense core vesicles (Larsen et al., 2006; Nemani et al., 2010; DeWitt and Rhoades, 2013). At dopaminergic terminals α-synuclein A30P mutation or overexpression can also impede vesicle release (Nemani et al., 2010; Janezic et al., 2013; Taylor et al., 2014) and in hippocampal neurons, alter vesicle tethering (Vargas et al., 2017). In contrast to these relatively modest effects, strong, repetitive stimulation in the presence of excess α-synuclein in a lamprey nerve preparation nearly eliminates normal endocytosis (Busch et al., 2014), and in mouse neurons, action potential dependent activity promotes α-synuclein secretion (Yamada and Iwatsubo, 2018). Thus, it may be that synucleins exert their greatest influence over particular kinds of synaptic activity, regulating trafficking events that are especially important for maintaining sustained, highly demanding activity as well as producing widespread responses (Diogenes et al., 2012; Shrivastava et al., 2015).
Mutant or overexpressed SNCA can also produce a blockade of ER to Golgi vesicle trafficking that appears to be a pathological gain in function because it is not consistent with results from SNCA knockout preparations and it targets cellular domains that extend beyond the normal expression pattern of SNCA, which is highly restricted to presynaptic terminals. In mammalian cells the blockade can be relieved by overexpression of Rab1, Rab3A or Rab8A, small GTPases that are important for directing and regulating vesicle traffic (Outeiro and Lindquist, 2003; Cooper et al., 2006; Gitler et al., 2008). Collectively, the data support a role for mutant or overexpressed SNCA in regulating vesicle traffic within and outside of synaptic terminals.
LRRK2 encodes a very large 286kDa multifunctional protein having interactive kinase and GTPase domains. PD causing mutations cluster in these two enzyme domains and most of them increase the phosphorylation of LRRK2 substrates (West et al., 2005; Greggio et al., 2006; Smith et al., 2006; West et al., 2007; Steger et al., 2016). Within the nervous system, LRRK2 is enriched in forebrain neurons (Gokce et al., 2016), with particularly high levels in cerebral cortex, dorsal and ventral striatum (Fig. 1). Expression levels are far lower in dopaminergic neurons in mice (West et al., 2014) and humans (Sandor et al., 2017). The regional distribution pattern displayed by LRRK2 mRNA is very similar to α-synuclein and more restricted than most PARK genes (Fig. 1), highlighting brain regions likely to be particularly important for PD symptoms. LRRK2 is also expressed at high levels in kidney and lung and in peripheral immune cells, including monocytes, dendritic cells and lymphocytes (Gardet et al., 2010; Hakimi et al., 2011; Thevenet et al., 2011). Significantly, its levels rise in immune cells, including brain microglia, upon stimulation (Gardet et al., 2010; Thevenet et al., 2011; Moehle et al., 2012). LRRK2 appears to act as a pro-inflammatory agent consistent with point mutations near or within LRRK2 that have been linked to inflammatory bowel disease and leprosy (Barrett et al., 2008). Since PD is associated with inflammation (Whitton, 2007; Dzamko et al., 2015), immune actions of LRRK2 are likely to be as relevant as neuronal mechanisms.
A growing body of research supports that LRRK2 kinase activity regulates vesicle trafficking (e.g. Piccoli et al., 2011; Matta et al., 2012; MacLeod et al., 2013; Beilina et al., 2014), but mechanistic detail is sparse. The most direct evidence for this role comes from the identification of several Rab family members as in vivo substrates for LRRK2-mediated phosphorylation (Steger et al., 2016). Phosphorylated Rabs have reduced affinity for most Rab effectors and would be predicted to negatively regulate traffic (Steger et al., 2016). Increased LRRK2-mediated Rab phosphorylation can also interfere with the generation of primary cilia (Dhekne et al., 2018; Madero-Perez et al., 2018). Functional studies of LRRK2 knockout/knockdown or loss of function support that LRRK2 can regulate synaptic vesicle recycling with several studies supporting that LRRK2 promotes endocytosis (Matta et al., 2012; Arranz et al., 2015) and others that it regulates exocytosis (Piccoli et al., 2011). Such differences may be reconciled by recent work showing that LRRK2 regulation of synaptic vesicle recycling is mediated differently according to neuron type (Pan et al., 2017). LRRK2 can also regulate postsynaptic expression of AMPA receptors (Parisiadou et al., 2014; Sweet et al., 2015) an activity that normally utilizes a likely LRRK2 substrate, Rab8 (Gerges et al., 2004). At early postnatal ages in mice that carry a Lrrk2-G2019S knockin mutation, the most common LRRK2 PD mutation (Paisan-Ruiz et al., 2013), spiny projection neurons (SPNs) in dorsal striatum show a substantial increase in glutamatergic activity and altered dendritic spine size (Beccano-Kelly et al., 2014; Matikainen-Ankney et al., 2016; Volta et al., 2017). Such excessive synaptic activity is driven in part by increased cortical activity (Beccano-Kelly et al., 2014; Matikainen-Ankney et al., 2016; Volta et al., 2017), and reflects mostly action potential based activity rather than spontaneous vesicle fusion (Matikainen-Ankney et al., 2016). The enhanced activity displayed by these circuits is transient, but is concurrent with synaptogenesis (Matikainen-Ankney et al., 2016; Volta et al., 2017), thus leading to the prediction that abnormalities in striatal synaptic activity, first detectable at early postnatal ages, could have lasting consequences for the function of basal ganglia circuits (Kozorovitskiy et al., 2012).
LRRK2 mutation, and in particular, its increased kinase activity, additionally appears to increase levels of cellular stress and to impact pathways that normally clear misfolded proteins and damaged organelles. LRRK2 overexpression can promote apoptosis (Skibinski et al., 2014) and reduce chaperone-mediated autophagy (Orenstein et al., 2013), effects that are exacerbated by the G2019S mutation (Orenstein et al., 2013; Skibinski et al., 2014). LRRK2 may also normally regulate macroautophagy since LRRK2-KO mice and mice exposed to LRRK2 kinase inhibitors for long periods accumulate protein aggregates and macroautophagy intermediates in kidney where LRRK2 is expressed at exceptionally high levels. Recent work using transgenes in Drosophila also suggests that dLRRK phosphorylation of EndoA may positively regulate macroautophagy of synaptic vesicles (Soukup et al., 2016). These activities are not well understood, but they have gained a substantial level of enthusiasm in the field because they place LRRK2 in pathways important for clearing α-synuclein aggregates and defective mitochondria in ways that could be synergistic with the pathways outlined for the Group I genes. Conversely, recent work shows that Parkin stabilizes endocytic zones and regulates surface expression of AMPA receptors (Cortese et al., 2016), suggesting certain Parkin activities may also converge on LRRK2-dependent trafficking pathways (Parisiadou et al., 2014).
VPS35 mutations underlie the most recently discovered autosomal dominant form of PD (Wider et al., 2008). Its expression is enriched in the forebrain, but broader than LRRK2 or α-synuclein (Fig. 2). VPS35 functions as part of the retromer complex, which together with sorting nexins and Rab7a directs cargo from endosomes to the plasma membrane or to the trans-Golgi (Bonifacino and Hurley, 2008; Williams et al., 2017). Maintaining appropriate levels of VPS35 appears to be crucial as either too much or too little can alter trafficking or impair lysosome function and produce mitochondrial fragmentation (Miura et al., 2014; Wang et al., 2014; Munsie et al., 2015; Tang et al., 2015; Wang et al., 2016). Such data suggest a possible link to Group I’s cellular stress pathways, but there is no evidence that altered levels of VPS35 play a role in PD (Ishizu et al., 2016). The most prevalent mutation, D620N, does not change protein stability (McGough et al., 2014; Tsika et al., 2014; Zavodszky et al., 2014; Munsie et al., 2015; Ishizu et al., 2016), but instead appears to interfere with retromer’s interaction with the WASH complex, which drives generation of F-actin patches at endosomes (McGough et al., 2014; Zavodszky et al., 2014). Perhaps most relevant to the disease are data suggesting that the impact of the D620N mutation may be specific to particular cargos (Choy et al., 2014; Follett et al., 2014; McGough et al., 2014; Tian et al., 2015; Temkin et al., 2017), including proteins relevant to autophagy suggesting a point of interaction with LRRK2 or Group I proteins (Zavodszky et al., 2014; Tang et al., 2015). Such specificity could explain the late onset and could account for anatomical and cell type specificity of the disease, although earlier changes conferred by VPS35 mutation have been described. For example, a recent study has shown that VPS35-D620N knockin mice already display altered striatal dopamine release and turnover by three months of age (Cataldi et al., 2018). Specificity may also be driven by the more restricted localization of LRRK2. Recent work supports that VPS35-D620N potentiates LRRK2 kinase activity suggesting the mutation is gain of function and in the same pathway as LRRK2 (Mir et al., 2018).
Mutations in Group III genes (Table 1), DNAJC6 and SYNJ1, cause autosomal recessive forms of PD that are early onset, severe, and accompanied by cognitive decline (Edvardson et al., 2012; Koroglu et al., 2013; Krebs et al., 2013; Quadri et al., 2013; Olgiati et al., 2014; Olgiati et al., 2016), combining aspects of neurodevelopmental and neurodegenerative disorders. Both Synaptojaninl and Auxilin (the product of DNAJC6) are broadly expressed throughout the brain, but levels are highest in cortical neurons beginning before birth (Fig. 1, 2) (Gokce et al., 2016). They both regulate the removal of clathrin coats from internalized presynaptic vesicles and PD-mutation interferes with this process. Mice expressing mutant SYNJ1 (R258Q knockin) accumulate clathrin coated vesicles in presynaptic terminals and show increased levels of Auxilin as well as Parkin (Cao et al., 2017). Data from Drosophila NMJ suggest that mutant Synaptogjaninl may also reduce presynaptic autophagy (Vanhauwaert et al., 2017). Mice lacking Auxilin (DNAJC6 mutations are complete loss of function) accumulate clathrin coated vesicles at synapses, similar to SYNJ1 mutants, and empty clathrin cages (Yim et al., 2010). Synaptojaninl and Auxilin act in the same pathway, but they do not compensate for one another: increased levels of Auxilin do not rescue mutant Synaptojaninl phenotypes and Synaptojaninl levels remain normal in the absence of Auxilin (Yim et al., 2010).
While these two forms of PD are accompanied by severe cognitive symptoms that are not observed in most PD, they highlight that endocytic trafficking, like mitochondrial health and clearance, is a common PD-sensitive pathway. Additionally, Parkin ubiquitinates (Trempe et al., 2009; Cao et al., 2014) and LRRK2 is suspected to phosphorylate Endophilin, Synaptojaninl and Auxilin (Matta et al., 2012; Arranz et al., 2015; Islam et al., 2016; Soukup et al., 2016; Nguyen and Krainc, 2018).
GBA1 is not classified as a PARK gene, but mutations in GBA1 are the most common genetic risk factor for PD and warrant special emphasis. It encodes an enzyme that is expressed throughout the brain and body and is found on lysosomal membranes where it breaks down glucocerebroside (Willemsen et al., 1987). Homozygous loss of function mutations produce Gaucher’s disease, a lysosomal storage disorder in which enzyme substrates accumulate and produce pathology in a wide variety of organs. Mutations (typically heterozygous) in GBA1 also appear in PD patients where they are five times more commonly observed than in healthy controls (reviewed in Sidransky and Lopez, 2012). Pathological GBA1 mutations all appear to decrease enzyme activity. A study of the distribution of active glucocerebrosidase suggests that activity normally varies considerably between brain regions and cell types (Herrera Moro Chao et al., 2015). This likely underlies particular symptoms and apparent differences in cellular vulnerability. GBA1 mutation can reduce α-synuclein clearance, and significantly, increased levels of α-synuclein appear to decrease the enzyme’s activity (reviewed in Wong and Krainc, 2016). How GBA1 mutations integrate with other causes of PD is not well understood, but they clearly point to the importance of normally functioning recycling and degradative pathways.
Brain circuits are affected by PARK genes throughout life
The late-life onset of dopamine neuron degeneration and subsequent motor symptoms of PD that dominate the clinical presentation have been somewhat tacitly viewed as a pathophysiological process that co-opts neuronal circuit function at the onset of the prodromal phase, usually later in life (Hawkes, 2008). However, as emphasized above, all of the PARK genes are expressed in the brain during development and throughout adulthood (Fig. 1, 2). Thus, PD-causing gene mutations are positioned to shape developing brain circuits, potentially changing fundamentally a variety of interconnected cell and synaptic networks throughout life, including and beyond the onset of neuronal degeneration. This is important, because it is well established that cortical and subcortical circuits display periods of heightened sensitivity to patterns and levels of synaptic activity during postnatal development that can permanently alter circuit structure and function (Wiesel and Hubei, 1963a; b). For example, during early postnatal life in mice, selectively silencing SPNs in dorsal striatum projecting directly to the SN (D1R expressing) leads to decreased synaptic input to both silenced SPNs as well as to neighboring unmanipulated SPNs projecting to the globus pallidus (D2R expressing). Selectively silencing pallidal-projecting SPNs has the opposite effect on both SPN subtypes. These effects are mediated through a multistep circuit and the consequences are long-lasting (Kozorovitskiy et al., 2012). Consequences can also be synapse specific. Embryonic day 9.5 deletion of Huntingtin in cerebral cortex, where it is most enriched, increases the pace of synaptogenesis in both cortical layer 5 neurons and in their dorsal striatum targets, but in adults, the resulting glutamatergic synapses are weaker in cortex and stronger in striatum than in control neurons (McKinstry et al., 2014). Even when the expression of mutant Huntingtin is restricted to development, ending at P21, at 9 months of age, mice show striatal neuron degeneration, motor deficits and altered corticostriatal plasticity similar to mice expressing mutant Huntingtin over the entire lifespan (Molero et al., 2016). Thus, by maturity, disease symptoms may emerge directly, as a cumulative consequence of these changes; indirectly, due to compensatory mechanisms; or they may arise independently, in response to an additional insult. Regardless, it is important to consider that PD-related symptoms may arise within an environment of cells, synapses and circuits that differ in important ways from an environment that had developed in the absence of mutation.
A consideration of LRRK2-G2019S knockin mice, where the mutant protein is expressed at normal physiological levels throughout life, illustrates these and related points. Elevated activity at glutamatergic synapses in striatum that is evident at three postnatal weeks is transient (as described in the section above), but synaptic responses in LRRK2-G2019S striatum remain functionally different than WT synapses into adulthood. In young adult G2019S mice, baseline AMPAR currents at glutamatergic synapses in nucleus accumbens, a part of the ventral striatum important for reward and motivation and implicated in the pathophysiology of depression (Carlezon et al., 2005; Bosch-Bouju et al., 2016; Han and Nestler, 2017), are mediated by a different composition of AMPAR subunits with fewer that are calcium permeable (CP) in comparison with WTs (Matikainen-Ankney et al., 2018). Dynamic trafficking of CP-AMPARs is an important, mechanistic component of persistent forms of synaptic plasticity (Plant et al., 2006; Ma et al., 2018; Zhou et al., 2018). Thus, baseline differences between genotypes in AMPAR stoichiometry would predict defects in lasting forms of synaptic plasticity. Corticostriatal synapses normally display bidirectional plasticity--long-term potentiation (LTP), a strengthening of synaptic signals which is postsynaptically mediated (Kreitzer and Malenka, 2008; Ma et al., 2018), and long-term depression (LTD), a weakening of synaptic strength that is pre-synaptically mediated (Calabresi et al., 1992; Choi and Lovinger, 1997; Kreitzer and Malenka, 2007). Consistent with the prediction, LRRK2-G2019S mice are unable to express corticostriatal LTP, a deficit evident in both D1R- and D2R-SPNs. This deficit is evident at three postnatal weeks and sustained into adulthood (Matikainen-Ankney et al., 2018). Additionally, D2R-SPNs abnormally display LTD following an LTP induction protocol. Thus, normally bidirectional striatal synaptic plasticity in WTs is instead abnormally unidirectional in G2019S striatum.
As might be predicted from impaired striatal synaptic plasticity, LRRK2-G2019S mice exhibit significant differences in striatal-dependent behavioral responses that depend on a full-range of synaptic modifications. Chronic social defeat stress (CSDS) is a validated rodent model of depression in which mice are subjected to brief, daily periods of physical subordination by a larger male aggressor mouse, then tested for their social interaction with a novel social target. WT mice that undergo CSDS exhibit one of two social behaviors when tested for social interaction--about half of the mice are "susceptible", meaning they display significant social avoidance and anhedonia-like behaviors that can be reversed by chronic antidepressants, while the rest are "resilient", meaning that despite defeat experience, they remain socially interactive (Golden et al., 2011). Susceptible WT mice acquire significant numbers of CP-AMPARs at synapses in the nucleus accumbens during social defeat, which in turn is thought to drive, at least in part, subsequent social avoidance behavior (Vialou et al., 2010). Resilient WT mice, in contrast, retain an AMPAR response profile that is more-or-less similar to unstressed control mice. Surprisingly, young adult LRRK2-G2019S mice are almost completely (~94%) resilient to CSDS (compared to 57% resilient WT mice), remaining highly socially interactive despite defeat experience, and concomitantly fail to acquire CP-AMPARs (Matikainen-Ankney et al., 2018). Thus, the G2019S mutation promotes resilience to chronic social stress in young adulthood which very likely reflects striatal synapses unable to display the full range of experience-dependent synaptic plasticity. In the absence of CSDS, G2019S and WT mice are indistinguishable on a variety of tasks across several behavioral domains, including motor coordination, anxiety, exploratory activity, self-care and anhe donia-like behaviors, consistent with other studies of different G2019S knockin lines of mice (reviewed in (Volta and Melrose, 2017). This could indicate that behavioral differences from WT mice become evident only under extreme, experience-dependent challenges, such as CSDS, and while the significance of a high degree of behavioral resilience to social stress in young adult LRRK2-G2019S mice is uncertain, it is possible this represents some kind of compensatory response. Since depression is a prominent comorbid non-motor symptom of PD (Ishihara and Brayne, 2006; Gaig et al., 2014), it is also possible that the behavioral resilience observed in young adult G2019S mice would give way to susceptibility later in life, an idea supported by transgenic mice overexpressing LRRK2-G2019S, which after 8 - 10 months of age show a profound deficit in hippocampal LTD (Sweet et al, 2015) and display anxiety and depression-like behaviors (Lim et al., 2018). It is unclear what would mediate the transition from adaptive resilience to maladaptive susceptibility and depression, but progressive changes in dopamine or other systems could play a role.
In addition to LRRK2-G2019S, several other mouse models of PD gene mutations have also yielded significant changes in normal brain synaptic plasticity in young adults. For example, mice lacking either Parkin or Pink1 show impaired striatal LTP and LTD (Kitada et al., 2007; Kitada et al., 2009). DJ-1 knockout mice show a selective loss of striatal LTD (Goldberg et al., 2005), and hippocampal neurons in mice expressing VPS35-D620N show deficits in LTP and reduced activity dependent incorporation of CP-AMPARs (Temkin et al., 2017). Taken together, these data strongly suggest that mechanisms of synapse plasticity that rely on AMPA receptor trafficking are particularly sensitive to PD gene mutation. They also support that PD gene mutations regulate brain development and function and most likely alter the landscape in which disease symptoms appear. Even more likely, they contribute to the disease, but in the absence of DA neuron degeneration, pursuit of these results has been less than aggressive and it remains an area of research that has great promise.
What’s Next?
Strong evidence supports that many of the PARK gene mutations change neural circuits and have a sustained influence on non-motor behaviors. This is likely to be true for all PARK gene mutations and similar to other brain disorders having genetic and experience dependent components (Caspi and Moffitt, 2006). Shared biological roles played by Group I genes justifies current focused efforts on understanding relationships between mitochondrial health and PD. At the same time, nearly all cells express Group I genes throughout the lifespan and nearly all Group I genes are multifunctional, having roles outside of mitochondrial health (and some of these roles are also shared). A broader view may reveal, for example, why most cells are resistant to PD pathology. Shared biological pathways and/or shared distribution patterns shown by Group II and Group III genes highlight the importance of membrane trafficking, endocytosis and mechanisms supporting sustained neural activity in cerebral cortex and dorsal striatum in addition to dopaminergic neurons. LRRK2 appears to have a highly restricted set of substrates, VPS35 may traffic an equally restricted set of proteins and work collaboratively with LRRK2, and α -synuclein and LRRK2 may selectively impact particular types of activity. These restricted roles may be important clues to cellular systems susceptible to cellular stress. The actions of Group III proteins for which disease onset is very early, underscore the relevance of endocytosis, and members of all three Groups as well as GBA1 are important for regulating autophagy and managing protein turnover. It is also worth highlighting that actions of PARK genes within the nervous system can be strongly influenced by interactions with other systems, and in particular, the immune system (Kannarkat et al., 2013; Dzamko et al., 2015). Additional work studying cross-talk will further the understanding of neural circuits and non-neural systems that contribute to or are impacted by PD and provide new insights into novel, targeted treatment strategies.
When and where are Parkinson’s disease (PARK) genes expressed in the brain? How do PARK gene mutations impact the development and function of neural circuits? Here we discuss how spatial and temporal expression data when coupled with emerging cellular mechanisms provide important clues for detecting, understanding and treating this disease.
Acknowledgments
We would like to thank Dr. Tim Ahfeldt and Dr. Bridget Matikainen-Ankney for critically reviewing an earlier version of this paper.
REFERENCES
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. 2000. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25(1):239–252. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MM, Wood NW. 2006. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 7(3):207–219. [DOI] [PubMed] [Google Scholar]
- Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, Deacon RM, Bannerman DM, Bolam JP, Chandra SS, Cragg SJ, Wade-Martins R, Buchman VL. 2011. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci 31(20):7264–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz AM, Delbroek L, Van Kolen K, Guimaraes MR, Mandemakers W, Daneels G, Matta S, Calafate S, Shaban H, Baatsen P, De Bock PJ, Gevaert K, Vanden Berghe P, Verstreken P, De Strooper B, Moechars D. 2015. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci 128(3):541–552. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Consortium NIG, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Belgian-French IBDC, Wellcome Trust Case Control C, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature genetics 40(8):955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ, Milnerwood AJ. 2014. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Frontiers in cellular neuroscience 8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, International Parkinson’s Disease Genomics C, North American Brain Expression C, Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans JM, Greggio E, Cookson MR. 2014. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 111(7):2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Sheng M. 2016. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free radical biology & medicine 100:210–222. [DOI] [PubMed] [Google Scholar]
- Bjorkblom B, Maple-Grodem J, Puno MR, Odell M, Larsen JP, Moller SG. 2014. Reactive oxygen species-mediated DJ-1 monomerization modulates intracellular trafficking involving karyopherin beta2. Molecular and cellular biology 34(16):3024–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A, Rosenblad C, Winkler C, Kirik D. 1997. Studies on neuroprotective and regenerative effects of GDNF in a partial lesion model of Parkinson’s disease. Neurobiology of disease 4(3–4):186–200. [DOI] [PubMed] [Google Scholar]
- Blesa J, Przedborski S. 2014. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Frontiers in neuroanatomy 8:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK. 2012. Living on the edge with too many mouths to feed: why dopamine neurons die. Movement disorders : official journal of the Movement Disorder Society 27(12):1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. 2008. Retromer. Curr Opin Cell Biol 20(4):427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch-Bouju C, Larrieu T, Linders L, Manzoni OJ, Laye S. 2016. Endocannabinoid-Mediated Plasticity in Nucleus Accumbens Controls Vulnerability to Anxiety after Social Defeat Stress. Cell reports 16(5):1237–1242. [DOI] [PubMed] [Google Scholar]
- Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, Abdelrahim M, Matikainen-Ankney B, Chao SH, Mrksich M, Rakic P, Fang G, Zhang B, Yates JR, 3rd, Gage FH. 2015. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Molecular psychiatry 20(3):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Melki R. 2017. Prying into the Prion Hypothesis for Parkinson’s Disease. J Neurosci 37(41):9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Kruger R, Surmeier DJ, Krainc D. 2017. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357(6357):1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H, Abramov AY, Hardy J, Wood NW, Whitworth AJ, Laman H, Plun-Favreau H. 2013. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16(9):1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Oliphint PA, Walsh RB, Banks Sm, Woods WS, George JM, Morgan JR. 2014. Acute increase of alpha-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol Biol Cell 25(24):3926–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. 1992. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci 12(11):4224–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. 2004. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A 101(24):9103–9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Milosevic I, Giovedi S, De Camilli P. 2014. Upregulation of Parkin in endophilin mutant mice. J Neurosci 34(49):16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA, Boassa D, Ellisman MH, Ryan TA, De Camilli P. 2017. Parkinson sac domain mutation in Synaptojanin 1 impairs Clathrin uncoating at synapses and triggers dystrophic changes in Dopaminergic axons. Neuron 93(4):882–896 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr., Duman RS, Nestler EJ. 2005. The many faces of CREB. Trends in neurosciences 28(8):436–445. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE. 2006. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 7(7):583–590. [DOI] [PubMed] [Google Scholar]
- Cataldi S, Follett J, Fox JD, Tatarnikov I, Kadgien C, Gustavsson EK, Khinda J, Milnerwood AJ, Farrer MJ. 2018. Altered dopamine release and monoamine transporters in Vps35 p.D620N knock-in mice. NPJ Parkinsons Dis 4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. 2012. A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 9(2):297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, Choi DJ, Yang H, Woo JH, Chang MY, Kim JY, Sun W, Park SM, Jou I, Lee SH, Joe EH. 2016. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Molecular brain 9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. 1997. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci U S A 94(6):2665–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA, von Zastrow M. 2014. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 82(1):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441(7097):1162–1166. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. 2006. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785):324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese GP, Zhu M, Williams D, Heath S, Waites CL. 2016. Parkin deficiency reduces hippocampal glutamatergic neurotransmission by impairing AMPA receptor endocytosis. J Neurosci 36(48):12243–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti O, Brice A. 2013. Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson’s disease. Curr Opin Neurobiol 23(1):100–108. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. 2006. Epidemiology of Parkinson’s disease. Lancet neurology 5(6):525–535. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. 2013. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A 110(19):E1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. 2012. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109(24):9611–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Yang Y, Ahmeti KB, Miller N, Huang C, Cheng L, Zhai H, Deng S, Nuytemans K, Corbett NJ, Kim MJ, Deng H, Tang B, Yang Z, Xu Y, Chan P, Huang B, Gao XP, Song Z, Liu Z, Fecto F, Siddique N, Foroud T, Jankovic J, Ghetti B, Nicholson DA, Krainc D, Melen O, Vance JM, Pericak-Vance MA, Ma yC, Rajput AH, Siddique T. 2016. Identification of TMEM230 mutations in familial Parkinson’s disease. Nature genetics 48(7):733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DC, Rhoades E. 2013. alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 52(14):2385–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhekne HS, Yanatori I, Gomez RC, Tonelli F, Diez F, Schule B, Steger M, Alessi DR, Pfeffer SR. 2018. A pathway for Parkinson’s Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Guedes L Correia, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V. 2009. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72(3):240–245. [DOI] [PubMed] [Google Scholar]
- Diogenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Nasstrom T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergstrom J, Ingelsson M, Quintas A, Sebastiao AM, Lopes LV, Outeiro TF. 2012. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci 32(34):11750–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Geczy CL, Halliday GM. 2015. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 302:89–102. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O. 2012. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PloS one 7(5):e36458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer MJ. 2006. Genetics of Parkinson disease: paradigm shifts and future prospects. Nature reviews Genetics 7(4):306–318. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Chesselet MF. 2004. Alpha-synuclein and transgenic mouse models. Neurobiology of disease 17(2):123–130. [DOI] [PubMed] [Google Scholar]
- Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, Collins BM, Bugarcic A, Teasdale RD. 2014. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic 15(2):230–244. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. 2005. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci 25(47):10913–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaig C, Vilas D, Infante J, Sierra M, Garcia-Gorostiaga I, Buongiorno M, Ezquerra M, Marti MJ, Valldeoriola F, Aguilar M, Calopa M, Hernandez-Vara J, Tolosa E. 2014. Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PloS one 9(10):e108982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. 2010. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol 185(9):5577–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges NZ, Backos DS, Esteban JA. 2004. Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J Biol Chem 279(42):43870–43878. [DOI] [PubMed] [Google Scholar]
- Giesert F, Hofmann A, Burger A, Zerle J, Kloos K, Hafen U, Ernst L, Zhang J, Vogt-Weisenhorn DM, Wurst W. 2013. Expression analysis of Lrrk1, Lrrk2 and Lrrk2 splice variants in mice. PloS one 8(5):e63778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. 2008. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A 105(1):145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokce O, Stanley GM, Treutlein B, Neff NF, Camp JG, Malenka RC, Rothwell PE, Fuccillo MV, Sudhof TC, Quake SR. 2016. Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-seq. Cell reports 16(4):1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. 2005. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45(4):489–496. [DOI] [PubMed] [Google Scholar]
- Golden SA, Covington HE, 3rd, Berton O, Russo SJ. 2011. A standardized protocol for repeated social defeat stress in mice. Nat Protoc 6(8):1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. 2006. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiology of disease 23(2):329–341. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. 2010. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A 107(45):19573–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. 2010. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468(7324):696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, LaVoie MJ, Gris D, Park DS, Angel JB, Shen J, Philpott DJ, Schlossmacher MG. 2011. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna) 118(5):795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Nestler EJ. 2017. Neural Substrates of Depression and Resilience. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 14(3):677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CH. 2008. The prodromal phase of sporadic Parkinson’s disease: does it exist and if so how long is it? Movement disorders : Official journal of the Movement Disorder Society 23(13):1799–1807. [DOI] [PubMed] [Google Scholar]
- Chao D Herrera Moro, Kallemeijn WW, Marques AR, Orre M, Ottenhoff R, van Roomen C, Foppen E, Renner MC, Moeton M, van Eijk M, Boot RG, Kamphuis W, Hol EM, Aten J, Overkleeft HS, Kalsbeek A, Aerts JM. 2015. Visualization of active Glucocerebrosidase in rodent brain with high spatial resolution following in situ Labeling with fluorescent activity based probes. PloS one 10(9):e0138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. 2011. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet 20(21):4209–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara L, Brayne C. 2006. A systematic review of depression and mental illness preceding Parkinson’s disease. Acta neurologica Scandinavica 113(4):211–220. [DOI] [PubMed] [Google Scholar]
- Ishizu N, Yui D, Hebisawa A, Aizawa H, Cui W, Fujita Y, Hashimoto K, Ajioka I, Mizusawa H, Yokota T, Watase K. 2016. Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum Mol Genet 25(20):4507–4517. [DOI] [PubMed] [Google Scholar]
- Islam MS, Nolte H, Jacob W, Ziegler AB, Putz S, Grosjean Y, Szczepanowska K, Trifunovic A, Braun T, Heumann H, Heumann R, Hovemann B, Moore DJ, Kruger M. 2016. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet 25(24):5365–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Nakamura K, Iijima M, Sesaki H. 2013. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol 23(2):64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. 1995. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14(2):467–475. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R. 2013. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A 110(42):E4016–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannarkat GT, Boss JM, Tansey MG. 2013. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis 3(4):493–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Karouani M, Haburcak M, Martella G, Tscherter A, Platania P, Wu B, Pothos EN, Shen J. 2009. Impaired dopamine release and synaptic plasticity in the striatum of parkin−/− mice. J Neurochem 110(2):613–621. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. 2007. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 104(27):11441–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. 2014. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet 23(11):2816–2833. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. 2008. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14(5):504–506. [DOI] [PubMed] [Google Scholar]
- Koroglu C, Baysal L, Cetinkaya M, Karasoy H, Tolun A. 2013. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism & related disorders 19(3):320–324. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. 2012. Recurrent network activity drives striatal synaptogenesis. Nature 485(7400):646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisan-Ruiz C. 2013. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34(9):1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner G 2015. Compensatory mechanisms in genetic models of neurodegeneration: are the mice better than humans? Frontiers in cellular neuroscience 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. 2007. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445(7128):643–647. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. 2008. Striatal plasticity and basal ganglia circuit function. Neuron 60(4):543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn K, Zhu XR, Lubbert H, Stichel CC. 2004. Parkin expression in the developing mouse. Brain research Developmental brain research 149(2):131–142. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. 2013. Cerebral organoids model human brain development and microcephaly. Nature 501(7467):373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. 2006. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci 26(46):11915–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschlager J, Kaminski CF, Schierle GS Kaminski. 2017. alpha-Synuclein - Regulator of exocytosis, endocytosis, or both? Trends Cell Biol 27(7):468–479. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson Cl, Varnam LR, Visel, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445(7124):168–176. [DOI] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, Erpapazoglou Z, Usenko T, Maurage CA, Sahbatou M, Liebau S, Ding J, Bilgic B, Emre M, Erginel-Unaltuna N, Guven G, Tison F, Tranchant C, Vidailhet M, Corvol JC, Krack P, Leutenegger AL, Nalls MA, Hernandez dG, Heutink P, Gibbs JR, Hardy J, Wood NW, Gasser T, Durr A, Deleuze JF, Tazir M, Destee A, Lohmann E, Kabashi E, Singleton A, Corti O, Brice A, French Parkinson’s Disease Genetics S, International Parkinson’s Disease Genomics C. 2016. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98(3):500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Bang Y, Choi JH, Han A, Kwon MS, Liu KH, Choi HJ. 2018. LRRK2 G2019S Induces Anxiety/Depression-like Behavior before the Onset of Motor Dysfunction with 5-HT1A Receptor Upregulation in Mice. J Neurosci 38(7):1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. 2012. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338(6109):949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth ES, Stavrovskaya IG, Bartels T, Kristal bS, Selkoe DJ. 2014. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289(31):21490–21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Cheng Y, Hellard E Roltsch, Wang X, Lu J, Gao X, Huang CCY, Wei XY, Ji JY, Wang J. 2018. Bidirectional and long-lasting control of alcohol-seeking behavior by corticostriatal LTP and LTD. Nat Neurosci 21(3):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. 2013. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77(3):425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madero-Perez J, Fdez E, Fernandez B, Ordonez AJ Lara, Ramirez M Blanca, Gomez-Suaga P, Waschbusch D, Lobbestael E, Baekelandt V, Nairn AC, Ruiz-Martinez J, Aiastui A, de Munain A Lopez, Lis P, Comptdaer T, Taymans JM, Chartier-Harlin MC, Beilina A, Gonnelli A, Cookson MR, Greggio E, Hilfiker S. 2018. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Molecular neurodegeneration 13(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraschi A, Ciammola A, Folci A, Sassone F, Ronzitti G, Cappelletti G, Silani V, Sato S, Hattori N, Mazzanti M, Chieregatti E, Mulle C, Passafaro M, Sassone J. 2014. Parkin regulates kainate receptors by interacting with the GluK2 subunit. Nature communications 5:5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. 1991. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain research Molecular brain research 11(3-4):335–343. [DOI] [PubMed] [Google Scholar]
- Martinez TN, Greenamyre JT. 2012. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxidants & redox signaling 16(9):920–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen-Ankney BA, Kezunovic N, Menard C, Flanigan M, Zhong Y, Russo SJ, Benson DL, Huntley GW. 2018. Parkinson’s Disease-linked LRRK2-G2019S mutation alters synaptic plasticity and promotes resilience to chronic social stress in young adulthood. J Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, Benson DL. 2016. Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J Neurosci 36(27):7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chavez-Gutierrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P. 2012. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75(6):1008–1021. [DOI] [PubMed] [Google Scholar]
- McGough IJ, Steinberg F, Jia D, Barbuti PA, McMillan KJ, Heesom KJ, Whone AL, Caldwell MA, Billadeau DD, Rosen MK, Cullen PJ. 2014. Retromer binding to FAM21 and the WASH complex is perturbed by the Parkinson disease-linked VPS35(D620N) mutation. Curr Biol 24(14):1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinstry SU, Karadeniz YB, Worthington AK, Hayrapetyan VY, Ozlu MI, Serafin-Molina K, Risher WC, Ustunkaya T, Dragatsis I, Zeitlin S, Yin HH, Eroglu C. 2014. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci 34(28):9455–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Rubinsztein DC. 2015. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16(6):345–357. [DOI] [PubMed] [Google Scholar]
- Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, Martinez TN, Chou MY, Howden AJM, Konig T, Hotzy C, Milenkovic I, Brucke T, Zimprich A, Sammler E, Alessi DR. 2018. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. The Biochemical journal 475(11):1861–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N, Geisler S, Tabuchi M, Oshima R, Kikuchi A, Baba T, Wada K, Nagai Y, Takeda A, Aoki M. 2014. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiology of disease 71:1–13. [DOI] [PubMed] [Google Scholar]
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. 2012. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32(5):1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero AE, Arteaga-Bracho EE, Chen CH, Gulinello M, Winchester ML, Pichamoorthy N, Gokhan S, Khodakhah K, Mehler MF. 2016. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington’s disease. Proc Natl Acad Sci U S A 113(20):5736–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovitz O, Ben-Nissan G, Fainer I, Pollack D, Mizrachi L, Sharon M. 2015. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nature communications 6:6609. [DOI] [PubMed] [Google Scholar]
- Munsie LN, Milnerwood AJ, Seibler P, Beccano-Kelly DA, Tatarnikov I, Khinda J, Volta M, Kadgien C, Cao LP, Tapia L, Klein C, Farrer MJ. 2015. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum Mol Genet 24(6):1691–1703. [DOI] [PubMed] [Google Scholar]
- Nakai M, Fujita M, Waragai M, Sugama S, Wei J, Akatsu H, Ohtaka-Maruyama C, Okado H, Hashimoto M. 2007. Expression of alpha-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochemical and biophysical research communications 358(1):104–110. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8(1):e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. 2010. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65(1):66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Krainc D. 2018. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc Natl Acad Sci U S A 115(21):5576–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgiati S, De Rosa A, Quadri M, Criscuolo C, Breedveld GJ, Picillo M, Pappata S, Quarantelli M, Barone P, De Michele G, Bonifati V. 2014. PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15(3):183–188. [DOI] [PubMed] [Google Scholar]
- Olgiati S, Quadri M, Fang M, Rood JP, Saute JA, Chien HF, Bouwkamp CG, Graafland J, Minneboo M, Breedveld GJ, Zhang J, International Parkinsonism Genetics N, Verheijen FW, Boon AJ, Kievit AJ, Jardim LB, Mandemakers W, Barbosa ER, Rieder CR, Leenders KL, Wang J, Bonifati V. 2016. DNAJC6 Mutations Associated With Early-Onset Parkinson’s Disease. Ann Neurol 79(2):244–256. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. 2013. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. 2003. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302(5651):1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Lewis PA, Singleton AB. 2013. LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3(2):85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PY, Li X, Wang J, Powell J, Wang Q, Zhang Y, Chen Z, Wicinski B, Hof P, Ryan TA, Yue Z. 2017. Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J Neurosci 37(47):11366–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H. 2014. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17(3):367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. 2014. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet 23(11):2802–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen K, Olesen OF, Mikkelsen JD. 1999. Developmental expression of alpha-synuclein in rat hippocampus and cerebral cortex. Neuroscience 91(2):651–659. [DOI] [PubMed] [Google Scholar]
- Piccoli G, Condliffe Sb, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W, Gloeckner CJ, Matteoli M, Sala C, Ueffing M. 2011. LRRK2 Controls Synaptic Vesicle Storage and Mobilization within the Recycling Pool. J Neurosci 31(6):2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci 9(5):602–604. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047. [DOI] [PubMed] [Google Scholar]
- Pompilio L, Kacelnik A. 2010. Context-dependent utility overrides absolute memory as a determinant of choice. Proc Natl Acad Sci U S A 107(1):508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G. 2015. MDS clinical diagnostic criteria for Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. 2007. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27(3):645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, Pappata S, Quarantelli M, Annesi G, Quattrone A, Chien HF, Barbosa ER, International Parkinsonism Genetics N, Oostra BA, Barone P, Wang J, Bonifati V. 2013. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34(9):1208–1215. [DOI] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. 2015. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends in biochemical sciences 40(4):200–210. [DOI] [PubMed] [Google Scholar]
- Sandor C, Robertson P, Lang C, Heger A, Booth H, Vowles J, Witty L, Bowden R, Hu M, Cowley SA, Wade-Martins R, Webber C. 2017. Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum Mol Genet 26(3):552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM. 2014. Parkin and PINK1: much more than mitophagy. Trends in neurosciences 37(6):315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation, Host Response to Injury LSCRP. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110(9):3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. 2004. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol 2(11):e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. 2000. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature genetics 25(3):302–305. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. 2011. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144(5):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, Liebmann T, Bousset L, Renner M, Lena C, Aperia A, Melki R, Triller A. 2015. alpha-Synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J 34(19):2408–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Lopez G. 2012. The link between the GBA gene and parkinsonism. Lancet neurology 11(11):986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton A, Gwinn-Hardy K. 2004. Parkinson’s disease and dementia with Lewy bodies: a difference in dose? Lancet 364(9440):1105–1107. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. 2003. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. 2014. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci 34(2):418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. 2006. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9(10):1231–1233. [DOI] [PubMed] [Google Scholar]
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, Geens A, De Strooper B, Verstreken P. 2016. A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 92(4):829–844. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. 1997. Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- Springer BD, Scott RD, Thornhill TS. 2006. Conversion of failed unicompartmental knee arthroplasty to TKA. Clin Orthop Relat Res 446:214–220. [DOI] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. 2016. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet ES, Saunier-Rebori B, Yue Z, Blitzer RD. 2015. The Parkinson’s disease-associated mutation LRRK2-G2019S impairs synaptic plasticity in Mouse hippocampus. J Neurosci 35(32):11190–11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro P, Burke RE. 2016. Retrograde axonal degeneration in Parkinson Disease. J Parkinsons Dis 6(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, Tang B, Mei L, Xiong WC. 2015. VPS35 in Dopamine neurons Is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-Synuclein degradation and prevention of pathogenesis of Parkinson’s Disease. J Neurosci 35(29):10613–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Potgieter D, Anwar S, Senior SL, Janezic S, Threlfell S, Ryan B, Parkkinen L, Deltheil T, Cioroch M, Livieratos A, Oliver PL, Jennings KA, Davies KE, Ansorge O, Bannerman DM, Cragg SJ, Wade-Martins R. 2014. Region-specific deficits in dopamine, but not norepinephrine, signaling in a novel A30P alpha-synuclein BAC transgenic mouse. Neurobiology of disease 62:193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin P, Morishita W, Goswami D, Arendt K, Chen L, Malenka R. 2017. The Retromer Supports AMPA Receptor Trafficking During LTP. Neuron 94(1):74–82 e75. [DOI] [PubMed] [Google Scholar]
- Thevenet J, Gobert R Pescini, van Huijsduijnen R Hooft, Wiessner C, Sagot YJ. 2011. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PloS one 6(6):e21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Tang FL, Sun X, Wen L, Mei L, Tang BS, Xiong WC. 2015. VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Molecular brain 8(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA. 2009. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate Parkin in synaptic ubiquitination. Molecular cell 36(6):1034–1047. [DOI] [PubMed] [Google Scholar]
- Tsika E, Glauser L, Moser R, Fiser A, Daniel G, Sheerin UM, Lees A, Troncoso JC, Lewis PA, Bandopadhyay R, Schneider BL, Moore DJ. 2014. Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum Mol Genet 23(17):4621–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D. 2014. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet 23(11):2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, Picillo M, Barone P, Munshi ST, de Vrij FM, Kushner SA, Gounko NV, Mandemakers W, Bonifati V, Meunier FA, Soukup SF, Verstreken P. 2017. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36(10):1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. 2014. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34(28):9364–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Schrod N, Davis T, Fernandez-Busnadiego R, Taguchi YV, Laugks U, Lucic V, Chandra SS. 2017. Synucleins have multiple effects on presynaptic architecture. Cell reports 18(1):161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Robison AJ, Laplant QC, Covington HE, 3rd, Dietz DM, Ohnishi YN, Mouzon E, Rush AJ, 3rd, Watts EL, Wallace DL, Iniguez SD, Ohnishi YH, Steiner MA, Warren BL, Krishnan V, Bolanos CA, Neve RL, Ghose S, Berton O, Tamminga CA, Nestler EJ. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci 13(6):745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, Yu I, Encarnacion M, Munsie LN, Tapia L, Gustavsson EK, Chou P, Tatarnikov I, Evans DM, Pishotta FT, Volta M, Beccano-Kelly D, Thompson C, Lin MK, Sherman HE, Han HJ, Guenther BL, Wasserman WW, Bernard V, Ross CJ, Appel-Cresswell S, Stoessl AJ, Robinson CA, Dickson DW, Ross OA, Wszolek ZK, Aasly JO, Wu RM, Hentati F, Gibson RA, McPherson PS, Girard M, Rajput M, Rajput AH, Farrer MJ. 2014. DNAJC13 mutations in Parkinson disease. Hum Mol Genet 23(7):1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingill S, Brockelt D, Lancelin C, Tatenhorst L, Dontcheva G, Preisinger C, Schwedhelm-Domeyer N, Joseph S, Mitkovski M, Goebbels S, Nave KA, Schulz JB, Marquardt T, Lingor P, Stegmuller J. 2016. Loss of FBXO7 (PARK15) results in reduced proteasome activity and models a parkinsonism-like phenotype in mice. EMBO J 35(18):2008–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. 2011. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72(1):57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta M, Beccano-Kelly DA, Paschall SA, Cataldi S, MacIsaac SE, Kuhlmann N, Kadgien CA, Tatarnikov I, Fox J, Khinda J, Mitchell E, Bergeron S, Melrose H, Farrer MJ, Milnerwood AJ. 2017. Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta M, Melrose H. 2017. LRRK2 mouse models: dissecting the behavior, striatal neurochemistry and neurophysiology of PD pathogenesis. Biochem Soc Trans 45(1):113–122. [DOI] [PubMed] [Google Scholar]
- Volta M, Milnerwood AJ, Farrer MJ. 2015. Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson’s disease. Lancet neurology 14(10):1054–1064. [DOI] [PubMed] [Google Scholar]
- Cl Wang, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC. 2012. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open 1(12):1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Toh J, Ho P, Tio M, Zhao Y, Tan EK. 2014. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Molecular brain 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X. 2016. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22(1):54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Cowell RM, Daher JP, Moehle MS, Hinkle KM, Melrose HL, Standaert DG, Volpicelli-Daley LA. 2014. Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J Comp Neurol.522: 2465–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. 2005. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102(46):16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. 2007. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16(2):223–232. [DOI] [PubMed] [Google Scholar]
- Westerlund M, Ran C, Borgkvist A, Sterky FH, Lindqvist E, Lundstromer K, Pernold K, Brene S, Kallunki P, Fisone G, Olson L, Galter D. 2008. Lrrk2 and alpha-synuclein are co-regulated in rodent striatum. Mol Cell Neurosci 39(4):586–591. [DOI] [PubMed] [Google Scholar]
- Whitton PS. 2007. Inflammation as a causative factor in the aetiology of Parkinson’s disease. British journal of pharmacology 150(8):963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider C, Skipper L, Solida A, Brown L, Farrer M, Dickson D, Wszolek ZK, Vingerhoets FJ. 2008. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism & related disorders 14(6):465–470. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. 1963a. Effects of Visual Deprivation on Morphology and Physiology of Cells in the Cats Lateral Geniculate Body. Journal of neurophysiology 26:978–993. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. 1963b. Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. Journal of neurophysiology 26:1003–1017. [DOI] [PubMed] [Google Scholar]
- Willemsen R, van Dongen JM, Ginns EI, Sips HJ, Schram AW, Tager JM, Barranger JA, Reuser AJ. 1987. Ultrastructural localization of glucocerebrosidase in cultured Gaucher’s disease fibroblasts by immunocytochemistry. Journal of neurology 234(1):44–51. [DOI] [PubMed] [Google Scholar]
- Williams ET, Chen X, Moore DJ. 2017. VPS35, the Retromer Complex and Parkinson’s Disease. J Parkinsons Dis 7(2):219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D. 2016. Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Movement disorders : official journal of the Movement Disorder Society 31(11):1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Iwatsubo T. 2018. Extracellular alpha-synuclein levels are regulated by neuronal activity. Molecular neurodegeneration 13(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. 2006. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A 103(28):10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YI, Sun T, Wu LG, Raimondi A, De Camilli P, Eisenberg E, Greene LE. 2010. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc Natl Acad Sci U S A 107(9):4412–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, Rubinsztein DC. 2014. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nature communications 5:3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Freed CR. 2005. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem 280(52):43150–43158. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Liu A, Xia S, Leung C, Qi J, Meng Y, Xie W, Park P, Collingridge GL, Jia Z. 2018. The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning. Nat Neurosci 21(1):50–62. [DOI] [PubMed] [Google Scholar]