

Summary
Tissue environment plays a powerful role in establishing and maintaining the distinct phenotypes of resident macrophages, but the underlying molecular mechanisms remain poorly understood. Here, we characterized transcriptomic and epigenetic changes in repopulating liver macrophages following acute Kupffer cell depletion as a means to infer signaling pathways and transcription factors that promote Kupffer cell differentiation. We obtained evidence that combinatorial interactions of the Notch ligand DLL4 and transforming growth factor-b (TGF-β) family ligands produced by sinusoidal endothelial cells and endogenous LXR ligands were required for the induction and maintenance of Kupffer cell identity. DLL4 regulation of the Notch transcriptional effector RBPJ activated poised enhancers to rapidly induce LXRα and other Kupffer cell lineage-determining factors. These factors in turn reprogrammed the repopulating liver macrophage enhancer landscape to converge on that of the original resident Kupffer cells. Collectively, these findings provide a framework for understanding how macrophage progenitor cells acquire tissue-specific phenotypes.
eTOC Blurb
Tissue environment plays a powerful role in establishing and maintaining the distinct phenotypes of resident macrophages, but the underlying molecular mechanisms remain poorly understood. Here, we characterized transcriptomic and epigenetic changes in repopulating liver macrophages following acute Kupffer cell depletion as a means to infer signaling pathways and transcription factors that promote Kupffer cell differentiation. We obtained evidence that combinatorial interactions of the Notch ligand DLL4 and transforming growth factor-b (TGF-β) family ligands produced by sinusoidal endothelial cells and endogenous LXR ligands were required for the induction and maintenance of Kupffer cell identity. DLL4 regulation of the Notch transcriptional effector RBPJ activated poised enhancers to rapidly induce LXRα and other Kupffer cell lineage-determining factors. These factors in turn reprogrammed the repopulating liver macrophage enhancer landscape to converge on that of the original resident Kupffer cells. Collectively, these findings provide a framework for understanding how macrophage progenitor cells acquire tissue-specific phenotypes.
Graphical Abstract
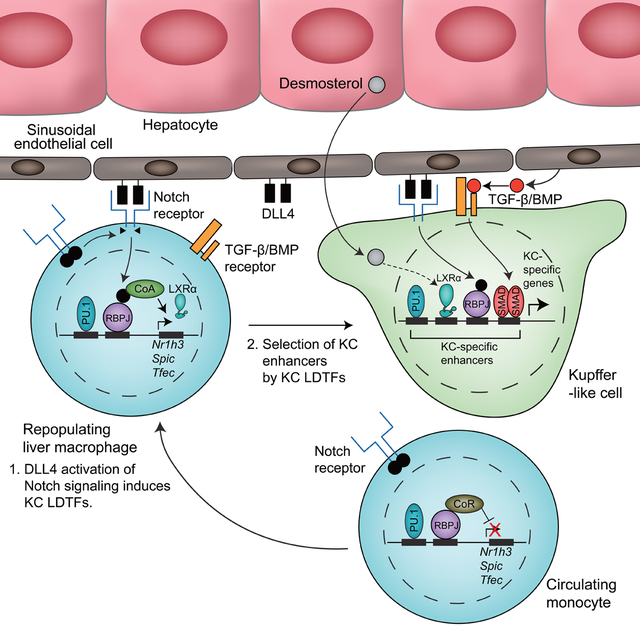
Introduction
Each tissue-resident macrophage population exhibits a distinct phenotype that is linked to the corresponding functions of that tissue. Well-studied examples include the role of Kupffer cells (KCs) (liver resident macrophages) in iron metabolism and clearance of gut-derived microbial products (Krenkel and Tacke, 2017). Investigation of molecular mechanisms underlying tissue-specific phenotypes indicated that different macrophage populations express distinct sets of transcription factors (TFs) and exhibit distinct patterns of enhancers (Gosselin et al., 2014; Lavin et al., 2014). KCs are embryonically-derived macrophages (Ginhoux and Guilliams, 2016; Perdiguero and Geissmann, 2016). Following entry into the fetal liver, embryonic macrophages rapidly upregulate Nr1h3 (encoding LXRα), Spic, Id1, Id3 and Irf7, suggesting roles in establishing KC-specific gene expression (Mass et al., 2016). The LXR motif is enriched in KC enhancers (Lavin et al., 2014) and loss of function of Nr1h3 in KCs indicates a key role in establishing KC identity (Scott et al., 2018).
Evidence supporting an instructive role of tissue environment in establishing tissue resident macrophage phenotypes has been provided by changes in enhancer activity and/or gene expression following transfer of microglia or peritoneal macrophages to a tissue culture environment (Gosselin et al., 2014; Gosselin et al., 2017) or following transfer of peritoneal macrophages to the alveolar airspaces (Lavin et al., 2014). However, the key signaling molecules and the mechanisms by which they drive the differentiation and specialized functions of most tissue resident macrophages are largely unknown.
Here we leveraged the anatomical relationship of KCs with liver sinusoidal endothelial cells (LSECs) and the power of KC depletion followed by repopulation in the mouse as a model system (Scott et al., 2016). KC ablation in mice expressing the diphtheria toxin receptor (DTR) specifically in KCs results in rapid colonization of the empty niche by circulating monocytes and their subsequent differentiation to Kupffer-like cells. Using this system, we found that liver-derived signals rapidly induce expression of KC lineage-determining TFs (LDTFs) within 24 h of monocyte entry by acting upon a pre-existing but poised enhancer landscape. The induction of these factors in turn drives the selection and function of additional enhancers that establish KC identity. We provide evidence that transforming growth factor-b (TGF-β) family ligands and DLL4 expressed by LSECs function in a combinatorial manner with liver-derived LXR ligands to initiate the KC differentiation program and maintain the KC phenotype.
Results
Rapid differentiation of recruited monocytes occurs following KC ablation
We first generated mice harboring Cre-T2A-nuclear localization signal-tagged tdTomato (tdTomato-NLS) in the 3’ UTR of the KC-specific Clec4f gene under translational control of an internal ribosome entry site (Figure S1A, B). While Lyz2-cre activity was observed in both KCs and hepatic CD11bHiF4/80Lo cells, Clec4f-cre-tdTomato mice show Cre activity and tdTomato expression specifically in KCs (Figure S1C). We then crossed these mice to mice in which the DTR was genetically targeted into the Rosa26 locus behind a Lox-Stop-Lox cassette (Figure 1A) (Buch et al., 2005), resulting in KC-specific expression of the DTR. Treatment of these animals with diphtheria toxin (DT) led to a near complete elimination of KCs from the liver by 12 h following injection (F4/80PosMHC II LoTim4PosClec4f-tdTomatoPos, Figure 1B), without affecting liver capsular macrophages (z-stack 1–5, F4/80PosMHCIIHiTim4Neg, Figure S1D–F), (Sierro et al., 2017). At 12h following DT treatment, adherent monocyte-derived cells (CD11bHiLy6CHiF4/80NegClec4f-tdTomatoNeg) were already present in the liver (Figure 1B), while neutrophil infiltration was not observed (Figure S1G). RNA-seq analysis was performed on circulating Ly6CHi monocytes, the recruited cells at 12h, 1d, 2d, 3d, 7d and 14d, and resident KCs. We refer to cells recovered within the first 12 h after DT as recruited liver monocytes and cells recovered from 24h to 14 days after DT as repopulating liver macrophages (RLMs).
Figure 1.

Recruited monocytes rapidly acquire expression of KC LDTFs followed by expression of a subset of KC-specific genes in KC-depleted livers
A. Experimental scheme: Clecf4-cre-tdTomato x Rosa26 LSL DTR +/− DT
B. Flow cytometry analysis of cell populations as a function of time following DT treatment.
C. MA plot of RNA-seq data comparing circulating monocytes and RLMs at 24 h. Data are from one or two experiments with n = 3–4 per group.
D. Genome-wide representation of DE genes in RLMs from 12h to 14 days in comparison to circulating monocytes and resident KCs. Data are from one or two experiments with n = 2–4 per group. DE genes are selected by DESeq2 (p-adj < 0.05).
E. PCA of 9568 detectable genes (at least 8 TPM in at least two samples) in circulating monocytes, recruited liver monocytes, RLMs and resident KCs.
F. Bar plots for expression of indicated genes. The significance symbols represent the p-adj from DESeq2 comparing to circulating monocytes respectively. ***p-adj < 0.001. Data are from one or two experiments with n = 2–4 per group. See also Figure S1.
RNA-seq analysis identified more than 3400 differentially expressed (DE) genes (FC > 2, p-adj < 0.05) in RLMs at 24h (Figure 1C). Even at this early time point, RLMs gained expression of 170 of 303 pre-defined KC identity genes (Figure S1H). RLMs progressively acquired a pattern of gene expression that converged with the KC gene signature over a 14 day time frame (Figure 1D, E). Cell cycle control genes were transiently upregulated at 2 and 3 days post DT injection, suggesting proliferation of recruited cells during this time frame (Figure S1I, J). Notably, genes encoding putative KC LDTFs, including Nr1h3, Spic and Id3, were strongly activated within the first 12h, while the monocyte-specific gene Ccr2 was strongly down-regulated (Figure 1F). Most KC genes exhibit a more delayed pattern. For example, Clec4f showed the strongest upregulation between 72h and 7d. Timd4, encoding a surrogate marker of many tissue macrophages of embryonic origin and/or prolonged tissue residence (Bain et al., 2016; Scott et al., 2016), was also upregulated during this time period, but did not approach its expression in resident KCs (Figure 1F), as previously reported (Scott et al., 2016).
Liver environment reprograms the recruited monocyte enhancer landscape
To investigate transcriptional mechanisms by which the liver environment induces a KC-like program of gene expression, we performed ATAC-seq to identify open chromatin in circulating monocytes, in RLMs at 1d and 2d post DT injection, and in resident KCs. Replicate samples were highly correlated and clustered according to condition (Figure S2A). Recruited cells exhibited marked changes in open chromatin that progressed towards the pattern observed in KCs (Figure 2A). De novo motif analysis of ATAC-seq peaks in RLMs at 48h indicated enrichment for motifs recognized by CREB and AP-1, MAFs, NFκB, TFEs, LXRs, RBPJ and SMADs (Figure 2B). In parallel, we identified putative KC-specific enhancers using histone H3 lysine 27 acetylation (H3K27ac), which is a dynamic histone modification that is highly correlated with transcriptional activity (Creyghton et al., 2010). KC-specific enhancers were defined as distal accessible chromatin regions with preferential enrichment of H3K27ac in KCs in at least 3/4 comparisons with resident peritoneal macrophages, microglia, monocytes, and bone marrow-derived macrophages (BMDMs) (Figure S2B). Enrichment analysis for de novo motifs returned a slightly larger but fully overlapping set of enriched motifs that included an IRF motif (Figure S2C). Motifs for signal-dependent TFs LXRs, SMADs, RBPJ, and NFκB are of particular interest because they could play roles in transmitting liver environmental signals (Bray, 2006; Heinz et al., 2015; Schmierer and Hill, 2007). The LXR response element and the motifs for MAF and TFE factors were consistent with the very rapid (12h-1d) upregulation of Nr1h3, Maf, Mafb, and Tfec mRNAs following monocyte retention in the liver (Figure 1F and 2C). Irf7, the most highly expressed member of the IRF family in KCs, was expressed in circulating monocytes and increased more slowly following recruitment to the liver (Figure S2D). Interpretation of enriched motifs for CREB, ATF, and AP-1 TFs was complex because there were high magnitude changes in the expression of those TFs in recruited monocytes that went in opposite directions. For example, Junb and Fosl2 were strongly downregulated, Jdp2 was strongly upregulated, and Jund was consistently highly expressed (Figure S2E).
Figure 2.

Rapid reprogramming of the RLM epigenetic landscape
A. Heat map of distal accessible chromatin regions defined by ATAC-seq in circulating monocytes, RLMs at 24 and 48h, and KCs. Each row is Z-score normalized tag counts for a peak. Data are from one or two experiments with n = 2–3 per group.
B. Enriched motifs in distal accessible chromatin regions defined by ATAC-seq of RLMs at 48 h using GC-matched genomic background.
C. Bar plots for expression of indicated genes in circulating monocytes (Circ Mo), RLMs, and resident KCs. Data are from one or two experiments with n = 2–4 per group. The significance markers represent the p-adj from DESeq2 comparing to circulating monocytes respectively. *p-adj < 0.05; ***p-adj < 0.001.
D. Scatter plot of distal ATAC-associated H3K27ac in RLMs at 24h vs circulating monocytes. Data are from one or two experiments with n = 2–3 per group. Color codes indicate significant changes (p-adj < 0.05 & FC > 2) in H3K27ac with or without significant changes in ATAC-seq peaks.
E. Genome browser tracks of ATAC-seq and H3K27ac ChIP peaks in the vicinity of the indicated loci in blood monocytes (Circ Mono), RLMs at 24 and 48 h and KCs. Yellow shading; pre-existing ATAC-seq peaks in circulating monocytes. Blue shading; regions of open chromatin acquired during RLM differentiation. See also Figure S2.
We next performed ChIP-seq for H3K27ac, in circulating monocytes, RLMs at 24h post DT injection, and in resident KCs to examine alterations in the activities of pre-existing regulatory elements. These experiments identified nearly 2000 upregulated H3K27ac peaks in recruited monocytes, ~2/3 of which were associated with pre-existing ATAC-seq peaks (Figure 2D). Sites gaining H3K27ac were enriched for LXR, MAF, MITF and RBPJ motifs (Figure S2F), consistent with rapid increases in the activities of these factors. Conversely, more than 2000 H3K27ac peaks were lost from circulating monocytes within the first 24 h following DT treatment, ~1/4 of which were associated with a loss of a corresponding ATAC-seq peak. Sites of reduced H3K27ac were enriched for motifs associated with KLF, C/EBP, RUNX, SP2 and bZIP motifs (Figure S2F), consistent with rapid down-regulation of their expressions and/or activities.
Composite ATAC-seq and H3K27ac ChIP-seq tracks are illustrated for Nr1h3, Mafb, Clec4f and Cd5l in Figure 2E. Pre-existing ATAC-seq peaks were observed at the putative regulatory elements of Nr1h3 and Mafb that exhibited increased H3K27ac in RLMs 24h in comparison to circulating monocytes (yellow shading, Figure 2E). These locations exhibited further H3K27ac in resident KCs. In contrast, Clec4f and Cd5l provide examples in which ATAC-seq peaks associated with putative regulatory elements in resident KCs do not become apparent until at least 48h after DT (light blue shading, Figure 2E). Collectively, these results support a model in which liver environmental signals act on pre-existing but poised regulatory elements to rapidly induce the expression of a set of TFs necessary for selection of additional enhancers that specify KC differentiation.
LXRα is a KC LDTF
LXRα and LXRβ are nuclear receptors that regulate cellular cholesterol and fatty acid homeostasis in response to sterol and oxysterol ligands (Calkin and Tontonoz, 2012; Spann and Glass, 2013). However, Nr1h3 is one of the most rapidly and highly induced genes in RLMs following KC depletion (Figure 1F), suggesting that its induction also plays a key role in driving the KC differentiation program. To address this question, we bred Clec4f-cre-tdTomato mice to Nr1h3fl/fl mice to generate a KC-specific genetic ablation. Flow cytometry indicated that expression of Clec4f-tdTomato was associated with a population of Tim4Pos KCs and an additional population of Tim4Neg cells (Figure S3A). The origin of Tim4Neg cells remains to be established. Reduced Tim4 expression could be due to it being a direct LXRα target gene. Alternatively, but not mutually exclusively, lack of LXRα could result in reduced embryonic KC survival and replacement by monocyte derived cells which express lower amounts of Tim4. Because of this ambiguity we analyzed both Clec4f-tdTomatoPosTim4Pos and Clec4f-tdTomatoPosTim4Neg cells from KC-Nr1h3−/− mice. Genetic ablation of Nr1h3 in both populations was confirmed by loss of specific exons of Nr1h3 gene (Figure S3B).
Comparison of the transcriptomes of Tim4Neg Nr1h3−/− KCs to control KCs revealed more than 250 DE genes (FC > 2, p-adj < 0.05, Figure 3A, Table S2). Taking the signature gene set defined by Lavin et al (Lavin et al., 2014), 22 KC-specific genes were among the 164 transcripts down-regulated in the KC-specific Nr1h3−/− cells. These down-regulated KC genes included Clec4f, Cd5l, Kcna2, Arg2 and Il18bp (Figure 3B). A similar pattern was observed in the Tim4Pos Nr1h3−/− KCs, although DE genes were less than observed for Tim4Neg cells (Figure S3C). Tim4Neg Nr1h3−/− KCs expressed more Ccr1 and Cx3cr1 than Tim4Pos Nr1h3−/− KCs (Figure S3D). These genes were highly expressed in monocytes, suggesting some of the Tim4Neg cells may be monocyte-derived cells with relatively brief tissue residence. In contrast, deletion of LXRα from BMDMs had almost no effect on basal gene expression (Figure S3E).
Figure 3.

LXRα is a KC LDTF
A. Scatter plot of mRNA expression in Control KCs vs Tim4Neg Nr1h3−/− KCs. Data are from one experiment with n = 2 per group. DE genes are colored (blue: down-regulated; purple: up-regulated in Nr1h3−/− KC). KC-specific genes are color-coded in red.
B. Bar plots for expression of the indicated genes in control or Tim4Neg Nr1h3−/− KCs. Data are from one experiment with n = 2 per group. The significance symbols represent the p-adj from DESeq2 comparing Nr1h3−/− KC to Control KC. ***p-adj < 0.001.
C. Scatter plot of IDR-defined ATAC-seq peaks in Control KCs vs. Tim4Neg Nr1h3−/− KCs. Significantly-changed ATAC-peaks are color-coded (blue: reduced; purple: gained in Nr1h3−/− KC). KC-specific enhancers identified in Figure S2B are color-coded in red.
D. Scatter plot of IDR-defined LXR ChIP-seq peaks in KCs against BMDMs. Data are from two experiments with n = 42 per group. Differential LXR-ChIP peaks (p-adj < 0.05 & FC > 2) are colored (Blue: KC-specific; Orange: BMDM-specific).
E. De novo motif enrichment analysis of KC-specific LXR peaks using a GC-matched genomic background
F. Overlaps of total and KC-specific LXR ChIP-seq peaks with lost ATAC-Seq peaks in Tim4Neg Nr1h3−/− KCs
G. Genome browser tracks of LXR ChIP-seq peaks in BMDMs and KCs aligned with ATAC-seq peaks in control and Nr1h3−/− KCs at the indicated loci. Yellow shading: common ATAC-seq peaks and LXR binding sites in BMDMs and KCs. Blue shading: KC-specific LXR binding sites associated with loss of ATAC peaks in Tim4Neg Nr1h3−/− KCs. See also Figure S3.
To investigate the role of LXRα in establishing the KC regulatory landscape, we performed ATAC-seq analysis of Nr1h3−/− KCs. Comparison of Tim4Neg Nr1h3−/− KCs to control KCs indicated more than 3700 differential ATAC-seq peaks, with loss of >2900 peaks in Tim4Neg Nr1h3−/− KCs (Figure 3C). More than half of the lost peaks correspond to KC-specific enhancers (from Figure S2B). Comparison of Tim4Pos Nr1h3−/− KCs to control KCs indicated ~800 differential ATAC-seq peaks, with loss of 725 peaks in Tim4Pos Nr1h3−/− KCs (Figure S3F). Again, more than half of the lost peaks correspond to KC specific enhancers.
The marked alterations in ATAC-seq peaks in Nr1h3−/− KCs suggested a direct role of LXRα in selection and maintenance of the KC-specific enhancer landscape. To generate a genome-wide profile of LXR binding in KCs, we took advantage of Clec4f-cre-tdTomato mice in which KC nuclei are specifically marked by tdTomato. This enabled fluorescence-activated sorting of crosslinked tdTomatoPos nuclei from total liver nuclei that were rapidly isolated from intact liver tissue (Figure S3G). This procedure eliminated the need for tissue digestion and resulted in improved ChIP efficiency as compared to experiments using sorted cells. Using this approach, we identified more than 22,000 reproducible LXR binding sites in KCs (Figure 3D). As current ChIP-protocols do not yet allow discrimination of LXRα from LXRβ, this cistrome corresponds to an aggregate of LXRα and LXRβ binding sites. Comparison of these sites to LXR binding sites in BMDMs indicated that a large fraction was specific to KCs (Figure 3D). Motif analysis of KC-specific LXR peaks indicated enrichment for the LXR binding site itself and co-enrichment for motifs for general macrophage LDTFs PU.1 and C/EBP. In addition, motifs for IRFs, TFEs, and MAF factors were all highly enriched (Figure 3E), suggesting a role for collaborative interactions between LXRs and these factors for establishing KC enhancers. Consistent with this possibility, intersecting LXR binding sites with KC ATAC-seq peaks indicated that LXR binding sites were associated with approximately half of ATAC-seq peaks that were lost in Tim4Neg Nr1h3−/− cells, including ATAC-seq peaks that are KC-specific (Figure 3F). A similar pattern was observed in comparison to Tim4Pos Nr1h3−/− KC ATAC-seq peaks (Figure S3H). Examples are provided for KC-specific LXR binding sites in the vicinities of the Cd5l, Kcna2 and Il18bp genes at which corresponding ATAC-seq peaks were lost in Tim4Pos or Tim4Neg Nr1h3−/− KCs (Blue shading in Figure 3G). In contrast, ATAC-seq peaks associated with LXR binding sites in the vicinity of the Abca1 locus were not lost in Nr1h3−/− KCs (yellow shading in Figure 3G). Of interest, RBPJ and SMAD binding motifs were enriched in the intersection of lost ATAC-seq peaks in the Tim4Pos Nr1h3−/− and KC-specific LXR binding sites (Figure S3I), suggesting co-occupancy of LXRs and these TFs at these locations. Collectively, these results provide evidence that LXRα functions as a KC LDTF by driving the selection and function of enhancers that regulate KC-specific gene expression.
SMAD4 regulates LXRα expression and KC identity
Having established rapid activation of enhancer landscapes associated with LXRα and other putative KC LDTFs in RLMs, we next sought to identify liver-derived signals responsible for these effects. The enrichment of motifs for SMAD and RBPJ in KC-specific enhancers (Figure 2B and Figure S2C) suggested potential roles of liver-derived TGF-β family ligands and activators of Notch signaling as candidate molecules. In the hepatic sinusoids, KCs reside in contact with LSECs (Dixon et al., 2013) which can be distinguished from other endothelial cells by intermediate to high amounts of STAB2 (Geraud et al., 2017). We therefore performed RNA-seq analysis of these cells (CD146PosCD31PosF4/80NegSTAB2Hi/Int) (Figure S4A). These studies demonstrated high expression of Tgfb1 and Bmp2 (Figure 4A, Table S3). In addition, Tgfb1 was expressed in RLMs and KCs, suggesting the possibility of autoregulation. Parallel evaluation of receptors for these ligands in KCs indicated high amounts of Tgfbr1, Acvr1b, Bmpr1a, Tgfbr2, and Bmpr2 (Figure 4B, Table S3). These findings are consistent with ligand-receptor pairings predicted from paired single cell sequencing (Halpern et al., 2018).
Figure 4.

TGF-β and/or BMP signaling regulates KC identity
A. Expression of TGF-β and BMP family members in endothelial cells.
B. Expression of TGF-β and BMP receptors in KCs.
C. Scatter plot of mRNA expression in control KCs vs Tim4Neg Smad4−/− KCs. Data are from one experiment with n = 2 per group. DE genes are colored (blue: down-regulated; green: up-regulated in Smad4−/− KC) KC-specific genes are color-coded in red.
D. Bar plots for expression of the indicated genes in control or Tim4Neg Smad4−/− KCs. Data are from one experiment with n = 2 per group. The significance symbols represent the p-adj from DESeq2 comparing Smad4−/− KC to Control KC. **p-adj < 0.01; ***p-adj < 0.001.
E. Scatter plot of IDR-defined ATAC-seq peaks in control or Smad4−/− KCs. Data are from one experiment with n = 2 per group. Significantly-changed ATAC-peaks are colored (blue: reduced; green: gained in Smad4−/− KC).
F. Browser track examples of ATAC-seq in control and Smad4−/− KCs, Chip-seq for SMAD4 in KCs vs BMDMs and ChIP-seq for LXRs in KCs at the indicated loci. Blue shading: Sites of SMAD4 binding exhibiting loss of corresponding ATAC-seq peak in Smad4−/− KCs.
G. Scatter plot of IDR-defined SMAD4 ChIP-seq peaks in KCs vs. BMDMs. Data are from two experiments with n = 2 per group. Differential LXR-ChIP peaks (p-adj < 0.05 & FC > 2) are colored (Blue: KC-specific; Orange: BMDM-specific).
H. De novo motif enrichment analysis of KC-specific SMAD4 ChIP peaks using a GC-matched genomic background.
I. Overlap of LXR and SMAD4 binding sites in KCs.
J. Overlap of SMAD4 ChIP-seq peaks in KCs and KC-specific SMAD4 peaks with lost ATAC-seq peaks in Smad4−/− KCs. See also Figure S4.
To investigate the potential for LSECs to communicate with KCs via TGF-β family signaling, we crossed Clec4f-cre-tdTomato mice with floxed Smad4 mice to generate a KC-specific deletion of Smad4. As SMAD4 functions as a co-SMAD required for transcriptional responses to both TGF-β and BMP signaling pathways (Meng et al., 2013), this strategy enabled assessment of their combinatorial importance at SMAD-dependent regulatory elements. Clec4f-tdTomato expression was substantially reduced in Smad4−/− KCs compared to KCs in Clec4f-Cre-tdTomato mice, suggesting the role of SMAD4 in Clec4f gene regulation and/or impaired survival of Clec4fPos KCs after Smad4 deletion (Figure S4C). However, efficient Smad4 deletion in both Clec4f-tdTomato-positive and negative populations were confirmed by qPCR targeting a floxed exon 8 (Figure S4B). Flow cytometry of Smad4−/− KCs also indicated that a subset of mice exhibited a substantial reduction of Tim4 expression, similar to the effects of Nr1h3 deletion (Figure S4C, S4D). However this phenotype was variable, with Tim4Pos cells being the predominant population in most KC specific Smad4−/− mice (Figure S4C, S4D). RNA-seq analysis of Tim4Neg and Tim4Pos Smad4−/− cells indicated a quantitatively greater effect of the genetically ablated cells on the Tim4Neg population (Figure 4C, S4E, Table S4), with ~240 DE genes in Tim4Neg Smad4−/− KCs compared to ~120 DE genes in Tim4Pos Smad4−/− KCs (FC > 2, p-adj < 0.05). Significantly down-regulated genes included Clec4f, Id3 and Nr1h3 (Figure 4D). As in the case of Nr1h3 deletion, DE genes between Tim4Neg and Tim4Pos Smad4−/− KCs (Figure S4F) might reflect replacement of embryonic KCs with monocyte-derived cells. Consistent with this, IPA functional analysis of DE genes in Smad4−/− KCs revealed the down-regulation of cell survival and cell viability and up-regulation of cell death and apoptosis (Figure S4G). Spic was significantly upregulated in Smad4−/− KCs. In addition, Cx3cr1, which was strongly down-regulated in RLMs following KC depletion, was upregulated in the Smad4−/− cells (Figure 4D). These results provide evidence that the TGF-β family signaling pathway is required for KC identity and that SMADs may also function to negatively regulate monocyte gene expression.
Next we performed ATAC-seq experiments comparing control and Smad4−/− KCs. In comparison to genetic ablation of Nr1h3, deletion of Smad4 had a more modest effect on open chromatin (Figure 4E). However, loss of Smad4 was associated with marked reductions in ATAC-seq peak amplitude at several of the most strongly down-regulated KC genes, exemplified for Clec4f and the leptin receptor Lepr (Blue shading, Figure 4F). To relate changes in open chromatin to SMAD4 binding, we isolated KC nuclei (Clec4f-cre-tdTomatoPos) and performed ChIP-Seq of SMAD4. These studies identified more than 15,000 reproducible SMAD4 binding sites in KCs (Figure 4G), including binding sites within putative regulatory elements of the Nr1h3 and Id3 genes (Figure S4H). Parallel ChIP-Seq experiments in BMDMs identified more than 14,000 SMAD4 binding sites. Comparison of these binding profiles indicated that substantial fractions were specific for KCs or BMDMs, respectively (Figure 4G). In addition to binding sites for the general macrophage LDTFs PU.1 and C/EBPs, and the SMAD motif itself, motif analysis also recovered motifs for LXRs and MAF among KC-specific binding sites (Figure 4H), suggesting that SMAD4 collaborates with these factors to achieve a KC specific binding pattern. Consistent with this, more than two thirds of the SMAD4 binding sites in KCs co-localized with LXR ChIP-seq peaks (Figure 4I), exemplified at the Clec4f and Lepr loci (Figure 4F). Overall, SMAD4 binding sites overlapped with 40% of the ATAC-seq peaks lost in Smad4−/− KCs (Figure 4J), suggesting direct roles in establishing a component of the KC transcriptional regulatory landscape.
Notch signaling induces expression of KC LDTFs
We next investigated the potential of TGF-β and BMP2 to activate KC LDTFs in vitro. Bone marrow (BM) cells were plated in the presence of M-CSF for three days to promote the proliferation and differentiation of macrophage progenitors. After three days, TGF-β or BMP2 were added and gene expression was evaluated by RNA-Seq one day later. Although TGF-β induced expression of known target genes, such as Tgfbi, no induction of Nr1h3 or Id3 was observed (Figure 5A, Table S6), despite high SMAD4 binding at the putative regulatory elements associated with these genes (Figure S4H). Similarly, no induction of these genes was observed in response to BMP2 treatment (not shown).
Figure 5.

Notch signaling activates KC LDTFs in BMDMs
A. Bar plots for expression of the indicated genes in BMDMs stimulated with or without TGF-β. Data are from one experiment with n = 2 per group. The significance symbols represent the p-adj from DESeq2 comparing TGF-β-stimulated BMDM to Control BMDM. ***p-adj < 0.001.
B. Expression of Notch ligands in endothelial cells
C. Expression of Notch receptors in KCs
D. Bar plots for expression of the indicated genes in control BMDMs or BMDMs stimulated with DLL4 alone or with DLL4 and DAPT. Data are from one experiment with n = 2 per group. The significance symbols represent the p-adj from DESeq2 comparing to Control BMDM respectively. **p-adj < 0.01; ***p-adj < 0.001.
E. MA plot of RNA-seq data comparing control BMDMs and BMDMs treated with DLL4 for 24h. DE genes are colored (purple: up-regulated; orange: down-regulated in DLL4-stimulated BMDMs) and KC-specific genes are color-coded (red: up-regulated; black: not up-regulated in DLL4-stimulated BMDMs). Data are from one experiment with n = 2 per group. Fisher’s exact test was used to determine the odds ratio between DLL4 stimulation and KC-specific genes.
F. Bar plots for expression of the indicated genes in circulating monocytes (Circ Mo), RLMs at 24 h, and BMDMs with or without DLL4 stimulation. Data are from one or two experiment with n = 2–4 per group. The significance symbols represent the p-adj from DESeq2 comparing DLL4-treated BMDM to control BMDM, and RLMs at 24 h to circulating monocytes respectively. ***p-adj < 0.001.
G. MA plot of RNA-seq data comparing control RLMs at 24 h and RLMs at 24 h treated with LY411575. DE genes are colored (orange: up-regulate; green: down-regulated in LY411575-stimulated RLMs) and genes up-regulated in RLM 24h compared to circulating monocytes are color-coded. (red: down-regulated; black: not down-regulated in LY411575-stimulated RLMs). Data are from one experiment with n = 2–3 per group. Fisher’s exact test was used to determine the odds ratio between LY411575 stimulation and genes up-regulated in RLM 24h.
H. Bar plots for expression of the indicated genes in RLMs at 24 h with or without LY411575 stimulation. Data are from one experiment with n = 2–3 per group. The significance symbols represent the p-adj from DESeq2 comparing LY411575-treated RLMs to control RLMs. *p-adj < 0.05. See also Figure S5.
Based on the enrichment of RBPJ motifs in KC specific enhancers, we next considered possible roles of Notch signaling in the induction of KC LDTFs. LSECs as defined above were found to express high amounts of the mRNA encoding the Notch ligand DLL4 (Figure 5B). Conversely, Notch1 and Notch2 were highly expressed in KCs (Figure 5C). We therefore stimulated BM progenitor cells with the Notch ligand by transferring them to tissue culture plates in which the surface was coated with DLL4. Cells were harvested for RNA-seq analysis one day later. In contrast to TGF-β, DLL4 strongly induced the expression of Nr1h3 and Spic and had lesser stimulatory effects on Tfec (Figure 5D, Table S6). Induction of these genes was blocked by co-treatment with DAPT, an inhibitor of gamma secretase, consistent with a requirement for cleavage of the Notch intracellular domain (NICD) for induction of gene expression (Figure 5D).
At a global scale, exposure of BM progenitor cells to DLL4 for 24h led to up-regulation of 259 mRNAs, 38 of which correspond to KC identity genes (Figure 5E). Of the induced genes, 105 were upregulated in RLMs 24h after KC depletion (Figure S5A). Examples of genes that were coordinately upregulated by DLL4 in BM progenitor cells and in RLMs 24h after KC depletion include Abcg3, Slc40a1, C1qa, Acp5 and P2ry13 (Figure 5F and S5B). A similar effect of DLL4 was also observed in BM monocytes, in which 90 KC identity genes were induced by DLL4 stimulation for 48 h (Figure S5C). Collectively, these findings indicate that DLL4 is capable of rapidly inducing a subset of KC LDTFs and other KC signature genes in BM progenitor cells. To obtain in vivo evidence for the involvement of Notch signaling in gene induction in RLMs, we acutely treated mice in which KCs had been depleted by DT with the gamma-secretase inhibitor LY411575. This resulted in down-regulation of 552 mRNAs in comparison to controls, 267 of which correspond to genes induced in the first 24 h in RLMs (Figure 5G). Genes suppressed by LY411575 in vivo included DLL4 activated genes in BM progenitor cells, such as Nr1h3, Spic, Tfec, Abcg3, Slc40a1, and C1qa (Figure 5H, Figure S5D). We also carried out experiments examining effects of neutralization of Notch ligands following KC ablation using antibodies targeting DLL1 and DLL4. This intervention resulted in fewer changes in gene expression, but exhibited substantial overlap with effects of LY411575 and preferentially inhibited genes upregulated in RLMs at 24h (p<0.001) and genes upregulated by DLL4 in vitro (p<0.001) (Figure S5E).
DLL4 induces KC LDTFs through a pre-existing enhancer landscape
To investigate the molecular mechanisms responsible for DLL4 activation of KC LDTFs, we performed ChIP-Seq for RBPJ in conventional BMDMs, in BM progenitor cells treated with DLL4 and in KC nuclei. We identified >60,000 reproducible RBPJ binding sites in BMDMs and DLL4-treated BM progenitor cells, whereas slightly less than 7000 reproducible binding sites were identified in KC nuclei (Figure 6A). The lower number of binding sites in KCs is consistent with lower Rbpj expression in these cells in comparison to BMDMs (not shown). This difference is also consistent with an ~75% down-regulation of Rbpj expression in monocytes following their recruitment into the liver (Figure 6B). In contrast to LXR and SMAD4, relatively few RBPJ bindings sites were identified that are specific to KCs (Figure 6A), suggesting that the impact of Notch signaling becomes progressively restricted following entry of monocytes into the KC-depleted liver. Significantly enriched motifs within the set of RBPJ peaks common to KCs and BMDMs include the binding sites recognized by the general macrophage LDTFs PU.1, AP-1, C/EBP and RUNX family members as well as binding sites for IRF and TFE factors, which are also present in KC-specific enhancers (Figure S6A).
Figure 6.

Notch signaling activates a pre-existing enhancer landscape in BM progenitor cells
A. Overlap of reproducible RBPJ ChIP-Seq peaks in KCs and BM progenitor cells
B. Bar plot for Rbpj expression in circulating monocytes (Circ Mo), RLMs, and resident KCs. Data are from one or two experiments with n = 2–4 per group. The significance symbols represent the p-adj from DESeq2 comparing to circulating monocytes respectively. ***p-adj < 0.001.
C. Scatter plot of IDR-defined distal ATAC-peaks in DLL4-treated BMDMs vs. control BMDMs. Data are from one experiment with n = 2 per group. Significantly-changed ATAC-peaks (p-adj < 0.05 & FC > 2) are colored (purple: gained; orange: reduced in DLL4-treated BMDM).
D. Scatter plot of distal ATAC-associated H3K27ac in DLL4-treated BMDMs vs control BMDMs. Data are from one experiment with n = 2 per group. Color codes indicate significant changes (p-adj < 0.05 & FC > 2) in H3K27ac.
E. Motif enrichment analysis of distal ATAC-seq peaks in DLL4-treated BMDMs that gain H3K27ac.
F. Browser tracks of ATAC-Seq, H3K27ac ChIP-seq and RBPJ ChIP-seq peaks in the vicinities of putative regulatory elements for the indicated genes (Yellow shading). Bar graphs illustrate H3K27ac normalized tag counts for the indicated genomic regions. See also Figure S6.
ATAC-Seq experiments in BM progenitor cells treated with DLL4 indicated fewer than 100 changes in open chromatin (Figure 6C), consistent with RBPJ being able to bind to its genomic targets in the presence or absence of Notch signaling (Bray, 2006). In contrast, ChIP-seq for H3K27ac identified more than 3400 sites of differential H3K27acetylation (Figure 6D). In addition to strong enrichment for the RBPJ motif, regions gaining H3K27ac following DLL4 treatment were also enriched for binding sites for IRF, MAF, TFE, SMADs and LXRs (Figure 6E), all of which are also present in KC-specific enhancers (Figure S2C). Examples of the relationships of RBPJ binding sites to putative regulatory elements for Nr1h3 and Spic exhibiting rapid increases in H3K27ac are illustrated in Figure 6F. Of interest, genomic regions exhibiting loss of H3K27ac were not enriched for RBPJ binding sites or motifs for KC LDTFs. Instead, these regions were enriched for AP-1, KLF and HLF motifs (Figure S6B) that are present in H3K27ac regions of circulating monocytes, but not in KCs (Figure S2F). Collectively, these findings provide evidence that Notch signaling induced by DLL4 directly activates a preexisting enhancer landscape to induce expression of Nr1h3, Spic and other genes that promote KC differentiation and indirectly suppresses activities of TFs that specify monocytic gene expression.
Combinations of liver factors induce KC-specific genes
Comparisons of the genome wide binding patterns of LXRs, SMAD4 and RBPJ indicated substantial overlap of these factors within KCs (Figure S7A), suggesting the potential for combinatorial interactions of the signaling pathways that regulate their activities. Examples of overlapping and distinct binding patterns in the vicinities of genes that were highly upregulated in RLMs are illustrated for Abca1 and C1qa in Figure S7B. To investigate functional consequences of simultaneous activation of SMAD and RBPJ, control and Smad4−/− BM progenitor cells were treated with TGF-β and DLL4 alone or in combination and RNA-seq analysis was performed 24h later (Figure 7A). DLL4 alone induced expression of Nr1h3 and Clec4f, whereas TGF-β alone had almost no effect. The combination of DLL4 and TGF-β had little additional effect on Nr1h3 expression, but increased expression of Clec4f. Notably, the TGF-β effect on Nr1h3 and Clec4f expression was reduced in Smad4−/− cells (Figure 7A). At a global level, the combination of TGF-β and DLL4 promoted a transition in the gene expression profile of BM progenitor cells towards that of KCs to a greater extent than TGF-β or DLL4 alone (Figure 7B).
Figure 7.

Combinatorial interactions of liver environmental signals
A. Effect of the combination of DLL4 and TGF-β on expression of indicated genes in control and Smad4−/− BMDMs. Data are from one experiment with n = 2 per group. The significance symbols represent the p-adj from DESeq2 comparing to control samples without stimulation in control and Smad4−/− respectively. ***p-adj < 0.001.
B. PCA of overall gene expression in BM progenitor cells stimulated with or without DLL4 and/or TGF-β, circulating monocytes (Circ Mo), RLMs, and resident KCs.
C. Bar plots for expression of the indicated genes in circulating monocytes (Circ Mo), RLMs, and resident KCs. Data are from one or two experiments with n = 2–4 per group. The significance markers represent the p-adj from DESeq2 comparing to circulating monocytes respectively. ***p-adj < 0.001.
D. Quantification of desmosterol, 24-, 25- and 27-OHC and 24,25-EC in mouse liver (left) and primary hepatocytes (right). Data are from two experiments with n = 4–6 per group.
E. Effects of the indicated combinations of DLL4, TGF-β and/or DMHCA on Abca1, Il18bp and Arg2 expression in BMDMs. Data are from two experiments with n = 2 per group. See also Figure S7.
In addition to SMAD and RBPJ, LXRα is also a signal-dependent TF that regulates gene expression in response to changes in sterol and oxysterol concentrations (Calkin and Tontonoz, 2012; Spann and Glass, 2013). Examination of genes involved in cholesterol homeostasis indicated marked upregulation of LXR target genes such as Abca1 (Figure 7C) and Scd1 (Figure S7C), and coordinate down-regulation of genes in the SREBP pathway, such as the genes encoding the LDL receptor (Ldlr) (Figure 7C) and HMG CoA reductase (Hmgcr) (Figure S7C), in RLMs. This pattern of gene expression could represent a cell-autonomous response to elevated intracellular cholesterol (Brown and Goldstein, 2009), which could occur following phagocytosis of apoptotic or necrotic KCs following DT ablation. However, an alternative possibility is that KCs and RLMs sense one or more liver-derived molecules that coordinately regulate the LXR and SREBP pathways. Naturally occurring molecules that function as LXR agonists and also suppress the SREBP pathway by binding to INSIGs or SCAP include oxysterols and desmosterol (Brown and Goldstein, 2009; Peet et al., 1998a; Radhakrishnan et al., 2004; Yang et al., 2006). To investigate the potential of these species to function as endogenous regulators of the LXR and SREBP pathways, the abundance of relevant oxysterols and unesterified sterols was quantified by targeted mass spectroscopy using deuterated internal standards. These studies identified desmosterol as the most abundant species in intact liver, being present at a concentration of ~5uM (Figure 7D). This concentration is close to the EC50 for coordinate LXR activation and SREBP repression (Muse et al., 2018; Spann et al., 2012; Yang et al., 2006) and is about ~10-times higher than its plasma concentration (Muse et al., 2018). Desmosterol thus represents a potential hepatocyte-derived environmental signal that could function with TGF-β family ligands and DLL4 to promote the KC phenotype.
Although exogenous desmosterol acutely activates LXR and suppresses SREBP in macrophages in vitro, it is also directly converted to cholesterol by the enzyme Dhcr24, limiting its duration of action. To reduce metabolic clearance as a confounding factor in time course experiments exceeding 12–16h, we used the synthetic LXR agonist DMHCA (Kratzer et al., 2009), which is a structural and functional mimetic of desmosterol (Muse et al., 2018) that cannot be converted to cholesterol. BM progenitor cells were treated with DLL4, DMHCA, TGF-β+DLL4 or TGF-β+DLL4+DMHCA for 24h or 72h followed by RNA-seq analysis (Figure 7E). To test the effect of desmosterol, we also stimulated BM monocytes with DLL4, desmosterol, TGF-β+DLL4 or TGF-β+DLL4+desmosterol for 48h by adding desmosterol to the media after the first 24 h (Figure S7D). These experiments revealed two classes of LXR target genes. The first class largely consisted of prototypic LXR target genes, such as Abca1, which were rapidly induced by DMHCA or desmosterol and were only modestly affected by DLL4 and/or TGF-β (Figure 7E, Figure S7D, S7E, Table S6). Of note, some of these genes, such as Abca1, were not affected by deletion of Nr1h3 (Table S2), suggesting compensation by LXRβ. LXR binding sites for these genes in KCs were largely at pre-existing regions of open chromatin (e.g., Figure 3G). The second class of target genes, exemplified by Il18bp and Arg2, consisted of genes that were induced late following recruitment into liver (Figure S7E) and were defined as LXRα target genes based on reduced expression in KC-specific Nr1h3−/− KCs (Figure 3B). These genes exhibited progressive activation by DLL4 and TGF-β from 24h to 72h and were less dependent on DMHCA for expression (Figure 7E). LXR binding sites in the vicinity of these genes were largely KC-specific and associated with induced enhancer like elements (Figure S7F). These results support a model in which LXRα induction of KC-specific genes involves collaborative interactions with TGF-β and Notch signaling pathways.
Discussion
The transcriptional mechanisms that enable myeloid progenitor cells to acquire the distinct functional characteristics of tissue resident macrophages are largely unknown. Here, we exploited the KC depletion to characterize the transcriptomes and epigenetic landscapes of RLMs as a function of time. While this method represents an artificial system, many aspects are similar to those observed during EMP to KC differentiation during development (Mass et al., 2016). Receptors for DLL4 and TGF-β family ligands were highly expressed in EMP cells and key KC LDTFs (including LXRα, MAF, and TFEC) were highly expressed in the fetal liver macrophages at the earliest time evaluated (E10.25) in comparison to EMP cells.
Our results provide evidence for a combinatorial and sequential model of differentiation of myeloid progenitor to KC involving the LXR, Notch and TGF-β family signaling pathways. A critical initial step is provided by the interaction of recruited monocytes with DLL4 on LSECs. DLL4 activation of Notch signaling converts RBPJ from a repressor to an activator by NICD-mediated exchange of co-repressor complexes with histone deacetylase activity for co-activator complexes with histone acetyl transferases (Borggrefe and Oswald, 2009). KC LDTFs including Nr1h3 and Spic were rapidly induced both in RLMs and DLL4-treated BMDMs. In addition, DLL4 treatment of BM progenitor cells resulted in down-regulation of monocyte specific genes. Suppressive effects of DLL4 on H3K27ac were not associated with local enrichment of RBPJ motifs, but associated with motifs associated with monocyte-specific enhancers, including AP-1 motifs, consistent with prior evidence that Notch signaling inhibits AP-1 activity in a manner involving the NICD (Chu et al., 2002; Nowell and Radtke, 2017). Thus, Notch signaling appears to simultaneously initiate a program of KC gene expression and suppress the expression of monocyte-specific genes.
Evidence that TGF-β family signaling is required for KC identity was provided by enrichment of SMAD motifs in RLM- and KC-specific enhancers and transcriptomic and epigenetic consequences of KC-specific deletion of Smad4. LSECs express high amounts of Tgfb1, and corresponding receptors were highly expressed on KCs. SMAD4 binding overlapped extensively with the KC-specific LXR cistrome, consistent with genomic location of SMADs being influenced by specific combinations of LDTFs (Mullen et al., 2011). In addition to functioning as a signal-dependent TF that regulates preexisting enhancers, our findings also indicate essential roles of SMAD4 in maintaining the open chromatin environment of a subset of KC identity genes. TGF-β alone was unable to induce Nr1h3 or many other KC-specific genes in BMDMs, but could stimulate expression of these genes in the presence of DLL4, suggesting that Notch signaling is permissive for a KC-specific differentiation role of the TGF-β pathway.
LXRα has recently emerged as a putative KC LDTF (Mass et al., 2016) (Lavin et al., 2014) (Scott et al., 2016). Here we established that LXRα was required for shaping the KC enhancer landscape. Previous studies of BMDMs indicate that genetic deletion of LXRs had no impact on binding of the macrophage LDTF PU.1, whereas PU.1 was required for the binding of LXRs at a subset of their genomic locations (Heinz et al., 2010). These observations suggested a hierarchical relationship in which PU.1 and other macrophage LDTFs primed cis-regulatory elements, providing access to signal-dependent factors. Here, deletion of Nr1h3 resulted in reduced chromatin access at a large fraction of KC specific enhancers. Whether this result is simply a consequence of higher LXRα expression or reflects an acquired chromatin remodeling function in KCs remains to be established.
These studies also provide evidence that LXR activity is regulated in an environment-dependent manner. The very rapid upregulation of LXR target genes and down-regulation of SREBP target genes in recruited cells strongly implies the presence of endogenous sterol and/or oxysterol regulators of these pathways. Based on lipidomic analysis, desmosterol is the most abundant lipid species that has the potential to both induce LXR target genes and repress the SREBP pathway. We recently reported the unexpected finding that while desmosterol and DMHCA effectively regulate both the LXR and SREBP pathways in macrophages, they have almost no activity in hepatocytes (Muse et al., 2018). Mechanisms responsible for cell-specific discrimination of desmosterol remain to be established. Notably, desmosterol concentration is highly dependent on the expression of Dhcr24 (Avigan et al., 1960; Muse et al., 2018), which is a direct target gene of SREBP (Spann et al., 2012). We speculate that desmosterol could function as a signal to other cell types in the liver reporting on the status of the SREBP pathway in hepatocytes.
The present studies focused on a subset of dynamic regulatory elements to infer roles of Notch, TGF-β family receptors, and LXR signaling pathways as mediators of the effects of liver environmental factors on KC differentiation and homeostasis. Several additional pathways are suggested by our studies, including signaling systems that converge on NFκB, IRF, and MAF TFs as well as additional nuclear receptors. For example, the enrichment of NFκB motifs in KC-specific enhancer may reflect exposure of KCs to gut-derived LPS present in portal blood. In conclusion, the ascertainment of transcriptomes and epigenetic landscapes of repopulating tissue macrophages provides a framework for understanding how a common macrophage progenitor cell acquires tissue specific phenotypes.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher K. Glass (ckg@ucsd.edu).
Mice
All animal procedures were approved by the University of California San Diego Institutional Animal Care and Use Committee in accordance with University of California San Diego research guidelines for the care and use of laboratory animals. The following mice were used in this study; C57BL/6J (The Jackson Laboratory, Stock No.000664), Clec4f-cre-tdTomato (generated by Glass Lab and transgenic core facility, University of California, San Diego) (The Jackson Laboratory, Stock No.033296), Rosa26-Lox-Stop-Lox-DTR mice (Buch et al., 2005)(The Jackson Laboratory, Stock No.007900), Rosa26-Lox-Stop-Lox-ZsGreen mice (The Jackson Laboratory, Stock No.007906), RosanT-nG mouse (Prigge et al., 2013)(The Jackson Laboratory, Stock No.023035), :Lyz2-crei(Clausen et al., 1999)(The Jackson Laboratory, Stock No.004781), Nr1h3fl/fl (developed by Chambon Lab), Nr1h3−/− (Peet et al., 1998b)(The Jackson Laboratory, Stock No. 013763), and Smad4fl/fl (Yang et al., 2002)(The Jackson Laboratory, Stock No.017462). Mice were used between 8 and 12 weeks of age. For the gamma secretase inhibitor treatment, female 8 weeks old Rosa26-Lox-Stop-Lox-DTR;Clecf4-cre-tdTomato+/− mice were injected with 10 mg/kg LY411575 (Sigma) intraperitoneally daily for 2 days. For neutralization of DLL1 and DLL4, male 8 weeks old Rosa26-Lox-Stop-Lox-DTR;Clec4-cre-tdTomato+/− mice were injected with 2 mg control IgG or 1 mg each of DLL1 and DLL4 antibodies (BioXcell) 2 days before sorting RLMs.
Cloning of the Clec4f targeting vector
The Clec4f targeting vector was cloned by modifying a pUC19 vector (NEB) using the In-Fusion HD Cloning Kit (Clontech). First, IRES-Cre-T2A was amplified from a pLV[Exp]-SYN1>mCherry:IRES:Cre:T2A:EGFP vector (VectorBuilder) with the forward primer IRES_Cre_T2A_F (5’-ATGCCTGCAGGTCGACCCCCTCTCCCTCCCCCCCCCCTAA-3’) and the reverse primer IRES_Cre_T2A_R (5’-GTGAATTCGAGCTCGGTACCGGGGCCGGGATTTTCCTCCACGTC-3’) and inserted into the pUC19 at the SalI/KpnI site. Second, a nuclear localization signal-tagged tdTomato (tdTomato-NLS) was amplified from the genomic DNA of a RosanT-nG mouse (The Jackson Laboratory, Stock No.023035) (Prigge et al., 2013) with the forward primer tdTomato_F (5’-CTAGAGGATCCCCGGGTACCATGGTGAGCAAGGGAGAGGAGGTC-3’) and the reverse primer tdTomato_R (5’-GTGAATTCGAGCTCGTTATGAACGTCTTCGTCGCCTATC-3’) and inserted into the KpnI site of pUC19-IRES-Cre-T2A. Third, a 2.5 kb left homology arm (LHA) was amplified from genomic DNA of a C57BL/6J mouse (The Jackson Laboratory) with the forward primer LHA_F (5’-ATGCCTGCAGGTCGACATGCTGAGAATCCCTGCTTCGCACAC-3’) and the reverse primer LHA_R (5’-ATCCTCTAGAGTCGACGACAGGAAGCCCTGAAGTGAGATCAAGG-3’) and inserted into the SalI site of the pUC19-IRES-Cre-T2A-tdTomato-NLS vector. Finally, a 2.5 kb right homology arm (RHA) was amplified from genomic DNA of a C57BL/6J mouse (The Jackson Laboratory) with the forward primer RHA_F (5’-TACCGAGCTCGAATTCCTGTATTGCCCTGTCGTTCTGCTATTT-3’) and the reverse primer RHA_R (5’-GACGGCCAGTGAATTCGCTGCGGCTGAGGACCATTCTGAGTT-3’) and inserted into the EcoRI site of the pUC19-LHA-IRES-Cre-T2A-tdTomato-NLS vector to make the Clec4f targeting vector, pUC19-LHA-IRES-Cre-T2A-tdTomato-NLS-RHA.
Generation of Clec4f-Cre-tdTomato mouse
30 ng/μl Cas9 protein (IDT), 0.61 pmol/μl Clec4f-crRNA (protospacer, ACGACAGGGCAATACAGGAC) (IDT), 0.61 pmol/μl tracrRNA (IDT), and 12ng/μl Clec4f targeting vector in IDTE pH7.5 (IDT) were injected into pronuclei of C57BL/6J one cell stage zygotes (Aida et al., 2015). Genetically-targeted mice were screened by PCR with KOD Xtreme Hot Start DNA polymerase (EMD Millipore) using three different pairs of primers, internal forward primer (Clec4f_IF, 5’-ACTGGAGACATAGGAACGGAGAGCG-3’) and internal reverse primer (Clec4f_IR, 5’-GTGCTGAGGGGACTCCAATGCAG-3’), left forward primer (Clec4f_LF, 5’-GCCAGGTCCAGTTTCCTGGTGATG-3’) and left reverse primer (Clec4f_LR, 5’-TCCAAGCGGCTTCGGCCAGTAAC-3’), and right forward primer (Clec4f_RF, 5’-GCCAGATAGGCGACGAAGACGTTCA-3’) and right reverse primer (Clec4f_RR, 5’-AGCCATTCCTGATACCTGGGGCC-3’). Clec4f_IF + Clec4f_IR amplified a 277-bp band from the WT allele and a 3652-bp band from the Clec4f-Cre-tdTomato allele. Clec4f_LF + Clec4f_LR amplified a 2668-bp band while Clec4f_RF + Clec4f_RR amplified a 2584-bp band from the Clec4f-Cre-tdTomato allele. Clec4f-Cre-tdTomato+/− mice were then crossed to C57BL/6J WT mice for at least for three generations.
Diphtheria toxin (DT)-mediated depletion of Kupffer cells
Mice expressing diphtheria toxin receptors (DTR) in Kupffer cells were produced by crossing Clec4f-cre-tdTomato mice to Rosa26-Lox-Stop-Lox-DTR mice (Rosa26iDTR, The Jackson Laboratory, Stock No.007900) (Buch et al., 2005). Rosa26-Lox-Stop-Lox-DTR +/− Clec4f-Cre-tdTomato +/− mice were depleted of Kupffer cells by a single intraperitoneal administration of 200ng DT (Sigma).
Flow cytometry to assess liver inflammatory response
Livers were perfused with 10ml PBS and then mechanically minced into small pieces and digested in PBS containing Collagenase D (1mg/ml), Dispase (2.4mg/ml) and DNAse (0.2mg/ml) for 30min at 37°C. Single cells su spensions were filtered through 100mm strainers, resuspended in 50 μL of FACS buffer (1X PBS, 0.5%BSA, 2mM EDTA) containing anti-mouse CD16/32 (1:100), 5% normal rat, 5% normal mouse and 5% normal rabbit serum for 15min at 4°C. Samples were stained with CD45-APC-Cy7(1:100), CD11b-PE-Cy7(1:200), F4/80-eF450(1:100), I-A/I-E-AF700(1:200), CD11c-BV605(1:200), Tim4-AF647(1:200), Ly6C-AF488(1:200), Ly6G-PE/Dazzle594(1:200), CD3 BV711(1:200), CD19 BV711(1:200), and Nkp46 BV711(1:200) antibodies mixes for 30min at 4°C. Data were acquired on an BD LSR Fortessa with Diva software and were analyzed by Flowjo software. The number of cells per gram of tissue was determined using a cell counter (GUAVA easyCyte HT).
Whole mount imaging
For whole mount immunofluorescence imaging, liver pieces were fixed in 4% paraformaldehyde (PFA) diluted in PBS for 30 min at room temperature with agitation. Samples were permeabilized with 1X PBS containing 0.3% TritonX-100, 4% BSA for 1 hour at room temperature and incubated with I-A/I-E-AF488(1:100), F4/80-eF450(1:100), and Tim4-AF647(1:100) antibodies mix for 2 hours at room temperature. Data were acquired using LSM880 Zeiss microscope for 30 to 40 Z-stacks with 1.4μm consecutive intervals and were analyzed using Imaris software.
Gene expression analysis by quantitative RT-PCR
Total RNA was isolated from sorted Kupffer cells with the use of a Direct-zol RNA MicroPrep Kit (Zymo Research). For quantitative RT-PCR analysis of Smad4, cDNA was synthesized from the total RNA extracted from sorted Kupffer cells with the use of a SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) and PCR was then performed with the use of a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific), KAPA SYBR FAST qPCR Master Mix (2X) (Kapa Biosystems), and primers Smad4_qF1 (5’-CAGCCTCCCATTTCCAATCATC-3’) and Smad4_qR1 (5’-CGAAGGATCCACATAGCCATCC-3’). Relative mRNA abundance was calculated by the standard curve method and was normalized by the corresponding amount of 18s rRNA using primers 18s_qF (5’-AATTCCCAGTAAGTGCGGGTCA-3’) and 18s_qR (5’-GATCCGAGGGCCTCACTAAACC-3’).
Sorting liver non-parenchymal cells
Mice were humanely euthanized by exposure to CO2 and whole liver pieces saved and liver non-parenchymal cells processed for fluorescence-activated cell sorting of Kupffer cells, liver recruited monocytes, repopulating liver macrophages (RLMs), and liver sinusoidal endothelial cells (LSECs) with modifications from published methodology (Mederacke et al., 2015; Seki et al., 2007). Livers were perfused in a retrograde fashion for 3 minutes at a rate of 5 ml/min through the inferior vena cava with HBSS without Ca or Mg (Gibco) supplemented with 1 μM flavopiridol (Sigma), 0.5 mM EGTA, 0.5 mM EDTA, and 20 mM HEPES (Gibco). Perfusions were then switched to 40 ml of digestion buffer comprised of HBSS with Ca and Mg (Gibco) supplemented with 0.033 mg/ml of Liberase TM (Roche), 20 μg/ml DNase I, 1 μM flavopiridol, and 20 mM HEPES. Livers were then excised, minced, and digested for an additional 20 minutes in vitro at 37°C with gentle rotation in 20 ml of fresh digestion buffer. After tissue digestion, cells were passed through a 70 μm cell strainer, and hepatocytes were removed by two low-speed centrifugation steps at 50 X G for 2 minutes. Non-parenchymal cells in the supernatant were further separated from debris by pelleting for 15 minutes at 600 X G in 50 ml of 20% isotonic Percoll (Sigma) at room temperature. Next, cells were washed from Percoll containing buffer and resuspended in 10 ml 28% OptiPrep (Sigma) and carefully underlaid beneath 3 ml of wash buffer. The resulting gradient was centrifuged at 1,400 X G for 25 minutes at 4°C with no brake and cells enriched at the interface were saved and subjected to RBC lysis (eBioscience). Enriched non-parenchymal cells were washed, suspended in PBS, then stained for 10 minutes with Zombie NIR (BioLegend) and purified anti-CD16/32 (93, BioLegend) to label dead cells and block Fc receptors. After that, cells were stained in wash buffer for an additional 20 minutes with the antibodies listed in the Key Resource Table. Stained cells were washed twice and strained through a 30 μm strainer, then subjected to cell sorting using a Beckman Coulter MoFlo Astrios EQ configured with 355 nm, 405 nm, 488 nm, 561 nm, and 642 nm lasers. Each cell population was hierarchically gated using Beckman Coulter Summit software. Kupffer cells were defined as CD45PosF4/80HiCD11bIntCD115PosClec4f-tdTomatoPosLy6CLoCD31NegCD146Neg. Liver recruited monocytes were defined as CD45PosF4/80LoCD11bHiCD115PosClec4f-tdTomatoNegLy6CHiCD31NegCD146Neg while RLMs at 24 hours, 48 hours, and 72 hours were defined as CD45PosF4/80Int~HiCD11bIntCD115PosClec4f-tdTomatoNegLy6CInt~LoCD31NegCD146Neg. RLMs at day 7 and day 14 were defined as CD45PosF4/80HiCD11bIntTim4NegClec4f-tdTomatoPosCD146Neg. Tim4Neg or Tim4Pos Kupffer cells were sorted as CD45PosF4/80HiCD11bIntTim4Neg or PosClec4f-tdTomatoNeg or PosCD146Neg. LSECs were defined as CD45NegF4/80NegCD11bNegCD31PosCD146PosSTAB2Int~Hi. Each cell population was further restricted to single particles by comparing height and area side scatter pulses, and dead cells were excluded by detecting the integration of the live/dead dye (Zombie NIR).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LXRα/β | Santa Cruz Biotechnology | Cat#sc-1000X; RRID: AB_632067 |
| LXRβ | Santa Cruz Biotechnology | Cat#sc-133221X RRID: AB_2154783 |
| LXRα/β | Santa Cruz Biotechnology | Cat#sc-271064X RRID: AB_10611071 |
| SMAD4 | Cell Signaling Technology | Cat#46535 RRID: AB_2736998 |
| SMAD4 | Cell Signaling Technology | Cat#38454 RRID: AB_2728776 |
| RBPJ | Abcam | Cat#ab25949 RRID: AB_778155 |
| CD115_PE | BioLegend | Cat#135506; RRID: AB_1937253 |
| CD115_PerCP-eFluor710 (clone AFS998) | eBioscience | Cat#46-1152-82; RRID: none |
| CD11b_BUV395 (clone m1/70) | BD Biosciences | Cat#565976; RRID: none |
| CD11b_PE/Cy7 | BD Biosciences | Cat#552850; RRID: AB_394491 |
| CD11b_PE/Cy7 | BioLegend | Cat#101216; RRID: AB_312799 |
| CD11c_BV605 | BioLegend | Cat#117334; RRID: AB_2562415 |
| CD14_PE | BioLegend | Cat#301806; RRID: AB_314188 |
| CD146_BUV395 (clone ME-9F1) | BD Biosciences | Cat#740330; RRID: none |
| CD16/32 | BioLegend | Cat#101302; RRID: AB_312801 |
| CD16/32 | BD Biosciences | Cat#553142; RRID: AB_394657 |
| CD19_BUV737 (clone 1D3) | BioLegend | Cat#564296; RRID: none |
| CD19_BV711 | BioLegend | Cat#563157; RRID: AB_2738035 |
| CD31_BUV737 (clone MEC 13.3) | BD Biosciences | Cat#565097; RRID: none |
| CD31_BV786 (clone MEC 13.3) | BD Biosciences | Cat#740870; RRID: none |
| CD45_Alexa488 | BioLegend | Cat#103122; RRID: AB_493531 |
| CD45_Alexa488 | BioLegend | Cat#304017; RRID: AB_389314 |
| CD45_Alexa647 | BioLegend | Cat#103124; RRID: AB_493533 |
| CD45_APC/Cy7 | BioLegend | Cat#103116; RRID: AB_312981 |
| CD45_BB515 | BD Biosciences | Cat#564590; RRID: none |
| CD90.2_BV785 | BioLegend | Cat#105331; RRID: AB_2562900 |
| CD3_BV711 | BioLegend | Cat#563123; RRID: AB_2687954 |
| F4/80_BV421 | BioLegend | Cat#123132; RRID: AB_11203717 |
| F4/80_PE | BioLegend | Cat#123110; RRID: AB_893486 |
| F4/80_eFluor450 | Thermo Fisher Scientific | Cat#48480182; RRID: AB_1548747 |
| Ly6C_APC/Cy7 | BioLegend | Cat#128026; RRID: AB_10640120 |
| Ly6C_BV785 | BioLegend | Cat#128041; RRID: AB_2565852 |
| Ly6C_Alexa488 | BioLegend | Cat#128022; RRID: AB_10639728 |
| Ly6G_BV650 | BioLegend | Cat#127641; RRID: AB_2565881 |
| Ly6G_FITC | BioLegend | Cat#127606; RRID: AB_1236494 |
| Ly6G_PE/Dazzle594 | BioLegend | Cat#127648; RRID: AB_2566319 |
| STAB2_Alexa488 (clone D317-A48)) | MBL | Cat#D317-A48; RRID: none |
| Tim4_Alexa647 | BioLegend | Cat#130008; RRID: AB_2271648 |
| Tim4_PE | BioLegend | Cat#130006; RRID: AB_2201843 |
| I-A/I-E_Alexa700 | BioLegend | Cat#107622; RRID: AB_493727 |
| I-A/I-E_Alexa488 | BioLegend | Cat#107616; RRID: AB_493523 |
| Nkp46_BV711 | BioLegend | Cat#137621; RRID: AB_2563289 |
| InVivoMAb anti-mouse DLL1 | BioXcell | Cat#BE0155; RRID: AB_10950546 |
| InVivoMAb anti-mouse DLL4 | BioXcell | Cat#BE0127; RRID: AB_10950366 |
| InVivoMAb polyclonal American hamster IgG | BioXcell | Cat#BE0091; RRID: AB_1107773 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Minimum Essential Medium Eagle | Sigma | Cat#M8042 |
| FBS | Biowest | Cat#S1620 |
| Penicillin-Streptomycin-Glutamine (100X) | Thermo Fisher Scientific | Cat#10378016 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15140122 |
| Amphotericin B | Thermo Fisher Scientific | Cat#15290018 |
| mouse M-CSF | Shenandoah Biotech | Cat#200-08 |
| KOD Xtreme Hot Start DNA polymerase | Sigma | Cat#71975 |
| KAPA SYBR FAST qPCR Master mix (2X) | Kapa Biosystems | Cat#07959427001 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat#10002D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat#10004D |
| SpeedBeads magnetic carboxylate modified particles | GE Healthcare | Cat#65152105050250 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596018 |
| Formaldehyde | Thermo Fisher Scientific | Cat#BP531-500 |
| Glycine | Thermo Fisher Scientific | Cat#BP3815 |
| DSG Crosslinker | ProteoChem | Cat#c1104-100mg |
| Oligo d(T)25 Magnetic Beads | NEB | Cat#S1419S |
| DTT | Thermo Fisher Scientific | Cat#P2325 |
| SUPERase-In | Ambion | Cat#AM2696 |
| Oligo dT primer | Thermo Fisher Scientific | Cat#18418020 |
| Agencourt RNA Clean XP Beads | Beckman Coulter | Cat#A63987 |
| 10 X Blue Buffer | Enzymatics | Cat#P7050L |
| DNA polymerase I | Enzymatics | Cat#P7050L |
| Random primers | Thermo Fisher Scientific | Cat#48190011 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080044 |
| 5 X first-strand buffer | Thermo Fisher Scientific | Cat#18080044 |
| Actinomycin D | Sigma | Cat#A1410 |
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat#1081058 |
| Alt-R CRISPR-Cas9 crRNA | Integrated DNA Technologies | N/A |
| Alt-R CRISPR-Cas9 tracrRNA | Integrated DNA Technologies | Cat#1072532 |
| IDTE pH 7.5 (1X TE Solution) | Integrated DNA Technologies | Cat#11-01-02-02 |
| Diphtheria Toxin from Corynebacterium diphtheriae | Sigma | Cat#D0564 |
| Flavopiridol hydrochloride hydrate | Sigma | Cat#F3055 |
| OptiPrep Density Gradient Medium | Sigma | Cat#D1556 |
| Liberase TM Research Grade | Sigma | Cat#5401127001 |
| Recombinant human DLL4 | R&D systems | Cat#1506-D4-050 |
| Human Transforming Growth Factor β1 (hTGF-β1) | Cell Signaling Technology | Cat#8915LC |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat#M0541L |
| DAPT | Cell Signaling Technology | Cat#15020S |
| LY411575 | Sigma | Cat#SML0506-25MG |
| Collagenase, Type 1 | Worthington | Cat# LS004196 |
| Hoechst 33342 Solution (20 mM) | Thermo Fisher Scientific | Cat# 62249 |
| Collagenase D | Sigma | Cat# 11088882001 |
| Dispase | Thermo Fisher Scientific | Cat# 17105-041 |
| DNAse | Sigma | Cat# DNA25-100mg |
| Critical Commercial Assays | ||
| Direct-zol RNA MicroPrep Kit | Zymo Research | Cat#R2062 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32851 |
| In-Fusion HD Cloning plus Kit | Clontech | Cat#638909 |
| Nextera DNA Library Prep Kit | Illumina | Cat#FC-121-1030 |
| NEBNext Ultra II DNA library prep kit | NEB | Cat#E7645L |
| NEXTflex DNA Barcodes | Bioo Scientific | Cat#NOVA-514104 |
| ChIP DNA Clean and Concentrator Kit | Zymo Research | Cat#D5205 |
| EasySep Mouse Monocyte Isolation Kit | STEMCELL Technologies | Cat#19861 |
| Deposited Data | ||
| RAW and analyzed data | GEO | GSE128662 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Clec4f-cre-tdTomato | Glass Lab | This study (JAX:033296). |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: Rosa26-Lox-Stop-Lox-DTR (Rosa26iDTR) | The Jackson Laboratory | RRID: IMSR_JAX:007900 |
| Mouse: Rosa26-Lox-Stop-Lox-ZsGreen (Ai6) | The Jackson Laboratory | RRID: IMSR_JAX:007906 |
| Mouse: RosanT-nG mouse | The Jackson Laboratory | RRID: IMSR_JAX:023035 |
| Mouse: Lyz2-cre | The Jackson Laboratory | RRID: IMSR_JAX:004781 |
| Mouse: Smad4fl/fl | The Jackson Laboratory | RRID: IMSR_JAX:017462 |
| Mouse: Nr1h3−/− | The Jackson Laboratory | RRID: IMSR_JAX:013763 |
| Mouse: Nr1h3fl/fl | Chambon Lab | N/A |
| Oligonucleotides | ||
| Smad4_qF1 | CAGCCTCCCATTTCCAATCATC | |
| Smad4_qR1 | CGAAGGATCCACATAGCCATCC | |
| 18s_qF | AATTCCCAGTAAGTGCGGGTCA | |
| 18s_qR | GATCCGAGGGCCTCACTAAACC | |
| Recombinant DNA | ||
| Clec4f targeting vector | Glass Lab | This study |
| Software and Algorithms | ||
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Irreproducibility Discovery Rate (IDR) | (Li et al., 2011) | https://www.encodeproject.org/software/idr/ |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| R package: DEseq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package: pheatmap | N/A | https://www.rdocumentation.org/packages/pheatmap/versions/1.0.10/topics/pheatmap |
| pygbrowse | N/A | https://github.com/phageghost/python-genome-browser |
| Ingenuity Pathway Analysis | N/A | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| GraphPad Prism | GraphPad Software, Inc., California | |
| FlowJo | FlowJo LLC, Ashland, Oregon | |
| Imaris | Bitplane | |
Sorting blood monocytes
Mice were humanely euthanized by exposure to CO2. Blood was collected from mice via cardiac puncture into K3EDTA tubes and subjected to RBC lysis (eBioscience). Cells were pelleted by centrifugation at 350 X G for 10 minutes at 4°C and washed, suspended in PBS, then stained for 10 minutes with Zombie NIR (BioLegend) and purified anti-CD16/32 (93, BioLegend) to label dead cells and block Fc receptors. Next, cells were stained in wash buffer for an additional 20 minutes with the antibodies listed in the Key Resource Table. Stained cells were washed twice and strained through a 30 m strainer, then subjected to cell sorting using a Beckman Coulter MoFlo Astrios EQ configured with 355 nm, 405 nm, 488 nm, 561 nm, and 642 nm lasers. Each cell population was hierarchically gated using Beckman Coulter Summit software. Ly6CHi monocytes were defined asCD45PosCD11bHiCD115PosCD19NegCD90.2NegLy6GNegLy6CHi. Ly6CHi monocytes were further restricted to single particles by comparing height and area side scatter pulses, and dead cells were excluded by detecting the integration of the live/dead dye (Zombie NIR).
Sorting crosslinked Kupffer cell nuclei
Livers of Clec4f-Cre-tdTomato mice were perfused in a retrograde fashion using a Masterflex multichannel peristaltic pump (Cole-Parmer) briefly at a rate of 5ml/min through the inferior vena cava with HBSS without Ca or Mg (Gibco) supplemented with 0.5mM EGTA, 0.5mM EDTA, and 20mM HEPES. Subsequently, they were fixed by perfusion with 3mM DSG (ProteoChem) in PBS for 30 minutes, and 1% formaldehyde for 10 minutes at a rate of 5ml/min. The reaction was quenched by perfusion with 20ml 0.125M glycine. After fixation, livers were excised, minced and washed twice with 20ml ice-cold NF1 buffer (10mM Tris-HCl pH8.0, 1mM EDTA, 5mM MgCl2, 0.1M Sucrose, 0.5% Triton X-100) and pelleted by centrifugation for 7 minutes at 1,200 X G at 4°C. Livers were resuspended in NF1 buffer and homogenized with 10 strokes using the loose pestle of a Dounce homogenizer and incubated on ice for 30 minutes, and then homogenized with 50–70 strokes using the tight pestle with periodic assessment for released nuclei by microscopy. The homogenized liver was then filtered with a 70 μm strainer into 50 ml tube and pelleted at 1,200g for 7 minutes at 4°C. Crude nuclei were then washed with 10ml PBS supplemented with 2mM EDTA and pelleted by centrifugation at 1,100 X G at 4°C for 5 minutes. The pellet was resuspended in PBS with 2mM EDTA and strained with a 40 μm strainer. Nuclei were purified by FACS using a Sony SH800 or Sony MA900 based on TdTomato expression and forward scatter. After sorting, nuclei were pelleted by centrifugation at 1,100 X G at 4°C for 5 minutes, and snap-frozen in liquid nitrogen and stored at −80°C until ready for ChIP-seq library preparation.
Bone marrow-derived macrophages (BMDM) culture
Femur, tibia and iliac bones from C57BL/6J mice, Nr1h3−/− mice, Smad4fl/fl mice, and Smad4fl/fl Lyz2-cre+/− mice were flushed with DMEM high glucose (Corning), and red blood cells were lysed using red blood cell lysis buffer (eBioscience).
For RNA-seq and ATAC-seq of Nr1h3 wild-type and genetically ablated, 20 million bone marrow cells were seeded per 15cm non-tissue culture plates in DMEM high glucose (60%) with 10% FBS (Biowest), 30% L929-cell conditioned laboratory-made media (as source of M-CSF), 100 U/ml penicillin-streptomycin (Thermo Fisher Scientific) and 2.5 μg/ml Amphotericin B (Thermo Fisher Scientific). After 4 days of differentiation, 16.7 ng/ml mouse M-CSF (Shenandoah Biotechnology) was added to the media. After an additional 2 days of culture, non-adherent cells were washed off with room temperature DMEM and macrophages were obtained as a homogeneous population of adherent cells which were scraped and subsequently seeded onto tissue culture-treated Petri dishes overnight in DMEM containing 10% FBS, 100 U/ml penicillin/streptomycin, 2.5 μg/ml Amphotericin B and 16.7 ng/ml M-CSF.
For RNA-seq, ATAC-seq, and ChIP-seq of bone marrow progenitor cells from C57BL/6J mice, Smad4fl/fl mice, or Smad4fl/fl Lyz2-Cre mice, 20 million bone marrow cells were seeded per 15cm non-tissue culture plates in MEM (90%) with 10% FBS (Biowest), 10ng/ml M-CSF (Shenandoah Biotechnology), 100 U/ml penicillin/streptomycin+L-glutamine (Thermo Fisher Scientific) and 2.5 μg/ml Amphotericin B (Thermo Fisher Scientific). For DLL4 stimulation, tissue culture-treated plates were coated with 1 μg/ml human recombinant DLL4 (R&D systems) overnight at 4°C. After 3 days of differentiation, non-adherent cells were washed off with room temperature MEM and adherent cells were scraped and subsequently seeded onto tissue culture-treated plates in which the surface was coated with or without DLL4, and cultured with or without 2ng/ml human recombinant TGF-β1 (Cell Signaling Technology) and/or 1 μM DMHCA, for 24 hours or 72 hours in DMEM containing 10% FBS, 100 U/ml penicillin/streptomycin, 2.5 μg/ml Amphotericin B, and 10 ng/ml M-CSF. Where indicated, cells were exposed to 10 μM DAPT (Cell Signaling Technology) for 24 hours.
Isolation and culture of bone marrow monocytes
Bone marrow monocytes were isolated from C57BL/6J mice using EasySep Mouse Monocyte Isolation Kit (STEMCELL technologies). They were seeded in tissue culture-treated plates in which the surface was coated with or without DLL4, and cultured with or without 2 ng/ml human recombinant TGF-β1 (Cell Signaling Technology) in RPMI1640 (Corning) containing 10% FBS, 100 U/ml penicillin/streptomycin, 2.5 μg/ml Amphotericin B, and 10 ng/ml M-CSF. Where indicated, 10 μM Desmosterol was added to the media when the culture is started and 24 hours after that.
Isolation and culture of mouse primary hepatocytes
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL/6J mice as described previously (Sakai et al., 2012). Mice were humanely euthanized by exposure to CO2. Livers were perfused in a retrograde fashion for 3 minutes at a rate of 5 ml/min through the inferior vena cava with HBSS with Ca or Mg (Gibco) supplemented with 10 mM HEPES (Gibco), then for 18 minutes with the same solution containing collagenase type I (32 mg per 100 ml, Worthington) and Protease Inhibitor Cocktail Complete–EDTA Free (Roche). The hepatocytes were harvested and purified by density gradient centrifugation with Percoll (Sigma) and plated on type I collagen–coated six-well plates (1 million cells per well) in Medium 199 (Gibco) supplemented with 5% FBS and 100 U/ml penicillin/streptomycin (Thermo Fisher Scientific).
Lipid Measurements
Livers and primary hepatocytes were processed at the University of Texas Southwestern Medical Center for oxysterol and lipid metabolite analysis by LC-MS as previously described in full (www.lipidmaps.org/protocols/index.html).
ATAC-seq library preparation
Approximately 50,000 sorted cells were washed once with PBS and once with cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Cells were then suspended in 50 μl 1X reaction buffer (25 μl Tagment DNA Buffer, 2.5 μl Tagment DNA enzyme I, and 22.5 μl water) (Nextera DNA Library Preparation Kit, Illumina) as previously described (Buenrostro et al., 2013). Transposase reactions were carried out at 37°C for 30 minutes and immediately DNA was purified using ChIP DNA Clean & Concentrator kits (Zymo Research). DNA was amplified using the Nextera primer Ad1 and a unique Ad2.n barcoding primer using NEBNext High-Fidelity 2XPCR Master Mix (NEB) for 14 cycles. Resulting libraries were size selected by gel excision to 175–225 bp, purified, and single end sequenced using a HiSeq 4000 or a NextSeq 500 (Illumina) for 51 cycles according to the manufacturer’s instructions.
RNA-seq library preparation
Total RNA was isolated from in vitro culture cells and purified using a Direct-zol RNA MicroPrep Kit (Zymo Research) according to the manufacturer’s instructions (Zymo Research). FACS purified cells were pelleted and put into 150 μl lysis/Oligo d(T) Magnetic Beads binding buffer (100mM Tris-HCl pH7.5, 500mM LiCl, 10mM EDTA pH8.0, 1% LiDS, 5mM DTT) and stored at −80°C until processing. mRNAs were enriched by incubation with Oligo d(T) Magnetic Beads (NEB, S1419S). Poly A enriched mRNA was fragmented, in 2x Superscript III first-strand buffer with 10mM DTT (Thermo Fisher Scientific), by incubation at 94°C for 9 minutes, then immediately chilled on ice before the next step. The 10 μL of fragmented mRNA, 0.5 μL of Random primers (3 μg/μL) (Thermo Fisher Scientific), 0.5 μL of Oligo dT primer (50 μM) (Thermo Fisher Scientific), 0.5 μL of SUPERase-In (Ambion), 1 μL of dNTPs (10 mM) and 1 μL of DTT (10 mM) were heated at 50°C for one minute. At the end of incubation, 5.8 μL of water, 1 μL of DTT (100 mM), 0.1 μL Actinomycin D (2 μg/μL), 0.2 μL of 1% Tween-20 (Sigma) and 0.5 μL of Superscript III (Thermo Fisher Scientific) were added and incubated in a PCR machine using the following conditions: 25°C for 10 minutes, 50°C for 50 minutes, and a 4°C hold. The product was then purified with RNAClean XP beads (Beckman Coulter) according to manufacturer’s instruction and eluted with 10 μL nuclease-free water. The RNA/cDNA double-stranded hybrid was then added to 1.5 μL of Blue Buffer (Enzymatics), 1.1 μL of dUTP mix (10 mM dATP, dCTP, dGTP and 20 mM dUTP), 0.2 μL of RNAse H (5 U/μL), 1.05 μL of water, 1 μL of DNA polymerase I (Enzymatics) and 0.15 μL of 1% Tween-20. The mixture was incubated at 16°C for 2.5 hours. The resulting dUTP-marked dsDNA was purified using 28 μL of SpeedBeads (GE Healthcare), diluted with 20% PEG8000, 2.5M NaCl to final of 13% PEG, eluted with 40 μL EB buffer (10 mM Tris-Cl, pH 8.5) and frozen at −80°C. The purified dsDNA (40 μL) underwent end repair by blunting, A-tailing and adapter ligation as previously described (Heinz et al., 2010) using barcoded adapters (NextFlex, Bioo Scientific). Libraries were PCR-amplified for 16 cycles, size selected by gel extraction, quantified using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and sequenced on a HiSeq 4000 or a NextSeq 500 (Illumina) for 51 cycles according to the manufacturer’s instructions.
Crosslinking in vitro culture cells for ChIP-seq
For H3K27ac ChIP-seq, culture media was removed and plates were washed once with PBS and then fixed with 1% formaldehyde (Thermo Fisher Scientific) in PBS for 10 minutes at room temperature. The reaction was quenched by adding glycine (Thermo Fisher Scientific) to 0.125M. For LXR, SMAD4, and RBPJ ChIP-seq, cells were crosslinked with 3mM DSG (ProteoChem) in PBS for 30 minutes at room temperature, and then fixed with 1% formaldehyde (Thermo Fisher Scientific) in PBS for 10 minutes at room temperature. The reaction was quenched by adding glycine (Thermo Fisher Scientific) to 0.125M. After fixation, cells were washed once with cold PBS and then pelleted at 700 X G for 5 minutes at 4°C. Crosslinked cells were stored at −80°C until ready for ChIP-seq library preparation.
ChIP-seq library preparation
Chromatin immunoprecipitation (ChIP) was performed in biological replicates as previously described (Heinz et al., 2018). Fixed cells were thawed on ice, resuspended in either ice-cold LB3 (10 mM Tris/HCl pH 7.5, 100 mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% Na-deoxycholate, 0.5% N-lauroylsarcosine, 1 X protease inhibitor cocktail, 1mM Na-Butyrate, for H3K27ac ChIP) or ice-cold RIPA-NR lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.4% Na-Deoxycholate, 0.1% SDS, 1% NP-40, 1x protease inhibitors, for LXR, SMAD4, and RBPJ ChIP). Frozen crosslinked nuclei were resuspended in wash buffer (10mM HEPES/KOH pH7.9, 85mM KCl, 1mM EDTA, 0.2% IGEPAL CA-630, 1x protease inhibitor cocktail (Sigma), 1 mM PMSF) for 5minutes on ice. Nuclei were spun down and then resuspended in RIPA-NR lysis buffer. Chromatin was sheared by sonication. Samples were sonicated in a 96 Place microTUBE Rack (Covaris cat#500282) using a Covaris E220 for 12 cycles (samples in LB3) or 18 cycles (samples in RIPA-NR buffer) with the following setting: time, 60 seconds; duty, 5.0; PIP, 140; cycles, 200; amplitude, 0.0; velocity, 0.0; dwell, 0.0. Samples were recovered and spun down at max speed, 4°C for 10 minutes. The LB3 lysate was diluted 1.1-fold with ice-cold 10% Triton X-100 after sonication. One percent of the lysate was kept as ChIP input. For each immunoprecipitation, aliquots of diluted lysate equivalent to 500,000 cells (for H3K27ac ChIP) or 2–3 million cells or nuclei (for LXR, SMAD4, or RBPJ ChIP), 30 μl of Dynabeads Protein A (for rabbit polyclonal antibodies) or Dynabeads protein A/G (for LXR ChIP) bound to specific antibodies for H3K27ac (2 μg, Active Motif, 39133), LXR (2 μg each of the indicated LXR specific antibodies, Santa Cruz Biotechnology: sc-1000X, sc-133221X, sc-271064X)), SMAD4 (1 μg each of Cell Signaling technology 46535 and 38454) or RBPJ (2 μg, Abcam, ab25949) were combined and rotated overnight at 4°C. For H3K27ac ChIP, beads were collected on a magnet and washed three times each with wash buffer I (20 mM Tris/HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA), wash buffer III (10 mM Tris/HCl pH 7.4, 250 mM LiCl, 1% Triton X-100, 0.7% Na-Deoxycholate, 1 mM EDTA) and twice with ice-cold TET (10 mM Tris/HCl pH7.5, 1 mM EDTA, 0.2% Tween-20). For LXR, SMAD4, or RBPJ ChIP, beads were washed three times with RIPA-NR buffer (20 mM Tris-HCl/pH7.5, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.4 % Na-Deoxycholate, 1 mM EDTA, 0.5mM EGTA, 0.5mM DTT), six times with RIPA-LiCl buffer (10 mM Tris-HCl/pH 7.5, 250 mM LiCl, 1% NP-40, 0.7% Na-Deoxycholate, 1 mM EDTA), three times with ice-cold TET (10 mM Tris/HCl pH7.5, 1 mM EDTA, 0.2% Tween-20), and one time with IDTE (10mM Tris-HCl pH 8.0, 0.1mM EDTA). Libraries were prepared with NEBNext Ultra II DNA library prep kit (NEB) reagents according to the manufacturer’s protocol on the beads suspended in 25 μL TT (10mM Tris/HCl pH7.5, 0.05% Tween-20), with reagent volumes reduced by half. DNA was eluted and crosslinks reversed by adding 4 μl 10% SDS, 4.5 μl 5 M NaCl, 3 μl EDTA, 1 μl proteinase K (20 mg/ml), 20 μl water, incubating for 1 h at 55°C, then 30 minutes to overnight at 65°C. DNA was purified using 2 μL of SpeedBeads (GE Healthcare), diluted with 20% PEG8000, 1.5M NaCl to final of 12% PEG, eluted with 25 μl TT. DNA contained in the eluate was then amplified for 12 cycles in 50 μl PCR reactions using NEBNext High-Fidelity 2X PCR Master Mix (NEB) and 0.5 mM each of primers Solexa 1GA and Solexa 1GB. ChIP input material (1 percent of sheared DNA) was treated with RNase for 15 minutes at 37°C in EB buffer (10 mM Tris pH 8, 0.5% SDS, 5 mM EDTA, 280 mM NaCl), then digested with Proteinase K for 1 h at 55°C and crosslinks reversed at 65°C for 30 minutes to overnight. DNA was purified using 2 μL of SpeedBeads (GE Healthcare), diluted with 20% PEG8000, 1.5M NaCl to final of 12% PEG, eluted with 25 μl TT and library prep and amplification were performed as described for ChIP samples. Resulting libraries were size selected by gel excision to 225–325 bp, purified, and single-end sequenced using a HiSeq 4000 or a NextSeq 500 (Illumina).
Data mapping
Libraries were sequenced either on a HiSeq 4000 (Illumina) or a NextSeq 500 (Illumina). Raw sequences/fastqs were obtained from Illumina Studio pipeline, and then some of them were trimmed according to their qualities. RNA-seq data was mapped to mm10 genome using STAR (Dobin et al., 2013) with default parameters. ChIP-seq and ATAC-seq data were mapped to custom genomes using bowtie2 (Langmead and Salzberg, 2012) with default parameters. Finally, HOMER (Heinz et al., 2010) tag directories were created for mapped samples. Mapping data for all sequencing studies is provided in Table S7.
RNA-seq analysis
The gene expression raw counts were quantified by HOMER’s analyzeRepeats with the option “-condenseGenes -count exons -noadj”. All genes shorted than 250bp were removed, and the TPM (transcript per kilobase million) were quantified for all genes matching accession number to the raw counts. Differentially expressed genes were assessed with DESeq2 (Love et al., 2014) at p-adj (adjusted p value) < 0.05 and FC (fold change) > 2 or FC > 1.5 where indicated.
IDR analysis
ChIP-seq experiments were performed in two replicates with corresponding input experiments. Peaks were called with HOMER for each tag directory with relaxed peak finding parameters “-L 0 -C 0 -fdr 0.9”) against the corresponding input directory. For ATAC-seq, no inputs were used, but the size was set to 200bp “-L 0 -C 0 -fdr 0.9 - minDist 200 -size 200”. IDR (Li et al., 2011) was used to test for reproducibility between replicates, and only peaks with IDR < 0.05 were used for downstream analysis. The pooled tag directory from two replicates was used for track visualization.
ATAC-seq and ChIP-seq analysis
To quantify the TF binding and chromatin accessibility, the raw tag counts at merged IDR peaks of different conditions by HOMER’s mergePeaks, were extracted by HOMER’s annotatePeaks with “-noadj”, “-size 200” for ATAC-Seq and TF ChIP-Seq, but “-size 2000” for H3K27ac ChIP on ATAC-associated peaks. Subsequently, DESeq2 (Love et al., 2014) was used to identify the differentially bound TF binding distal sites (3000bp away from known TSS/transcription starting sites) or the differential distal chromatin accessibilities with p-adj < 0.05 and FC > 2.
Motif analysis
To identify motifs enriched in peak regions over the background, HOMER’s motif analysis (findMotifsGenome.pl) including known default motifs and de novo motifs was used (Heinz et al., 2010). The background peaks used either from random genome sequences or from peaks in comparing condition were indicated throughout the main text and in the figure legends.
Data visualization
Heatmap of RNA expression or tags of ATAC-Seq peaks were generated by pheatmap, an R package. Significance indicated by “*” in bar-plot represents the p-adj (after multiple-testing correction). MA-plot was used to demonstrate the differentially expressed genes for RNA-Seq data with log2fold change against expression value TPM, additionally with the sizes of dots representing the significant p-values. Scatter plots were used for direct comparison of two conditions, either the gene TPM or normalized tag counts were used for RNA-Seq, ChIP-/ATAC-Seq respectively. Browser tracks for ATAC-Seq and ChIP-Seq were generated by open source pygbrowse (https://github.com/phageghost/python-genome-browser).
Data and software availability
The accession number for the raw data and processed data files reported in this paper is GEO: GSE128662.
Supplementary Material
Table S1. Summary of mRNA expression, Related to Figure 1
Gene expression measured by RNA-seq (TPM) for circulating monocytes, recruited liver monocytes, RLMs and resident KCs.
Table S2. Summary of mRNA expression, Related to Figure 3, S3
Gene expression measured by RNA-seq (TPM) for control KCs (Clec4fNegTim4Pos), LXRα KCs (Clec4fPosTim4Neg and Clec4fPosTim4Pos), control BMDM, and Nr1h3−/− BMDM.
Table S3. Summary of mRNA expression (EC, KC), Related to Figure 4, 5
Gene expression measured by RNA-seq (TPM) for Stab2Lo, Stab2Int, Stab2Hi endothelial cells and KCs.
Table S4. Summary of mRNA expression (Smad4−/−), Related to Figure 4, S4
Gene expression measured by RNA-seq (TPM) for control KCs (Tim4Pos) and Smad4−/− KCs (Tim4Neg and Tim4Pos).
Table S5. Summary of mRNA expression (RLMs at 24h), Related to Figure 4, S5
Gene expression measured by RNA-seq (TPM) for RLMs at 24h isolated from Rosa26-Lox-Stop-Lox-DTR;Clec4f-cre-tdTomato+/− mice injected with or without LY411575 or DLL1 and DLL4 antibodies.
Table S6. Summary of mRNA expression (BMDM, BM monocytes, BMDM time course), Related to Figure 5, 7, S5, S7
Gene expression measured by RNA-seq (TPM) for control and Smad4−/− BM progenitor cells stimulated with or without DLL4, TGF-β, or DAPT for 24 h, BM monocytes stimulated with or without desmosterol, DLL4, or TGF-β for 48 h, and BM progenitor cells stimulated with or without DMHCA, DLL4, or TGF-β for 24 h or 72 h
Table S7. Alignment stats, Related to Figures 1–7 and S1–S7
The number of total and unique mapped reads of RNA-seq, ATAC-seq, and ChIP-seq samples.
Highlights.
Determinants of Kupffer cell identity are inferred from dynamic enhancer landscapes
DLL4 activates poised enhancers to induce Kupffer cell lineage determining factors
LXRα induced by DLL4 drives subsequent activation of Kupffer cell enhancers
TGF-β and desmosterol regulate SMADs and LXRs to maintain Kupffer cell identity
Acknowledgements
These studies were supported by NIH grants DK091183, HL088093 and GM085764 and a Leducq Transatlantic Network Grant. MS was supported by the Manpei Suzuki Diabetes Foundation of Tokyo, Japan, and the Osamu Hayaishi Memorial Scholarship for Study Abroad, Japan. TDT was supported by P30 DK063491, T32DK007044, and NRSA T32CA009523. JSS was supported by American Heart Association Fellowship 16PRE30980030 and NIH Predoctoral Training Grant 5T32DK007541. JGM is supported in part by NIH HL20948. YA was supported by Japan Society for the Promotion of Science Overseas Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, and Tanaka K (2015). Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biology 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avigan J, Steinberg D, Thompson MJ, and Mosettig E (1960). Mechanism of Action of MER-29, An inhibitor of cholesterol biosynthesis. Biochemical and biophysical research communications 2, 63–65. [DOI] [PubMed] [Google Scholar]
- Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, Mack M, Joshi A, Guilliams M, Mowat AM, et al. (2016). Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat Commun 7, ncomms11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T, and Oswald F (2009). The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci 66, 1631–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ (2006). Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7, 678–689. [DOI] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (2009). Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. Journal of lipid research 50 Suppl, S15–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, and Waisman A (2005). A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods 2, 419–426. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, and Tontonoz P (2012). Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Jeffries S, Norton JE, Capobianco AJ, and Bresnick EH (2002). Repression of activator protein-1-mediated transcriptional activation by the Notch-1 intracellular domain. J Biol Chem 277, 7587–7597. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Forster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon LJ, Barnes M, Tang H, Pritchard MT, and Nagy LE (2013). Kupffer cells in the liver. Compr Physiol 3, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, Leibing T, Demory A, Ulbrich F, Diett M, et al. (2017). GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest 127, 1099–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, and Guilliams M (2016). Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 44, 439–449. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, Massasa EE, Medgalia C, David E, Giladi A, Moor AE, et al. (2018). Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 36, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. (2018). Transcription Elongation Can Affect Genome 3D Structure. Cell 174, 1522–1536 e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, Miyazaki M, Miyazaki-Anzai S, Ebner B, Chandak PG, Kadam RS, et al. (2009). Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res 50, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, and Tacke F (2017). Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17, 306–321. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel J (2011). Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics 5, 1752–1779. [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, Jacome-galarza CE, Händler K, Klughammer J, Kobayashi Y, et al. (2016). Specification of tissue-resident macrophages during organogenesis. Science 4238, epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederacke I, Dapito DH, Affo S, Uchinami H, and Schwabe RF (2015). High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 10, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Chung AC, and Lan HY (2013). Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) 124, 243–254. [DOI] [PubMed] [Google Scholar]
- Mullen AC, Orlando DA, Newman JJ, Loven J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, and Young RA (2011). Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, Seidman JS, Henke A, Roland JT, Ozeki KA, et al. (2018). Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc Natl Acad Sci U S A 115, E4680–E4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell CS, and Radtke F (2017). Notch as a tumour suppressor. Nat Rev Cancer 17, 145–159. [DOI] [PubMed] [Google Scholar]
- Peet DJ, Janowski BA, and Mangelsdorf DJ (1998a). The LXRs: a new class of oxysterol receptors. Curr Opin Genet Dev 8, 571–575. [DOI] [PubMed] [Google Scholar]
- Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, and Mangelsdorf DJ (1998b). Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 93, 693–704. [DOI] [PubMed] [Google Scholar]
- Perdiguero EG, and Geissmann F (2016). The development and maintenance of resident macrophages. Nat Immunol 17, 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge JR, Wiley JA, Talago EA, Young EM, Johns LL, Kundert JA, Sonsteng KM, Halford WP, Capecchi MR, and Schmidt EE (2013). Nuclear double-fluorescent reporter for in vivo and ex vivo analyses of biological transitions in mouse nuclei. Mamm Genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, and Goldstein JL (2004). Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol Cell 15, 259–268. [DOI] [PubMed] [Google Scholar]
- Sakai M, Matsumoto M, Tujimura T, Yongheng C, Noguchi T, Inagaki K, Inoue H, Hosooka T, Takazawa K, Kido Y, et al. (2012). CITED2 links hormonal signaling to PGC-1alpha acetylation in the regulation of gluconeogenesis. Nat Med 18, 612–617. [DOI] [PubMed] [Google Scholar]
- Schmierer B, and Hill CS (2007). TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8, 970–982. [DOI] [PubMed] [Google Scholar]
- Scott CL, T’Jonck W, Martens L, Todorov H, Sichien D, Soen B, Bonnardel J, De Prijck S, Vandamme N, Cannoodt R, et al. (2018). The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 49, 312–325 e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G, et al. (2016). Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nature Communications 7, 10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner D.a., and Schwabe RF (2007). TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature medicine 13, 1324–1332. [DOI] [PubMed] [Google Scholar]
- Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, Beattie L, Walters SB, Tay SS, Lu B, et al. , A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment, Immunity 47, 2017, 374–388.e6. [DOI] [PubMed] [Google Scholar]
- Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, et al. (2012). Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151, 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann NJ, and Glass CK (2013). Sterols and oxysterols in immune cell function. Nat Immunol 14, 893–900. [DOI] [PubMed] [Google Scholar]
- Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC, et al. (2006). Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. The Journal of biological chemistry 281, 27816–27826. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, and Deng CX (2002). Generation of Smad4/Dpc4 conditional knockout mice. Genesis 32, 80–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of mRNA expression, Related to Figure 1
Gene expression measured by RNA-seq (TPM) for circulating monocytes, recruited liver monocytes, RLMs and resident KCs.
Table S2. Summary of mRNA expression, Related to Figure 3, S3
Gene expression measured by RNA-seq (TPM) for control KCs (Clec4fNegTim4Pos), LXRα KCs (Clec4fPosTim4Neg and Clec4fPosTim4Pos), control BMDM, and Nr1h3−/− BMDM.
Table S3. Summary of mRNA expression (EC, KC), Related to Figure 4, 5
Gene expression measured by RNA-seq (TPM) for Stab2Lo, Stab2Int, Stab2Hi endothelial cells and KCs.
Table S4. Summary of mRNA expression (Smad4−/−), Related to Figure 4, S4
Gene expression measured by RNA-seq (TPM) for control KCs (Tim4Pos) and Smad4−/− KCs (Tim4Neg and Tim4Pos).
Table S5. Summary of mRNA expression (RLMs at 24h), Related to Figure 4, S5
Gene expression measured by RNA-seq (TPM) for RLMs at 24h isolated from Rosa26-Lox-Stop-Lox-DTR;Clec4f-cre-tdTomato+/− mice injected with or without LY411575 or DLL1 and DLL4 antibodies.
Table S6. Summary of mRNA expression (BMDM, BM monocytes, BMDM time course), Related to Figure 5, 7, S5, S7
Gene expression measured by RNA-seq (TPM) for control and Smad4−/− BM progenitor cells stimulated with or without DLL4, TGF-β, or DAPT for 24 h, BM monocytes stimulated with or without desmosterol, DLL4, or TGF-β for 48 h, and BM progenitor cells stimulated with or without DMHCA, DLL4, or TGF-β for 24 h or 72 h
Table S7. Alignment stats, Related to Figures 1–7 and S1–S7
The number of total and unique mapped reads of RNA-seq, ATAC-seq, and ChIP-seq samples.
Data Availability Statement
The accession number for the raw data and processed data files reported in this paper is GEO: GSE128662.