

Abstract
Exosomes show potential as ideal vehicles for drug delivery because of their natural role in transferring biological cargo between cells. However, current methods to engineer exosomes without negatively impacting their function remain challenging. Manipulating exosome-secreting cells is complex and time-consuming, while direct functionalization of exosome surface proteins suffers from low specificity and low efficiency. We demonstrate a rapid, versatile and scalable method with oligonucleotide tethers to enable diverse surface functionalization on both human and murine exosomes. These exosome surface modifiers that range from reactive functional groups and small molecules to aptamers and large proteins, can readily and efficiently enhance native exosome properties. We show that cellular uptake of exosomes can be specifically altered with a tethered AS1411 aptamer, and targeting specificity can be altered with a tethered protein. We functionalize exosomes with an immunomodulatory protein, FasL, and demonstrate their biological activity both in vitro and in vivo. FasL-functionalized exosomes, when bioprinted on collagen matrix allows spatial induction of apoptosis in tumor cells, and when injected in mice, suppresses proliferation of alloreactive T-cells. This oligonucleotide tethering strategy is independent of the exosome source and further circumvents the need to genetically modify exosome-secreting cells.
Keywords: drug delivery, extracellular vesicle, exosome, DNA, aptamer, membrane engineering, immunomodulation
Extracellular vesicles (EVs), including exosomes and microvesicles, are bilayer lipid membrane-bound vesicles containing proteins and nucleic acids such as miRNAs, mRNAs, and DNA. EVs are released from many, if not all, cell types in the body and play a key role in intercellular communication in autocrine, paracrine and telecrine pathways.1–3 This ability of EVs to selectively transport proteins, lipids and nucleic acids to cells is especially important in the drug delivery arena,4,5 where efficient and targeted delivery of biomolecules is desired but otherwise challenging.3,6–10 Multiple studies have shown that the use of EVs for therapeutic purposes is feasible,11,12 and EVs have been applied in Phase I clinical trials.4,9 Of particular interest are exosomes, which are EVs in the size range of 30 - 200 nm. Exosomes have many unique desirable characteristics for certain therapies, such as the ability to cross the blood brain barrier.13,14 Exosomes distinctively originate from the endocytic compartment and their molecular content reflects, at least in part, that of the parental cell. Unfortunately, native exosomes may also possess undesirable properties that limit their applications as drug delivery vehicles. For example, their natural bioactive payloads may counteract the desired therapeutic effects, and a lack of desired targeting specificity may result in uptake by non-targeted, healthy cells. Multiple recent reports have shown that exosomes can be engineered to include specific cargo or express targeting ligands to improve their drug delivery potential.14–20 These targeting strategies to engineer exosomes have been mainly based on genetic manipulation of exosome-sourced cells, resulting in the fusion of targeting ligands with exosome membrane proteins, such as Lamp2b.14,18,19 However, the function of exosome membrane proteins (e.g. fusion with cellular membranes or immune regulation6,21) may be compromised upon fusion with targeting ligands. Furthermore, targeting ligands have been described to undergo premature degradation instead of functional display on exosomes.22 Scalability can also be an issue when dealing with engineering exosome-secreting cells.23 To avoid such issues and circumvent the need to modify EV producing cells, others have explored strategies to functionalize exosome surfaces after their secretion.20,22,24–27 However, such modifications may also compromise the functionality of crucial exosome components for exosome-cell interactions and cargo delivery.28
To address these challenges, we have developed a general method to rapidly and efficiently engineer exosomes with functional groups, nucleic acids and bioactive proteins via DNA tethers. A single-stranded DNA (ssDNA) with a conjugated cholesterol moiety can be tethered to the exosome lipid bilayer, and the ssDNA itself or complementary DNA can be exploited to readily attach other functionalities – from a reactive chemical functional group and small molecules such as dyes and biotin to large aptamer (oligonucleotide), antibody or other protein cargo. The use of DNA tethers include the following advantages: the negatively charged DNA helps to ensure that the membrane anchored DNA is predominantly externally oriented; the DNA can be easily quantified permitting improved estimation of cargo attached to the exosome surface; DNA complementarity allows for non-covalent attachment of cargo via a hybridizing strand; DNA length can be controlled thereby permitting precise placement of cargo above the exosome surface; and, DNA tethered cargo is releasable from the exosome surface under highly controlled conditions permitting controlled temporal release of cargo.
This general DNA tether approach preserves the native cell-exosome binding interactions as assessed using on-bead flow cytometry and confocal microscopy-based cell uptake studies. We show this versatile method can be used to readily functionalize exosomes membranes to carry bioactive cargo and demonstrate how this method can be used with a membrane surface active protein in a specific and effective way to modulate targeted immune pathways in vivo.
Results and Discussion
DNA tethers for exosomes
As a versatile platform method to functionalize exosome membranes, we first investigated the tethering efficiency of cholesterol modified oligonucleotides. We used an 18-mer DNA tether with a 5’- tetra(ethylene glycol) (TEG) spacer and cholesterol modification (Chol-DNA; Supplementary Table 1) at varied concentrations to treat exosomes isolated from THP1 cells (Supplementary Fig. 1). This was accomplished by simply vortexing the exosomes with Chol-DNA in buffered solution at ambient room temperature for five minutes (Fig. 1a). DLS measurements showed an approximately 5 nm increase in the diameter at all DNA tether concentrations and a decrease in zeta potential with increasing Chol-DNA concentration, suggesting successful tethering of DNA onto the exosome membrane (Supplementary Table 2). Analysis by flow cytometry using a Cy5-labeled DNA tether (Chol-DNA-Cy5) showed a proportional increase in Cy5 signal with increasing tether concentrations upto 20 μM (Fig. 1b; see also Supplementary SI Figs. 2–5). There was a concentration dependent increase of Chol-DNA tethers per exosome ranging from approximately 1800 to 6900 tethers when the total concentration of the Chol-DNA in the vortexing mixture was varied from 0.5 μM to 20 μM (Supplementary Fig. 5). Higher stock concentrations of the Chol-DNA strand to enable greater than 20 μM tethers on the exosomes, led to aggregation and micelles and reduced DNA tether insertion.
Figure 1.
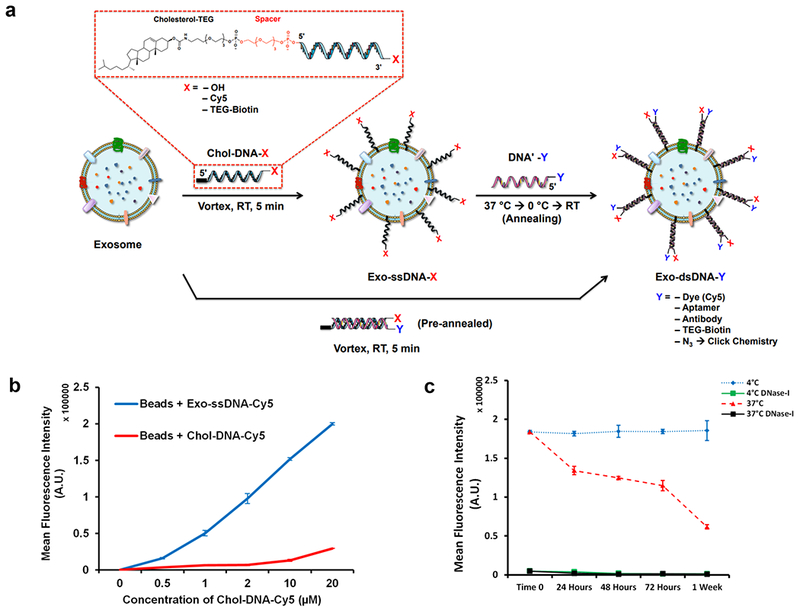
DNA tethering of exosomes. (a) Schematic of cholesterol-oligonucleotide tethering method on exosome membrane. Cholesterol on the 5’ end of TEG-DNA-X embeds into the exosome membrane (Exo-ssDNA-X) and a complementary strand, DNA’-Y, can hybridize to Chol-DNA-X for a duplex oligonucleotide tethered to the exosome membrane (Exo-dsDNA-Y). Pre-annealed Chol-DNA-X and DNA’-Y tethers can also be embedded into the exosome membrane for functionalization (X and Y) (b) Graph of fluorescence intensity for flow cytometry assessment of exosomes with ssDNA tethers using CD63 conjugated magnetic beads (see also supplementary Figs. 2 and 4). The Chol-DNA concentration was varied between 0 and 20 μM and incubation with 20 μg of exosome leads to a corresponding increase in fluorescence intensity. Bars indicate relative mean fluorescence intensities ± SEM (n=3 independent experiments). (c) The stability of ssDNA (see supplementary Fig. 6 for dsDNA) tethered on the exosome membrane assessed by on-bead flow cytometry at 4 °C and 37 °C. The DNA tethers can be readily removed with DNase-I (see supplementary Fig. 3 for confocal images). Bars indicate relative mean fluorescence intensities ± SEM (n=3 independent experiments).
To assess the bioavailability of DNA tethered to an exosome surface membrane for further functionalization, we used a Cy5-labeled DNA strand (DNA’-Cy5) that was complementary to a Chol-DNA tethered exosome (Supplementary Fig. 6a). Annealing the complementary strand, simply by warming to 37 °C and cooling, yields an exosome with a duplex DNA (Exo-dsDNA-Cy5) with a loading efficiency of approximately 80 % compared to the Exo-ssDNA-Cy5 that are directly labeled with Cy5. Pre-annealing the Chol-DNA and DNA’-Cy5 strands before tethering onto exosomes shows the same labeling efficiency as directly labeled single-stranded DNA tethers (Supplementary Fig. 6b–c).
Next, we assessed the stability of the Chol-DNA tethers at 4 °C in PBS buffer and at 37 °C in simulated body fluid. Both Exo-ssDNA-Cy5 and Exo-dsDNA-Cy5 were incubated and analyzed by flow cytometry at different time points. No decrease in the mean fluorescent intensity (MFI) was observed at 4 °C for both systems, indicating a high stability of the DNA tethers (Fig. 1c and Supplementary Fig. 6d). At 37 °C, a drop in MFI was observed, likely due to protein shedding of CD63 from the exosome membrane29 that would affect the binding to anti-CD63 magnetic beads and thereby reduce the overall signal, rather than due to the loss of DNA tethers. Complete removal of the DNA tethers was observed after treatment with DNase enzyme, highlighting the reversibility of the DNA modification of exosomes (Fig. 1c and Supplementary Fig. 6d).
Click chemistry for rapid functionalization of exosomes
To demonstrate the versatility of the exosome functionalization method, we used both copper and copper-free click chemistry to attach a diverse array of molecules via the DNA tether. Exo-dsDNA-N3 were prepared by hybridizing DNA’-N3 to Exo-ssDNA. Strain-promoted azide-alkyne cycloaddition (SPAAC) was performed using dibenzo-bicyclo-octyne (DBCO) functionalized SF488 dye. SF488-clicked exosomes were confirmed by flow cytometry analysis using 488 nm channels, while no significant signal was observed for control experiments (Supplementary Fig. 7). Additionally, Cu-click functionalization was performed with Cy5-alkyne (Supplementary Fig. 7).
Cell uptake of DNA-tethered exosomes
To investigate whether the presence of negatively charged DNA affects the internalization efficiency of the exosomes, we performed cell uptake studies with human embryonic kidney (HEK293) cells. HEK293 cells were incubated with native exosomes as well as Exo-ssDNA-Cy5 and Exo-dsDNA-Cy5. These cell internalization trials were also performed in the presence of two inhibitors, heparin and methyl-β-cyclodextrin, which partially inhibit exosome internalization by blocking heparin sulfate proteoglycans and lipid-raft mediated processes, respectively.30 We observed similar internalization rates for native exosomes, Exo-ssDNA-Cy5 and Exo-dsDNA-Cy5 (Fig. 2a–b). Time-dependent internalization of native exosomes, Exo-ssDNA-Cy5 and Exo-dsDNA-Cy5 is shown in Supplementary Figs. 8–13. In the presence of inhibitors, we observed a significant drop in internalized fluorescence. Complete inhibition was not observed in the presence of inhibitors, presumably due to other potential pathways for cell internalization of exosomes.2
Figure 2.
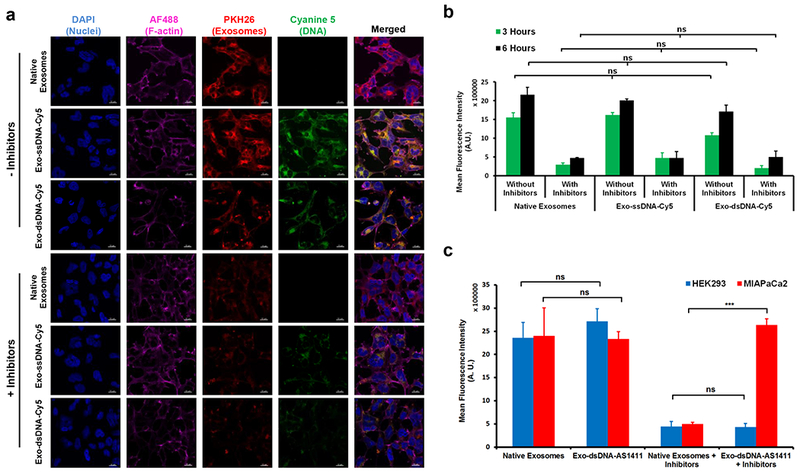
Assessment of cellular uptake of DNA tethered exosomes. (a) Confocal micrographs show uptake of PKH26 labeled DNA tethered exosomes and native exosomes. Scale bar = 20 μM. (b) Quantification of exosome uptake from confocal micrographs of three independent experiments. To inhibit uptake, cells were pretreated with a combination of 10 μg/ml heparin and 1 μM methyl-β-cyclodextrin for 1 hr at 37 °C. Both native and DNA tethered exosomes behave similarly. There is a linear increase in DNA-tethered exosome uptake from 3 to 6 hrs, whereas in the presence of inhibitors, uptake is reduced to 29.4 (± 8.4) % at 3 hrs and to 23.7 (± 8.5) % compared to native exosome control. Bars represent relative mean fluorescence intensities ± SEM (n=3 independent experiments), ns: no significant difference. (c) Rescue of uptake inhibition in MIAPaCa2 cells with AS1411 aptamer tethered onto exosome surface. AS1411 binds to nucleolin on MIAPaCa2 (but not on HEK293) cell surface and is thereby able to overcome the inhibitory effects of heparin and mβCD to exosome uptake in MIAPaCa2 cells. Bars represent relative mean fluorescence intensities ± SEM (n=3 independent experiments), ns: no significant difference, ***p=0.0002.
Functionalization of exosomes with AS1411 aptamer
To take advantage of the rapid DNA functionalization of exosomes, we investigated if cell uptake of exosomes could be altered with an AS1411 aptamer. The AS1411 aptamer is an oligonucleotide sequence designed to bind nucleolin. This sequence can be directly displayed on exosomes using a ‘tail’ that binds to the Chol-DNA strand. These studies were performed in two cell lines (HEK293 and human pancreatic cancer cells (MiaPaCa2). Cancerous MiaPaCa2 cells are known to have high levels of nucleolin protein on the cell membrane,31–33 which mediates internalization, whereas normal HEK293 cells do not have membrane associated nucleolin,34 thus serving as a negative control. Exosomes prepared with and without AS1411 were incubated with the cells and imaged after 6 hrs. Exosome internalization in the presence of inhibitors (heparin and methyl-β-cyclodextrin), which inhibits the two major pathways for exosome internalization, showed that exosomes with AS1411 can be internalized by nucleolin-expressing MiaPaCa2 cells, even in the presence of inhibitors, while low internalization efficiency was observed in HEK293 cells (Fig. 2c). In contrast, native exosomes showed very low internalization in the presence of inhibitors, with no difference between the two cell lines in the absence of inhibitors (Supplementary Fig. 14). These results highlight the effect of AS1411 aptamer and demonstrate that exosome internalization pathways can be readily altered using the DNA tethering approach to functionalize exosomes.
Inducing apoptosis in Jurkat cells via FasL functionalized exosomes
Immunomodulatory agents such as FasL and PDL-1 expressed on tumor exosomes (TEX) have been reported to contribute to spontaneous T-cell apoptosis.35,36 Fas and FasL interaction plays an important role in activation-induced cell death and in immune homeostasis.37 Inspired by this, THP1 exosomes were functionalized with a novel form of FasL chimeric with modified form of streptavidin (SA-FasL)38 using biotin-modified DNA tethers, and their biological activity was evaluated using Fas receptor-expressing Jurkat cells (Fig. 3a). A dose-dependent interaction resulted in a corresponding dose-dependent increase in Jurkat cell apoptosis as evaluated by Annexin V - PI staining (Fig. 3b). At the lowest concentration, 1 μg exosome protein resulted in 17.5 % (± 0.9) apoptosis, whereas the higher concentration, 20 μg exosome protein resulted in 99.3 % (± 0.03) (Fig. 3c). In contrast, native THP1 exosomes and 100 ng of soluble SA-FasL did not induce significant apoptosis. Similar apoptosis was also observed with Exo-dsDNA-SA-FasL. This data suggests that biologically active response modifiers can be conjugated using DNA-tethering technology.
Figure 3.
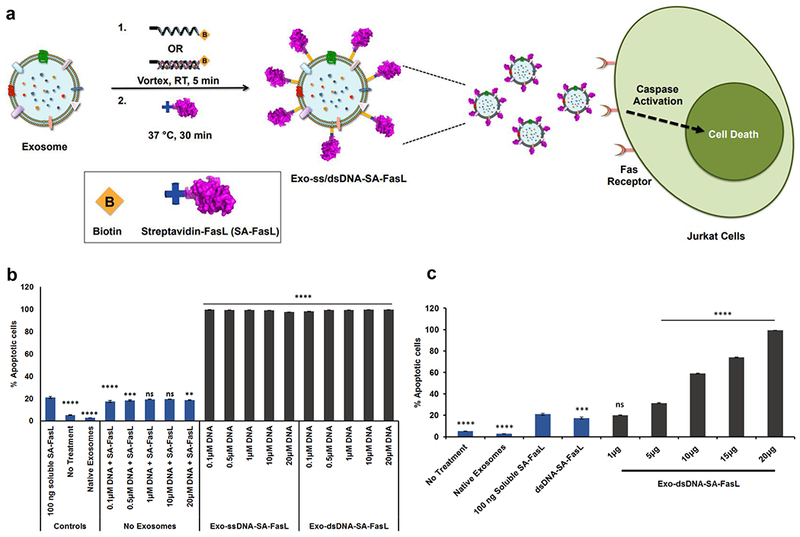
In vitro assessment of SA-FasL tethered exosomes. (a) Schematic diagram showing exosomes with tethered SA-FasL binding to Fas Receptor on Jurkat T cells resulting in their apoptosis. (b) Graph of Jurkat cell apoptosis as evaluated by flow cytometry. The plot shows treatments with 20 μg of exosomes tethered with varying concentrations of Chol-ssDNA-Biotin and 100 ng of soluble SA-FasL. The lowest tether concentration i.e., 0.1 μM resulted in 99.3 % (± 0.03) % apoptosis whereas 100 ng of soluble SA-FasL, native exosomes or ssDNA-SA-FasL did not result in any significant apoptosis. Bars indicate mean ± SEM (n=3 independent experiments), ns: no significant difference, **p=0.003, ***p=0.0003, ****p<0.0001, vs soluble SA-FasL treatment. (c) Titration of SAFasL tethered exosomes on Jurkat cells showed a dose-dependent increase in apoptosis. The dosage consisted of varying concentrations of exosomes with 0.1 μM Chol-dsDNA-biotin with 100 ng of SA-FasL. The lowest concentration i.e., 1 μg/ml exosome-dsDNA-SA-FasL resulted in 17.46 (± 0.86) % apoptosis whereas 20 μg exosome-dsDNA-SA-FasL resulted in 99.3 (± 0.03)%, while native exosomes, 100 ng of soluble SA-FasL, or ds-DNA-SA-FasL did not result in any significant apoptosis. Bars indicate mean ± SEM (n=3 independent experiments), ns: no significant difference, ***p=0.003, ****p<0.0001 vs soluble SA-FasL treatment.
Bioprinted exosomes induce spatially controlled apoptosis in cancer cells
Although FasL is considered to be useful for immunotherapy, major side effects have precluded its systemic use.39 One way to mitigate off-target effects is localized delivery of FasL immobilized on scaffold materials.40 Hence, we hypothesized that using bioprinting technology,41–45 spatially controlled exosome-microenvironments could be created46 to locally modify cell behavior, thereby limiting off-target responses. As a proof-of-concept, bioink containing Exo-ssDNA-Cy5 was printed onto collagen type-1 coated coverslips to create persistent ‘solid-phase’ patterns as shown in Supplementary Fig. 15. The deposited concentrations of exosomes were modulated using an overprinting strategy as previously described,42,47 where ‘OP’ (referred to in sections below) signifies the number of overprints (OPs) of dilute bioinks deposited at each substrate location to modulate delivered bioink dosage. The tethering of DNA onto exosomes did not hamper the ability of membrane associated integrins to interact with collagen binding domains. To assess whether bioprinted oligonucleotide-tethered exosomes are biologically active, Exo-ssDNA-SA-FasL was printed and, following overnight rinsing to remove unbound exosomes, PCI13 cells were seeded onto the coverslips. The Exo-ssDNA-SA-FasL pattern resulted in spatially restricted apoptosis in PCI13 cells (Fig. 4a–b). There was no significant apoptosis on native exosome patterns and on off-pattern regions, suggesting that Exo-ssDNA-SA-FasL are biologically active when presented in the solid-phase. Furthermore, we also printed overlapping counter gradients of native exosomes and Exo-ssDNA-SA-FasL on collagen type-I coated coverslips and evaluated its effects on PCI-13 cells. The region with high concentration (50 OPs) of Exo-ssDNA-SA-FasL resulted in significant number of dead cells as evaluated by live-dead staining (Fig. 4c–d).
Figure 4.
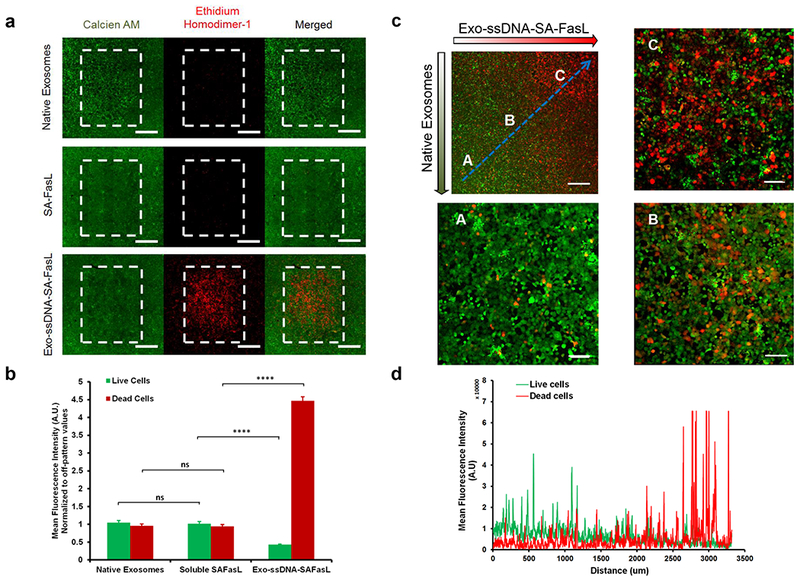
Bioprinting of FasL-functionalized Exosomes. (a) Florescence images show live/dead (calcien AM/ethidium bromide) staining of PCI-13 cells post 24 hrs on bioprinted patterns of native exosomes, SA-FasL and Chol-ssDNA-SA-FasL tethered exosomes on collagen type-1 coated coverslips. Scale bar = 600 μM (b) Quantification of live/dead stains of (a) showed selective apoptosis in presence of Exo-ssDNA-FasL, whereas native exosomes and soluble SA-FasL showed no significant effect. Bars indicate mean ± SEM (n=3 independent experiments), ns: no significant difference, ****p<0.0001. (c) Fluorescence image of live/dead staining of PCI-13 cells post 24 hrs on bioprinted counter gradient patterns of exosome-ssDNA-SA-FasL and native exosomes. An increasing concentration of immobilized SA-FasL tethered exosomes resulted in a corresponding increase in dead cells along the gradient. Scale bar = 500 μM (d) Quantification of live/dead staining along the blue line in (c). Plot indicates mean fluorescence intensity quantification along the blue line.
Exo-ssDNA-SA-FasL eliminate T cells responding to alloantigens in vivo
To provide evidence that exosomes engineered with biologics are viable drug candidates, we targeted the Fas death pathway as a model system since it plays a critical role in T cell homeostasis. We have previously shown that SA-FasL can be positionally and transiently displayed on the surface of cells or tissues.48,49 Transplantation of the engineered cells and tissues into allogeneic recipients induces tolerance via apoptosis in responding alloreactive T cells through engagement of Fas receptor.49 We then used an in vivo MLR assay in a parent-to-F1 mouse model to assess activity of the Exo-ssDNA-SA-FasL on donor T cells proliferation. The spleen cells from donor mice (C57BL/6) were labelled with carboxyfluorescein succinimidyl ester (CFSE) and adoptively transferred into F1 (C57BL/6-DTRxBalb/c) recipients followed by two separate administrations of Exo-ssDNA-SA-FasL via systemic intraperitoneal (i.p.) injections at 2 hrs and 24 hrs post-cell infusion. We observed a significant reduction in the percentage of proliferating donor CD3+ and CD4+ T cells in the spleen and lymph nodes of F1 mice treated with Exo-ssDNA-SA-FasL over control groups including Exo-ssDNA-biotin at 72 hrs post cell infusion (Fig. 5). The decrease in proliferating cell percentages resulted in significantly less absolute cell numbers of CD3+ and CD4+ T cells in the spleen and CD4+ T cells in lymph nodes (Supplementary Fig. 16). Interestingly, CD4+ T cell proliferation was significantly inhibited in mice that received Exo-ssDNA-SA-FasL treatment but not in mice that received the same dose of soluble SA-FasL or native exosomes. These results showed that SA-FasL activity is significantly enhanced when immobilized on exosomes rather than used as freely soluble (‘liquid-phase’) proteins. Collectively, the targeted delivery of SA-FasL via exosomes could substantially increase the therapeutic effect of SA-FasL protein while minimizing potential off-target effects caused by soluble injections of readily diffusible soluble SA-FasL.
Figure 5.
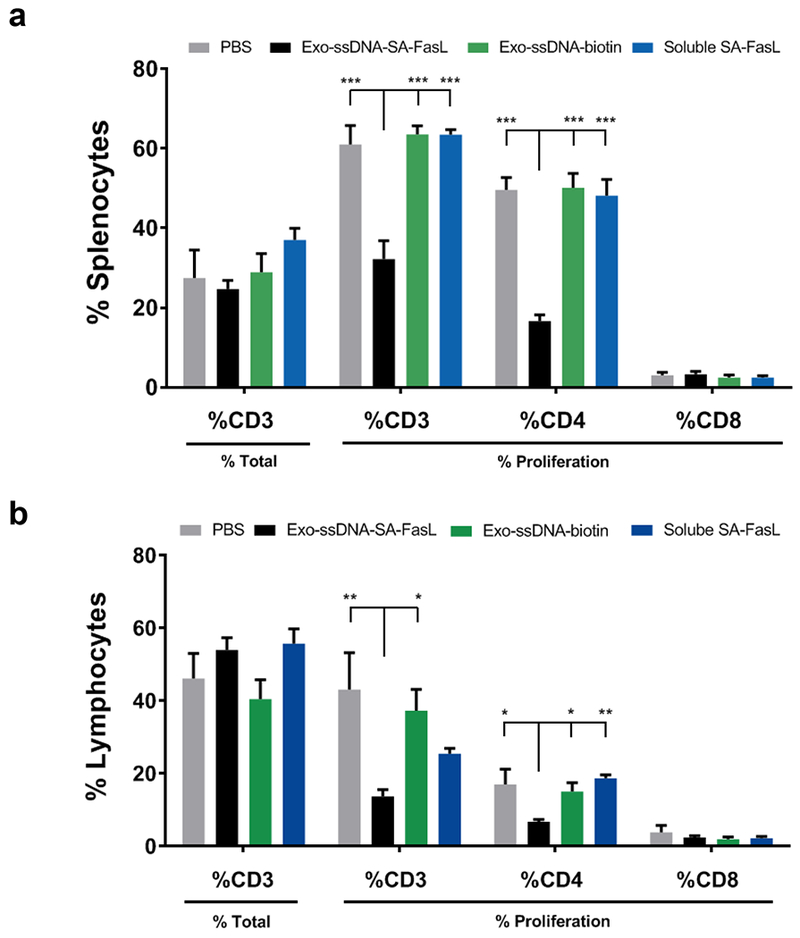
Systemic delivery of SA-FasL tethered exosomes blocks the proliferation of donor T cells in vivo. (a) schematic of CFSE labeled cell infusion and Exo-ssDNA-SA-FasL i.p. treatment in mice, following which the percentages of donor CD3 positive T cells were assessed by gating on donor (H2Kd negative) cells (% CD3) in treatment and control groups in (b) spleen and (c) mesenteric lymph nodes. The proliferation of donor CD3+, CD4+ and CD8+ T cells were measured by CFSE dilution using an LSR II and Diva software (BD Biosciences). ***P=0.0001,**P=0.003 and *P<0.05.
In this work we demonstrate a versatile method for rapid exosome membrane functionalization using hydrophobically modified oligonucleotide tethers. The use of hydrophobically modified oligonucleotides to insert into lipid membranes was first demonstrated by Pfeiffer and Höök.50 This work formed the basis of several studies ranging from cell membrane functionalization51,52 to specific DNA-based nanosensors as well as the recent use of siRNA and RNA nanostructures with exosomes.53–59 Here we provide a more generalizable approach with DNA tethers to rapidly functionalize exosome surfaces. The DNA tether approach offers the advantage of easily quantitated inserts via the DNA UV absorbance, as well as a providing a versatile functionalizable moiety. Our data indicate a very high number of DNA strands/exosome suggesting that oligonucleotide tethering results in densely tethered exosome-DNA hybrids. We hypothesize that the high lipid content in the exosome membrane60 and the relatively high membrane rigidity61 compared to cells and liposomes make the tethers less vulnerable to release from the exosome membrane. The features of our approach that distinguish it from prior methods for exosome surface functionalization include its rapid processing and potential for scalability. As both the synthetic DNA with surface modifier and exosomes can be produced separately and combined, robustness and reversibility through DNase treatment shown here or other chemically labile linkages that can be incorporated in synthetic DNA. Moreover, oligonucleotide tethering provides a unique advantage of functionalizing exosomes by exploiting the non-covalent interactions through complementary strand pairing.
Surface tethering of oligonucleotides did not alter cell uptake of tethered exosomes. Immobilization of biologically active payloads onto the exosome surface was mediated via these oligonucleotide tethers enabling payload surface modification independent of the exosome cell source. We demonstrated that two independent cargos, AS1411 and SA-FasL, immobilized onto exosomes via oligonucleotide tethers were biologically active in vitro. We further demonstrated the ability of exosome tethered SA-FasL to modulate immune responses in vivo. This tethering technique can also be potentially used to tether other targeting moieties such as antibodies (Supplementary Fig. 18).
Compared to synthetic nanoparticle delivery systems used to treat a broad range of pathologies,62–66 exosomes hold promise as potentially ideal delivery vehicles because they are naturally occurring nanovesicles, evolved explicitly for intercellular communication.5,24,67 Although the mechanisms of exosome signaling and delivery of molecular cargo remain to be elucidated, our data suggests that DNA-tethered extracellular vesicles may represent a unique way to engineer designer exosomes with versatile surface functionalities, expanding the existing arsenal of drug-delivery systems. Further, synergistic interactions of membrane-tethered cargo with native endogenous exosome co-cargo constituents represent a potential advantage over current artificial delivery strategies.
Membrane proteins, including integrins, enzymes and ligands exhibiting surface membrane bioactivities, naturally occur on exosomes23 and FasL expressed on exosomes has been implicated in the suppression of lymphocyte function68 and immune tolerance.69 Additionally, exosomes with surface immunosuppressive cargo such as PDL-1 and CTLA-4 have been implicated in the interference of cellular immunotherapy in cancer patients.36 Such intrinsic exosome surface cargoes have motivated recent strategies to both use such innate cargo and to develop strategies to controllably engineer surface cargo for translational applications. For example, vaccine efficacy is improved by triggering antigen-specific immunogenicity using exosomes carrying MHC-peptide complexes.70,71 As targeted delivery of exosomes loaded with exogenous drugs and RNA-based therapeutics is enabled using exosomes with engineered surface ligands,14,19,26 we envision that our method of rapid surface functionalization can readily be used together with methods to load and deliver cargo in the exosome lumen.
Here, we used FasL as a proof-of-concept therapeutic membrane cargo for delivery via DNA tethered exosomes. We demonstrate the immunoregulatory activity of FasL in vivo. FasL is one of the key ‘death’ ligands involved in immune homeostasis, tolerance to self-antigens, and immune evasion by various tumor types.72 Because of these attributes, there has been a significant interest in targeting the FasL pathway for immunomodulation. However, the effective direct use of FasL as a biologic for immunomodulation is limited by its off-target effects that cause significant toxicity.36 To overcome this obstacle, we generated the SA-FasL recombinant protein for transient display on biologic and non-biologic surfaces for targeted, localized immunomodulation to control allograft rejection and autoimmunity, with demonstrated therapeutic efficacy.40,49,72–74 We showed that exosomes can be engineered to positionally display SA-FasL on their surface, and such engineered exosomes have a robust apoptotic efficacy on human Jurkat cells expressing the Fas receptor. The SA-FasL-tethered exosomes were also effective in eliminating alloreactive T cells in vivo. Importantly, the in vivo efficacy of SA-FasL on exosomes was 10-fold higher than soluble injection of SA-FasL. The robust activity from SA-FasL-tethered exosomes could be attributed to the dense tethering of DNA strands on exosome membrane, which in turn results in a more tethering of FasL per exosome. This could result in greater engagement of multiple Fas receptors on recipient cells resulting in a robust FasL/Fas-mediated apoptosis. On similar lines, Meckes et. al. has previously reported that activation of Toll-like receptor 9 on a reporter macrophage cell line by spherical nucleic acids depends on the density of DNA strands.75 This is highly significant not only for FasL but it is expected to be relevant for delivery of different peptide/protein hormones and other exosome surface constituents that can alter biodistribution, cell trafficking and/or interaction with the extracellular matrix.
Conclusion
Overall, surface modification of exosomes via membrane tethering of oligonucleotides represents an ‘all-purpose’ strategy to include biological signaling cargo to exosome surfaces. Such cargo can potentially modify not only cell function but also alter intercellular trafficking. This method represents a straightforward, practical approach that is independent of the exosome’s cell origin. Therefore, exosomes from any biological source, including those harvested from natural or engineered cells or found secreted in tissues and body fluids, may be readily augmented using the described oligonucleotide tethering method. This versatility in combination with its other attributes of DNA tethering scalability, control of spacing of cargo from the exosome surface, processing speed and reversibility are critical enabling features that will aid in translating exosome-based drug delivery therapies to the clinic.
Experimental
Cell culture
THP1 (ATTC®TIB-202™) and J774A.1 (ATTC®TIB-67™) cells were cultured in heat-inactivated fetal bovine serum (HI-FBS; ThermoFisher Scientific, Waltham, MA) that had been depleted of exosomes. HI-FBS was centrifuged at 100,000 xg for 3 hrs and the exosome depleted supernatant was collected (ED-HI-FBS). The final media for THP1 cells consisted of RPMI-1640 (ThermoFisher Scientific, Waltham, MA) supplemented with 10 % ED-HI-FBS and 1 % Penicillin-Streptomycin (PS; ThermoFisher Scientific, Waltham, MA). Jurkat cells (ATTC®TIB-152™) were grown in RPMI-1640 supplemented with 10 % ED-HI-FBS and 1 % PS. HEK293, MIAPaCa2 and PCI13 cells were cultured and maintained in Delbecco’s modified eagle media (DMEM; ThermoFisher, Waltham, MA) supplemented with 10 % ED-HI-FBS and 1 % PS. All the cell lines were regularly tested for mycoplasma contamination and were negative. Additionally, J774A.1 cells used for in vivo studies were certified by IDEXX BioResearch (Columbia, MO) to be free of bacteria, virus and mycoplasma.
Exosome isolation and characterization
Exosomes were isolated from THP1 and J774A.1 cells using the mini-SEC method as previously described.30,35,36,76 Briefly, conditioned media (minimum of 48 hrs in cell culture) were differentially centrifuged (2500 xg for 10 min at 4 °C and 10,000 xg for 30 min at 4 °C), followed by ultrafiltration (0.22 μm filter; Millipore-Sigma, Billicera, MA) and then size-exclusion chromatography on an A50 cm column (Bio-Rad Laboratories, Hercules, CA) packed with Sepharose 2B (Sigma-Aldrich, St. Louis, MO). Protein concentrations of exosome fractions were determined using a BCA Protein Assay kit as recommended by the manufacturer (Pierce, ThermoFisher Scientific, Waltham, MA). Further characterization of exosomes was done with dynamic light scattering, tunable resistive pulse sensing (TRPS), western blotting and transmission electron microscopy (TEM). See Supplemental document for detailed procedures.
DNA tethering of exosomes (Exo-ssDNA) and quantitation
20 μg of isolated THP1 exosomes were gently vortexed (600 RPM, Scientific Industries Vortex-Genie 2 Vortex Mixer) with different concentrations of cholesterol-DNA (Chol-DNA; 0.1 μM to 20 μM) for 5 min at room temperature in 100 µL PBS buffer (final exosome concentration= 0.2 μg/μL). Samples were then washed with Amicon Ultra Centrifugal Filters (100k MWCO, Millipore Sigma, St. Louis, MO), followed by reverse spin to get ssDNA-tethered exosomes (Exo-ssDNA). Quantification of Chol-ssDNA-Cy5 tethers on exosomes was done by analyzing the absorbance at 650 nm using TECAN spectrophotometer (Männedorf, Switzerland) and using the Beer Lambert’s law assuming the extension coefficient of Cy5 as 250000. To quantitate the DNA tethers on exosomes, we used Chol-ssDNA-Cy5 and exosomes from THP1 cells (SI Fig 5). Quantitation of the number of Chol-ssDNA-Cy5 strands tethered onto exosomes was based on the Cy5 absorbance at 650 nm (SI Fig. 5b). When 20 μM Chol-ssDNA-Cy5 was vortexed with 50 μg of exosomes, shows a loading of 6908 ± 242 tethers/exosome. This number is fairly close to a theoretical maximal accommodation of 7850 DNA tethers - assuming 4 nm2/tether in 100 nm diameter exosomes (SI Fig 5a) for which the surface area of an exosome would be ~ 31,400 nm2.
Flow cytometry studies
Exo-ssDNA-Cy5 were prepared using Cy5-conjugated Chol-DNA (Cholesterol and Cy5 on the 5’ and 3’ end respectively) using the tethering protocol mentioned above. Exo-ssDNA-Cy5 (6 μg protein) were gently vortexed overnight at 4°C with anti-CD63 conjugated magnetic streptavidin beads. For the control experiments, beads were incubated with the Chol-DNA-Cy5 to determine non-specific binding. Beads were washed with 1X PBS buffer three times (5 min each wash) to remove any unbound exosomes, followed by flow cytometry analysis on an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) connected to an Intellicyt HyperCyt autosampler (IntelliCyt Corp., Albuquerque, NM) using Cy5 channel (649 nm). Data were processed and interpreted using FlowJo® software (Flowjo LLC, Ashland, Oregon).
Exo-ssDNA stability assessment
120 μg of Exo-ssDNA-Cy5 (20 μM ssDNA tether concentration) were prepared using Chol-DNA-Cy5. Triplicate samples were incubated at 4 °C in 1X PBS buffer and 37 °C in simulated body fluid (10 % FBS, 0.1 % NaN3, 100 mM HEPES in DMEM) for 24h, 48h, 72h, and 1 week. At each time point, samples were incubated with anti-CD63 beads, rinsed three times (5 min each wash), followed by flow cytometry studies as described above using the Cy5-channel. Please see supplemental document for detailed procedures.
Click chemistry on exosomes (preparation of exosome-dsDNA-N3)
10 nmole of Chol-DNA were incubated with 10 nmole of N3-modified complementary DNA strand (DNA’-N3) in PBS buffer for annealing (37°C (15 min) → 0 °C (10 min) → RT (30 min)). Preannealed Chol-dsDNA-N3 was vortexed with 100 μg of exosomes in 500 μL PBS buffer to prepare Exo-dsDNA-N3 (20 μM azide concentration). The sample was concentrated to 200 μM azide concentration using ultra centrifugal filters (MWCO = 100k) for click reaction. For details regarding Cu-click and Cu-free click please see supplemental document.
AS1411 functionalized exosomes
DNA’-AS1411 with complementary region to Chol-DNA (5’- GGTGGTGGTGGTTGTGGTGGTGGTGGTTAGCTATGGGATCCAACTGCAGT-3’) was preannealed to Chol-DNA using standard annealing conditions. The preannealed Chol-dsDNA-As1411 was vortexed with exosomes to prepare Exo-dsDNA-AS1411 (20 μM dsDNA tether concentration).
Preparation of exosome-ssDNA-biotin
20 μg of exosomes were vortexed with different concentrations of Chol-DNA-Biotin (0.1 μM to 20 μM) for 5 min at room temperature in 100 μL PBS buffer. Samples were then washed with 100k MWCO filters, followed by reverse spin to get Exo-ssDNA-biotin.
Preparation of exosome-dsDNA-biotin
Chol-DNA and DNA’-Biotin were preannealed as described above. 20 μg of exosomes were gently vortexed with preannealed solution for tethering (20 μM dsDNA tether concentration). Samples were washed with 100k MWCO filters, followed by reverse spin to get Exo-dsDNA-biotin. Exosome with 10μM biotinylated DNA as described above. 20 μg exosomes were incubated with 100ng SA-FasL for 30 min at 37°C. Post incubation, Exo-dsDNA-SA-FasL were isolated by the miniSEC method described in the exosome isolation protocol. Jurkat cells (206/mL) were cultured in freshly prepared RPMI-1640 medium supplemented with 10 % ED-HI-FBS for 48 hrs. 20 μg exosomes decorated with 100ng (dsDNA)SA-FasL were added to the media and incubated for 12 hrs. Native exosomes (non-engineered exosomes) and Exo-dsDNA-biotin were used as controls. Apoptosis of Jurkat cells was measured by flow cytometry using an Annexin V assay (Beckman Coulter, Brea, CA).
Bioprinting exosomes for solid-phase presentation
Exosomes were inkjet-printed as previously described.46 Briefly, 100 μg/ml of Exo-ssDNA-SA-FasL exosome solution containing a final concentration of 10 % glycerol was used as the bioink. 50 overprints (OPs) of exosome bioink was printed on collagen type-1 coated coverslips to create patterns of 1.25 mm X 1.75 mm corresponding to a total deposited exosome protein concentration of 76 ng. Post overnight rinsing in PBS to wash-off unbound exosomes, PCI13 cells were seeded at a density of 2.5×103 cells/cm2. After 24 hrs, cells were stained with live/dead cell viability assay for mammalian cells (ThermoFisher Scientific, Waltham, MA) according to manufacturer’s instruction. This kit uses Calcien AM and ethidium bromide to differentiate between live and dead cells. Post staining, imaging was performed on ZEISS LSM 880 confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). Quantification of mean fluorescence intensities post background subtraction was performed on NIH Image J software by selecting a region of region corresponding to the printed exosome pattern.
In vivo studies
Animals
C57BL/6 and C57BL/6-Tg (Foxp3-DTR-eGFP; referred to as C57BL/6-DTR in the manuscript) and BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME), F1 mice were produced by crossing C57BL/6-DTR and BALB/c, and all mice were maintained under standard conditions in the Institute for Cellular Therapeutics barrier facility. The animals were cared for according to the University of Louisville and National Institutes of Health animal care and use guidelines. Six to eight weeks old female C57BL/6 (H-2b) and F1 (C57BL/6-DTR×BALB/c, H-2b×d) mice were used as splenocytes donors and recipients, respectively.
Preparation of CFSE labeled splenocytes
Spleens were collected from naïve C57BL/6 female mice, processed into single-cell suspension, and red blood cells were lysed using ACK (ammonium-chloride-potassium lysis buffer) solution. Splenocytes were passed through sterile nylon mesh strainers with 100 μm pores, centrifuged, and washed several times with PBS (Gibco, Gaithersburgh, MD). Cells were incubated with 2.5 μM CFSE in PBS for 7 minutes at room temperature and labeling reaction was stopped by the addition of an equal volume of FBS (fetal bovine serum, RMBIO). CFSE labeled cells were then washed twice with PBS and each female F1 mouse was injected through tail vein with 5x106 cells in 600 μl PBS.
Treatment and in vivo tracking of donor cells in F1 recipients
F1 mice were divided into four groups and subjected to two i.p. treatments with 40 μg of exosomes engineered with SA-FasL (Exo-ssDNA-SA-FasL) at 2 and 24 hrs after CFSE labeled splenocytes injection. An equal amount of Exo-ssDNA-biotin, the same dose of soluble SA-FasL used for exosome engineering, and saline (PBS) were used as controls. Cells from mesenteric LNs and spleens of treatment and control groups were harvested at 72 hrs post cell injection, erythrocytes were lysed with ACK lysis buffer, and cells were washed with PBS. Cells were incubated for 15 min at room temperature with anti-mouse CD16/CD32 (Mouse FC block, BioLegend, San Diego, CA) antibody to block Fc receptors. Samples were then stained with antibodies to mouse CD3-V500, CD4-Alexa Flour 700, CD8-APC Cy7 (BD Biosciences) and MHC class I (H2Kd)-APC (Bio legend, San Diego, CA) molecules for 25 min at 4°C and washed with PBS prior to analysis. The cells were run on the LSR II (BD Biosciences, San Jose, CA) flow cytometry and the data was analyzed by BD FACSDiva™ software and graphed using GraphPad Prism. Representative gating of splenocytes is shown in Supplementary Fig. 16.
Statistical analysis
Data are presented as the average ± SEM (n=3 independent experiments). One-way analysis of variance (ANOVA) was used for data analysis to determine any statistically significant differences between two and multiple groups with Tukey’s post-hoc analysis where appropriate using GraphPad Prism (v8.0) software. P ≤ 0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported in part by grants NIH 1U01AI132817-01 and R01AI121281-01A1 (H.S., E.S.Y.), NSF DMR 1501324 (K.M.) and the Dowd Fellowship at Carnegie Mellon University (S.S.Y.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare the following competing financial interest(s): H.S. and E.S.Y. are co-inventors on patents for SA-FasL and hold equity in FasCure Therapeutics, LLC, which has an option to license the SA-FasL technology from the University of Louisville. Other authors declare no competing financial interests.
REFERENCES
- (1).Colombo M; Raposo G; Théry C Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol 2014, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- (2).Yanez-Mo M; Siljander PR; Andreu Z; Zavec AB; Borras FE; Buzas EI; Buzas K; Casal E; Cappello F; Carvalho J; Colás E Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Van Dommelen SM; Vader P; Lakhal S; Kooijmans SAA; Van Solinge WW; Wood MJA; Schiffelers RM Microvesicles and Exosomes: Opportunities for Cell-Derived Membrane Vesicles in Drug Delivery. J. Control. Release 2012, 161, 635–644. [DOI] [PubMed] [Google Scholar]
- (4).György B; Hung ME; Breakefield XO; Leonard JN Therapeutic Applications of Extracellular Vesicles: Clinical Promise and Open Questions. Annu. Rev. Pharmacol. Toxicol 2015, 55, 439–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Armstrong JPK; Holme MN; Stevens MM Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano. 2017, 11, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kooijmans SAA; Vader P; van Dommelen SM; van Solinge WW; Schiffelers RM Exosome Mimetics: A Novel Class of Drug Delivery Systems. Inter. J. of Nanomed 2012, 7, 1525–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Van Der Meel R; Fens MHAM; Vader P; Van Solinge WW; Eniola-Adefeso O; Schiffelers RM Extracellular Vesicles as Drug Delivery Systems: Lessons from the Liposome Field. J. Control. Release 2014, 195, 72–85. [DOI] [PubMed] [Google Scholar]
- (8).EL Andaloussi S; Mäger I; Breakefield XO; Wood MJA Extracellular Vesicles: Biology and Emerging Therapeutic Opportunities. Nat. Rev. Drug Discov 2013, 12, 347–357. [DOI] [PubMed] [Google Scholar]
- (9).Lener T; Gimona M; Aigner L; Börger V; Buzas E; Camussi G; Chaput N; Chatterjee D; Court FA; del Portillo HA; O’Driscoll L Applying Extracellular Vesicles Based Therapeutics in Clinical Trials - An ISEV Position Paper. J. Extracell. Vesicles 2015, 4, 30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Batrakova EV; Kim MS Using Exosomes, Naturally-Equipped Nanocarriers, for Drug Delivery. J. Control. Release 2015, 219, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fuhrmann G; Serio A; Mazo M; Nair R; Stevens MM Active Loading into Extracellular Vesicles Significantly Improves the Cellular Uptake and Photodynamic Effect of Porphyrins. J. Control. Release 2015, 205, 35–44. [DOI] [PubMed] [Google Scholar]
- (12).Fuhrmann G; Chandrawati R; Parmar PA; Keane TJ; Maynard SA; Bertazzo S; Stevens MM Drug Delivery: Engineering Extracellular Vesicles with the Tools of Enzyme Prodrug Therapy. Adv. Mater 2018, 30, 1706616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wood MJA; O’Loughlin AJ; Lakhal S Exosomes and the Blood-Brain Barrier: Implications for Neurological Diseases. Therapeutic Del. 2011, 2, 1095–1099. [DOI] [PubMed] [Google Scholar]
- (14).Alvarez-Erviti L; Seow Y; Yin H; Betts C; Lakhal S; Wood MJA Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol 2011, 29, 341–345. [DOI] [PubMed] [Google Scholar]
- (15).Tang K; Zhang Y; Zhang H; Xu P; Liu J; Ma J; Lv M; Li D; Katirai F; Shen GX; Zhang G Delivery of Chemotherapeutic Drugs in Tumour Cell-Derived Microparticles. Nat. Commun 2012, 3, 1282. [DOI] [PubMed] [Google Scholar]
- (16).Gujrati V; Kim S; Kim SH; Min JJ; Choy HE; Kim SC; Jon S Bioengineered Bacterial Outer Membrane Vesicles as Cell-Specific Drug-Delivery Vehicles for Cancer Therapy. ACS Nano. 2014, 8, 1525–1537. [DOI] [PubMed] [Google Scholar]
- (17).Ohno SI; Takanashi M; Sudo K; Ueda S; Ishikawa A; Matsuyama N; Fujita K; Mizutani T; Ohgi T; Ochiya T; Gotoh N Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor Microrna to Breast Cancer Cells. Mol. Ther 2013, 21, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cooper JM; Wiklander PBO; Nordin JZ; Al-Shawi R; Wood MJ; Vithlani M; Schapira AHV; Simons JP; El-Andaloussi S; Alvarez-Erviti L Systemic Exosomal siRNA Delivery Reduced Alpha-Synuclein Aggregates in Brains of Transgenic Mice. Mov. Disord 2014, 29, 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tian Y; Li S; Song J; Ji T; Zhu M; Anderson GJ; Wei J; Nie G A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials. 2014, 35, 2383–2390. [DOI] [PubMed] [Google Scholar]
- (20).Kooijmans SAA; Fliervoet LAL; Van Der Meel R; Fens MHAM; Heijnen HFG; Van Bergen En Henegouwen PMP; Vader P; Schiffelers RM PEGylated and Targeted Extracellular Vesicles Display Enhanced Cell Specificity and Circulation Time. J. Control. Release 2016, 224, 77–85. [DOI] [PubMed] [Google Scholar]
- (21).Mulcahy LA; Pink RC; Carter DRF Routes and Mechanisms of Extracellular Vesicle Uptake. J. of Extracell. Vesicles 2014, 3, 24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Smyth T; Petrova K; Payton NM; Persaud I; Redzic JS; Graner MW; Smith-Jones P; Anchordoquy TJ Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem 2014, 25, 1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yang Y; Hong Y; Cho E; Kim GB; Kim IS Extracellular Vesicles as a Platform for Membrane-Associated Therapeutic Protein Delivery. J. of Extracell. Vesicles 2018, 7, 1440131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Armstrong JPK; Stevens MM Strategic Design of Extracellular Vesicle Drug Delivery Systems. Adv. Drug Del. Rev 2018, 130, 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).O’Loughlin AJ; Mäger I; de Jong OG; Varela MA; Schiffelers RM; El Andaloussi S; Wood MJA; Vader P Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther 2017, 25, 1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Didiot MC; Hall LM; Coles AH; Haraszti RA; Godinho BMDC; Chase K; Sapp E; Ly S; Alterman JF; Hassler MR; Echeverria D Exosome-Mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther 2016, 24, 1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gao X; Ran N; Dong X; Zuo B; Yang R; Zhou Q; Moulton HM; Seow Y; Yin HF Anchor Peptide Captures, Targets, and Loads Exosomes of Diverse Origins for Diagnostics and Therapy. Sci. Transl. Med 2018, 10, eaat0195. [DOI] [PubMed] [Google Scholar]
- (28).Christianson HC; Svensson KJ; van Kuppevelt TH; Li J-P; Belting M Cancer Cell Exosomes Depend on Cell-Surface Heparan Sulfate Proteoglycans for Their Internalization and Functional Activity. Proc. Natl. Acad. Sci 2013, 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lee M; Ban JJ; Im W; Kim M Influence of Storage Condition on Exosome Recovery. Biotechnol. Bioprocess Eng 2016, 21, 299–304. [Google Scholar]
- (30).Ludwig N; Yerneni SS; Razzo BM; Whiteside TL HNSCC-Derived Exosomes Promote Angiogenesis through Reprogramming of Endothelial Cells In Vitro and In Vivo. Mol. Cancer Res 2018, 16, 1798–1808. [DOI] [PubMed] [Google Scholar]
- (31).Bates PJ; Laber DA; Miller DM; Thomas SD; Trent JO Discovery and Development of the G-Rich Oligonucleotide AS1411 as a Novel Treatment for Cancer. Exp. and Mol. Path 2009, 86, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang C; Liu B; Xu X; Zhuang B; Li H; Yin J; Cong M; Xu W; Lu A Toward Targeted Therapy in Chemotherapy-Resistant Pancreatic Cancer with a Smart Triptolide Nanomedicine. Oncotarget 2016, 7, 8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Park JY; Cho YL; Chae JR; Moon SH; Cho WG; Choi YJ; Lee SJ; Kang WJ Gemcitabine-Incorporated G-Quadruplex Aptamer for Targeted Drug Delivery into Pancreas Cancer. Mol. Ther. - Nucleic Acids 2018, 12, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ai J; Xu Y; Lou B; Li D; Wang E Multifunctional AS1411-Functionalized Fluorescent Gold Nanoparticles for Targeted Cancer Cell Imaging and Efficient Photodynamic Therapy. Talanta 2014, 118, 54–60. [DOI] [PubMed] [Google Scholar]
- (35).Theodoraki M-N; Yerneni SS; Hoffmann TK; Gooding WE; Whiteside TL Clinical Significance of PD-L1 + Exosomes in Plasma of Head and Neck Cancer Patients. Clin. Cancer Res 2018, 24, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hong CS; Sharma P; Yerneni SS; Simms P; Jackson EK; Whiteside TL; Boyiadzis M Circulating Exosomes Carrying an Immunosuppressive Cargo Interfere with Cellular Immunotherapy in Acute Myeloid Leukemia. Sci. Rep 2017, 7, 14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Maher S; Toomey D; Condron C; Bouchier-Hayes D Activation-Induced Cell Death: The Controversial Role of Fas and Fas Ligand in Immune Privilege and Tumour Counterattack. Immun. and Cell Bio 2002, 80, 131–137. [DOI] [PubMed] [Google Scholar]
- (38).Yolcu ES; Askenasy N; Singh NP; Cherradi SEL; Shirwan H Cell Membrane Modification for Rapid Display of Proteins as a Novel Means of Immunomodulation: FasL-Decorated Cells Prevent Islet Graft Rejection. Immunity 2002, 17, 795–808. [DOI] [PubMed] [Google Scholar]
- (39).Peter ME; Hadji A; Murmann AE; Brockway S; Putzbach W; Pattanayak A; Ceppi P The Role of CD95 and CD95 Ligand in Cancer. Cell Death and Diff. 2015, 22, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Headen DM; Woodward KB; Coronel MM; Shrestha P; Weaver JD; Zhao H; Tan M; Hunckler MD; Bowen WS; Johnson CT; Shea L Local Immunomodulation with Fas Ligand-Engineered Biomaterials Achieves Allogeneic Islet Graft Acceptance. Nat. Mater 2018, 17, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Campbell PG; Weiss LE Tissue Engineering with the Aid of Inkjet Printers. Expert Opin. Biol. Ther 2007, 7, 1123–1127. [DOI] [PubMed] [Google Scholar]
- (42).Phillippi JA; Miller E; Weiss L; Huard J; Waggoner A; Campbell P Microenvironments Engineered by Inkjet Bioprinting Spatially Direct Adult Stem Cells Toward Muscle-and Bone-Like Subpopulations. Stem Cells. 2008, 26, 127–134. [DOI] [PubMed] [Google Scholar]
- (43).Miller ED; Fisher GW; Weiss LE; Walker LM; Campbell PG Dose-Dependent Cell Growth in Response to Concentration Modulated Patterns of FGF-2 Printed on Fibrin. Biomaterials. 2006, 27, 2213–2221. [DOI] [PubMed] [Google Scholar]
- (44).Ceren T; Kostas V; Lee W; Phil C Crosstalk between Substance P and Calcitonin Gene-related Peptide during Heterotopic Ossification in Murine Achilles Tendon. J. Orthop. Res 2018, 36, 1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang W; Gorantla VS; Campbell PG; Li Y; Yang Y; Komatsu C; Weiss LE; Zheng XX; Solari MG Biopatterned CTLA4/Fc Matrices Facilitate Local Immunomodulation, Engraftment, and Glucose Homeostasis after Pancreatic Islet Transplantation. Diabetes. 2016, 65, 3660–3666. [DOI] [PubMed] [Google Scholar]
- (46).Yerneni SS; Whiteside TL; Weiss LE; Campbell PG Bioprinting Exosome-Like Extracellular Vesicle Microenvironments. Bioprinting. 2019, 13, e00041. [Google Scholar]
- (47).Campbell PG; Miller ED; Fisher GW; Walker LM; Weiss LE Engineered Spatial Patterns of FGF-2 Immobilized on Fibrin Direct Cell Organization. Biomaterials 2005, 26, 6762–6770. [DOI] [PubMed] [Google Scholar]
- (48).Yolcu ES; Gu X; Lacelle C; Zhao H; Bandura-Morgan L; Askenasy N; Shirwan H Induction of Tolerance to Cardiac Allografts Using Donor Splenocytes Engineered to Display on Their Surface an Exogenous Fas Ligand Protein. J. Immunol 2008, 181, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yolcu ES; Zhao H; Bandura-Morgan L; Lacelle C; Woodward KB; Askenasy N; Shirwan H Pancreatic Islets Engineered with SA-FasL Protein Establish Robust Localized Tolerance by Inducing Regulatory T Cells in Mice. J. Immunol 2011, 187, 5901–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pfeiffer I; Höök F Bivalent Cholesterol-Based Coupling of Oligonucletides to Lipid Membrane Assemblies. J. Am. Chem. Soc 2004, 126, 10224–10225. [DOI] [PubMed] [Google Scholar]
- (51).Selden NS; Todhunter ME; Jee NY; Liu JS; Broaders KE; Gartner ZJ Chemically Programmed Cell Adhesion with Membrane-Anchored Oligonucleotides. J. Am. Chem. Soc 2012, 134, 765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Weber RJ; Liang SI; Selden NS; Desai TA; Gartner ZJ Efficient Targeting of Fatty-Acid Modified Oligonucleotides to Live Cell Membranes through Stepwise Assembly. Biomacromolecules 2014, 15, 4621–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).You M; Lyu Y; Han D; Qiu L; Liu Q; Chen T; Sam Wu C; Peng L; Zhang L; Bao G; Tan W DNA Probes for Monitoring Dynamic and Transient Molecular Encounters on Live Cell Membranes. Nat. Nanotechnol 2017, 12, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rabuka D; Forstner MB; Groves JT; Bertozzi CR Noncovalent Cell Surface Engineering: Incorporation of Bioactive Synthetic Glycopolymers into Cellular Membranes. J. Am. Chem. Soc 2008, 130, 5947–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Modi S; Swetha MG; Goswami D; Gupta GD; Mayor S; Krishnan Y A DNA Nanomachine That Maps Spatial and Temporal pH Changes inside Living Cells. Nat. Nanotechnol 2009, 4, 325–330. [DOI] [PubMed] [Google Scholar]
- (56).Saha S; Prakash V; Halder S; Chakraborty K; Krishnan Y A pH-Independent DNA Nanodevice for Quantifying Chloride Transport in Organelles of Living Cells. Nat. Nanotechnol 2015, 10, 645–651. [DOI] [PubMed] [Google Scholar]
- (57).Pi F; Binzel DW; Lee TJ; Li Z; Sun M; Rychahou P; Li H; Haque F; Wang S; Croce CM; Guo B Nanoparticle Orientation to Control RNA Loading and Ligand Display on Extracellular Vesicles for Cancer Regression. Nat. Nanotechnol 2018, 13, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Banga RJ; Chernyak N; Narayan SP; Nguyen ST; Mirkin CA Liposomal Spherical Nucleic Acids. J. Am. Chem. Soc 2014, 136, 9866–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).He F; Liu H; Guo X; Yin BC; Ye BC Direct Exosome Quantification via Bivalent-Cholesterol-Labeled DNA Anchor for Signal Amplification. Anal. Chem 2017, 89, 12968–12975. [DOI] [PubMed] [Google Scholar]
- (60).Skotland T; Hessvik NP; Sandvig K; Llorente A Exosomal Lipid Composition and the Role of Ether Lipids and Phosphoinositides in Exosome Biology. J. Lipid Res 2019, 60, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Laulagnier K; Motta C; Hamdi S; Roy S; Fauvelle F; Pageaux J-F; Kobayashi T; Salles J-P; Perret B; Bonnerot C; Record M Mast Cell- and Dendritic Cell-Derived Exosomes Display a Specific Lipid Composition and an Unusual Membrane Organization. Biochem. J 2004, 380, 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Torchilin VP Recent Advances with Liposomes as Pharmaceutical Carriers. Nat. Rev.Drug Disc 2005, 4, 145–160. [DOI] [PubMed] [Google Scholar]
- (63).Allen TM; Cullis PR Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- (64).Sercombe L; Veerati T; Moheimani F; Wu SY; Sood AK; Hua S Advances and Challenges of Liposome Assisted Drug Delivery. Front. in Pharm 2015, 6, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Zylberberg C; Matosevic S Pharmaceutical Liposomal Drug Delivery: A Review of New Delivery Systems and a Look at the Regulatory Landscape. Drug Deliv. 2016, 23, 3319–3329. [DOI] [PubMed] [Google Scholar]
- (66).Kauscher U; Holme MN; Björnmalm M; Stevens MM Physical Stimuli-Responsive Vesicles in Drug Delivery: Beyond Liposomes and Polymersomes. Adv. Drug Deliv. Rev 2019, 138, 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Fuhrmann G; Herrmann IK; Stevens MM Cell-Derived Vesicles for Drug Therapy and Diagnostics: Opportunities and Challenges. Nano Today. 2015, 10, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ludwig S; Floros T; Theodoraki MN; Hong CS; Jackson EK; Lang S; Whiteside TL Suppression of Lymphocyte Functions by Plasma Exosomes Correlates with Disease Activity in Patients with Head and Neck Cancer. Clin. Cancer Res 2017, 23, 4843–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Lundy SK; Klinker MW; Fox DA Killer B Lymphocytes and Their Fas Ligand Positive Exosomes as Inducers of Immune Tolerance. Front. in Immunol 2015, 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Hartman ZC; Wei J; Glass OK; Guo H; Lei G; Yang XY; Osada T; Hobeika A; Delcayre A; Le Pecq JB; Morse MA Increasing Vaccine Potency through Exosome Antigen Targeting. Vaccine 2011, 29, 9361–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Andre F; Chaput N; Schartz NEC; Flament C; Aubert N; Bernard J; Lemonnier F; Raposo G; Escudier B; Hsu D-H; Tursz T Exosomes as Potent Cell-Free Peptide-Based Vaccine. I. Dendritic Cell-Derived Exosomes Transfer Functional MHC Class I/Peptide Complexes to Dendritic Cells. J. Immunol 2004, 172, 2126–2136. [DOI] [PubMed] [Google Scholar]
- (72).Askenasy N; Yolcu ES; Yaniv I; Shirwan H Induction of Tolerance Using Fas Ligand: A Double-Edged Immunomodulator. Blood. 2005, 105, 1396–1404. [DOI] [PubMed] [Google Scholar]
- (73).Askenasy N; Yolcu ES; Wang Z; Shirwan H Display of Fas Ligand Protein on Cardiac Vasculature as a Novel Means of Regulating Allograft Rejection. Circulation. 2003, 107, 1525–1531. [DOI] [PubMed] [Google Scholar]
- (74).Singh NP; Yolcu ES; Askenasy N; Shirwan H ProtEx™: A Novel Technology to Display Exogenous Proteins on the Cell Surface for Immunomodulation. Ann. of the New York Acad. of Sci. 2005, 1056, 344–358. [DOI] [PubMed] [Google Scholar]
- (75).Meckes B; Banga RJ; Nguyen SBT; Mirkin CA Enhancing the Stability and Immunomodulatory Activity of Liposomal Spherical Nucleic Acids through Lipid-Tail DNA Modifications. Small 2018, 14, 1702909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Theodoraki M-N; Yerneni SS; Brunner C; Theodorakis J; Hoffmann TK; Whiteside TL Plasma-Derived Exosomes Reverse Epithelial-to-Mesenchymal Transition after Photodynamic Therapy of Patients with Head and Neck Cancer. Oncoscience 2018, 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.