

Summary
Innate lymphocytes maintain tissue homeostasis at mucosal barriers, with group 2 innate lymphoid cells (ILC2s) producing type 2 cytokines and controlling helminth infection. While the molecular understanding of ILC2 responses has advanced, the complexity of microenvironmental factors impacting ILC2s is becoming increasingly apparent. Herein, we used single cell analysis to explore the diversity of gene expression among lung lymphocytes during helminth infection. Following infection, we identified a subset of ILC2s that preferentially expressed Il5 encoding interleukin (IL)-5, together with Calca encoding calcitonin gene related peptide (CGRP) and its cognate receptor components. CGRP in concert with IL-33 and neuromedin-U (NMU) supported IL-5 but constrained IL-13 expression and ILC2 proliferation. Without CGRP signaling, ILC2 responses and worm expulsion were enhanced. Collectively, these data point to CGRP as a context dependent negative regulatory factor that shapes innate lymphocyte responses to alarmins and neuropeptides during type 2 innate immune responses.
Keywords: Host defense, immunoregulation, innate lymphoid cells, neuropeptides, cytokines, single cell RNA-seq
Graphical Abstract
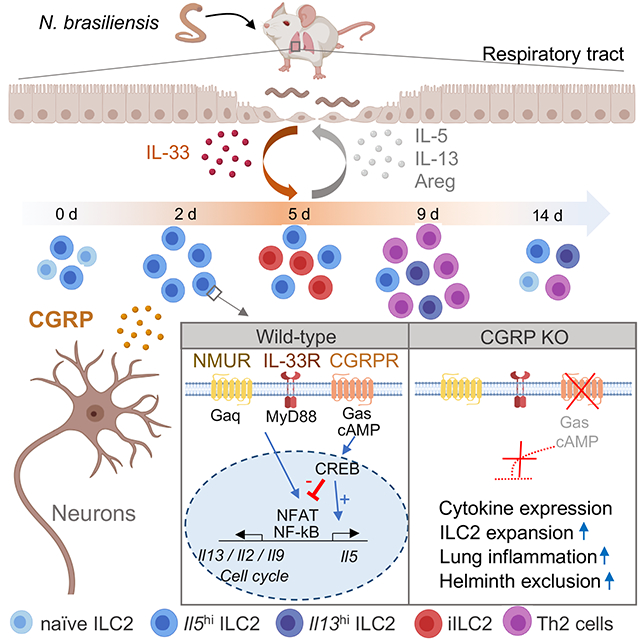
eTOC Blurb
Neuronal and immune systems coordinately orchestrate responses at mucosal barriers. Nagashima et al applied scRNA-seq technology to track type 2 immune responses in worm infection, identifying neuropeptide CGRP as a factor that modulates inflammation. The study suggests that CGRP may be a useful target in type 2 inflammation.
Introduction
The vertebrate immune system comprises innate and adaptive arms that use distinct modalities to perceive and respond to microbial insults. The former employs genetically conserved, hardwired pathogen recognition receptors whereas the latter employs clonotypic antigen receptors generated by DNA rearrangement. More recently, the innate lymphoid cell (ILCs) family has been identified and shown to act in concert with conventional T and B cells to orchestrate host defense, barrier integrity, homeostasis and tissue repair (Artis and Spits, 2015; Diefenbach et al., 2014; Eberl et al., 2015; Morita et al., 2016; Sonnenberg and Artis, 2015; Spits and Cupedo, 2012).
The major difference between ILCs and T helper (Th) cells is their readiness to produce effector cytokines prior to encountering microbial pathogens (Robinette et al., 2015; Shih et al., 2016). While ILC precursors acquire their distinct accessibility of regulatory elements to drive cytokine expression in a developmental, stepwise fashion, naive Th cells exhibit markedly ‘inactive’ chromatin landscapes at effector loci, and dramatically acquire accessibility only after activation and final differentiation. This implies that tissue ILCs are conditioned via microenvironmental factors to rapidly respond to local demands in the tissue in which they reside.
Indeed, these tissue-specific environmental signals have a substantial impact on lymphocyte gene expression profiles beyond lineage per se (Rankin and Artis, 2018). Recently, many reports point to neural regulation of local immune responses at barrier tissues where lymphocytes reside in close proximity to dense neuronal networks. For example, ILC2s in the mouse gastrointestinal tract co-localize with cholinergic neurons that express the neuropeptide neuromedin U (NMU) and selectively express the NMU receptor 1 (NMUR1) (Cardoso et al., 2017; Klose et al., 2017; Wallrapp et al., 2017). Acting with the alarmin interleukin (IL)-33 and IL-25, NMU induces ILC2 proliferation and secretion of the type 2 cytokines, and promotes lung inflammation or expulsion of the gastrointestinal nematode Nippostrongylus brasiliensis (N. brasiliensis). Engagement of the β-adrenergic receptor on ILC2s counteracts ILC2 activation induced by helminth and fungi, serving as a cell-intrinsic negative regulator of ILC2 responses (Moriyama et al., 2018). These emerging findings of neural-immune crosstalk are collectively referred to as neuroimmune cell units (Veiga-Fernandes and Pachnis, 2017).
One such neuropeptide reported to regulate immune cells is α-CGRP, a 37 amino acid neuropeptide produced as an alternatively spliced product of the calcitonin (Calca) gene (Holzmann, 2013; Russell et al., 2014). Secreted by pulmonary neuroendocrine cells (PNECs) in the lung, CGRP exacerbates experimentally induced asthma in mice via production of IL-5 in ILC2s (Sui et al., 2018). In contrast, CGRP produced by nociceptor neurons, termed transient receptor potential vanilloid (TRPV)1+ neurons, attenuates severity of Staphylococcus aureus-induced pneumonia by limiting neutrophils and γδ T cells in the lung (Baral et al., 2018). These results argue for both pro- and anti-inflammatory effects of CGRP on immune responses in the lung depending on the context of inflammation.
In the present study, we examined dynamic transcriptomic programs of lung lymphocytes during helminth infection using single cell RNA sequencing (scRNA-seq) to decipher microenvironmental signals received by lymphocytes. This analysis revealed that multiple subsets of innate and adaptive lymphocytes emerged with different kinetics during infection, including subsets of ILCs that preferentially expressed IL-5 versus IL-13. We identified the expression of the neuropeptide CGRP (Calca) and its cognate receptor enriched within an Il5hi subpopulation of ILC2s. CGRP, which mediated cyclic AMP (cAMP) production, antagonized many of the actions of NMU and IL-33 such as promotion of cell proliferation and IL-13 production, while selectively promoting IL-5 production. Without CGRP signaling, ILC2 responses were enhanced, facilitating worm expulsion in vivo. Collectively, CGRP is a crucial factor that coordinately shape the magnitude and complexity of type 2 innate response with other tissue signals. The complex interplay among neuropeptides, alarmin and cytokines may well be relevant to the clinical use of CGRP antagonists and could offer insights into therapeutic opportunities.
Results
Diverse populations of lung ILCs and T helper cells emerge during helminth infection
To gain insights into the type 2 responses developing during a model helminth infection, we inoculated mice with infective N. brasiliensis larvae, collected multiple fractions of Th cells (CD3+ TCRβ+ CD4+) and ILCs (Lin− CD3− TCRβ− Thy1+) from the lung, and analyzed gene expression by scRNA-seq. We also analyzed gene expression from pooled populations of Th2 cells (CD3+ TCRβ+ CD4+ ST2+) and ILC2s (Lin− CD3− TCRβ− CD4− Thy1+ CD127+ KLRG1+) from the same mice (Figure 1A–B and S1A–D) in which approximately 90% of ST2+ Th cells were transcription factor GATA3hi Foxp3− Th2 cells (Figure S1B). For single cell data, we aggregated all data points (Figure S1E) and identified 10 clusters in an unbiased manner based on differentially expressed genes (DEG) (Figure 1C), and inferred cluster identities based on DEG and marker gene expression (Figure 1C–D, S1F–J and Table S1). For T helper clusters, two overlapping populations of naïve T cells could be discerned (C0, C2) and a distinct population of active Th2 cells was readily apparent (C6). A population of T cells with transcriptomes shared by both naïve and active Th2 cells was noted and designated as “intermediate” cells (C5). Analysis of ILC populations revealed two different ILC2 populations; natural ILC2 (nILC2) (C1, 3 and 4) defined as Il1rl1 (ST2)+ KLRG1low ILC2 and inflammatory ILC2 (iILC2) (C7) defined as ST2− KLRG1hi ILC2 (Figure 1D and S1F) (Huang et al., 2015). nILC2 were further classified into resting (C1), Il13hi (C3) and Il5hi (C4) cells. The proportion of the different populations varied during the course of N. brasiliensis infection. While the naïve clusters declined, the active Th2 cell population peaked at day 9 and lasted until day 14 (Figure 1E). By contrast, nILC2 expressed the type 2 cytokine IL-5 at steady state; more than 50% of nILC2 were Il5hi cells. Following infection, Il5hi nILC2s increased, peaking early on day 2. This was followed by transient emergence of iILC2 (day 5) and later, expansion of Il13hi cells (day 9) (Figure 1F–G). The dynamic transition of ILC2 subpopulations and the differential time course of Il5 and Il13 expression measured by scRNA-seq was in line with previous studies using cytokine reporter mice (Huang et al., 2015; Ricardo-Gonzalez et al., 2018). Following infection, the proportion of innate versus adaptive populations of type 2 lymphoid cells shifted, with Th2 cells becoming dominant at day 9 (Th2: ~2x105 cells, ILC2s: ~3x104 cells) (Figure 1G–H and S1C–D). Furthermore, lung cell preparations included lymphatic endothelial cells (Shinoda et al., 2016) (C8) and NK or ILC1 (C9) (Figure S1I–J). Collectively, our single cell analysis reveals a heterogeneous type 2 immune response and dynamic transitions both in ILCs and Th cells during helminth infection.
Figure 1. Diverse populations of lung ILCs and T helper cells emerge during helminth infection.

(A and B) Experimental design for single cell RNA-seq (scRNA-seq) and bulk mRNA-seq of Th cells and ILCs from lungs of N. brasiliensis infected mice. Time course of N. brasiliensis infection experiment (A) and cell markers (B) for isolating total Th cells and total ILCs for scRNA-seq, or Th2 cells and ILC2s for bulk mRNA-seq. (C) Ten gene expression clusters (#0 - 9) projected on the t-Distributed Stochastic Neighbor Embedding (tSNE) plot of the scRNA-seq libraries from mice during N. brasiliensis infection as in A. Clusters, depicted by color, were identified by Seurat based on gene expression profiles in an unbiased manner, with manually labelling inferred cluster “identities”. For additional details, see Figure S1E–J. (D) Gene expression heatmap showing unbiased generation of top 10 differentially expressed genes for each cluster. Genes picked for multiple clusters are marked with asterisk. (E and F) Proportion of each cluster in the scRNA-seq library of Th cells (E) or ILCs (F) during the course of infection (day 0 to 14). (G-H) Expression of Il5 and Il13 evaluated by bulk mRNA-seq in ILC2s (G) and naïve Th cells and Th2 cells (H) isolated as shown in B. Data are from one experiment. See also Figure S1 and table S1.
CGRP and its cognate receptor are induced in ILC2s during helminth infection
Seven transmembrane (7TM) receptors are a large class of molecules whose ligands have profound effects on ILCs (Rankin and Artis, 2018). Using bulk mRNA-seq data, we evaluated the expression of 7TM receptors that might be relevant to the regulation of ILC2s, including receptors for chemokines, lipid metabolites, and neuropeptides (Figure S2A). Nmurl and Calcrl encode neuropeptide receptors Neuromedin U (NMU) receptor 1 and Calcitonin receptor like receptor (CLR), whose ligands, NMU and CGRP respectively, have been reported to promote ILC2 activation (Cardoso et al., 2017; Klose et al., 2017; Sui et al., 2018; Wallrapp et al., 2017). CLR forms a heterodimer with an accessory protein Ramp1, and Calcrl and Ramp1 genes were expressed in nILC2 clusters (C1, C3, C4) but not in inflammatory ILC2s (C7); whereas, Nmur1 was minimally detectable at single cell level (Figure 2A). CGRP receptor (Calcrl and Ramp1) expression increased at a later time point following infection (day 9), whereas Nmur1 expression in ILC2s was downregulated (Figure 2B).
Figure 2. CGRP and its cognate receptor are induced in ILC2s during helminth infection.

(A) Single-cell expression of receptors for NMU and CGRP in each cell cluster defined as in Figure 1C. (B) Kinetics of gene expression during N. brasiliensis infection in ILC2s for genes encoding receptors for NMU and CGRP using bulk mRNA-seq as in Figure 1B. (C-E) Calca expression in Th cells and ILCs using scRNA-seq (C-D) and confirmation by bulk mRNA-seq (E), as in A and B. (F) Immunohistochemical staining of lungs from Calca+/gfp mice 7 days after N. brasiliensis infection. The arrows depict ILC2s (CD3− KLRG1+) that express Calca (GFP+). The scale bar is 100 μm. (G) Calca expression in ILC2s from lung, mesentery and mesenteric LNs (mLNs) of Calca+/gfp mice before (0) and after N. brasiliensis infection (days 2 and 7). (H) Calca-driven expression of GFP in ILC2s from indicated tissues of Calca+/gfp mice, assessed by flow cytometry. (I) Accessibility of indicated loci in various cell types, as measured by ATAC-seq (data from Shih H et al. 2016: GSE77695). Data are from one experiment (A-E) or representative from three independent experiments with similar results (F-H). See also Figure S2.
Not only we observed the regulated expression of CLR and Ramp1, we also found that the gene encoding CGRP (Calca), the ligand for this receptor, was highly expressed and dynamically regulated in ILC2s. At single cell levels, Calca was enriched in Il5hi ILC2 (C4), and to a lesser extent in activated Th2 cell fraction (C6) and in other ILC2 clusters (C1 and C3) (Figure 1D and 2C–D). With bulk mRNA-seq, we confirmed high expression of Calca mRNA (~500 FPKM) even in steady state ILC2s and peaking at day 2 after infection (~2000 FPKM). Calca expression waned quickly thereafter (Figure 2E), mirroring Il5 transcription more closely than Il13 (Figure 1D and 1F–G).
Using Calca reporter mice, we evaluated populations of hematopoietic cells expressing Calca in vivo. Approximately 50% of Calca expressing cells were ILC2s in the lung in the steady state; although other non-ILC2 populations (e.g. TCRβ+ T cells) were detected within GFP-positive cells (Figure S2B). Calca expression in ILC2s was further upregulated by intranasal administration of IL-33 or N. brasiliensis infection (Figure 2F–G and S2C–D). ILC2s from tissues other than the lung also expressed Calca including the small intestine, colon, and visceral adipose tissue (VAT). Calca expression was barely detectable in mesenteric ILC2s (Figure 2H), indicating that Calca expression in ILC2s is differentially controlled in a tissue-specific manner.
To gain insight into regulation of the Calca gene, we analyzed prior ATAC-seq data generated by our group (Shih et al., 2016) to evaluate accessibility of the Calca locus during ILC2 development or in N. brasiliensis-infected Th2 cells. The Calca locus acquired accessibility at the ILC2 precursor stage similar to Il5 and but was inaccessible in ILC1s, ILC3s and naïve T cells (Figure 2I). The Calca locus became accessible in Th2 cells induced by N. brasiliensis infection. The results indicate that Calca expression is selectively regulated in type 2 immune cells; as with other ILC loci, accessibility occurs in a developmental manner prior to acute activation in ILC2.
CGRP and NMU have divergent effects on ILC2 gene expression
To evaluate the functional outcome of neuropeptide signaling, we stimulated isolated lung ILC2s with the alarmin IL-33 alone or in combination with NMU or CGRP, and assessed IL-5 and IL-13 proteins by flow cytometry (Figure 3A–B). NMU cooperatively enhanced IL-5 and IL-13 expression with IL-33. On the other hand, CGRP exerted contrasting effects on IL-5 and IL-13, enhancing IL-5 and repressing IL-13 production mediated by IL-33. These results are in line with scRNA-seq data showing differential expression of IL-5 and IL-13 in ILC2s following helminth infection (Figure 1C–G), suggesting that CGRP may take part in controlling cytokine expression in vivo.
Figure 3. Differential effects of CGRP and NMU on ILC2 gene expression.

(A) Flow cytometric assessment of intracellular IL-5 and IL-13 expression in lung ILC2s in the absence of stimulation (−) or following stimulation with IL-33 alone, or in combination with CGRP or NMU for 4 hr with inclusion of Brefeldin A. (B) Pooled data showing percentage of IL-13 positive (left) or IL-5 positive (right) ILC2s cultured with indicated neuropeptide and/or cytokine as in A. (C) Principal component (PC) analysis of gene expression using bulk mRNA-seq of lung ILC2s cultured under the indicated 7 conditions for 4 hr. Two replicates were analyzed per condition. (D) Heatmap depicting 958 differentially expressed genes (DEG) (> 3 log2 fold change and > 10 FPKM expression in at least one sample) in samples shown in C. Four groups of genes were defined using hierarchical clustering. (E) Comparison of gene expression in ILC2s stimulated with IL-33+NMU versus IL-33+CGRP. Representative DEGs are depicted. (F) Expression of selected cytokine genes across various conditions. (G) Isolated ILC2s (5 x 103) were cultured under the indicated culture conditions for 5 days and cell counts were recorded. (H and I) The concentration of the indicated cytokines in the supernatant from cultures as in G, assessed by Legendplex (H) or ELISA (I). (J) Transcript expression of Calca and receptors for neuropeptides across conditions, quantitated by mRNA-seq (dataset shown in Figure 3C–D). Statistical significance are depicted as *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test). Data are from two (C and D) or average (E, F and J) from two independent experiments or representative (A, G, H, I) or pool (B) from three independent experiments with similar results. (Average ± SD from three experiments (B) or triplicated samples (G, H and I)). See also Figure S3 and table S2.
To gain a more comprehensive view of the direct effects of CGRP and other factors on ILC2, we treated isolated lung ILC2s in vitro with neuropeptides and IL-33 and assessed transcriptomic changes using mRNA-seq (Figure 3C–F). IL-33, NMU and CGRP had very distinct global impacts on transcriptomic output (Figure 3C), with a total of 958 genes being differentially regulated by IL-33, CGRP and NMU, classified into 4 groups based on expression profiles (Figure 3D and Table S2). In line with the reported ability of CGRP to activate ILC2s (Sui et al., 2018), we identified 116 genes that were selectively induced by CGRP (group 3). On the other hand, a suppressive action of CGRP was also evident on genes otherwise induced by NMU and IL-33 (97 and 265 genes in group 1 and 4, respectively). We also identified a group of 350 genes whose expression was higher before stimulation and downregulated with each stimulation tested (group 2).
We next analyzed the combinatorial effects of CGRP, NMU and IL-33 on effector cytokines in more detail (Figure 3F). We found that while CGRP inhibited Il2, Il9, Il4, Il6 and Il13 expression induced by IL-33 or IL-33 + NMU, CGRP enhanced Il5 in combination with IL-33 (Figure 3F), consistent with discordant regulation of IL-5 and IL-13 protein by CGRP (Figure 3A–B). Areg was another gene strongly induced with IL-33 + CGRP and it was further enhanced by addition of NMU (Figure 3F).
Since CGRP has previously been reported to inhibit T cell proliferation (Umeda et al., 1988), we tested the effects of IL-33, CGRP and NMU on ILC2 proliferation. Whereas the combination of IL-33 and NMU strongly induced proliferation, CGRP suppressed proliferation induced by IL-33 and the combination of IL-33 and NMU with both in vitro expanded ILC2 (Figure 3G and S3A) and fresh ex vivo ILC2 (Figure S3B). Even though CGRP induced IL-5 on a per cell basis (Figure 3A–B and 3F), the total amount of IL-5 in supernatants of cultures of ILC2s (5 day) was reduced by CGRP because of its anti-proliferative effect (Figure 3H); this was also the case for amphiregulin (Figure 3I). Although NMU did not induce Areg mRNA in the setting of short stimulation (4h), 5 days culture with NMU strongly enhanced production of amphiregulin protein, suggesting that NMU elicited indirect positive control over Areg through induction of several STAT5 transcription factor activating cytokines such as IL-2 and IL-9, all of which strongly enhance type 2 cytokine expression (Figure 3F and 3I) (Moro et al., 2010; Wilhelm et al., 2011). Notably, CGRP strongly induced Calca transcript compared with IL-33 alone. Moreover, NMU promoted the expression of CGRP receptor components (Calcrl and Ramp1), whereas it negatively regulated its own receptor, Nmur1 (Figure 3J), consistent with the in vivo observation during N. brasiliensis infection (Figure 2B). Collectively, our data indicate that NMU is a robust activator of ILC2 proliferation that cooperatively activates gene expression with IL-33, and especially impacts pro-inflammatory cytokines including IL-2, IL-6, IL-13 and IL-17F. In contrast, CGRP counteracts NMU by suppressing cell proliferation and gene expression of inflammatory cytokines. At the same time, CGRP dominantly activates a select gene module including Il5 and Areg, leading to a unique type 2 cytokine expression profile in ILC2s.
CGRP and NMU have differential effects on ILC2 regulatory elements
Having observed differential actions of CGRP and NMU on ILC2 gene expression and cytokine production, we next evaluated how genomic regulatory elements were controlled by these two neuropeptides using ATAC-seq (Figure 4). More than half of accessible regions (57% of peaks) were present before stimulation, but a large fraction of peaks (43%) were induced by neuropeptide or alarmin stimulation. Similar fractions (~10% of peaks) were uniquely affected by CGRP or NMU (Figure 4A). We next examined the frequency of opposing or independent actions of CGRP and NMU on chromatin accessibility (Figure 4B). We found that more NMU-induced peaks were suppressed by CGRP (n=4390 peaks, cluster A in Figure 4B) than the converse, i.e. CGRP-induced peaks suppressed by NMU (n=2291 peaks, cluster C). Under the conditions employed, this is consistent with a dominant effect of CGRP in counteracting the effect of NMU on transcription (Figure 3D).
Figure 4. Differential effects of CGRP and NMU on ILC2 regulatory elements.

(A) Venn diagram demonstrating percentages of 54,355 chromatin accessible regions identified by FastATAC-seq among ILC2s treated with or without IL-33, CGRP, and NMU for 4 hr. (B) Heatmap illustrating the chromatin accessibility among the dynamic regions shown in A, highlighting 4 categories of differentially accessible regions. Categories A and B represent NMU targeted regions that are independent (A) of, or antagonized (B) by CGRP, respectively. Categories C and D represent CGRP targeted regions that are independent (C) of or counteracted (D) by NMU, respectively. (C) Genomic track view of Th2 cytokine loci showing distinct regulation of chromatin accessibility of Il5 and Il13 loci across different conditions. (D) Heatmap showing relative enrichment of TF motifs within chromatin regions categorized in B. Data are from representative (C) of two independent experiments with similar results.
Given the opposing regulation of Il5 and Il13 by CGRP and NMU (Figure 2A–B and 2F), we examined the extended type 2 cell-associated cytokine loci for chromatin accessible regions. Individually, IL-33 and the neuropeptides had minor effects on Il5 and Il13, whereas the combination of IL-33 and NMU increased accessibility of both loci (Figure 4C). In contrast, the combination of IL-33 and CGRP selectively increased accessibility of Il5 without impacting Il13. Moreover, the combination of IL-33, NMU and CGRP reduced Il13 accessibility while preserving Il5 accessibility, in line with transcript or protein expression data for the two cytokines (Figure 2A–B and 2F).
Having observed key differences in the accessibility landscape induced by CGRP and NMU, we next sought potential transcriptional regulators of these differences. We identified several families of transcription factors whose motifs were enriched in chromatin regions selectively opened by NMU (bZIP, NFkB(RHD), Zinc finger, NFAT) versus CGRP (ETS, GATA, RUNX) (Figure 4D). This analysis of neuropeptide-dependent modulation of chromatin accessibility identified distinct transcription factors that likely underlie both the differential (Ill3) and coordinate (Il5) regulation of key regulated genes.
Signaling downstream of CGRP involves cAMP to shape unique gene expression profile manifested by IL-5
We next sought to clarify the modes of intracellular signaling downstream of CGRP and NMU in ILC2s. IL-33 induces activation of NF-κB, p38 and Erk, and NMU further promotes NFAT activation (Cardoso et al., 2017; Furusawa et al., 2013; Nagashima et al., 2018). We also observed that an NF-κB inhibitor completely abrogated cytokine production induced by IL-33 and neuropeptides (Figure S4A). A p38 inhibitor strongly suppressed the actions of IL-33 with or without NMU on cytokines, but had no effect on CGRP regulation of IL-5. Furthermore, CGRP had no effect on the nuclear translocation of NF-κB (p65) and NFAT1 transcription factors induced by IL-33 and NMU, indicating that CGRP-mediated repression was not mediated by interfering with these transcription factors (Figure S4B–C).
cAMP, a known CGRP second messenger in other cells (Russell et al., 2014), was highly induced in ILC2s by CGRP, but not by IL-33 and NMU (Figure 5A). To assess whether production of cAMP was sufficient to explain CGRP’s actions in ILC2s, we used a cell membrane permeable cAMP analogue. We found that dibutyryl-cAMP recapitulated the effects of CGRP on cell proliferation, and discordant regulation of IL-5 and IL-13 production induced by IL-33. (Figure 5B–D). One known downstream mediator of cAMP action is the transcription factor cAMP response element binding protein (CREB) (Russell et al., 2014). We observed that CREB phosphorylation was induced by CGRP and dibutyryl-cAMP but also by IL-33 and NMU (Figure 5E).
Figure 5. Signaling downstream of CGRP is mediated by cAMP to shape unique ILC2 gene expression profiles, including in vivo IL-5 expression.

(A) cAMP concentration in the cell lysates of lung ILC2s without stimulation (−) or stimulated with IL-33, CGRP or NMU (20 min) as assessed by ELISA. (B) Isolated ILC2 (5 x 103) were cultured under the indicated conditions for 5 days after which cell counts were recorded. cAMP = Dibutyryl-cAMP (C) Concentration of cytokines in the supernatant from cultures depicted in Figure 5B, assessed by Legendplex. (D) Expression of Il5 and Il13 in lung ILC2s cultured under the indicated conditions, confirmed by bulk mRNA-seq. Two replicates were analyzed per condition. (E) Phospho-CREB (Ser133) was measured by flow cytometric analysis of lung ILC2s following the indicated stimulations for 20 min. (F) Global transcriptomic similarities between the indicated conditions was evaluated by Pearson correlations calculated based on log2 (FPKM+1) from the average of replicates. Gene expression data for IL-33, IL-33+CGRP, IL-33+NMU or IL-33+cAMP conditions are from data depicted in Figure 3C and 5D. Gαqi = Gαq inhibitor (G) Inferred transcriptional modules in ILC2s regulated by IL-33 and neuropeptides. See also Figure S5A–B. (H) Single cell scores of representative transcriptional modules (IL-33, NMU, CGRP) in ILC2s from mice with N. brasiliensis infection as in Figure 1A. The gene modules were generated using bulk RNA-seq data from ILC2s treated with IL-33, NMU, or CGRP compared to unstimulated cells. IL-33, NMU, and CGRP module scores were projected onto separate tSNE representations of ILCs (from Figure 1C). Scores are comparable within a module across timepoints, but not across modules. (I) The ratio of Il5 to Il13 abundance (log-space) in all cells is shown as a function of module score thresholds. Statistical significance are depicted as *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test). Data are representative (A – C) from three independent experiments or representative (E) from two independent experiments with similar results. (Average ± SD from triplicated well in A – C). See also Figure S4–S5 and Table S3.
To evaluate the role for Gαq and CREB downstream of NMU and CGRP receptor, we next treated cells with inhibitors for these molecules. As anticipated, a Gαq inhibitor efficiently abrogated NMU-but not CGRP-dependent IL-5 and IL-13 production (Figure S4D–E). We also observed that a CREB inhibitor strongly suppressed CGRP induction of IL-5 and a modest effect on IL-33 and IL-33+NMU mediated induction. These data suggest that the CGRP-cAMP axis enhanced IL-5 production by activating CREB, which may be a shared component downstream of IL-33 and NMU that induces IL-5 expression (Figure S4D–E). However, NMU also acts through Gαq to activate NFAT, which in turn acts on the Il5, l13 and other relevant loci (Cardoso et al., 2017; Klose et al., 2017). Thus, consistent with ligand-induced chromatin landscape alterations, it appears that distinct pathways converge to positively regulate the Il5 locus.
To elucidate the global effects of cAMP and Gαq pathways downstream of CGRP and NMU, we compared the effect of neuropeptides on ILC2 transcriptome with cAMP or a Gαq inhibitor. We found that the dibutyryl-cAMP transcriptome and the CGRP transcriptome were relatively similar based on the Pearson correlation calculation (0.974) (Figure 5F). Similarly, the transcriptomic comparison between IL-33 alone and IL33+NMU+Gαq inhibitor was nearly identical (0.997), suggesting that most of NMU’s effects were completely abrogated by the Gαq inhibitor (Figure 5F).
To gain further insight underlying the biological processes distinctively regulated by CGRP and NMU, we performed pathway analysis on the modules selectively induced by these factors (Figure S5A). The enriched biological processes as well as representative genes associated with each module included: inflammatory (IL-17) response (group 1: Il17f), cell cycle regulation (group 2: Ccmd, Cdkn), immunoregulatory response (group 3: Areg, Ifngrl, Il10ra) and inflammatory (Th2) response (group 4: Il5, Il13, Il6) (Figure 3D and S5A). CREB was predicted to be the main regulator of the group 3 genes (Figure S5B). On the other hand, NF-κB and NFATc2 were enriched in the group 4 module cooperatively activated by NMU and IL-33 and constrained by CGRP. These effects are consistent with the view that CGRP acts predominantly via Gas and cAMP whereas NMU acts via Gαq (Klose et al., 2017; Russell et al., 2014).
Having defined the transcriptional modules regulated by NMU and CGRP that act through distinct signaling pathways (Figures 3 and 5), we sought to use this in vitro information to interrogate the in vivo dynamics of ILC2 responses as measured by scRNA-seq following helminth infection. Specifically, we scored each GATA3-expressing ILCs based on its expression of the IL-33, NMU (Gαq) and CGRP (cAMP) modules (Table S3) and used these scores as a proxy for signals sensed by cells (Figure S5C). The IL-33 and NMU (Gαq) modules both developed and peaked at day 5 following infection. In contrast, the CGRP (cAMP) module rapidly developed and peaked at day 2 (Figure S5C).
Next, we evaluated phenotypic heterogeneity across the ILC2 single-cell clusters by visualizing their expression of the IL-33, NMU and CGRP signature modules (Figure 5H). Both IL-33 (left panel) and NMU (Gαq) (middle panel) signatures were distributed across active ILC2 subsets, though both were modestly enriched in inflammatory ILC2. In contrast, the CGRP signature (which approximated a cAMP signature) (right panel) was highly enriched in the Il5hi ILC2 subset, suggesting that CGRP was most relevant in modulating this subset following helminth infection.
Having demonstrated the divergent regulation of IL-13 and IL-5 by CGRP versus NMU or IL-33 in vitro (Figure 3 and 4), we next examined the balance of IL-5 and IL-13 expression in ILC2s in vivo upon infection. Cells receiving IL-33 (Figure 5I: left panel) or NMU (Figure 5I: middle panel) signals maintained equivalent expression of Il5 and Il13 (shown in Y-axis). In contrast, cells receiving a stronger CGRP signal exhibited higher Il5 expression (Figure 5I: right panel). These results were in line with the kinetics of Il5 and Il13 expression as detected by single cell and bulk mRNA-seq (Figure 1F–G). Collectively, our data argue for a dynamic role of the CGRP-cAMP axis in ILC2 responses following helminth infection and in particular, the selective modulation of IL-5 and IL-13 and tuning type 2 immune responses during infectious challenge.
CGRP suppresses IL-33-mediated pulmonary inflammation and worm expulsion of N. brasiliensis
We next explored whether our in vitro observations were relevant to pulmonary host defense in vivo. To test this, we challenged Rag1−/− or Rag2−/− mice with intranasal administration of IL-33 to activate ILC2s and induce lung inflammation (Figure 6A). As expected, IL-33 induced severe lung pathology as evidenced by enhanced cell infiltration and goblet cell hyperplasia (Figure 6B–C), as well as increased numbers of eosinophils and ILC2s in the lung and bronchoalveolar lavage fluid (BALF). We found that CGRP significantly reduced IL-33-mediated inflammation as measured by all these parameters (Figure 6D–F).
Figure 6. CGRP is required for negatively regulating ILC2s and type 2 responses in vivo.

(A-F) Effect of CGRP in IL-33-induced pulmonary inflammation. (A) Rag1−/− or Rag2−/− mice were administered PBS (n=8), IL-33 (n=10), or IL-33+CGRP (n=10) intranasally for four consecutive days, with the following analyses of lung pathology one day after the last administration: (B) Periodic acid-Schiff (PAS) staining, (C) Pathology score, (D) Cell count of total cells, eosinophils (CD11c− CD11b+ Siglec-F+) and ILC2s (Lin− Thy1+ CD127+ GATA3+), (E and F) Cell counts (E) and cytokine production (F) in BALF. The scale bar in B is 100 μm. (G-I) Impact of genetic deletion of CGRP on helminth infection: Experimental scheme (G) and worm count of small intestine (H) or cell numbers of eosinophils, ILC2s and Th cells (I) in mLNs from aCGRP+/+ (n=5) or aCGRP−/− (n=5) mice infected with N. brasiliensis and analyzed at day 7 post infection. (J-M) Impact of genetic deletion of CGRP receptor subunit Ramp1 in transferred hematopoietic cells following helminth infection-experimental scheme (J). Fecal egg counts (K), small intestine worm counts (L) numbers of eosinophils, ILC2s and Th cells (M) in lungs were assessed in irradiated chimeric Rag2−/− gc−/− mice reconstituted with the bone marrow from either wild-type (n=5) or Ramp1 −/− (n=5) mice infected with N. brasiliensis on day 9 post infection. Each symbol represents an individual mouse (C-F, H-I, K-M). Statistical significance is depicted as *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test). Data are from representative (B, H, I) or pool (C-F) of two experiments with similar results or from one experiment (K-M). See also Figure S6.
Next, we challenged wild type and Calcagfp/gfp mice (which lack transcripts of both of Calcitonin and aCGRP) with IL-33 to assess the role of endogenous CGRP to control IL-33 induced lung inflammation (Figure S6A–C). Although Calcagfp/gfp mice had comparable numbers of eosinophils, ILC2s and Th cells at the steady state (Figure S6A), Calcagfp/gfp mice showed higher accumulation of ILC2s and Th cells in BALF following IL-33 intranasal administration (Figure S6B–C). Although there was a trend towards increased accumulation of eosinophils in Calcagfp/gfp mice, the difference with wild type mice did not attain significance (p=0.07) (Figure S6B–C). Collectively, these data indicated that CGRP is required to constrain IL-33-mediated ILC2 responses and subsequent inflammatory cell infiltration in vivo.
Next, we challenged wild type and aCGRP-deficient (aCGRP−/−) mice with N. brasiliensis to evaluate the role of endogenous CGRP to control helminth infection (Figure 6G). aCGRP−/− mice showed enhanced intestinal worm expulsion with increased numbers of ILC2s in mLNs (Figure 6H–I). The results indicate that in response to helminth infection endogenous production of CGRP is a negative regulator of ILC2 responses, consistent with our in vitro observations and challenge with an exogenous alarmin (IL-33).
Having observed that endogenous tissue CGRP acts to constrain ILC2 activation induced by helminth infection, we next asked whether genetic interference with CGRP signaling within hematopoietic cell compartments affects type 2 immune response. To this end, we used mice in which Ramp1, a component of CGRP receptor complex, was deleted. Irradiated Rag2−/− gc−/− mice were reconstituted with bone marrow from wild type or Ramp1−/− mice to specifically eliminate CGRP sensing on hematopoietic cells in the host. The reconstituted hosts were then challenged with helminth infection (Figure 6J). In mice reconstituted with Ramp1−/− bone marrow, we observed reduced numbers of eggs in the feces, as well as reduced worm burden in the small intestine along with enhanced ILC2 responses, but reduced Th2 cell response following N. brasiliensis infection (Figure 6K–M). Collectively, our results demonstrate that sensing of environmental CGRP signals by ILC2s leads to limited magnitude of their responses to infection in vivo.
Discussion
In this study, we used single cell transcriptomic analysis and identified heterogeneous populations of ILC2s, preferentially expressing either Il5 (Il5hi ILC2s) or Il13 (Il13hi ILC2s); both populations emerged during helminth infection, but with distinct kinetics. We also identified a neuropeptide, CGRP, which antagonized many actions of the neuropeptide NMU and the alarmin IL-33, but promoted IL-5 production. Accordingly, CGRP suppressed type 2 immune responses mediated by IL-33 or helminth infection, suggesting context dependent immunoregulatory effect of CGRP on ILC2 responses in vivo.
The neuro-immune interactions at peripheral barrier tissues are increasingly appreciated clinically and experimentally (Klose and Artis, 2018; Pavlov and Tracey, 2017; Veiga-Fernandes and Mucida, 2016) and led to the term, neuroimmune cell units (NICUs) to denote their functional interaction (Veiga-Fernandes and Pachnis, 2017). Interest in neuronal-lymphocyte crosstalk has been augmented by recent reports that neuropeptides directly impact tissue-resident mucosal innate immune cells. (Veiga-Fernandes and Artis, 2018). Specifically, cholinergic neurons adjacent to intestinal and pulmonary ILC2s produce the neuropeptide NMU, which acts via NMU receptor 1 (NMUR1) selectively expressed by ILC2s to induce the production of type 2 cytokines (Cardoso et al., 2017; Klose et al., 2017; Wallrapp et al., 2017). Sympathetic neurons also innervate these areas and their product, β adrenergic agonists, counteract the action of IL-33 and dampen ILC2 responses (Moriyama et al., 2018).
The data presented herein help to resolve some of the apparent contradictory effects of CGRP in the lung. CGRP induces very distinct ILC2 transcriptomic and epigenomic programs compared to NMU in the presence or absence of the alarmin, IL-33, with the dominant outcome being opposition of IL-33 and NMU effects. Specifically, CGRP inhibited the production of many proinflammatory cytokines produced by ILC2s. In part, the negative regulatory effect of CGRP on ILC2s can be explained by its inhibitory effect on cell growth, an effect of CGRP that has been noted for more than 25 years (Boudard and Bastide, 1991; Umeda et al., 1988). CGRP has been shown to exert its effect through Gas to induce production of cAMP and we confirmed that this was the case in ILC2s. Therefore, the effect of CGRP on ILC2s is consistent with the general view that cAMP production limits lymphocyte proliferation. Globally, our data showed that exposing ILC2s to a cell-permeant form of cAMP mimicked many of the transcriptomic effect of CGRP. By contrast, NMU exerts its effect via Gαq. Accordingly, administration of CGRP limited IL-33-induced pulmonary immunopathology, while CGRP-deficient mice showed severe symptoms and pathology. In addition, CGRP-deficient mice exhibited enhanced intestinal worm expulsion following helminth infection along with enhanced ILC2 expansion. This outcome on ILC2s was phenocopied by hematopoietic reconstitution of immunodeficient mice with Ramp1−/− bone marrow which eliminated CGRP signaling in ILC2 and other blood cells. Taken together, the observed repressive effects of CGRP on ILC2 responses are consistent with the view that CGRP suppresses lymphocyte proliferation through cAMP generation. It is of note that we observed discordant effects of Ramp1 deletion on T cells and ILC2 regarding cell numbers. Additional studies using cell type specific deletion of CGRP or its cognate receptors will be needed to further decipher the relative importance of the CGRP-cAMP axis in different cell types in vivo.
While predominantly suppressive effect of CGRP on ILC2s is evident, the promotion of IL-5 production is a notable exception that exemplifies the proinflammatory action of CGRP. Although type 2 responses collectively refer to the production of the cytokines IL-4, IL-5, and IL-13 whose genes are clustered together on the same chromosome, these cytokines can be discoordinately regulated. For instance, IL-5-producing Th2 cells represent a highly differentiated or pathogenic subset (Nakayama et al., 2017; Upadhyaya et al., 2011). In fact, our single cell data also shows distinct subsets of activated ILC2s separable as Il5hi ILC2 and Il13hi ILC2, supporting the notion that distinct subsets of type 2 immune cells might reflect different pathogenic potential. It is also conceivable that the reason why eosinophilia was not affected by the lack of CGRP signaling in helminth infection may be attributable to CGRP’s complex effects on IL-5 and ILC2 homeostasis.
Another finding in our report is that in the setting of helminth infection, a relevant immune cell population (ILC2s) expresses Calca, which encodes CGRP. CGRP has been previously found in non-neuronal cells, such as endothelial cells and adipocytes, and there is some evidence that it is also produced by several immune cell types, including activated B lymphocytes, mitogen activated peripheral blood mononuclear cells, and macrophages (Russell et al., 2014). Our mRNA-seq and ATAC-seq data (Shih et al., 2016) are notable in that ILC2s, but not other ILC subsets, express Calca. Thus, the accessibility of Calca locus is programmed at the ILC2 precursor stage, allowing ILC2s to rapidly produce this neuropeptide upon activation. Elucidating the significance of lymphoid-derived versus neural-derived CGRP will be important.
Corresponding to its role as a potent vasodilator, CGRP and its receptor have been clinically targeted by a variety of monoclonal antibodies, which are efficacious in the prevention and treatment of migraine (Mitsikostas and Reuter, 2017; Walter and Bigal, 2015). The reported side effects of CGRP blockade include infection, consistent with findings of Baral et al (Baral et al., 2018). However, given the complexity of the effects of CGRP and NMU, much is still to be learned about the consequences of blocking these neuropeptides to various immune responses and disorders characterized by dysregulated inflammatory cytokines. The action of CGRP to tilt the balance between IL-5 and IL-13 will become quite consequential in situations such as pulmonary arterial hypertrophy in which IL-5-producing ILC2 and eosinophils play pivotal roles (Ikutani et al., 2017), or tissue fibrosis in which IL-13 is an important driver (Gieseck et al., 2018). Understanding the contribution of ILCs, their effector cytokines and the role of CGRP in tissue damage and repair is clearly an important area for future research. The current clinical use of CGRP antagonists, both monoclonal antibodies and small molecules, indicates that advances in understanding the role of CGRP in lung immunopathology and other settings will be rapidly translated.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John O’Shea (john.oshea@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal experiments were performed in the AAALAC-accredited animal housing facilities at NIH. All animal studies were performed according to the NIH guidelines for the use and care of live animals and were approved by the Institutional Animal Care and Use Committee of NIAMS. Mice of 6 −12 weeks old were used in all experiments. For sample size, see corresponding figure legends.
METHOD DETAILS
Mice
Wild-type C57BL/6J mice, Rag1−/− mice, Rag2−/− mice were purchased from the Jackson Laboratory. Rag2−/− gc−/− mice were purchased from ENVIGO. Calcagfp/gfp mice were from MMRRC. aCGRP−/− mice were provided by Dr. Anne Luebke (Rochester University) (Lu et al., 1999).
For generation of bone marrow chimera mice, Rag2−/− gc−/− mice were irradiated (600 rads x 2), and were injected intravenously with 5 millions bone marrow cells from either Ramp1−/− mice (Li et al., 2014) or background matched wild-type mice (provided by Kathleen M. Caron). Bone marrow reconstituted mice were placed on antibiotics in drinking water for 2 weeks followed by additional 6 weeks with normal water supply before starting experiment.
Preparation of cell suspensions from tissues
BALF cells were collected from the lung by gently washing with 1 ml of ice cold PBS with 18G plastic cannula and 1 ml syringes. For isolation of cells from lung, mesentery, VAT, small intestine and colon, mice were perfused with 20 ml of PBS for the remove of blood from tissues. The isolated lungs, mesentery and VAT were cut into several small pieces, then were placed in RPMI-1640 containing 2% (vol/vol) FCS (Thermo Fisher Scientific), 50 μg/ml Liberace TM (Roche, 05401127001), 10 μg/ml DNase I (Sigma-Aldrich, DN25) (4 ml per mouse), and incubated for 45 min at 37°C. After that, tissues were further proceeded with the gentleMACS Dissociators (Miltenyi) using gentleMACS C Tubes (Miltenyi), then all cell suspensions were passed 70 μm cell strainers, and then cell strainers were washed twice with PBS to recover cells. Small intestines and colons were harvested and the contents were emptied. Peyer’s patches were removed and then the intestines were opened longitudinally, cut into small pieces and shaken at 37 °C for 30 min in HEPES media containing 5 mM EDTA to dissociate intraepithelial cells. The remained fragments were washed twice with HBSS and then were digested at 37 °C for 45 min in HBSS media containing 250 μg/ml Liberase TL (Roche, 5401020001) and 10 μg/ml DNase I. The digested tissues were passed 70 μm cell strainers, and then cell strainers were washed twice with PBS to recover cells. The total single cell suspension was enriched with 30% Per coll (GE Healthcare) by centrifugation at 400 g for 20 min, and isolated cells were washed twice before further analysis.
Flow cytometry
Lung ILC2s were identified as lineage markers (CD3ε, CD4, CD8α, CD19, CD11c, CD11b, γδTCR, FcεR1, Gr1, DX5, Ter119) and additional markers as indicated in figure legends. Eosinophils were identified as Siglec-F+ CD11b+ CD11c− cells. All cells were incubated with anti-CD16/CD32 (2.4G2) to block non-antigen-specific binding of immunoglobulins to the Fcγ receptors before being stained with the appropriate antibodies. For Live/Dead staining, Zombie Violet™ Fixable Viability Kit (Biolegend) was used. For staining of intracellular cytokines and transcription factors, cells were fixed and permeabilized with Fixation Buffer (Biolegend) or Foxp3 staining buffer (Thermo Fisher Scientific) according to the manufacturer’s instructions. Data were acquired on a FACSCanto II (BD Biosciences) or LSR Fortessa (BD Biosciences) and were analyzed with FlowJo software (Tree Star).
In vitro ILC2 culture
For purification of ILC2s from lung of wild-type mice, lung cells were stained with biotin conjugated anti-linage markers (CD8α, CD19, CD11c, CD11b, γδTCR, FcεR1, Gr1, DX5, Ter119) and anti-Biotin MicroBeads (Miltenyi Biotec #130-090-485), followed by sorting linage negative cells by MACS manual separator (Miltenyi). Linage negative cells were further stained with fluorochrome conjugated antibodies against CD3ε, TCRβ, Thy1, CD127, and KLRG1 together with APC-R700 Streptavidin (BD Biosciences), followed by sorting ILC2s (Lin− CD3ε− TCRβ− Thy1+ CD127+ KLRG1+) on FACS Aria IIIu or Aria Fusion cell sorter (BD Bioscience). Lung ILC2s were sorted into 96-well plates at the density of 10,000 cells per well and were cultured in 10% RPMI (RPMI1640 medium (Thermo Fisher Scientific) containing 10% (vol/vol) FCS, 50 μM 2-mercaptoethanol (Sigma-Aldrich), 100 U/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific) in the presence of 100 U/ml of IL-2 and 10 ng/ml of IL-7. After 2 to 4 weeks, IL-2 and IL-7 were removed from ILC2 culture and additionally cultured for 4 hr before use for further analysis. For cytokine production and cell proliferation assays, lung ILC2s were seeded into 96-well round-bottomed tissue culture plates at the density of 5,000 cells per well. For bulk mRNA-seq, lung ILC2s were seeded at the density of 30,000 cells per well. For ATAC-seq, lung ILC2s were seeded at the density of 10,000 cells per well. Cells were cultured in 200 to 300 μl of 10% RPMI with various stimulants, including IL-33 (10 ng/ml), NMU (10 ng/ml), CGRP (10 ng/ml), and Dibutyryl-cAMP (100 μM) in the presence or absence of chemical inhibitors, YM-254890 (10 μM), 666-15 (1 μM), BAY11-7082 (10 μM), SB203580 (10 μM), and PD98059 (10 μM). For intracellular cytokine staining, GolgiPlug (Brefeldin A) (BD Biosciences) was added into the culture from the beginning (4 hr stimulation). Production of cytokines in culture supernatant was measured by LEGENDplex Mouse Th Cytokine Panel (Biolegend, #740005) after 5 days culture. Cell proliferation was assessed by trypan blue staining of cells after 5 days culture, or by BrdU uptake of cells cultured for 24 h, followed by addition of 10 μM BrdU for 1.5 hr, then detection of incorporated BrdU by flow cytometry according to the manufacturer’s instructions (BD Biosciences, #552598).
Nippostrongylus brasiliensis infection
Mice were given subcutaneous injection of 300 or 500 infective third-stage N. brasiliensis larvae as described previously (Huang et al., 2015). Worm burden in small intestines were measured on day 7 or 9. Lung cells were harvested on day 2, 5, 9 and 14 after infection for scRNA-seq and bulk mRNA-seq. Before cell sorting, we confirmed comparable viability of cells for all time points tested (93% - 95%).
IL-33-mediated pulmonary inflammation in vivo
Mice were anesthetized with isoflurane and treated intranasally with 30 μl of IL-33 (500 ng per mouse) with or without CGRP (1 μg per mouse) in PBS for consecutive 4 days. Treated mice were analyzed 1 day after last administration.
Single-cell RNA sequencing (scRNA-seq)
Freshly sorted ILCs and Th cells (13,000 cells each, cell viability >98%) were encapsulated into droplets, and libraries were prepared using Chromium Single Cell 3’ Reagent Kits v2 according to manufacturer’s protocol (10X Genomics). The generated scRNA-seq libraries were sequenced using a 50 cycle (single read) with a HiSeq 3000 (Illumina).
Bulk mRNA sequencing
Approximately 20,000–50,000 cells (cell viability >98%) freshly sorted from mice or stimulated with various conditions in vitro were lysed with TRIzol (Life Technologies) and total RNAs were extracted using Direct-zol™ RNA MicroPrep (Zymo Research). Total RNAs were subsequently processed to generate an mRNA-seq library using a NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490S), NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, E7530S) and NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) (NEB, E7335S) according to protocol. The libraries were sequenced for 50 cycles (single read) with a HiSeq 3000 (Illumina).
FastATAC sequencing
FastATAC-seq was performed according to a published protocol (Corces et al., 2016) with minor modification. Ten thousand cells were pelleted and washed with 50 μl 1× PBS. After pelleting the nuclei by centrifuging at 500 × g for 10 min, the pellets were resuspended in 50 μl transposase mixture (25 μl of 2x TD buffer, 2.5 μl of TDE1, 0.5 μl of 1% digitonin, 22 μl of nuclease-free water) (#FC-121-1030, Illumina; #G9441, Promega). The reaction was incubated at 37°C with shaking at 300 rpm for 30 min. The fragmentalized DNAs were then purified using a QIAGEN MinElute kit and amplified with 10 or 11 cycles of PCR based on the amplification curve. Once the libraries were purified using a QIAGEN PCR cleanup kit, they were further sequenced for 50 cycles (paired-end reads) on a HiSeq 3000 (Illumina).
Imagestream
Cells stained for DAPI (CST, #4083), anti-NFAT1-AF488, anti-NF-kB p65-AF647, and anti-CD45-PE/Cy7 were run on an ImageStreamXMKII (Amnis, Seattle, WA) 2 camera imaging flow cytometer at 60X magnification using INSPIRE software. DAPI was detected in channel 7 with a 405 laser power of 30 mW, anti-NFAT1-AF488 was detected in channel 2 with a 488 laser power of 200 mW, anti- CREB -PE , and anti-CD45- PE/Cy7 were detected in channels 3 and 6, respectively, at a 561 laser power of 100 mW, and anti-NF-kB p65-AF647 was detected in channel 11 at a laser power of 150 mW. The 758 laser was turned off for these experiments. 5000 single, in focus cells were acquired. Single color controls were acquired with the Brightfield and 758 lasers off. IDEAS software (Amnis, Seattle, WA) was used to create a compensation matrix and analyze data. Cells with nuclei which were in focus were gated on a histogram of channel 7 Gradient RMS (root mean square). Next, single cells having focused nuclei were gated on an Area_Brightfield versus Aspect Ratio_Brightfield dot plot. DAPI-positive and CD45-positive cells were gated on an Intensity_CD45 versus Intensity_DAPI dot plot gated on single cells with focused nuclei. All subsequent plots were gated on DAPI-positive and CD45-positive cells. DAPI-positive and NFAT1-positive cells were gated on an Intensity_DAPI versus Intensity_NFAT1 dot plot. DAPI-positive and NF-kB p65 -positive cells were gated on an Intensity_DAPI versus Intensity_NF-kB p65 dot plot. The Similarity feature, which is a pixel by pixel comparison of the two images, was created to measure co-localization of NFAT1 and NF-kB p65, respectively, with the nucleus.
Measurement of cytokines and cAMP
Cytokine concentrations in BALF and supernatant of in vitro ILC2 cultures were analyzed by the LegendPlex Mouse Th Cytokine Panel (13-plex) (BioLegend) on a FACS Cant II (BD Biosciences) according to the manufacturer’s instructions. For cAMP measurement, 40,000 of ILC2s were stimulated with a variety of stimuli as indicated above, followed by lysis of cells with 50 μl of lysis buffer (Cell signaling technology, 9803) and measurement of cAMP concentration using Cyclic AMP XP® Chemiluminescent Assay Kit (Cell signaling technology, 8019S). Relative Light Units (RLU) at 425nm were measured by Multimode Microplate Reader (TriStar2 LB942).
Histological analysis
Lungs were fixed for at least 24 h with 10% formalin (Sigma-Aldrich, #HT5012) and were embedded in paraffin. Cross-sectional lung tissues were stained with Periodic acid-Schiff (PAS) or hematoxylin and eosin. Pathological score was evaluated by an observer masked to treatment group for the following parameters: peribronchiolar inflammation and cuffing (0–4), perivascular inflammation and cuffing (0–4), goblet cell hyperplasia (0–4) and interstitial infiltrate (0–3).
Immunofluorescence microscopy
Lung was fixed in 2% PFA for 4 h on ice and incubated in 30% sucrose at 4°C overnight, followed by embedding in OCT (Leica) for cryocutting. 8-μm tissue slices on slides were washed 3 x in ice-cold PBS, blocked in 10% donkey serum in PBS and stained with: hamster anti-CD3ε-APC (145-2C11, eBioscience), hamster anti-KLRGl-PE (2F1, eBioscience). Nuclei were counterstained with DAPI (Thermo Fisher Scientific). Images were captured with an inverted Nikon Eclipse Ti microscope (Nikon).
QUANTIFICATION AND STATISTICAL ANALYSIS
Bulk mRNA-seq analysis
Raw sequencing data were processed with CASAVA 1.8.2 (Illumina; (Bentley et al., 2008)) to generate FastQ files. Sequence reads were mapped onto mouse genome build mm10 using TopHat 2.1.0 (Trapnell et al., 2012). Gene expression values (FPKM; fragments per kilobase exon per million mapped reads) were calculated with Cufflinks 2.2.1 (Trapnell et al., 2012). BigWig tracks were generated from Bam files and converted into bedGraph format using bedtools (Quinlan and Hall, 2010). These were further reformatted with the UCSC tool bedGraphToBigWig. Pathway analysis and prediction of upstream gene were performed with Ingenuity pathway analysis (IPA). Scatter plot was created with Partek Genomics Suite (Partech). The Pearson correlation analysis was done by corr() function in R 3.0.1 (R_Core_Team, 2018) and the heat map and clustering tree was plotted by “pheatmap” package in R using clustering method “ward”. The heat map showing gene expression was created with CIMminer (Weinstein et al., 1994).
ATAC-seq analysis
FastATAC-seq reads from two biological replicates for each sample were mapped to the mouse genome (mm9 assembly) using Bowtie 0.12.8 (Langmead et al., 2009). In all cases, redundant reads were removed using FastUniq (Xu et al., 2012), and customized Python scripts were used to calculate the fragment length of each pair of uniquely mapped paired-end (PE) reads. The fragment sizes distribute similar to previously published data (data not shown). Only one mapped read to each unique region of the genome that was less than 175 bp was kept and used in peak calling. Regions of open chromatin were identified by MACS (version 1.4.2) (Zhang et al., 2008) using a p-value threshold of 1 × 10−5. Only regions called in both replicates were used in downstream analysis. Peak annotation and motif analysis were performed with the Hypergeometric Optimization of Motif EnRichment program (HOMER) version 4.9 (Heinz et al., 2010) using the “annotatePeaks.pl peak_file mm9 -size 1000 -hist 40 -ghist” and “findMotifsGenome.pl peak_file mm9 motif_folder -size given -preparsedDir tmp 2>out”. The heatmap were generated by “pheatmap” package in R 3.0.1 (R_Core_Team, 2018).
scRNA-seq analysis
Sequence reads from all samples were processed and aggregated using Cell Ranger. Aggregated data was further analyzed by Seurat (Macosko et al., 2015). Specifically, we log-normalized the expression matrix, regressed the data against the total number of UMI’s detected per cell, performed PCA analysis, used PCA dimensions 1-10 to find clusters, and finally found marker genes for these clusters. We visualized single cell gene-expression as tSNE overlays, marker gene heatmaps, and violin plots. We used the normalized expression matrix, tSNE coordinates, and clustering produced by Seurat for the transcription module analysis described below.
Analyzing transcriptional modules
To explore cytokine-induced transcriptional modules in our scRNA-seq data, we first defined modules by calling differentially expressed genes in our bulk samples, and then scored each single cell based on their expression of these genes. Specifically, we used kallisto v 0.43.0 to quantify abundance (Bray et al., 2016) and sleuth v 0.29.0 (Pimentel et al., 2017) to define differentially expressed genes (q-value < 0.05; fold change >= 1.5; higher than 10 TPM expression in at least one sample) in bulk samples treated with IL-33, NMU, CGRP, or cAMP compared to no stimulation. Next, we row-normalized the single cell gene expression matrix produced by Seurat to obtain Z-scores. To calculate a module score for each single cell, we added the Z-scores of up-regulated genes and subtracted those of down-regulated genes, then normalized by the total number of module genes. To partially overcome the dropout artifact of 10X sequencing, we only considered module genes that expressed at least 300 TPM in at least one bulk sample. In our hands, genes expressed at this level are detected by 10X sequencing in approximately 50% of single cells. We performed module analysis using ILC cells with detectable Gata3 expression.
Statistical analysis
Statistical significance was assessed with non-paired two-tailed Student’s t-test.
DATA AND SOFTWARE AVAILABILITY
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE131996.
Supplementary Material
Table S1. Gene expression of top 10 genes that are differentially expressing across clusters, Related to Figure 1D.
Table S2. Gene list of group 1 (97), group 2 (350), group 3 (116), and group 4 (265), Related to Figure 3D.
Table S3. Gene list of modules for IL-33, NMU, CGRP and cAMP, Related to Figure 5H.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin-anti-mouse CD8a | Biolegend | 100704 |
| Biotin-anti-mouse CD19 | Biolegend | 115504 |
| Biotin-anti-mouse CD11b | Biolegend | 101204 |
| Biotin-anti-mouse CD11c | Biolegend | 117304 |
| Biotin-anti-mouse TCRγ/δ | Biolegend | 118103 |
| Biotin-anti-mouse Gr1 | Biolegend | 108404 |
| Biotin-anti-mouse CD49b | Biolegend | 108904 |
| Biotin-anti-mouse TER-119 | Biolegend | 116204 |
| Biotin-anti-mouse FcεR1α | Biolegend | 134304 |
| Biotin-anti-mouse CD3ε | Biolegend | 100304 |
| PerCP/Cy5.5-anti-mouse CD11b | Biolegend | 101227 |
| APC-anti-mouse CD11c | Biolegend | 117309 |
| PE-anti-mouse TCRβ | Biolegend | 109208 |
| PE/Cy7-anti-mouse KLRG1 | Biolegend | 138416 |
| APC-anti-mouse IL-5 | Biolegend | 504306 |
| PerCP/Cy5.5-anti-mouse CD4 | Biolegend | 100540 |
| PerCP/Cy5.5-anti-mouse CD3ε (145-2C11) | Invitrogen | 45-0031-82 |
| PerCP/Cy5.5-anti-mouse CD5 (53-7.3) | Invitrogen | 45-0051-82 |
| PerCP-eFluor 710-anti-mouse FcεRI (CRA1) | Invitrogen | 46-5898-82 |
| APC-Cy7-anti-mouse B220 (RA3-6B2) | Invitrogen | 17-0455-81 |
| APC-Cy7-anti-mouse CD11b (M1/70) | Invitrogen | 47-0112-82 |
| APC-Cy7-anti-mouse CD11c (N418) | Invitrogen | 47-0114-82 |
| PerCP/Cy5.5-anti-mouse NK1.1 (PK136) | Invitrogen | 45-5941-82 |
| FITC-anti-mouse KLRG1 (2F1) | Invitrogen | 11-5893-82 |
| BV650-anti-mouse CD45 (30-F11) | Biolegend | 103151 |
| PE/Cy7-anti-mouse CD127 (A7R34) | Biolegend | 135014 |
| PE-anti-mouse SiglecF (E50-2440) | BD Bioscience | 552126 |
| AF700-anti-mouse CD90.2 (53-2.1) | Biolegend | 105320 |
| BV421-anti-mouse ST2 (DIH9) | Biolegend | 145309 |
| BUV395-anti-mouse CD4 (GK1.5) | BD Bioscience | 563790 |
| APC-anti-CD25 | Invitrogen | 17-0251-82 |
| PE-anti-mouse KLRG1 | Invitrogen | 12-5893-82 |
| APC-anti-mouse CD3ε (145-2C11) | Invitrogen | 17-0031-83 |
| APC-Cy7-anti-mouse CD3 | BD Bioscience | 560590 |
| V500-anti-mouse CD90.2 (Thy1.2) | BD Bioscience | 561616 |
| PE-anti-mouse Siglec-F | BD Bioscience | 552126 |
| PE-CF594-anti-mouse CD127 | BD Bioscience | 562419 |
| BUV395-anti-mouse GATA3 | BD Bioscience | 565448 |
| FITC-anti-mouse Foxp3 | Thermo Fisher Scientific | 11-5773-82 |
| eFluor450-anti-mouse IL-13 | Thermo Fisher Scientific | 48-7133-82 |
| APC-anti-mouse ST2 | Thermo Fisher Scientific | 17-9335-82 |
| Alexa Fluor 647-anti-NFkB p65 | Cell Signaling Technology | 8801S |
| Alexa Fluor 488-anti-NFAT1 | Cell Signaling Technology | 14324S |
| PE-Phospho-CREB (Ser133) | Cell Signaling Technology | 14228S |
| Bacterial and Virus Strains | ||
| Nippostrongylus brasiliensis | Huang et al., 2015 | N/A, generated in house |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant mouse IL-7 | R&D systems | 407-ML |
| Recombinant mouse IL-33 | R&D systems | 3626-ML |
| Recombinant mouse IL-33 | Biolegend | 580508 |
| Rat CGRP | R&D systems | 1161 |
| Rat Neuromedin U | R&D systems | 1917 |
| 666-15 | R&D systems | 5661 |
| BAY11-7082 | Sigma-Aldrich | B5556 |
| SB203580 | Sigma-Aldrich | S8307 |
| PD98059 | Sigma-Aldrich | P215 |
| N6,2′-O-Dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt | Sigma-Aldrich | D0260 |
| YM-254890 | Wako Pure Chemical | 257-00631 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Reagent Kits v2 | 10X Genomics | PN-120237 |
| NEBNext Ultra RNA Library Prep Kit for Illumina | New England BioLabs | E7530S |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England BioLabs | E7490S |
| NEBNext Multiplex Oligos for Illumina | New England BioLabs | E7335S |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE131996 |
| ATAC-seq samples for lymphoid cells (related to Figure 2) | Shih et al., 2016 | GEO: GSE77695 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | #000664 |
| B6 (Cg)-Rag2tm1.1Cgn/J (Rag2−/−) | The Jackson Laboratory | #008448 |
| B6.129S7-Rag1tm1Mom/J (Rag1−/−) | The Jackson Laboratory | #002216 |
| B6.129S(Cg)-Calcatm1.1(EGFP/HBEGF)Mjz/Mmnc (Calcagfp/gfp) | MMRRC | 036773-UNC |
| Ramp1−/− | Kathleen M. Caron | PMID: 25010197 |
| alpha CGRP −/− | Dr. Anne Luebke | PMID: 10532808 |
| B6;129-Rag2tm1FwaII2rgtm1Rsky/DwlHsd (Rag2−/− cg−/−) | ENVIGO | # 2103F |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| TopHat 2.1.0 | Trapnell et al., 2012 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks 2.2.1 | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| bedtools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| Bowtie 0.12.8 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| FastUniq | Xu et al., 2012 | https://sourceforge.net/projects/fastuniq/ |
| MACS (version 1.4.2) | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/index.html |
| Hypergeometric Optimization of Motif EnRichment program (HOMER) version 4.9 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/motif/ |
| R 3.0.1 | R Core Team, 2014 | https://www.r-project.org |
| Seurat | Macosko et al., 2015 | http://satijalab.org/seurat/ |
| Kallisto v 0.43.0 | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| sleuth v 0.29.0 | Pimentel et al., 2017 | https://github.com/pachterlab/sleuth/ |
| Ingenuity pathway analysis (IPA) | Qiagen | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis |
| CIMminer | Weinstein et al., 1994) | https://discover.nci.nih.gov/cimminer/home.do |
| The Integrative Genomics Viewer (IGV) | Thorvaldsdottir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| Parteck Genomics Suite | Parteck | http://www.partek.com/pgs |
| Other | ||
| N/A | ||
Highlights.
Single cell analysis reveals heterogeneity of ILC2 responses to N. brasiliensis
Il5hiILC2s express CGRP and its receptor following helminth infection
CGRP modulates type 2 cytokine production by ILC2s induced by alarmin and NMU
CGRP constrains the magnitude of innate type 2 responses following helminth infection
ACKNOWLEDGMENTS
We thank members of Molecular Immunology and Inflammation Branch (NIAMS) for input and support. We thank S. Dell’Orso, G. Gutierrez-Cruz and F. Naz (Genome Analysis Core Facility, NIAMS); J. Simone, J. Lay, and K. Tinsley (Flow Cytometry Section, NIAMS); H-W. Sun and S.R. Brooks (Biodata Mining and Discovery Section, NIAMS); E. Ralston and A. Kenea (Light Imaging Section, NIAMS) and L. Samsel and P. J. McCoy (Flow Cytometry Core, NHLBI) for their technical support. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH. This work was supported by the Intramural Research Programs of NIAMS and NIAID. Research in the Artis laboratory is supported by the National Institutes of Health (AI074878, AI095466, AI095608 and AI102942), the Burroughs Wellcome Fund, the Crohn’s & Colitis Foundation, Cure for IBD and the Rosanne H. Silberman Foundation. H.N. was supported by the JSPS Research Fellowship for Japanese Biomedical and Behavioural Researchers at NIH. T.M. was supported by the Crohn’s & Colitis Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
D.A. has contributed to scientific advisory boards at MedImmune, Pfizer, FARE and the KRF.
REFERENCES
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, Wei Y, Zhou Y, Kuchroo VK, Burkett PR, et al. (2018). Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 24, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudard F, and Bastide M (1991). Inhibition of mouse T-cell proliferation by CGRP and VIP: effects of these neuropeptides on IL-2 production and cAMP synthesis. J Neurosci Res 29, 29–41. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, Shah K, Barbosa-Morais NL, Harris N, and Veiga-Fernandes H (2017). Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach A, Colonna M, and Koyasu S (2014). Development, differentiation, and diversity of innate lymphoid cells. Immunity 41, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, and McKenzie AN (2015). Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348, aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa J, Moro K, Motomura Y, Okamoto K, Zhu JF, Takayanagi H, Kubo M, and Koyasu S (2013). Critical Role of p38 and GATA3 in Natural Helper Cell Function. Journal of Immunology 191, 1818–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieseck RL 3rd, Wilson MS, and Wynn TA (2018). Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 18, 62–76. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann B (2013). Modulation of immune responses by the neuropeptide CGRP. Amino Acids 45, 1–7. [DOI] [PubMed] [Google Scholar]
- Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, Williamson PR, Urban JF Jr., and Paul WE (2015). IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nature immunology 16, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikutani M, Tsuneyama K, Kawaguchi M, Fukuoka J, Kudo F, Nakae S, Arita M, Nagai Y, Takaki S, and Takatsu K (2017). Prolonged activation of IL-5-producing ILC2 causes pulmonary arterial hypertrophy. JCI Insight 2, e90721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CS, and Artis D (2018). Neuronal regulation of innate lymphoid cells. Curr Opin Immunol 56, 94–99. [DOI] [PubMed] [Google Scholar]
- Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, Monticelli LA, Moriyama S, Putzel GG, Rakhilin N, et al. (2017). The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wetzel-Strong SE, Hua X, Tilley SL, Oswald E, Krummel MF, and Caron KM (2014). Deficiency of RAMP1 attenuates antigen-induced airway hyperresponsiveness in mice. PLoS One 9, e102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JT, Son YJ, Lee J, Jetton TL, Shiota M, Moscoso L, Niswender KD, Loewy AD, Magnuson MA, Sanes JR, et al. (1999). Mice lacking alpha-calcitonin gene-related peptide exhibit normal cardiovascular regulation and neuromuscular development. Mol Cell Neurosci 14, 99–120. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsikostas DD, and Reuter U (2017). Calcitonin gene-related peptide monoclonal antibodies for migraine prevention: comparisons across randomized controlled studies. Curr Opin Neurol 30, 272–280. [DOI] [PubMed] [Google Scholar]
- Morita H, Moro K, and Koyasu S (2016). Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol 138, 1253–1264. [DOI] [PubMed] [Google Scholar]
- Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, Yudanin NA, Monticelli LA, Putzel GG, Rodewald HR, et al. (2018). beta(2)-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061. [DOI] [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, and Koyasu S (2010). Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544. [DOI] [PubMed] [Google Scholar]
- Nagashima H, Okuyama Y, Fujita T, Takeda T, Motomura Y, Moro K, Hidaka T, Omori K, Sakurai T, Machiyama T, et al. (2018). GITR cosignal in ILC2s controls allergic lung inflammation. J Allergy Clin Immunol. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, Tumes DJ, and Okamoto Y (2017). Th2 Cells in Health and Disease. Annual review of immunology 35, 53–84. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, and Tracey KJ (2017). Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci 20, 156–166. [DOI] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R_Core_Team (2018). R: A language and environment for statistical computing. .
- Rankin LC, and Artis D (2018). Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 173, 554–567. [DOI] [PubMed] [Google Scholar]
- Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, Vaka D, Eckalbar WL, Molofsky AB, Erle DJ, et al. (2018). Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 19, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, and Immunological Genome C. (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell FA, King R, Smillie SJ, Kodji X, and Brain SD (2014). Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev 94, 1099–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, Urban JF Jr., Davis FP, Kanno Y, and O’Shea JJ (2016). Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda K, Hirahara K, Iinuma T, Ichikawa T, Suzuki AS, Sugaya K, Tumes DJ, Yamamoto H, Hara T, Tani-Ichi S, et al. (2016). Thy1+IL-7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation. Proc Natl Acad Sci U S A 113, E2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, and Artis D (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, and Cupedo T (2012). Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual review of immunology 30, 647–675. [DOI] [PubMed] [Google Scholar]
- Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, Lashua A, Yu C, Klein BS, Locksley RM, et al. (2018). Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda Y, Takamiya M, Yoshizaki H, and Arisawa M (1988). Inhibition of mitogen-stimulated T lymphocyte proliferation by calcitonin gene-related peptide. Biochem Biophys Res Commun 154, 227–235. [DOI] [PubMed] [Google Scholar]
- Upadhyaya B, Yin Y, Hill BJ, Douek DC, and Prussin C (2011). Hierarchical IL-5 expression defines a subpopulation of highly differentiated human Th2 cells. J Immunol 187, 3111–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Artis D (2018). Neuronal-immune system cross-talk in homeostasis. Science 359, 1465–1466. [DOI] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Mucida D (2016). Neuro-Immune Interactions at Barrier Surfaces. Cell 165, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, and Pachnis V (2017). Neuroimmune regulation during intestinal development and homeostasis. Nat Immunol 18, 116–122. [DOI] [PubMed] [Google Scholar]
- Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, et al. (2017). The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S, and Bigal ME (2015). TEV-48125: a review of a monoclonal CGRP antibody in development for the preventive treatment of migraine. Curr Pain Headache Rep 19, 6. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Myers T, Buolamwini J, Raghavan K, van Osdol W, Licht J, Viswanadhan VN, Kohn KW, Rubinstein LV, Koutsoukos AD, et al. (1994). Predictive statistics and artificial intelligence in the U.S. National Cancer Institute’s Drug Discovery Program for Cancer and AIDS. Stem Cells 12, 13–22. [DOI] [PubMed] [Google Scholar]
- Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, and Stockinger B (2011). An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol 12, 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, and Chen S (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7, e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene expression of top 10 genes that are differentially expressing across clusters, Related to Figure 1D.
Table S2. Gene list of group 1 (97), group 2 (350), group 3 (116), and group 4 (265), Related to Figure 3D.
Table S3. Gene list of modules for IL-33, NMU, CGRP and cAMP, Related to Figure 5H.
Data Availability Statement
The data discussed in this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE131996.