

Abstract
Metastasis is the leading cause of mortality in patients with solid tumors. In this regard, we previously reported that Pseudopodium-Enriched Atypical Kinase One (PEAK1) is necessary for non-canonical Transforming Growth Factor β (TGFβ) signaling and TGFβ/fibronectin-induced metastasis. Here, we demonstrate that inhibition of DHPS-dependent eIF5A1/2 hypusination blocks PEAK1 and E-Cadherin expression, breast cancer cell viability and TGFβ/fibronectin-induced PEAK1-dependent breast cancer metastasis. Interestingly, TGFβ stimulation of high-grade metastatic breast cancer cells increases and sustains eIF5A1/2 hypusination. We used a suite of bioinformatics platforms to search biochemical/ functional interactions and clinical databases for additional control points in eIF5A1/2 and PEAK1-Epithelial to Mesenchymal Transition (EPE) pathways. This effort revealed that interacting EPE genes were enriched for TP53 transcriptional targets and were commonly co-amplified in breast cancer patients harboring inactivating TP53 mutations. Taken together, these results suggest that combinatorial therapies targeting DHPS and protein activities elevated in TP53-mutant breast cancers may reduce systemic tumor burden and improve patient outcomes.
Keywords: DHPS, eIF5A1/2, Breast cancer, TGFβ-mediated metastasis, TP53
1. Introduction
The 5-year survival for breast cancer patients who develop or are diagnosed with metastatic disease drops sharply from over 90% to less than 20% [1]. Thus, there is a critical and urgent need to identify targetable mechanisms that support breast cancer cell dissemination to and expansion within the metastatic niche. Research over the past decade has uncovered critical connections between increased cell state plasticity and improved fitness of malignant cells within solid tumor types [2]. Transforming growth factor beta (TGFβ)-induced Epithelial to Mesenchymal Transition (EMT) is one mechanism by which breast cancer cells increase in plasticity [3] to promote metastasis.
Previous work from our group has established an essential role for the cytoskeleton-associated kinase PEAK1 (pseudopodium-enriched atypical kinase one) during switching of TGFβ signaling outcomes from tumor-suppressive to tumor-promoting (as assayed by proliferation/survival, metastasis and EMT) [4–6]. In parallel, we have linked PEAK1 protein translation and its protumorigenic activities to post-translational activation of the eukaryotic Initiation Factor five A, isoforms one and two (eIF5A1/2) [7,8]. This work demonstrated that deoxyhypusine synthase (DHPS)/deoxyhypusine hydroxylase (DOHH)-dependent hypusination of eIF5A1/2 was necessary for PEAK1 translation and PEAK1-mediated pancreatic cancer progression [9]. In agreement with these data, eIF5A1/2 protein and hypusination pathway machinery predict reduced survival across a number of cancers and represent promising therapeutic targets [10].
We hypothesized that blocking the eIF5A1/2 hypusination pathway would result in impaired PEAK1 protein expression and TGFβ/fibronectin-induced metastatic capabilities in breast cancer cells. Our data demonstrate that GC7 inhibition of DHPS reduces eIF5A1/2 hypusination in human and mouse breast cancer cells, concomitant with decreased cell survival and/or proliferation, and expression of epithelial markers and PEAK1. Further, we demonstrate that GC7 treatment completely blocked PEAK1-dependent TGFβ/fibronectin-induced metastasis of human breast cancer cells. Bioinformatic analyses of the eIF5A1/2-PEAK1-EMT (EPE) axis revealed elevated levels of DHPS expression and/or activity as a potential vulnerability in breast cancer patients with TP53 genomic alterations. Taken together, these studies identify a new therapeutically-targetable axis that may be harnessed/to thwart the protumorigenic effects of TGFβ signaling and improve outcomes in breast cancer patients with genomic alterations in TP53.
2. Results
2.1. Breast Cancer Cells Are Sensitive to GC7-Mediated Inhibition of Survival and Hypusination
We first examined whether eIF5A protein was detectable in its hypusinated form in a panel of breast cells. Protein lysates from non-tumorigenic human MCF10A; tumorigenic MCF10A derivatives MCF10AT1K, MCF10CA1h and MCF10CA1a; human MDA-MB-231 and -468; and mouse tumorigenic 4T1 and 67NR cells showed detectable levels of hypusinated eIF5A1/2 (Fig. 1A–B). Quantification of the abundances of eIF5A1 and eIF5A2 transcripts using real-time PCR analysis showed consistently higher levels of the eIF5A2 transcript in human cells compared to mouse cells (Fig. 1C).
Fig. 1.

Breast Cancer Cells Are Sensitive to GC7-Mediated Inhibition of Survival and Hypusination. (A) Protein lysates were collected 24 h after plating MCF10A cells, malignant H-Ras transformed breast cancer cells (MCF10AT1K, MCF10CA1h and MCF10CA1a), triple negative breast cancer cells (MDA-MB-231 and MDA-MB-468), and mouse malignant breast cancer cells (4T1 and 67NR) and immunoblotted for hypusine (Hyp-eIF5A), total eIF5A, and β-actin as loading control. (B) Quantification of band intensities of hypusinated-to-total eIF5A determined for each cell line using densitometric analysis of blots in panel A, normalized to the ratio in MCF10A. The pattern of the predominant eIF5A isoform mRNA expression profile (see (C)) is indicated. (C) qPCR of eIF5A isoforms in the same cells as in (A), normalized to the level of eIF5A1 separately for each cell. (D) Cytotoxicity response showing cell viability following 72-h treatments with GC7 or CPX (0.1 nM–1 mM) relative to control treatment. Data are reported as the triplicate mean ± standard error. (E) Phase-contrast micrographs for the indicated cell lines were taken after 48 h of control or 100 μM GC7 treatment (Scale bar: 100 μm). (F) Western blot and band densitometry quantification of hypusinated-to-total eIF5A1/2 levels following 48 h of control or 100 μM GC7 treatment.
Since eIF5A1/2 are the only proteins that have been reported to be post-translationally hypusinated, we aimed to test whether pharmacological inhibition of hypusination could impair breast cancer cell survival using the DHPS-specific inhibitor GC7 and the non-specific iron chelating agent ciclopirox olamine (CPX, an inhibitor of DOHH). We observed dose-dependent inhibition of breast cell survival following 72 h treatments with either CPX or GC7. IC50 values for GC7 in the human cells ranged from 50 to 200 μM, while mouse 4T1 and 67NR cells were more sensitive (Fig. 1D). Notably, CPX was as potent and efficacious as GC7 across all cells tested (Fig. 1D). The increased potency of CPX in the MCF10AT1k and the MCF10CA1h cells may be due to DOHH-independent effects of CPX iron chelation. Interestingly, both GC7 and CPX displayed a reduced efficacy (~50% reduction in viable cell number) in the human triple-negative MDA-MB-468 and -231 breast cancer cells.
Based upon the dose-response curves, we selected 100 μM GC7 as the lowest sub-maximal dose above the IC50s in GC7-responsive breast cancer cells. Treatment of the cell lines with 100 μM GC7 for 48 h resulted in reduced cell number compared to control treatment in all cell except MDA-MB-468 and -231, as assessed by phase-contrast brightfield imaging (Fig. 1E). Of note, breast cancer cells also exhibited changes in cellular morphology following GC7 treatment – more specifically, cells that did change became more mesenchymal, elongated and less tightly packed (Fig. 1E). Finally, we observed a significant decrease in hypusinated:total eIF5A1/2 levels following 48 h of GC7 treatment across all breast cells except the MDA-MB-468 and -231 lines (Fig. 1F).
2.2. Serum or TGFβ Stimulation Limits Potency/Efficacy of GC7-Dependent inhibition of eIF5A1/2
Since the high-grade metastatic (MCF10CA1a and 4T1) and non-metastatic (67NR) breast cancer cells were sensitive to GC7-mediated reduction of cell number and hypusinated eIF5A1/2 levels, we continued in vitro work with these. We first tested a dose regimen ranging from 0.1 to 100 μM GC7 on the morphology and survival/proliferation of these cells. Consistent with data presented in Fig. 1, all three lines showed reduced cell numbers following 10–100 μM GC7 treatment (Fig. 2A). In parallel, we probed cell lysates following 48 h GC7 treatment for hypusinated and total eIF5A1/2, PEAK1 and E-Cadherin expression. This confirmed a dose-dependent decrease in eIF5A1/2 hypusination following GC7 treatment (Fig. 2B). GC7 treatment also reduced PEAK1 and E-Cadherin protein levels across all cell lysates. Interestingly, however, both the high-grade human and mouse metastatic breast cancer cells maintained PEAK1 and E-Cadherin expression at 10 μM GC7 in spite of hypusinated eIF5A1/2 being nearly undetectable. In contrast, the non-metastatic 67NR cells exhibited concomitant reduction in PEAK1, E-Cadherin and hypusinated eIF5A1/2 levels at 10 μM GC7 (Fig. 2B).
Fig. 2.

Serum or TGFβ Stimulation Limits Potency/Efficacy of GC7-Dependent Inhibition of eIF5A1/2. (A) Phase-contrast images of 4T1, 67NR, and MCF10CA1a cells 48 h after treatment with control or GC7 (Scale bar: 100 μm). (B) Western blot and band densitometry quantification for hypusinated eIF5A1/2, total eIF5A1/2, E-Cadherin, PEAK1 and β-actin in 4T1, 67NR and MCF10CA1a cell lysates after 48 h treatment with control or 100 μM GC7. (C) Cytotoxicity response showing cell viability of 4T1, 67NR and MCF10CA1a cells following 72 h treatment with GC7 (0.1 nM–1 mM) on plastic or fibronectin in media with or without serum. Data are reported as the triplicate mean ± standard error. (D) 4T1, 67NR, and MCF10CA1a lines were treated with 0.1% BSA control or TGFβ (2.5 ng/mL) for 48 h and immunoblotted against E-cadherin and β-actin as a loading control (top). E-cadherin protein bands were quantified relative to β-actin and normalized to control (bottom). Phase-contrast micrographs (right) of cells were obtained prior to collection of protein lysates for all three cell lines (Scale bar: 100 μm) (E) Western blots and band densitometry quantification for hypusinated-to-total eIF5A1/2 in cell lysates following 10,30 or 120 min treatment with control or TGFβ (2.5 ng/mL). (F) Protein lysates of 4T1 cells treated with 10 μM GC7 or control and 2.5 ng/mL TGFβ or control for 72 or 96 h were assessed for total protein content using Ponceau S staining.
We next sought to assess the effects of serum and/or exogenous fibronectin on the efficacy and potency of GC7. As reported in Fig. 2C, removing serum from culture media rendered GC7 nearly 8-fold more potent in both 4T1 and MCF10CA1a cells. Notably, fibronectin further increased the GC7 potency by 2-fold in these cells. In contrast, while serum did make the 67NR cells less sensitive to GC7 treatment, the GC7 efficacy was reduced in the absence of serum (Fig. 2C).
Finally, we sought to evaluate the role of TGFβ in regulating eIF5A1/2 hypusination and response to GC7 inhibition of eIF5A1/2-dependent protein translation. First, we establish that 4T1, 67NR and MCF10CA1a cells are all responsive to TGFβ and undergo both an EMT morphology shift and/or downregulation of E-Cadherin (Fig. 2D). Since maximal canonical and non-canonical TGFβ signaling activities occur on the order of 5–60 min in most cell types, we next tested whether TGFβ stimulation could affect basal levels of hypusinated eIF5A1/2. As shown in Fig. 2E, both high-grade, metastatic 4T1 and MCF10CA1a cells responded positively to TGFβ stimulation with modest increases in hypusinated eIF5A1/2 levels. In contrast, the non-metastatic 67NR cells failed to elicit this same response profile (Fig. 2E). As a result, we reasoned that TGFβ may be able to counter the translation inhibitory effects of GC7 treatment. Indeed, Fig. 2F demonstrates that pre- and concurrent-treatment of 4T1 cells with TGFβ modestly rescues protein levels in cell populations treated with a sub-maximal GC7 dose for 96 h.
2.3. Inhibition of eIF5A Hypusination Reduces TGFβ/Fibronectin-Induced Metastatic Dissemination
We previously reported on the use of the Chicken Chorioallantoic Membrane (CAM) assay as a means to quantitatively evaluate cross-talk between TGFβ and the extracellular matrix in driving metastatic spread of breast and pancreatic cancers [11,12] (Fig. 3A). Furthermore, this work demonstrated that MCF10CA1h cells could be induced to undergo metastasis in this model by pre-treatment/incubation with TGFβ and exogenous fibronectin [4]. Using this model, we have observed that, while GC7 treatment of TGFβ/fibronectin-MCF10CA1h cell xenografts did not affect primary tumor mass (Fig. 3B), metastatic dissemination of these cells to lung tissues was completely blocked (Fig. 3C).
Fig. 3.

Inhibition of eIF5A Hypusination Reduces TGFβ/Fibronectin-Induced Metastatic Dissemination. (A) Schematic of the chicken embryo chorioallantoic membrane (CAM) assay for quantitative detection of MCF10CA1h breast cancer metastasis. (B) Primary tumors were isolated and weighed to obtain tumor mass in eggs treated with GC7 or water control. (C) Lung and liver tissues were analyzed for metastatic dissemination of primary tumor cells via real-time PCR against human Alu repeats in relation to background chicken gapdh signal. Representative images of primary tumors, chicken lungs, and chicken livers after dissection and prior to analysis. *indicates p-value <0.05. (Scale bars: 0.5 cm for primary tumor, 1.0 cm for lung and liver).
2.4. The DHPS-EIF5A1/2-PEAK1-EMT axis is associated with Diminished Survival in Breast Cancer Patients Harboring Genomic Alterations in TP53
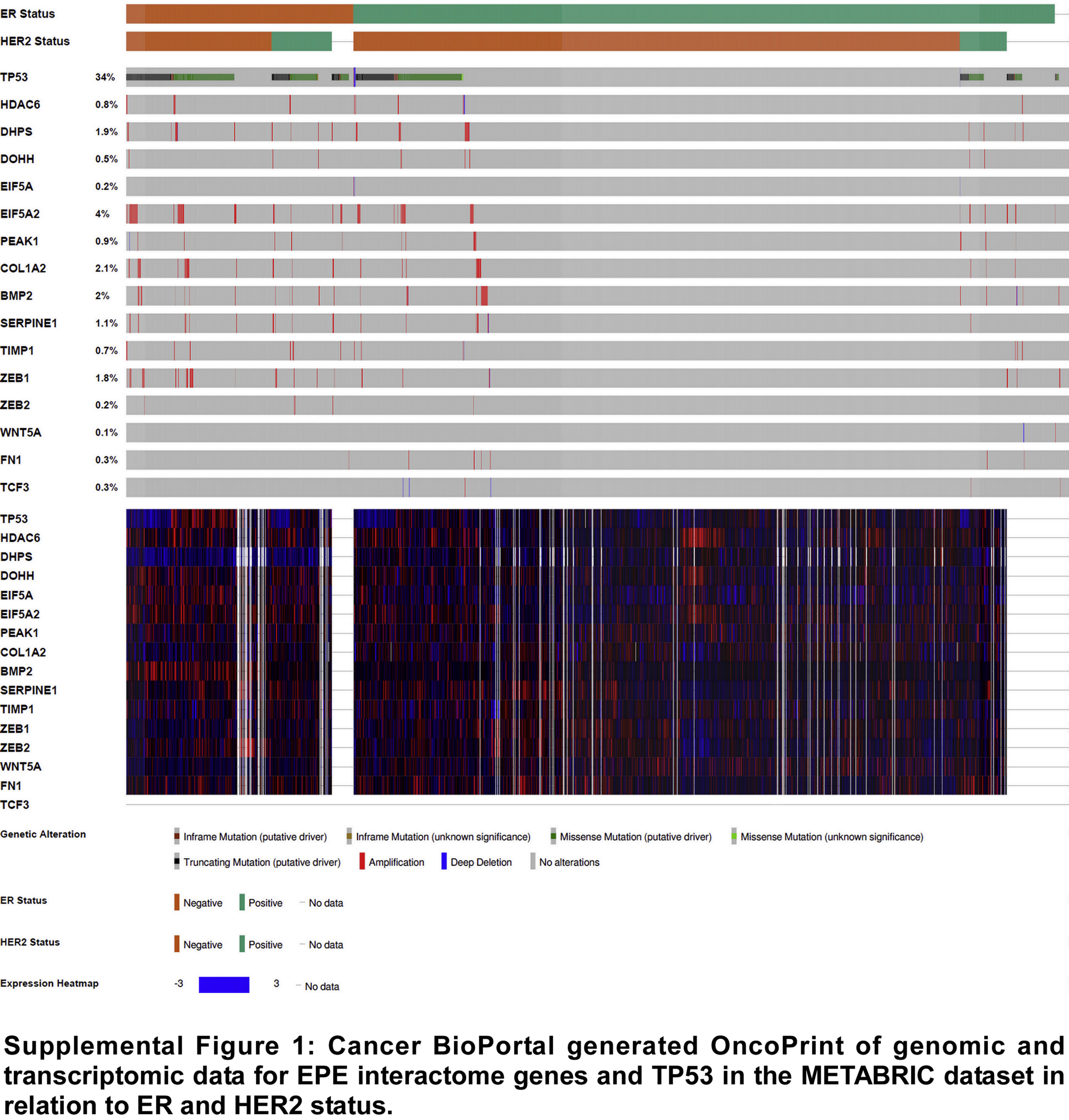
To identify potential regulatory networks across eIF5A1/2 and/or PEAK1-EMT pathways, we generated a literature-based list (termed “EPE Genes”) of these pathway members (Fig. 4A). EPE Genes were used to generate a literature-based interactome using Cytoscape Agilent Literature Search module (Fig. 4B). EPE list member present in the interactome (Fig. 4A) were then analyzed in concert for predicted transcription factor regulation enrichment using the DAVID bioinformatics resource (Fig. 4C). Using Cancer BioPortal, we further analyzed genetic mutations in patient samples that co-occur with genomic alterations in these EPE interactome genes. Notably, EPE interactome gene amplification events were significantly enriched in patients harboring TP53 mutations – a genomic hallmark for poor prognosis (Fig. 4D and Supplemental Fig. 1) [13]. We further observed a significant decrease in median survival of patients harboring amplification of the DHPS gene and/or TP53 mutations versus patients harboring simply DHPS amplifications (Fig. 4E) and noted nuclear localization of TP53 in breast cancer patient tissues with elevated DHPS expression (Fig. 4F) [14].
Fig. 4.

The DHPS-EIF5A1/2-PEAK1-EMT Axis is Associated with Diminished Survival in Breast Cancer Patients Harboring Genomic Alterations in TP53. (A) Schematic representing the bioinformatics work flow for identifying new regulatory axes/nodes and related clinical significance of eIF5A-PEAK1-EMT (EPE) pathway genes. (B) Cytoscape interactome using the EPE gene set with interacting query genes highlighted in yellow. Relevant mutations in PI3KCA and TP53 – see (D) below – are emphasized as enlarged nodes. (C) The top-10 transcription factors predicted to regulate EPE genes present in the interactome – identified using the DAVID platform (B). (D) Genomic alterations associated with EPE interactome genes in breast cancer patients - * indicates significance of q < 0.05. (E) Survival of breast cancer patients with and without alterations in the indicated genes obtained from the METABRIC dataset available on Cancer BioPortal. (F) IHC stains against the indicated proteins in breast tumor samples obtained from the Human Protein Atlas Pathology Atlas (Scale bars: 100 μm; inset, 50 μm).
3. Discussion
Herein, we have identified a new targetable axis that may be harnessed to thwart the protumorigenic effects of TGFβ signaling, reduce metastatic burden and improve outcomes in breast cancer patients with genomic alterations in TP53. Previous work characterizing mechanisms by which TGFβ signaling activates tumorigenic activities has been primarily focused on SMAD-dependent and -independent kinase signaling cascades upstream of either transcriptional [15] and/or cytoskeleton [16] regulatory nodes. More recently, several noteworthy molecular switches (including PEAK1 by our group) have been implicated in converting TGFβ signaling from tumor suppressive to tumor promoting [6]. Still, little is known about whether TGFβ can regulate translation machinery, and if so, how that may occur in the context of inactivating TP53 alterations [17]. In this regard, we report evidence that TGFβ can induce post-translational hypusination/activation of eIF5A1/2 (Fig. 2E–F) and that inhibition of eIF5A1/2 hypusination is an efficacious method for abrogating TGFβ-induced breast cancer metastasis (Fig. 3). It will be relevant for future work to define the molecular mechanism by which TGFβ induces eIF5A1/2 hypusination in normal and disease states.
Recent work has highlighted the importance of epithelial-mesenchymal plasticity and transition states as essential for tumor cells to navigate the varied microenvironments they encounter throughout the metastatic cascade [3]. Recently, E-Cadherin – traditionally associated with an epithelial, non-metastatic cell state – has been demonstrated to be required for metastasis [18]. Interestingly, we observed that GC7-mediated inhibition of DHPS-dependent hypusination/activation of eIF5A1/2 was able to more effectively reduce PEAK1 and E-Cadherin expression levels in the non-metastatic 67NR breast cancer cells – at doses paralleling those that effectively inhibited hypusinated eIF5A1/2 (Fig. 2B). This suggests that DHPS-eIF5A1/2 signaling and subsequent PEAK1/E-Cadherin expression may have more robust compensatory mechanisms within metastatic cancer cells that support cell state plasticity needed for metastasis.
Taken together with our identification of prognostically-significant clinical links between DHPS, eIF5A1/2, PEAK1/EMT and TP53 mutations, this work collectively supports future studies aimed at determining potential tumor suppressor activities that may be enhanced by targeting the DHPS-eIF5A1/2 pathway as well as identifying additional PEAK1-dependent vulnerabilities in mutant TP53 cancer types.
4. Materials and methods
4.1. Cell culture
MCF10A, MCF10AT1k, MCF10AC1h and MCF10CA1a cells were purchased from the Karmanos Cancer Institute (Detroit, MI). MDA-MB-231, MDA-MB-468, 4T1 and 67NR cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). All cells were cultured according to source instructions. Dulbecco’s Modified Eagle’s/Ham’s Nutrient Mixture F-12 (DMEF12) growth media (Genesee Scientific, San Diego, CA), horse serum (GE Healthcare Life Sciences, Logan, UT), insulin (Thermo Fisher Scientific, Waltham, MA), human recombinant EGF (Corning, Bedford, MA), hydrocortisone (BD Biosciences, Bedford, MA), cholera toxin (Enzo Life Sciences, Farmingdale, NY), penicillin/streptomycin (Thermo Fisher Sci.) and gentamycin (GE Healthcare Life Sciences) were used as previously indicated.
4.2. Cell treatments
Ciclopirox olamine (CPX), an iron chelator that inhibits the hypusination enzyme deoxyhypusine hydroxylase was obtained from Santa Cruz Biotechnology (Dallas, TX) and diluted in dimethyl sulfoxide (DMSO). N1-guanyl-1,7-diamineheptane (GC7), a spermidine analog that inhibits the activity of the hypusination enzyme deoxyhypusine synthase, was obtained from Biosearch Technologies (Petaluma, CA) and diluted in sterile water. Recombinant human TGFβ1 was diluted in 0.1% BSA (Thermo Fisher Scientific, Waltham, MA) and used at a working concentration of 2.5 ng/mL. Control treatments used DMSO (in place of CPX), sterile water (in place of GC7) or 0.1% BSA (in place of TGFβ). Fibronectin (Corning, Bedford, MA) coating was performed by treating plates with 3 μg/mL fibronectin in sterile PBS at 37 °C for 1 h, followed by one wash with sterile PBS prior to cell plating.
4.3. Chicken Chorioallantoic Membrane assay (CAM)
Rhode Island Red eggs were obtained from Meyer Hatchery (Polk, OH) and incubated in Digital Incubator & Automatic Egg Turner chambers (Incubators NZ) at 38 °C and 60% humidity for 10 days. MCF10CA1h cells were plated on fibronectin-coated plates and one day later, TGFβ1 (PeproTech, Rocky Hill, NJ) was added at a working concentration of 2.5 ng/mL. Four days after plating, cells were trypsinized and pelleted prior to re-suspension in Matrigel® (Corning) at 1E+06 cells/20 μL Matrigel on ice using chilled pipette tips. Drug treatments were added to Matrigel resuspensions just prior to xenografting. Cell xenografting onto the chicken CAM was performed on day 10 after dropping the CAM and exposing the CAM by cutting a 2.5 cm × 2.5 cm window into the egg shell. Ice-chilled cell resuspensions in Matrigel® were added to the CAM following addition of drugs and sterile surgical tape was used to cover the egg shell window. Eggs were replaced with window facing up into the egg chamber, in which the egg turner function had been disabled. On day 17, primary tumors were removed, weighed, and immediately flash frozen using dry ice-ethanol. Chickens were extracted from the eggs, decapitated, and liver and lung tissues were excised, weighed and flash frozen. Tissue samples were stored at −80 °C until further processing for genomic DNA extraction. Genomic DNA was isolated from 20 mg of each tissue using the GeneJet Genomic DNA Purification kit (Thermo Fisher Sci.). Homogenization was performed using micro-homogenizers (Claremont BioSolutions, Upland, CA) in digestion solution, followed by heat incubation at 56 °C for 3.5 h gDNA concentration was measured using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Sci.) and stored at −20 °C until qPCR analysis.
4.4. Cell proliferation/survival assay
Cells were plated at 5E+03 cells/mL with 200 μL/well in 96-well plates and allowed to attach overnight. 24 h after plating, cells were treated with serially diluted drugs as indicated. 72 h after plating, 40 μL of CellTiter 96® AQueous One Solution (Promega, Madison, WI) was added to each well and absorbance readings at 490 nm wavelength were taken on a Spectra Max 190 plate reader (Molecular Devices, San Jose, CA) at 1.5,2.0 and 3.0 h after AQueous One addition. Data were imported into GraphPad Prism (v. 7.04) and used to generate survival plots and determine IC50.
4.5. Quantitative polymerase chain reaction
Cell lines were plated in 6-well plates (Thermo Fisher Sci.) at le5 cells/mL and allowed to attach overnight. At the indicated time points, cells were trypsinized (0.25% trypsin (Thermo Fisher Sci.) in PBS), collected and centrifuged to obtain cell pellets. RNA was isolated using the GeneJET RNA Purification Kit (Thermo Fisher Sci.) and reverse transcribed to cDNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Sci.). Primers for EIF5A1, EIF5A2, HPRT1, and POLR2A were obtained from Integrated DNA Technologies (IDT, San Jose, CA). HPRT1 and POLR2A were used as house-keeping genes. Quantitative PCR (qPCR) reactions were set up using the Maxima SYBR Green/ROX qPCR kit (Thermo Fisher Sci.) and Abgene SuperPlate 96-well plates (Thermo Fisher Sci.). qPCR reactions were performed on the ABI 7300 system (Thermo Fisher Sci.) running SDS software (vl.3.1) using the following stages: stage 1 at 50 °C for 2 min for one cycle; stage 2 at 95 °C for 10 min for one cycle; stage 3 at 95 °C for 15 s and 62 °C for 1 min repeated for 40 cycles. Cycle threshold (Ct) values were obtained using the SDS software and used to determine mRNA expression levels relative to house-keeping genes via the Relative Quantification (RQ) method. For gDNA samples from CAM tissues, 56.25 ng of gDNA was used as template for detection of human Alu repeats to detect metastasis and chicken GAPDH as internal control. Primers against human Alu and chicken GAPDH were purchased from IDT and used at working concentration of 500 nM.
4.6. Western blot
Cells were plated on 6-well plates at 1E+05 cells/mL and one day later treated as indicated. Total protein lysates were collected using RIPA buffer with Pierce™ Protease and Phosphatase Inhibitor Mini Tablets (Thermo Fisher Sci.) added. Lysis was performed under rotation at 4 °C for 3 h. Lysates were then cleared by centrifugation at 12 000 RPM at 4 °C for 10 min. Protein concentrations were determined using the Coomassie Protein Assay Reagent (Thermo Fisher Sci.) via Bradford Assay at 595 nm absorbance. 20 μg of total protein was loaded for each sample into NuPAGE™ 4–12% Bis-Tris gels (Thermo Fisher Sci.) and separated electrophoretically in NuPage LDS Running Buffer (Thermo Fisher Sci.). Spectra™ Multicolor Protein Ladder (Thermo Fisher Sci.) was loaded as protein size standards. Gels were then transferred using Pierce® Transfer Buffer (Thermo Fisher Sci.) onto Amersham™ Protran™ nitrocellulose membranes (GE Healthcare Life Science) at 30 V and 4 °C for 1 h. Following transfer, total protein was visualized using PonceauS staining (Sigma-Aldrich, St. Louis, MO), blocked using 5% non-fat milk powder in TBST and exposed to primary antibody solutions diluted in blocking solution at 4 °C overnight: PEAK1 (Millipore, Burlington, MA; 1:400), E-cadherin (Cell Signaling Technology, Danvers, MA; 1:1000), Hypusine (Thermo Fisher Sci.; 1:1000), eIF5A1/2 (Thermo Fisher Sci.; 1:1000) and β–actin (Pro-Sci Inc., Poway, CA; 1:1000). HRP-conjugated goat anti-mouse/rabbit Pierce® secondary antibodies (Thermo Fisher Sci.) were diluted 1:10 000 in blocking solution and applied at room temperature for 1 h. Protein bands were visualized using Pierce™ ECL Western Blotting Substrate kit (Thermo Fisher Sci.) and autoradiography film (Genesee Sci.) with exposures ranging from 10 s to 30 min. Films were developed using a Medical Film Processor SRX-101 A (Konica Minolta, Inc., Tokyo, Japan) and scanned on an Officejet Pro 8600 (HP, Palo Alto, CA) for protein band quantification in Fiji (v.1.51n).
4.7. Phase-contrast imaging
Cells were plated as described above for cell proliferation/survival assays and imaged at the indicated time points using a Leica DMI-6000 B inverted phase-contrast microscope. A minimum of three biological replicates were analyzed and representative images were selected to represent relevant conclusions.
4.8. Bioinformatics
We generated a list of genes known to be involved in eIF5A1/2 signaling and PEAK1-EMT regulators. The resulting list of eIF5A-PEAK1-EMT (EPE) genes were used to generate an interactome map using Cytoscape. Interactome EPE genes were evaluated for regulation by transcription factors using DAVID and genomic amplification co-occurrence with gene mutations in breast cancer patients using the largest breast cancer study data set (METABRIC) available on Cancer BioPortal. Using the same cBioPortal data set, correlations between DHPS alterations alone or in combination with TP53 mutations and patient survival were also analyzed via Kaplan-Meier survival plots.
4.9. The Human Protein Atlas
Images of immunohistochemically stained tumor tissue sections were obtained from the Human Protein Atlas using indicated patients.
4.10. Statistical analyses
All statistical analyses were performed in GraphPad Prism with a p-value threshold of 0.05 for significance, adjusted for multiple testing where appropriate and as indicated.
Supplementary Material
{kind=link}
Acknowledgements
We also thank members of the Developmental Oncogene Laboratory at California State University, Northridge for their input on this manuscript. This work was funded in part by the California State University Northridge College of Science and Mathematics, the Sidney Stern Memorial Trust, the Sutter family, NIH NIGMS SC1GM121182 (to J.A.K.), NIH NIGMS T34GM008395/R25GM063787 (to M.E.Z. for J.M. and S.L.K.), and NIH NIGMS 2UL1GM118976 (to C.L.S et al. for L.K).
Footnotes
Conflicts of interest
None.
Transparency document
Transparency document related to this article can be found online at https://doi.org/10.1016/j.bbrc.2019.09.075.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbrc.2019.09.075.
References
- [1].Waks AG, Winer EP, Breast cancer treatment: a review, J. Am. Med. Assoc 321 (2019) 288–300. [DOI] [PubMed] [Google Scholar]
- [2].Yuan S, Norgard RJ, Stanger BZ, Cellular plasticity in cancer, Cancer Discov. 9 (7) (2019. July) 837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kroger C, Afeyan A, Mraz J, et al. , Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 7353–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Agajanian M, Campeau A, Hoover M, et al. , PEAK1 acts as a molecular switch to regulate context-dependent TGFbeta responses in breast cancer, PLoS One 10 (2015), e0135748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Agajanian M, Runa F, Kelber JA, Identification of a PEAK1/ZEB1 signaling axis during TGFβ/fibronectin-induced EMT in breast cancer, Biochem. Biophys. Res. Commun 465 (2015) 606–612. [DOI] [PubMed] [Google Scholar]
- [6].Yeh HW, Lee SS, Chang CY, et al. , A new switch for TGFbeta in cancer, Cancer Res. 79 (2019) 3797–3805. [DOI] [PubMed] [Google Scholar]
- [7].Strnadel J, Choi S, Fujimura K, et al. , eIF5A-PEAK1 signaling regulates YAP1/TAZ protein expression and pancreatic cancer cell growth, Cancer Res. 77 (2017) 1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fujimura K, Wright T, Strnadel J, et al. , A hypusine-eIF5A-PEAK1 switch regulates the pathogenesis of pancreatic cancer, Cancer Res. 74 (22) (2014. November 15) 6671–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Runa F, Adamian Y, Kelber JA, Ascending the PEAK1 toward targeting TGFβ during cancer progression: recent advances and future perspectives, Cancer. Cell. Microenviron 3 (1) (2016), e1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nakanishi S, Cleveland JL, Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer, Amino Acids 48 (2016) 2353–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gharibi A, La Kim S, Molnar J, et al. , ITGA1 is a pre-malignant biomarker that promotes therapy resistance and metastatic potential in pancreatic cancer, Sci. Rep 7 (2017) 10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kelber JA, Reno T, Kaushal S, et al. , KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer, Cancer Res. 72 (2012) 2554–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pereira B, Chin SF, Rueda OM, et al. , The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes, Nat. Commun 7 (2016) 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Uhlen M, Oksvold P, Fagerberg L, et al. , Towards a knowledge-based human protein Atlas, Nat. Biotechnol 28 (2010) 1248–1250. [DOI] [PubMed] [Google Scholar]
- [15].Hao Y, Baker D, Ten Dijke P, TGF-beta-Mediated epithelial-mesenchymal transition and cancer metastasis, Int. J. Mol. Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lamouille S, Xu J, Derynck R, Molecular mechanisms of epithelial-mesenchymal transition, Nat. Rev. Mol. Cell Biol 15 (2014) 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Morrison CD, Schiemann WP, Tipping the balance between good and evil: aberrant 14-3-3zeta expression drives oncogenic TGF-beta signaling in metastatic breast cancers, Breast Cancer Res. 17 (2015) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Padmanaban V, Krol I, Suhail Y, et al. , E-cadherin is required for metastasis in multiple models of breast cancer, Nature 573 (7774) (2019. September) 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.