

Abstract
Purpose:
The goal of this study was to characterize the activity of the covalent CDK7 inhibitor THZ1 in multiple myeloma (MM) models.
Experimental Design:
MM lines were exposed to varying THZ1 concentrations alone or with carfilzomib or ABT-199, after which apoptosis was monitored by flow cytometry, protein expression by western blot analysis, mRNA by RT-PCR. Analogous studies were performed in cells ectopically expressing c-MYC, MCL-1, or BCL-XL, or CRISPER-Cas CDK7 sgRNA knock-out. Primary MM cells were exposed to THZ1 ± carfilzomib or ABT-199. In vivo effects of THZ1 were examined in a systemic U266 xenograft model.
Results:
THZ1 markedly diminished MM cell proliferation and survival despite bortezomib or stromal cell resistance in association with G2M arrest, inactivation of CTD RNA Pol II, dephosphorylation of CDKs 7 as well as 1, 2, and 9, and MCL-1, BCL-xL, and c-MYC mRNA or protein down-regulation. Ectopic MCL-1, c-MYC, or BCL-XL expression significantly protected cells from THZ1 lethality. Both THZ1 and CRISPR-Cas CDK7 knock-out sharply diminished MM cell proliferation and significantly increased carfilzomib and ABT-199 lethality. Parallel effects and interactions were observed in primary CD138+ (N=22) or primitive MM cells (CD138−/CD19+/CD20+/CD27+; N=16). THZ1 administration (10 mg/kg ip qd, 5 days/week) significantly improved survival in a systemic MM xenograft model with minimal toxicity and induced similar events observed in vitro e.g., MCL-1 and c-MYC down-regulation).
Conclusions:
THZ1 potently reduces MM cell proliferation through transcriptional down-regulation of MCL-1, BCL-XL, and c-MYC in vitro and in vivo. It warrants further attention as a therapeutic agent in MM.
Introduction
Cyclin-dependent kinases (CDKs) catalyze the phosphorylation of cyclins, proteins intimately involved in the progression of cells through the cell cycle. However, in addition to their cell cycle regulatory activities, CDKs exert several functions unrelated to cell cycle control. For example, CDK9 acts as a transcriptional regulator as a participant in the p-TEFb (positive-transcription elongation factor b) complex which phosphorylates the carboxy-terminal domain (CTD) of RNA Pol II (1). Indeed, CDK9 antagonists have been shown to down-regulate the expression of short-lived proteins such as the anti-apoptotic BCL-2 family member MCL-1 (2).
CDK7 represents a component of the TFIIH (general transcription factor IIH) multi-protein complex which cooperates with p-TEFb to regulate RNA Pol II transcriptional activity (1). In addition to its transcriptional regulatory role, TFIIH has also been implicated in DNA repair (1). Moreover, CDK7 regulates the phosphorylation and activity of most CDKs through its participation in the CAK (cyclin-activating kinase) complex (3). The transcriptional addiction of many tumor types prompted the development of CDK7 inhibitors as anti-cancer agents. THZ1 is a covalent inhibitor of CDK7 which potently represses transcription and down-regulates several short-lived proteins such as MYC (4,5). In this regard, THZ1 has shown activity in multiple MYC-driven tumor types including neuroblastoma (4), breast cancer (6), and small cell lung cancer (7,8). In contrast, THZ1 activity in T-cell acute lymphoblastic leukemia has been related to disruption of the RUNX1 transcription factor (9).
Multiple myeloma (MM) is an accumulative disorder of mature plasma cells that despite the introduction and approval of multiple novel agents (e.g., proteasome inhibitors, immunomodulatory agents, and antibodies (10) is in most cases incurable. Consequently, new and more effective approaches are urgently needed, particularly in the case of relapsed or refractory disease. Notably, several short-lived proteins e.g., MCL-1 and MYC have been implicated in myelomagenesis as well as resistance to established therapies (11,12). The potential dependence of MM cells on these proteins raised the possibility that a transcriptional CDK7 inhibitor like THZ1 might be particularly effective in this disease. Currently, the impact of CDK7 interruption has not yet been assessed in MM models. Here we report that THZ1 potently inhibits MM cell proliferation and survival in a MYC, MCL-1, and BCL-XL -dependent manner, and potentiates the activity of proteasome inhibitors (carfilzomib, bortezomib) and BH3-mimetics (venetoclax) in both cell lines and primary patient samples. It also significantly improves survival in a MM xenograft model with minimal toxicity. Together, these findings argue that CDK7 inhibitors like THZ1 warrant attention as therapeutic agents in MM.
Materials and Methods
Cell lines and reagents
Human NCI-H929, U266, OPM2, and RPMI8226 cells were all from ATCC and maintained as described previously (13). Btz-resistant cells, U266/PS-R and 8226/V10R were established and maintained as described previously (14). Revlimid-resistant (R10R) RPMI8226 sublines were maintained as before (15). U266/MCL-1, U266/MYC and 8226/BCL-XL were established by stably transfecting full-length human MCL-1, MYC and BCL-XL cDNA separately as described previously (13). KMS28-BM, and KMS28-PE were from Japanese Cancer Research Resources Bank (JCRB) (Tokyo, Japan).
All experiments utilized logarithmically growing cells (3–5×105 cells/ml). MycoAlert (Lonza, Allendale, NJ) assays were performed, demonstrating that all cell lines were free of mycoplasma contamination.
THZ1 was purchased from Medchem Express (Monmouth Junction, NJ). Bortezomib (Btz), Carfilzomib (Cfz), and Venetoclax (ABT-199) were purchased from ChemieTek (Indianapolis, IN). The caspase inhibitor Z-VAD-FMK was obtained from Enzo Life Sciences, Inc., Farmingdale, NY. All drugs were dissolved in DMSO, aliquoted, and stored at −80°C. In all experiments, final DMSO concentrations did not exceed 0.1%.
CRISPR/Cas9 plasmids and Virus Infection
Construction of lenti-CRISPR/Cas9 vectors targeting CDK7 was performed following the protocol associated with the backbone vector (#45, Addgene) (16). The following sequences were chosen from the published literature (6).
sgGFP (fwd: CACCGGGGCGAGGAGCTGTTCACCG;
rv: AAACCGGTGAACAGCTCCTCGCCCC),
sgCDK7-1 (fwd: CACCGGAAGCTGGACTTCCTTGGGG
rv: AAACCCCCAAGGAAGTCCAGCTTCC);
sgCDK7-2 (fwd: CACCGATCTCTGGCCTTGTAAACGG
rv: AAACCCGTTTACAAGGCCAGAGATC).
Virus infection were performed as described previously (13)
Cell proliferation Assay
For clonogenic cell growth assays, cells in which lenti-CRISPR vectors were introduced, following puromycin selection for 5 days, were harvested and seeded into round-bottom 96 wells at 250–500 cells/well diluted by an equal volume of RPMI1640 with 10% FBS, incubated for varying intervals (e.g., 2, 4, 6, and 8 days) after seeding, and images captured using an Olympus IX71 Inverted System Microscope.
For 96-well plate assays, cells as described above were plated at a density of 2,000 cells per well, and CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS) Kit (Promega, Madison, WI, USA) was used to monitor cell viability at 2, 4, 6, and 8 days according to the manufacturer’s instructions.
For I.C.50 determinations, MM cells were seeded in 96-well plates at a density of 1.0 × 104 cells per well. Each experiment was performed utilizing 3 independent wells. After 24 hr, cell viability was determined using a CellTiter Kit. Absorbance was measured at 490 nm using a microplate spectrophotometer (Thermo, Waltham, MA, USA OR Promega, Madison, WI, USA). To determine the half maximal inhibitory concentration (IC50), 6 concentration points (corresponding to two-fold increases) were chosen for each cell line. Experiments were performed 2–3 times independently.
Animal studies
All animal studies were IACUC approved and performed in accordance with AAALAC, USDA, and PHS guidelines. For the orthotopic murine model, NOD/SCID-γ mice were injected i.v. with either 5×106 U266 stably transfected with constructs encoding luciferase (U266/Luc). For the flank murine model, NOD/SCID-γ mice were inoculated s.c. with 5×106 PS-R/Luc cells. Control animals received equal volumes of vehicle (10% DMSO in D5W, 5% dextrose in water).
Tumors were measured in two dimensions using manual calipers. Tumor volume was calculated using the formula: V = 0.5 × length × width2. Animal with tumor established (mean tumor volume of ~200 mm3) were randomly divided into two groups, which were then treated with vehicle (10% DMSO in D5W), THZ1 (3 mg/ml, prepared in vehicle solutions) at the dose of 10 mg/kg via intraperitoneally (i.p.) twice daily, 5 days a week.
Mice were monitored for tumor growth with an IVIS 200 imaging system (Xenogen Corporation, Alameda, CA), and tumor volume was monitored every 2–3 days. Measurement of animal body weight was performed every other day throughout the study to monitor toxicity. Upon harvesting, tumors were dissected into small pieces, and frozen in liquid nitrogen for preparation of lysates and immunoblotting.
Statistical analysis
Values represent the means ± SD for at least three independent experiments performed in triplicate. The significance of differences between experimental variables was determined using the Student’s t test or One-way ANOVA with Tukey-Kramer Multiple Comparisons Test. The significance of P values are * < 0.05, ** < 0.01, or *** < 0.001 wherever indicated. Analysis of synergism was performed by Median Dose Effect analysis using the software Calcusyn (Biosoft, Ferguson, MO). Kaplan-Meier analysis of mouse survival performed with GraphPad Prism 6 software (La Jolla, CA).
Cell cycle analysis
Cell cycle analysis by propidium iodide (PI) staining was performed by flow cytometry (FCM) using the Modfit LT2.0 software (Verity Software House, Topsham, ME, USA) as described previously (17).
See Supplementary Methods for transfection, Quantitative real-time PCR, immunoblot analysis, immunofluorescence, Chromatin IP, isolation of primary MM cells, analysis of cell death and cell viability assay.
Results
Exposure (24 hr) of multiple MM cell lines (OPM2, RPMI8226, H929, U266, PS-R, KMS28-BM, and KMS28-PE) to increasing concentrations of THZ1 variably reduced cell survival (Fig 1A), but in all cases I.C.50 values were in the low nM range (e.g., <300 nM; Supplemental Table 1). Low nM THZ1 concentrations also killed bortezomib- or revlimid-resistant 8226 cells (15) (1B). Concordant findings were obtained when PARP and caspase-3 cleavage were monitored (Fig 1C), including in bortezomib-resistant U266 cells (PS-R), previously shown to exhibit MCL-1 up-regulation and BIM down-regulation (18). THZ1 also induced expression of γH2A.X, an indicator of DNA double-strand breaks in multiple cell lines (Fig 1C), consistent with the role of CDK7 in the DNA repair response (19).
Figure 1. THZ1 inhibits proliferation and induces apoptosis in parental and drug-resistant MM cells.
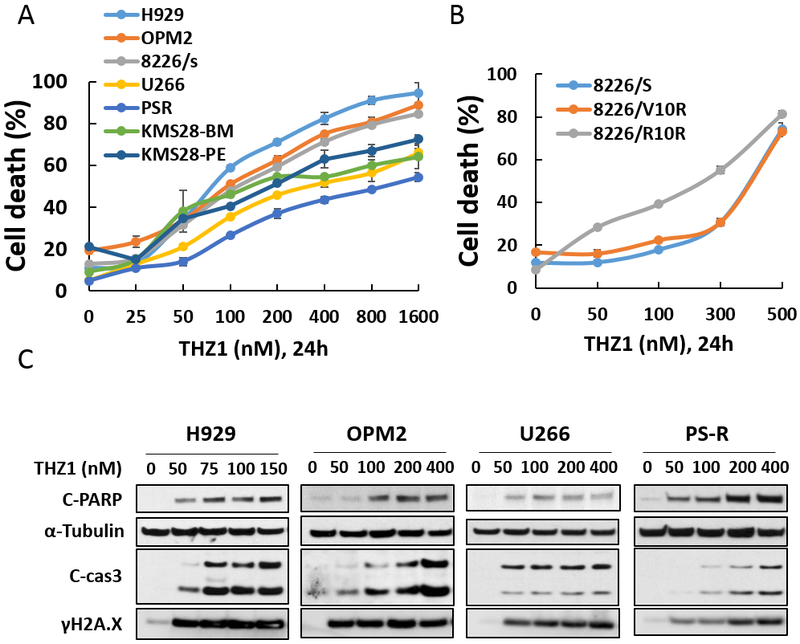
(A-B) H929, OPM2, RPMI8226, 8226/V10R, 8226/R10R, U266, PS-R, and KMS28-BM/PE were exposed to the indicated concentrations of THZ1 for 24 hr, followed by flow cytometric analysis of cell death after staining with 7-AAD. (C) H929, OPM2, U266 and PS-R cells were incubated with THZ1 for 24 hr, after which γH2A.X, and cleavage of caspase-3 and PARP were monitored by immunoblotting analysis. α-Tubulin was assayed to ensure equivalent loading and transfer. Results are representative of 3 separate experiments.
Cell cycle analysis revealed that sub-micromolar THZ1 concentrations (e.g., 50–150 nM; 16 hr) moderately but significantly increased the G2M and diminished the S-phase fraction in H929 and U266 cells (Supplemental Fig 1A-1C). These effects were associated with diminished T161 CDK1 and T160 CDK2 phosphorylation (Supplemental Fig 1D), consistent with the cyclin-activating kinase activity of CDK7 (20).
Studies in U266, their bortezomib-resistant counterparts PS-R (15) and OPM2 MM cells demonstrated that low nM THZ1 concentrations (50–400 nM, 24 hr) potently diminished phosphorylation of RNA Pol II at both Ser2 and Ser5 sites, while also dephosphorylating CDK9 at the T186 activation site (Fig 2A). In contrast, the BET inhibitor JQ1, known to disrupt the transcriptional regulatory apparatus (21), administered at a higher concentration (500 nM), had little effect on these events. Time course studies in U266 cells showed discernible dephosphorylation of Pol II and CDK9 at 3 hr, which became more pronounced over the ensuing 24 hr (Fig 2B). Finally, studies in U266 and PS-R cells demonstrated that these changes persisted despite THZ1 washout (Fig 2C), consistent with the covalent nature of THZ1/CDK7 interactions (22).
Figure 2. THZ1 inhibits general transcription irreversibly in MM cells.
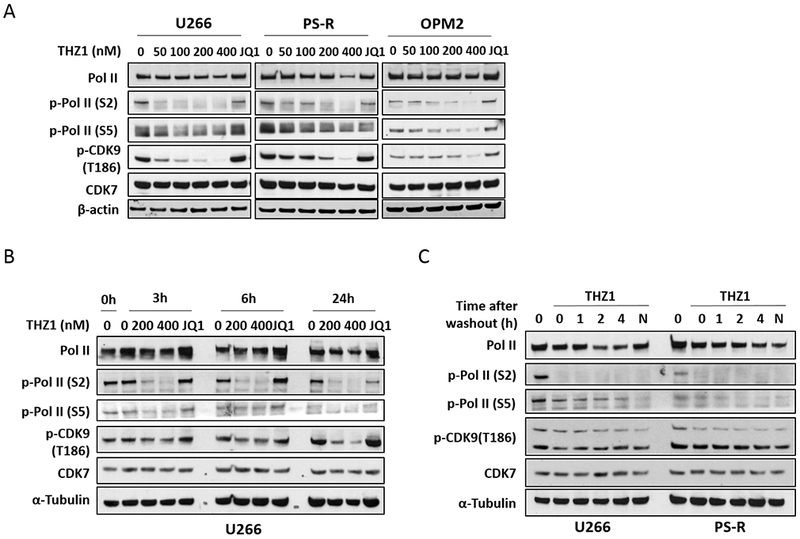
(A) U266, PS-R and OPM2 cells were treated with the indicated concentrations of THZ1 or BET inhibitor JQ1 (500 nM) for 24 hr, after which Pol II, p-Pol II (S2/S5), p-CDK9 (T186) and CDK7 were monitored by immunoblotting analysis. α-Tubulin was assayed to ensure equivalent loading and transfer. (B) U266 cells were exposed to the indicated concentrations of THZ1 and the BET inhibitor JQ1 (500 nM) for 3, 6 and 24 hr. Immunoblotting analysis was then performed as (A). (C) U266 and PS-R cells were treated for 2 hr with THZ1 (400 nM), followed by washing, transfer to fresh medium, and collection at the indicated time points. Immunoblotting analysis was then performed as in (A).
Exposure of U266 cells to low nM concentrations of THZ1 (50–400 nM, 24 hr) led to down-regulation of c-MYC, MCL-1, and BCL-XL protein levels (Fig 3A). JQ1 also down-regulated c-MYC, consistent with previous reports (21), as well as BCL-XL, but not MCL-1. Virtually identical findings were obtained in H929 cells (Supplemental Fig 2A). Time course analysis of the latter showed discernible c-MYC, MCL-1, and BCL-XL protein down-regulation associated with PARP cleavage after 6 hr of THZ1 exposure, which was considerably more pronounced down-regulation at 24 hr (Supplemental Fig 2B). Notably, co-administration of the caspase inhibitor Z-VAD-fmk blocked THZ1-mediated PARP and caspase cleavage but not MCL-1, BCL-XL, or c-MYC down-regulation (Supplemental Fig 2C), arguing against the possibility that these events represented secondary caspase-dependent phenomena.
Figure 3. THZ1 downregulates c-MYC, MCL-1 and BCL-XL transcriptional and overexpression of c-MYC, MCL-1, or BCL-XL significantly diminishes apoptosis induced by THZ1.
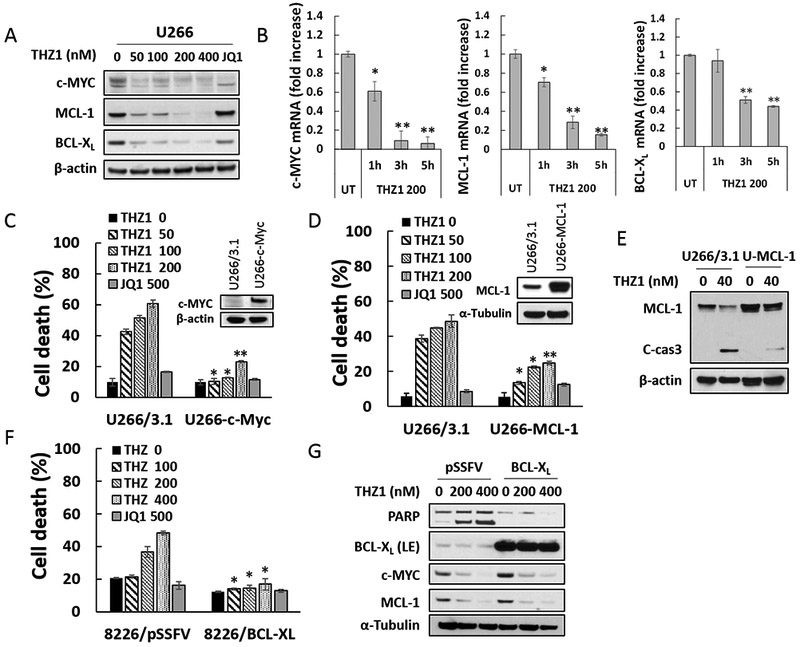
(A) U266 cells were treated with THZ1 or the BET inhibitor JQ1 (500 nM) for 24 hr, after which c-MYC, MCL-1 and BCL-XL proteins were monitored by immunoblotting analysis. β-actin was assayed to ensure equivalent loading and transfer. (B) U266 cells were exposed for 1, 3, or 5 hours to THZ1 (200 nM), after which RNA was extracted from the cells. Expression of c-MYC, MCL-1 and BCL-XL were analyzed by real-time RT-PCR, and results are presented relative to GAPDH expression in the un-treated control group. (C and D) U266 cells were stably transfected with pcDNA3.1, pcDNA3.1-c-MYC, or pcDNA3.1-MCL-1. Cells were treated with THZ1 or the BET inhibitor JQ1 (500 nM) for 24 hr, after which cell death was analyzed by flow cytometry following staining with 7-AAD. Values represent the means ± SD for at least three independent experiments performed in triplicate. *P<0.05; **P<0.01 (E) Transfected U266 cells were treated with the indicated concentration of THZ1 for 24 hr. MCL-1 and caspase-3 cleavage were monitored by immunoblotting analysis. β-actin was assayed to ensure equivalent loading and transfer. (F and G) 8226 cells stably transfected with pSSFV (empty vector) or BCL-XL, were treated with THZ1 or the BET inhibitor JQ1 (500 nM) for 24 hr. Cell death was analyzed by flow cytometry following staining with 7-AAD. Expression of PARP, BCL-XL, c-MYC, and MCL-1 were monitored by immunoblotting analysis. α-Tubulin was assayed to ensure equivalent loading and transfer.
To elucidate mechanisms underlying THZ1 actions, RT-PCR analysis was performed. RT-PCR demonstrated that 200 nM THZ1 significantly diminished c-MYC, MCL-1, and BCL-XL mRNA levels in U266 cells as early as 1–3 hr after drug exposure (Fig 3B). Comparable MCL-1, BCL-XL, and c-MYC mRNA down-regulation was observed in H929 cells (Supplemental Fig 2D).
To assess the functional significance of transcriptional repression of these proteins in the anti-myeloma activity of THZ1, lines were generated ectopically expressing c-MYC, MCL-1, or BCL-XL. U226 cells ectopically expressing c-MYC (Fig 3C, inset) were significantly less sensitive to 50–200 nM THZ1-induced cell death than empty-vector controls, but equally sensitive to 500 nM JQ1 (Fig 3C). Analogously, ectopic expression of MCL-1 (Fig 3D, inset) protected U266 cells from low concentrations of THZ1 but not JQ1 (Fig 3D). The ability of MCL-1 to block THZ1-mediated caspase-3 cleavage is shown in Fig 3E. Concordant results were obtained in cells ectopically expressing BCL-XL (Fig 3F). Of note, while enforced expression of BCL-XL clearly blocked PARP degradation in U266 cells exposed to THZ1, it did not prevent MCL-1 or c-MYC down-regulation (Fig 3G). Collectively, these findings argue that transcriptional down-regulation of c-MYC, MCL-1, and BCL-XL contribute functionally to THZ1-induced cell death in MM cells.
It has been recently reported that THZ1 also inhibits CDK12 and CDK13 kinase activity (23), which control the expression of DNA damage response (DDR) genes. Therefore, the effects of THZ1 on DDR genes were examined in MM lines.THZ1 dramatically reduced the expression of most DNA repair proteins (CtIP, FANCD2, RAD51, BRAC1, and ERCC1) examined (Supplemental Fig 3A), and also down-regulated their mRNA levels (Supplemental Fig 3B), accompanied by increased expression of γH2A.X, indicative of double-stranded DNA breaks. These findings are concordant with the notion that THZ1 inhibits both CDK7 and CDK12 activities, consistent with what has been reported in the literature for other cell types (23). They are also consistent with the concept that down-regulation of DNA repair genes may also contribute to THZ1 activity in MM cells.
The effects of stromal cells on THZ1-mediated cell death was then examined. When luciferase-labeled U266 cells were exposed to sub-μM concentrations of THZ1 in the presence of HS-5 stromal cells, a modest reduction in cell death was observed (Supplemental Fig 4A). No reductions in bortezomib sensitivity were observed. Interestingly, HS-5 cell co-culture failed to protect cells from THZ1 lethality in highly bortezomib-resistant PS-R cells (Supplemental Fig 4B). Fluorescent microscopic images revealed the robust dose-dependent induction of cell death (7-AAD positivity) by THZ1 in the presence of HS-5 co-culture, particularly in PS-R cells (Supplemental Fig 4C).
The functional significance of CDK7 inhibition by THZ1 or CDK7 knock-out on MM cell growth and responses to targeted agents was then examined. To this end, two OPM-2 CRISPR-Cas CDK7 knock-out cell lines (sg_CDK7–1 and −2) were generated which exhibited markedly reduced or virtually absent CDK7 expression (Fig 4A, inset). Cell viability was monitored by the Titer-Glo assay, which reflects both cell death as well loss of cell proliferation. The knock-out lines displayed dramatically reduced viability and proliferation compared to control lines, which may reflect both THZ1’s anti-proliferative and pro-apoptotic actions (Fig 4A). Light-microscopic images of cells plated at 250 cells/well and incubated for 6 days are shown in Fig 4B. Similar growth inhibition was observed in H929 CDK7 knock-down cells (Supplemental Fig 5A). The effects of CDK7 knock-down on the response of MM cells to the clinically relevant agents bortezomib, carfilzomib, or ABT-199 were then investigated. Both of the knock-down lines were significantly more sensitive to these agents than controls, which demonstrated only a modest reduction in cell death (7-AAD uptake) (Fig 4C). Parallel studies were then performed employing THZ1. Treatment of U266 cells with 50 or 100 nM THZ1 significantly increased the lethal effects of all three agents compared to untreated controls (*P<0.05 in each case; Fig 4C). Equivalent results were obtained in bortezomib-resistant PS-R cells (Supplemental Fig 5B). Isobologram analysis in U266 cells revealed CI ratios substantially less than 1.0 for each agent, indicating synergistic interactions (Fig 4E). Synergistic interactions were also observed in H929 cells (Supplemental Fig 5C). These findings indicate that pharmacologic disruption of CDK7 mimics the effects of genetic CDK7 interruption in increasing the susceptibility of MM cells to PIs and BH3-mimetics. Given the pleiotropic actions of THZ1 (23), we examined the effects of CDK7 deficiency on c-MYC, MCL-1 and BCL-XL protein expression. c-MYC, MCL-1 and BCL-XL expression were clearly reduced in CDK7 knock-out OPM2 cells (Supplemental Fig 6A). To determine whether c-MYC, MCL-1 or BCL-XL could be rescued by CDK7 in knock-out cells, HA-CDK7 (24) was reintroduced in CDK7 knock-down cell expressing shRNA targeting 3-UTR. Notably, wild-type CDK7 effectively rescued c-MYC, MCL-1 and BCL-XL expression in CDK7 knock-down cells (Supplemental Fig 6B). Significantly, however, CDK7 did not reverse down-regulation of these proteins following THZ1 treatment (Supplemental Fig 6C). As THZ1 targets both CDK7 and CDK12/13 (23,25), it is possible that both CDK7 and CDK12/13 inhibition may be involved in down-regulation of MYC, MCL-1 and BCL-XL by THZ1 in these cells, and thus not rescued by CDK7 alone. The effects of THZ1, alone or in combination, were then examined in primary MM cells. As shown in the fluorescence photomicrographs in Fig 5A, exposure (16 hr) of CD138+ cells from two patients with MM to 300 nM THZ1 sharply increased expression of activated caspase-3. In a series of primary MM patient samples (N =17) exposed to 300 nM THZ1; 16 hr, a very significant reduction in survival (Annexin V/7-AAD staining) was observed in both the CD138+ fraction as well as in the more primitive CD138−, CD19+, CD20+, CD27+ population (26) (N=11; P<0.01 in each case; Fig 5B, left panel). In marked contrast, an identical THZ1 exposure was minimally toxic to normal CD34+ cord blood cells (Fig 5B, right panel). In addition, co-administration (16 hr) of THZ1 (25 nM) and bortezomib (2 nM) very significantly reduced survival compared to individual treatment in both of these MM cell populations (Fig 5C and 5D), but had little effect on the survival of normal cord blood CD34+ cells (Fig 5I). Similar results were obtained when THZ1 was combined with carfilzomib (3 nM; Fig 5E and 5F; 5J) or ABT-199 (200 nM; Fig 5G and H; 5K).
Figure 4. Genetic or pharmacologic CDK7 inhibition diminish MM cell cell proliferation and potentiate the anti-myeloma effects of PIs and BH3-mimetics.
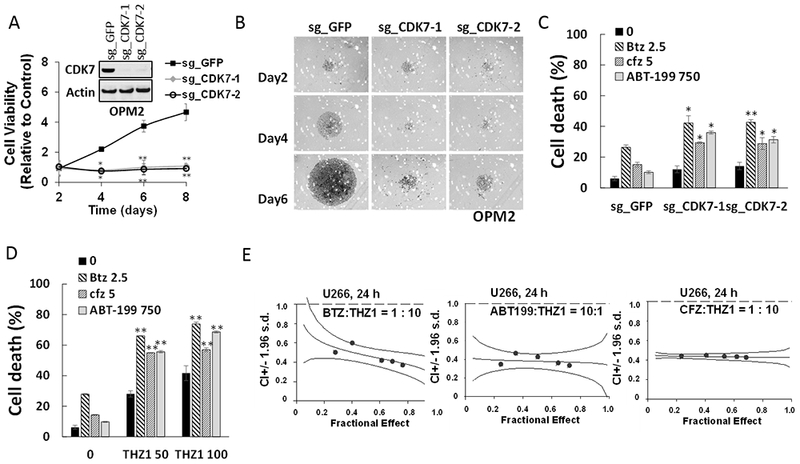
(A) OPM2 cells were infected with lentivirus encoding Cas9 and sgRNA targeting GFP or CDK7. After infection for 48 hr and selection with puromycin (0.2 μg/ml) for 5 days, cells were collected and the expression of CDK7 was monitored by immunoblotting. Cell vitality was determined by the cell titer-glo assay at day 2, 4, 6 and 8. **P<0.01. (B) Cells were seeded in a 48-well round-bottom plates (250 cells per well), and images were obtained on day 2, 4, 6 and 8 with an IX71-Olympus research inverted system microscope at 40× magnification. (C) OPM2 cells were infected with lentivirus encoding Cas9 and sgRNA targeting GFP or CDK7 for 48 hr without selection, and then treated with the indicated concentration (nM) of Btz (bortezomib), Cfz (carfilzomib), or ABT-199 (venetoclax) for 24hr, after which cell death was analyzed by flow cytometry following staining with 7-AAD. *P<0.05, **P<0.01. (D) U266 cells were incubated with THZ1 ± Btz, Cfz, or ABT-199 for 24 hr, after which cell death was analyzed by flow cytometry after staining with 7-AAD. **P<0.01. (E) U266 cells were exposed (24 hr) to varying concentrations of Btz ± THZ1 at a fixed ratio (1:10), ABT ± THZ1 at a fixed ratio (10:1) or Cfz ± THZ1 at a fixed ratio (1:10), after which apoptosis was determined by 7-AAD staining. Combination Index (CI) values less than 1.0 denote a synergistic interaction. The results are representative of three separate experiments.
Figure 5. THZ1 exhibits marked activity and significantly enhances proteasome inhibitor or BH3 mimetic lethality in human primary CD138+ MM cells but displays minimal toxicity towards normal CD34+ cells.
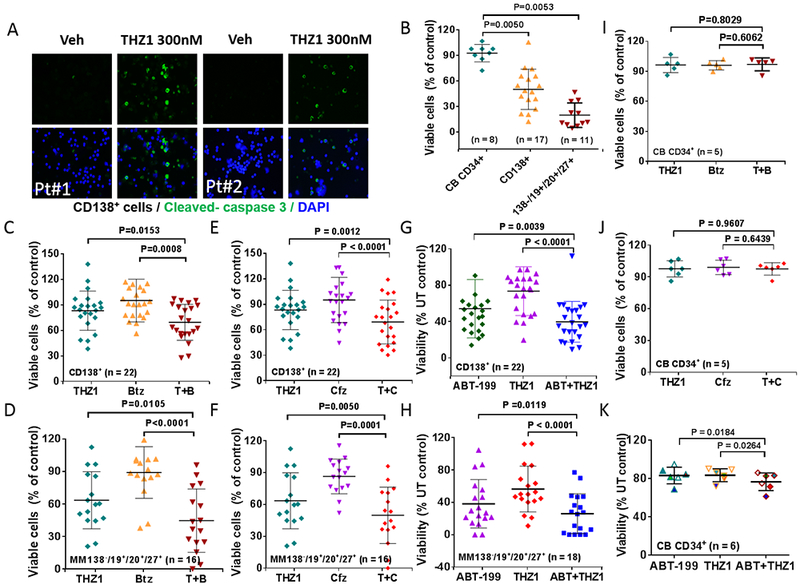
(A) Isolated CD138+ MNC cells from primary MM cells harboring high-risk cytogenetic aberration (#1: del 13q (13q14.3, 13q34), 14 del/rearrangement, del 17p (17p13.1); #2: del 13q (13q14.3, 13q34), 14 rearrangement, del 17p (17p13.1)), were treated with THZ1 300nM for 16 hr, after which slides were prepared and stained with antibodies against activated caspase-3, followed by Alexa Fluor 488-conjugated secondary antibody with DAPI counterstaining (×200 magnification). (B) Parallel experiments were carried out with primary samples, treated with THZ1 300nM. (C, E and G) Isolated mononuclear cells were treated with THZ1 ± Btz (N = 22), Cfz (N = 22), or ABT-199 (N = 22) for 16 hr, after which cells were stained with CD138-PE and Annexin V-FITC. The percentages of Annexin V+ cells were determined by flow cytometry. Lines indicate means and SD. (D, F and H) Parallel experiments were performed with 50 primary samples (THZ1 ± Btz (N = 16), Cfz (N = 16), or ABT-199 (N = 18). Viability of the CD138-/CD19+/CD20+/CD27+ cell population, which is enriched for MM progenitor cells, was determined by 7-AAD staining and analyzed by multicolor flow cytometry. (J and K) Normal cord blood cells were exposed (16 hr) to THZ1 ± Cfz (n=5), or Btz (N = 6), and viability determined by flow cytometry (7-AAD).
It has been reported that CDK7 inhibition can disrupt super-enhancers (25), which are frequently associated with genes like MYC, MCL-1, BCL-XL, and CCND2 (27) that figure prominently in MM biology. Consequently, chromatin IP of histone H3K27 acetylation, a mark of an active enhancer, was therefore performed. Super-enhancer-associated genes such as c-MYC, MCL-1 and BCL-XL displayed diminished H3K27 acetylation with THZ1 treatment (Supplemental Fig 7), raising the possibility that MYC, MCL1 and BCL-XL expression may be regulated by histone H3K27 acetylation, which can in turn be disrupted by THZ1 in myeloma cells.
Finally, the in vivo activity of THZ1 was examined in systemic and flank MM models. NSG mice were injected intravenously with 1×106 luciferase-labeled U266 cells, and treated with 10 mg/kg THZ1 (10 mg/kg ip qd, 5 days/week). As shown in Fig 6A, THZ1 treatment sharply reduced the luciferase signal from days 19 to 61 compared to controls, without significantly reducing body weight (Fig 6B). Kaplan-Meier analysis revealed that THZ1 also very significantly prolonged survival of mice compared to untreated controls (P<0.0069; Fig 6C). Parallel studies employing a luciferase-labeled bortezomib-resistant PS-R cell flank model showed a sharp decrease in tumor size and weight following THZ1 treatment (Fig 6D) which were both statistically significant (Fig 6E and 6F). As in the systemic model, THZ1 administration did not lead to significant weight loss (Fig 6G). Finally, as observed in vitro, Western blot analysis of excised tumors revealed down-regulation of MYC, MCL-1, and BCL-XL in tumors obtained from two representative THZ1-treated animals (Fig 6H), analogous to results obtained in vitro. Additionally, T161 CDK1 and T160 CDK2 phosphorylation were modestly decreased in tumors obtained from THZ1-treated animals compared to untreated controls (Fig 6H), raising the possibility that THZ1 might exert its anti-tumor effects in vivo through transcriptional inhibition as well as anti-proliferative actions.
Figure 6. THZ1 reduces tumor burden and significantly enhances survival in flank and orthotopic xenograft mouse MM models with minimal toxicity.
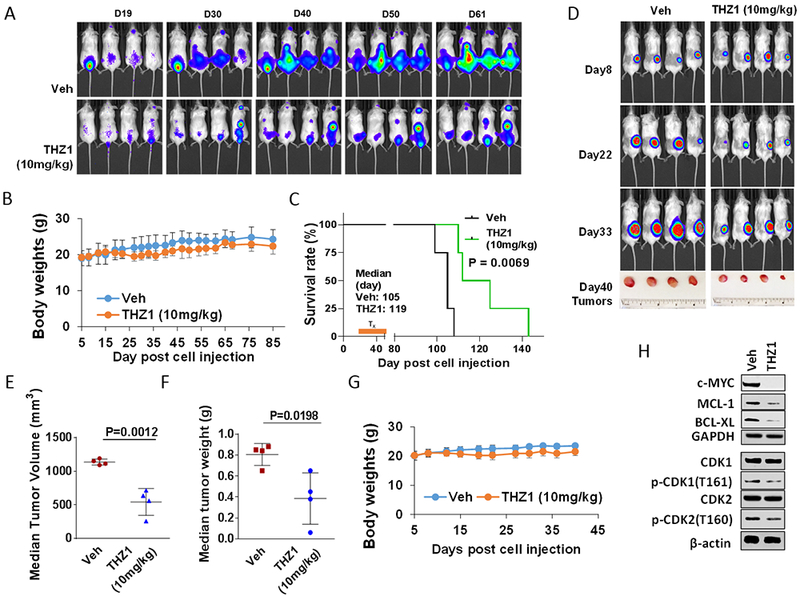
(A-C) NOD/SCID-γ (NSG) mice were injected intravenously via tail vein with 5×106 U266 cells stably expressing luciferase. After signals were visible (10 days after injection of tumor cells), THZ1 (10 mg/kg, i.p.) was administered daily for 5 days/week for an additional 4 weeks; n = 4 per group. Control animals were treated with equal volumes of vehicle. (A) Tumors were monitored every other day after i.p. injection with 150 mg/kg luciferin using the IVIS 200 imaging system. Veh, vehicle. (B) Survival of the animals was determined by Kaplan–Meier analysis. Inset, median survival days. P-values indicate significant difference between groups. (D-H) NSG mice were subcutaneously (s.c.) inoculated in the right rear flank with 5×106 luciferase-expressing U266 cells. Treatment was initiated after a luciferase signal was detected. THZ1 (10 mg/kg, i.p.) was administered daily for 5 days a week for an additional 4 weeks; n = 4 per group. Control animals were administered equal volumes of vehicle. (D) Tumors were monitored every other day after i.p. injection with 150 mg/kg luciferin using the IVIS 200 imaging system. Veh, vehicle. (E and F) Tumors were removed and weighed at day 40 after cell injection. Tumor volume was then plotted against actual tumor weight for each individual mouse. (C and G) Mice did not display significant body weight loss (>20% of initial weight) or other signs of toxicity due to the treatment. (H) Western blot analysis was performed to monitor the indicated candidate pharmacodynamic markers, identified from in vitro experiments, in tumors excised from representative mice.
Discussion
While the activity of CDKs may reflect inhibition of cell cycle progression, other actions are likely to contribute to the anti-tumor activity of this class of agents. For example, CDK9 inhibitors disrupt the function of the pTEFb complex and by extension, RNA Pol II, leading to transcriptional repression of short-lived proteins necessary for tumor cell survival (28). Similar observations have been reported in the case of CDK7, which participates in the function of the TFIIH transcription factor (29), implicated in transcriptional regulation, among other functions (30). Consistent with previous findings in other tumor types, THZ1 blocked phosphorylation of the CTD of RNA Pol II at serine 2 and 5 sites (31,32), and diminished transcription of several key anti-apoptotic proteins in MM cells. Given the pleiotropic actions of TFIIH (29), it might be anticipated that a CDK7 inhibitor such as THZ1 would act through multiple mechanisms to diminish MM cell proliferation and survival. In this regard, the CAK-inhibitory activity of THZ1 (3) was associated with diminished phosphorylation/activation of CDK9, but also CDK1 and 2, accompanied by reductions in S-phase progression as well as G2M arrest. Moreover, TFIIH has also been implicated in the DNA damage response (29,30), and CDKs are known to participate in DNA repair (1,19), particularly CDK12/13, which potentially accounts for the increase in γH2A.X expression and the diminished expression of DDR genes (e.g., CtIP, FANCD2, RAD51, BRCA1, and ERCC1) in THZ1-treated cells. The latter phenomenon takes on added significance in light of recent evidence that MM cells may be particularly susceptible to replicative stress and DNA damage (33). It is tempting to speculate that each of these actions e.g., down-regulation of pro-survival proteins, inhibition of cell cycle progression, and promotion of DNA damage cooperate to reduce MM cell proliferation and promote cell death.
The c-MYC oncogene is involved in diverse oncogenic pathways, including those related to cell proliferation, survival, metabolism, and immune surveillance, among others (34). It plays a critical role in several tumor types, including neuroblastoma (35), certain lymphoid leukemia (36), and lymphomas, e.g., double-hit DLBCL (37). De-regulation of c-MYC has also been implicated in myelomagenesis and c-MYC is considered a validated target in this disease (12). However, the lack of a druggable binding pocket has made direct pharmacologic disruption of c-MYC challenging, and it has been described, along with KRAS, as an undruggable target (38,39). Nevertheless, there have been multiple attempts to target c-MYC indirectly, either by inhibiting upstream pathways or disrupting complexes in which it participates. Agents reported to interfere with c-MYC function or expression include inhibitors of β-catenin (40), aurora kinase inhibitors (41), and bromodomain extra-terminal domain (BET) inhibitors (42), which interfere with c-MYC transcription. More recently, CDK7 inhibitors, including THZ1, have been shown to be potent antagonists of c-MYC expression (4,32). In accord with these findings, THZ1 robustly reduced c-MYC mRNA levels and protein expression in MM cells. Notably, on a molar basis, THZ1 was considerably more potent than the BETi JQ1 in down-regulating c-MYC. The observation that ectopic expression of c-MYC significantly diminished the anti-proliferative and –survival actions of THZ1 argues that transcriptional down-regulation of c-MYC plays an important role in the anti-MM activity of this agent.
Exposure of MM cells to THZ1 also down-regulated transcription and expression of MCL-1 and BCL-XL, both of which have been identified as determinants of both proteasome inhibitor and BH3-mimetic responsiveness (43,44). Due to a short half-life (e.g., 2–4 hr) (45), MCL-1 has been shown to be an important target of transcriptional repressive CDKIs such as alvocidib (46) and more recently, THZ1 (4,7,34). While it is conceivable that down-regulation of these proteins could reflect caspase-dependent cleavage occurring in apoptotic cells, the finding that down-regulation persisted in cells exposed to a broad caspase inhibitor argues against this possibility. Notably, CRISPR-Cas knock-down of CDK7 sharply reduced MM cell proliferation and survival, and significantly increased the sensitivity of MM cells to several anti-MM agents, including proteasome inhibitors and the BCL-2 antagonist venetoclax. Furthermore, genetic disruption of CDK7 was phenocopied by the pharmacologic agent THZ1 which potentiated the activity of each of these agents, supporting the notion that THZ1-mediated CDK7 inhibition was responsible for the observed synergism. In this context, we have recently reported that the CDK9 inhibitor alvocidib promoted cell death induction in high-risk MM cells exhibiting high low BCL-2 to MCL-1 ratios (e.g., t(4;14) MM) and venetoclax resistance (14). In light of its ability to down-regulate MCL-1 as well as BCL-XL, it is likely that THZ1 would exert a similar activity, and efforts to confirm this possibility are currently underway. Finally, the ability of ectopic expression of c-MYC, MCL-1, and BCL-XL to ameliorate the lethal effects of THZ1 alone or in combination with other agents argues that transcriptional repression of these proteins plays a key functional role in THZ1 anti-MM actions.
It is important to note that THZ1 administered alone and particularly in combination with PIs or BH3-mimetics resulted in significant increases in cell death in primary, patient-derived MM cells (CD138+) as well as in a population putatively enriched for MM stem cell-like cells (CD138−, CD19+, CD20+, CD27+) (47), but was relatively non-toxic to normal CD138− and CD34+ cord blood cells. However, synergism between THZ1 and other agents (e.g., venetoclax, carfilzomib) was not as dramatic in primary myeloma cells as in their continuously cultured counterparts. This may reflect the greater susceptibility of cell lines to diverse agents compared to primary myeloma cells due to a various factors e.g., greater cytokinetic activity versus quiescent primary CD138+ cells. The basis for the observed selectivity of CDKIs toward MM cells is not clear, but has been attributed to the intrinsic vulnerability of transformed cells to cell cycle deregulation compared to their normal counterparts (48). The increased susceptibility of MM cells to replicative stress (49) and the requirement for active transcription to circumvent this phenomenon could also contribute. Alternatively, the presence of super-enhancers at MYC and other key genes e.g., MCL-1 and BCL-XL associated with MM led us to test the hypothesis that super-enhancers may be particularly sensitive to CDK7 inhibitors like THZ1 in MM cells. This appeared to be the case. However, to determine whether this phenomenon contributes to the selectivity of THZ1 toward transformed cells will require additional studies, including ChIP-seq analysis and comparisons in normal hematopoietic cells. In addition to these possibilities, several alternative mechanisms have been implicated in interactions between CDKIs and other agents in MM cells, including up-regulation of pro-apoptotic proteins (50,51), induction of ER stress (52), and in the case of BH3-mimetics, disruption of cytoprotective autophagy (53). Whether any of these mechanisms contributes to the preferential targeting of MM cells by THZ1 alone or in combination remains to be determined.
Consistent with its relative sparing of normal hematopoietic cells, THZ1 administration displayed little in vivo toxicity, but significantly increased animal survival in a MM xenograft model, while exerting minimal toxicity. CDK and checkpoint (e.g., CHK1) inhibitors often have only modest single-agent activity, but are most effective when combined with other agents. In addition, the xenograft studies demonstrated recapitulation of several of the in vitro pharmacodynamic observations. Importantly, THZ1 reduced MM cell expression of several proteins e.g., c-MYC, MCL-1 shown to be down-regulated in in vitro studies. Deregulation of both c-MYC (12) and MCL-1 (11) have been implicated in myeloma pathogenesis, raising the possibility that CDK7 inhibitors such as THZ1 may be particularly appropriate in this disease. We also found that p-CDK1 and p-CDK2 levels were modestly reduced in THZ1-treated tumors compared to untreated controls (Fig 6H), at least at the 40 day interval. However, it is possible that different results might be obtained at alternative time intervals. Given these results, we cannot conclusively determine whether the in vivo growth suppressive effects of THZ1 stem predominantly from transcriptional repression, cell cycle effects, or a combination of the two. Whether a CDK7 inhibitor like THZ1 will prove superior to other CDK inhibitors e.g., the pan CDKi alvocidib remains to be determined, although the ability of THZ1 to disrupt the THFII complex (31) may have therapeutic implications. The recent entry of CDK7 inhibitors (e.g. SY-1365; ) into the clinical arena (7), along with their activity against MM cell lines and primary MM cells, in vivo efficacy, and synergistic interactions with PIs and BH3-mimetics argue that these agents warrant attention in MM. Successor studies examining in vivo interactions between THZ1 and other agents, ideally in MM PDX models as they become available, would be a logical plan. Efforts to test this notion are currently being explored.
Supplementary Material
Translational relevance: This study provides a theoretical foundation for incorporating CDK7 inhibitors, alone or in rational combinations, into the therapeutic armamentarium for patients with relapsed/refractory multiple myeloma.
Acknowledgments
This work was supported by awards CA205607 and UH2TR001373 (SG) from National Cancer Institute (NCI), and R6508–18 from Leukemia and Lymphoma Society (LLS). Services and products in support of the research project were generated by the Virginia Commonwealth University Cancer Mouse Models Core Laboratory, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Rimel JK, Taatjes DJ. The essential and multifunctional TFIIH complex. Protein Sci 2018;27:1018–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Zhou L, Leng Y, Dai Y, Orlowski RZ, Grant S. Positive transcription elongation factor b (P-TEFb) is a therapeutic target in human multiple myeloma. Oncotarget 2017;8:59476–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J Biol Chem 2018;293:10009–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014;159:1126–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Posternak V, Cole MD. Strategically targeting MYC in cancer. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015;163:174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014;26:909–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augert A, MacPherson D. Treating transcriptional addiction in small cell lung cancer. Cancer Cell 2014;26:783–4. [DOI] [PubMed] [Google Scholar]
- 9.Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K, et al. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood 2017;130:2326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reece DE, Phillips MJ. Infusion Reactions With Monoclonal Antibody Therapy in Myeloma: Learning From Experience. J Oncol Pract 2018;14:425–6. [DOI] [PubMed] [Google Scholar]
- 11.Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017;129:1969–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jovanovic KK, Roche-Lestienne C, Ghobrial IM, Facon T, Quesnel B, Manier S. Targeting MYC in multiple myeloma. Leukemia 2018;32:1295–306. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol 2009;29:6149–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou L, Zhang Y, Sampath D, Leverson J, Dai Y, Kmieciak M, et al. Flavopiridol enhances ABT-199 sensitivity in unfavourable-risk multiple myeloma cells in vitro and in vivo. Br J Cancer 2018;118:388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai Y, Landowski TH, Rosen ST, Dent P, Grant S. Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood 2002;100:3333–43. [DOI] [PubMed] [Google Scholar]
- 16.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou L, Zhang Y, Chen S, Kmieciak M, Leng Y, Lin H, et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015;29:807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pei XY, Dai Y, Felthousen J, Chen S, Takabatake Y, Zhou L, et al. Circumvention of Mcl-1-dependent drug resistance by simultaneous Chk1 and MEK1/2 inhibition in human multiple myeloma cells. PLoS One 2014;9:e89064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenall SA, Lim YC, Mitchell CB, Ensbey KS, Stringer BW, Wilding AL, et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017;6:e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell 2007;25:839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SK, Hong SH, Kim HS, Shin CY, Nam SW, Choi WS, et al. JQ1, an inhibitor of the epigenetic reader BRD4, suppresses the bidirectional MYC-AP4 axis via multiple mechanisms. Oncol Rep 2016;35:1186–94. [DOI] [PubMed] [Google Scholar]
- 22.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014;511:616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng M, Kwiatkowski NP, Zhang T, Nabet B, Xu M, Liang Y, et al. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 2003;22:5707–11. [DOI] [PubMed] [Google Scholar]
- 25.Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol 2016;12:876–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raja KR, Kovarova L, Hajek R. Review of phenotypic markers used in flow cytometric analysis of MGUS and MM, and applicability of flow cytometry in other plasma cell disorders. Br J Haematol 2010;149:334–51. [DOI] [PubMed] [Google Scholar]
- 27.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natoni A, Coyne MR, Jacobsen A, Rainey MD, O’Brien G, Healy S, et al. Characterization of a Dual CDC7/CDK9 Inhibitor in Multiple Myeloma Cellular Models. Cancers (Basel) 2013;5:901–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coin F, Egly JM. Revisiting the Function of CDK7 in Transcription by Virtue of a Recently Described TFIIH Kinase Inhibitor. Mol Cell 2015;59:513–4. [DOI] [PubMed] [Google Scholar]
- 30.Compe E, Egly JM. Nucleotide Excision Repair and Transcriptional Regulation: TFIIH and Beyond. Annu Rev Biochem 2016;85:265–90. [DOI] [PubMed] [Google Scholar]
- 31.Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol Cell 2015;59:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat Commun 2017;8:14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrero AB, Gutierrez NC. Targeting Ongoing DNA Damage in Multiple Myeloma: Effects of DNA Damage Response Inhibitors on Plasma Cell Survival. Front Oncol 2017;7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caforio M, Sorino C, Iacovelli S, Fanciulli M, Locatelli F, Folgiero V. Recent advances in searching c-Myc transcriptional cofactors during tumorigenesis. J Exp Clin Cancer Res 2018;37:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang XH, Tang F, Shin J, Cunningham JM. A c-Myc-regulated stem cell-like signature in high-risk neuroblastoma: A systematic discovery (Target neuroblastoma ESC-like signature). Sci Rep 2017;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohanian M, Rozovski U, Kanagal-Shamanna R, Abruzzo LV, Loghavi S, Kadia T, et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk Lymphoma 2018:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Zhang X, Zhang T, Song Z, Hu G, Li W, et al. Prognostic Significance of BCL-2 and BCL-6 Expression in MYC-positive DLBCL. Clin Lymphoma Myeloma Leuk 2018;18:e381–e9. [DOI] [PubMed] [Google Scholar]
- 38.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 2017;17:502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol 2016;56:23–40. [DOI] [PubMed] [Google Scholar]
- 40.Rennoll S, Yochum G. Regulation of MYC gene expression by aberrant Wnt/beta-catenin signaling in colorectal cancer. World J Biol Chem 2015;6:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, et al. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res 2011;71:3912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolani B, Gopalakrishnan R, Punj V, Matta H, Chaudhary PM. Targeting Myc in KSHV-associated primary effusion lymphoma with BET bromodomain inhibitors. Oncogene 2014;33:2928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nencioni A, Hua F, Dillon CP, Yokoo R, Scheiermann C, Cardone MH, et al. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood 2005;105:3255–62. [DOI] [PubMed] [Google Scholar]
- 44.Harper MT, Poole AW. Bcl-xL-inhibitory BH3 mimetic ABT-737 depletes platelet calcium stores. Blood 2012;119:4337–8. [DOI] [PubMed] [Google Scholar]
- 45.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol 1996;166:523–36. [DOI] [PubMed] [Google Scholar]
- 46.Ma Y, Cress WD, Haura EB. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther 2003;2:73–81. [PubMed] [Google Scholar]
- 47.Boucher K, Parquet N, Widen R, Shain K, Baz R, Alsina M, et al. Stemness of B-cell progenitors in multiple myeloma bone marrow. Clin Cancer Res 2012;18:6155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015;125:443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyashita K, Fujii K, Suehiro Y, Taguchi K, Uike N, Yoshida MA, et al. Heterochronous occurrence of microsatellite instability in multiple myeloma - an implication for a role of defective DNA mismatch repair in myelomagenesis. Leuk Lymphoma 2018:1–6. [DOI] [PubMed] [Google Scholar]
- 50.Komina O, Nosske E, Maurer M, Wesierska-Gadek J. Roscovitine, a small molecule CDK inhibitor induces apoptosis in multidrug-resistant human multiple myeloma cells. J Exp Ther Oncol 2011;9:27–35. [PubMed] [Google Scholar]
- 51.Semenov I, Akyuz C, Roginskaya V, Chauhan D, Corey SJ. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk Res 2002;26:271–80. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen TK, Grant S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol Cancer Ther 2014;13:662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Zhou L, Zhang Y, Leng Y, Pei XY, Lin H, et al. Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol Cell Biol 2014;34:3435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.