

Summary
Air‐side stripping without a prior solid–liquid phase separation step is a feasible and promising process to control ammonia concentration in thermophilic digesters. During the process, part of the anaerobic biomass is exposed to high temperature, high pH and aerobic conditions. However, there are no studies assessing the effects of those harsh conditions on the microbial communities of thermophilic digesters. To fill this knowledge gap, the microbiomes of two thermophilic digesters (55°C), fed with a mixture of pig manure and nitrogen‐rich co‐substrates, were investigated under different organic loading rates (OLR: 1.1–5.2 g COD l−1 day−1), ammonia concentrations (0.2–1.5 g free ammonia nitrogen l−1) and stripping frequencies (3–5 times per week). The bacterial communities were dominated by Firmicutes and Bacteroidetes phyla, while the predominant methanogens were Methanosarcina sp archaea. Increasing co‐substrate fraction, OLR and free ammonia nitrogen (FAN) favoured the presence of genera Ruminiclostridium, Clostridium and Tepidimicrobium and of hydrogenotrophic methanogens, mainly Methanoculleus archaea. The data indicated that the use of air‐side stripping did not adversely affect thermophilic microbial communities, but indirectly modulated them by controlling FAN concentrations in the digester. These results demonstrate the viability at microbial community level of air side‐stream stripping process as an adequate technology for the ammonia control during anaerobic co‐digestion of nitrogen‐rich substrates.
Introduction
Anaerobic digestion (AD) is a microbial process based on the combined and syntrophic activities of a wide range of microorganisms from hydrolytic and fermentative bacteria to methanogenic archaea (Stams and Plugge, 2009). The microbial community of AD reactors is mainly affected by substrate composition (Regueiro et al., 2012; Zhang et al., 2014) and operational conditions (Vanwonterghem et al., 2015; Regueiro et al., 2016; Amha et al., 2018). One of the most important factors that drastically affects AD microbial communities is temperature as it imposes a selection pressure on the communities shifting the abundance and activity of specific populations in digesters treating different kinds of waste like beet molasses, maize, slaughterhouse or fish residues, municipal solid wastes and swine or pig manures (Regueiro et al., 2014; Vrieze et al., 2015; Lin et al., 2016). Thermophilic (45–60°C) communities are regarded as less diverse and even than mesophilic (30–45°C) ones. Under high temperatures, microbial communities are usually dominated by Firmicutes phylum and hydrogenotrophic methanogens, whereas Bacteroidetes and acetoclastic archaea are the prevalent groups under mesophilic conditions in reactors treating sewage sludge or co‐digesting food waste and slaughterhouse residues (Sundberg et al., 2013). In these thermophilic communities, syntrophic oxidizing bacteria and hydrogenotrophic methanogens play a significant role. Particularly, hydrogen‐producing bacteria from the genus Clostridium, together with syntrophic bacteria of the genera Pelotomaculum, Syntrophomonas and Coprothermobacter, associated with Methanoculleus, Methanosarcina or Methanobrevibacter hydrogenotrophic methanogens have been observed in anaerobic reactors fed with biowaste and sewage sludge (Ritari et al., 2012), food waste (food waste; Guo et al., 2014), cattle manure (Moset et al., 2015) and maize silage (Pap et al., 2015).
The organic loading rate (OLR) is another important factor shaping AD microbial communities. Organic overloading causes process imbalance, and thus volatile fatty acid (VFA) accumulation, and a shift in the microbial community (Guo et al., 2014; Regueiro et al., 2015). Overloading events are also associated with an increase in free ammonia nitrogen (FAN) concentrations in the digesters when the substrate used is rich in nitrogen compounds such as manure (De Francisci et al., 2015; Regueiro et al., 2016; Sun et al., 2016) and some types of food waste (Gao et al., 2015; Yun et al., 2016). High levels of ammonia can inhibit microbial activities (Liu and Sung, 2002; Zhang et al., 2012; Westerholm et al., 2018) although it has been demonstrated that acclimatization of microbial communities can alleviate this inhibition (Yan et al., 2019). It is well known that acetoclastic methanogens are less tolerant to ammonium than hydrogenotrophic archaea (Angelidaki and Ahring, 1993; Vrieze et al., 2012; Westerholm et al., 2012, 2018; Yan et al., 2019), and consequently, increasing ammonium concentration shifts methanogenesis from acetoclastic to hydrogenotrophic pathway (Lü et al., 2013; Werner et al., 2014; Regueiro et al., 2016). In contrast, less is known about the effects of high ammonium concentrations on bacterial organisms. Recent studies have reported the dominance of Bacteroidetes group under low‐ammonium conditions, while under high concentrations the phylum Cloacimonetes disappears and the Firmicutes phylum, in particular Clostridiales and Lactobacillales orders, dominates the community (Werner et al., 2014; Vrieze et al., 2015; Regueiro et al., 2016; Westerholm et al., 2018). When high ammonia levels inhibit obligate acetoclastic methanogens, the microbial community shifts towards syntrophic acetate oxidation pathway (Westerholm et al., 2016) although the keystone taxa responsible for the function are unclear (Werner et al., 2014; Theuerl et al., 2019).
Some strategies have been developed to avoid ammonia AD inhibition (Krakat et al., 2017). Among them, side‐stream ammonia stripping has modest reagent costs and easy operation and allows the recovery of nitrogen as ammonium sulfate (Zhang et al., 2012; Pedizzi et al., 2017). During this process, the digester content (digestate) is treated in a separate column where it is exposed to increased pH and temperature causing the transfer of dissolved ammonia to gas phase which is removed through the application of a gas flow, usually air. Then, the stripped digestate is recirculated back into the reactor (Bousek et al., 2016; Pedizzi et al., 2017). When the stripping process lacks a previous solid–liquid separation step, the complete digestate from the reactor enters the stripping column, and therefore, a fraction of the reactor microbial community is exposed to the harsh conditions of the stripping process. It is well established that the conditions applied during the stripping process (high temperature, high pH and the presence of oxygen at saturation) alter AD microbial communities. High‐pH conditions inhibit methanogenic archaea (Yuan et al., 2006; Chen et al., 2017), and high oxygen concentrations modify microbial composition by removing Clostridium senso stricto 1, which negatively impacts the capacity for hydrogen production (Yang and Wang, 2018). Finally, high temperature correlates with lower microbial community diversity and complexity (Karakashev et al., 2005; Westerholm et al., 2018). But, to the best of our knowledge, the combination of high temperature, pH and oxygen levels on AD microbiome has not been assessed previously. The aim of this work was to study the effects of applying an air side‐stream stripping column without a previous solid–liquid separation step on the thermophilic microbial community of anaerobic digesters under different ammonia concentrations, stripping frequencies and organic loading rates.
Results and discussion
Reactor performance during microbiome analysis
The operation of the two reactors (R1 and R2) throughout the periods during which anaerobic microbiome was monitored is shown in Fig. 1. A detailed description of reactor operation and of the stripping unit can be found in Pedizzi et al. (2017). In brief, after the initial start‐up and acclimation to thermophilic conditions (Period 1, from day 0 to day 79), co‐digestion was started in both reactors with the addition of maize silage and Ecofrit® (Period 2: 80–299 days in R1 and 80–329 in R2). The OLR was increased from 1.0 to 3.0 g chemical oxygen demand (COD) l−1 day−1, whereas FAN concentration rose from 0.2 to 0.6 g N‐FAN l−1 (Fig. 1A and B). In Period 3 of R1 (300–365 days), the stripping process was applied by treating and recirculating 21% of the reactor volume three times per week. This lowered FAN concentration from 0.6 to 0.2 g N‐FAN l−1, while both OLR and methane production remained stable (Fig. 1A). In R2, Period 3 (330–439 days) corresponded to the sensible increase of FAN concentration from 0.6 to 1.3 g N‐FAN l−1 throughout a time span of almost 100 days, while the OLR was kept constant, leading to a lack of negative effects on methane production (Fig. 1B). During Period 4 of R2 (440–494 days), the stripping process was applied under the same conditions as in Period 3 of R1, but with a frequency of five times per week, decreasing FAN levels from 1.3 to 0.2 g N‐FAN l−1, whereas OLR lowered slightly to 2.5–2.8 g COD l−1 day−1 (Fig. 1B) and methane production remained stable. After a maintenance interval of 2 months (Period 5 of R2), in Period 6 of R2 (580–639 days) the stripping process was used again three times per week to maintain FAN concentration close to 0.6 g N‐FAN l−1 while the OLR was increased from 2.5 to 5.0 g COD l−1 day−1 by incrementing the co‐substrate fraction in the feeding (Fig. 1C). Consequently, methane production rose from 1.6 to 3.0 g COD l−1 day−1 resulting in a COD removal efficiency of 65%. During Period 7 of R2 (640–707 days), side‐stream stripping was stopped and FAN rose sharply from 0.6 to 1.5 g N‐FAN l−1 (OLR remained at 5.1 g COD l−1 day−1), causing VFA accumulation and leading to final digestion collapse (Fig. 1C).
Figure 1.
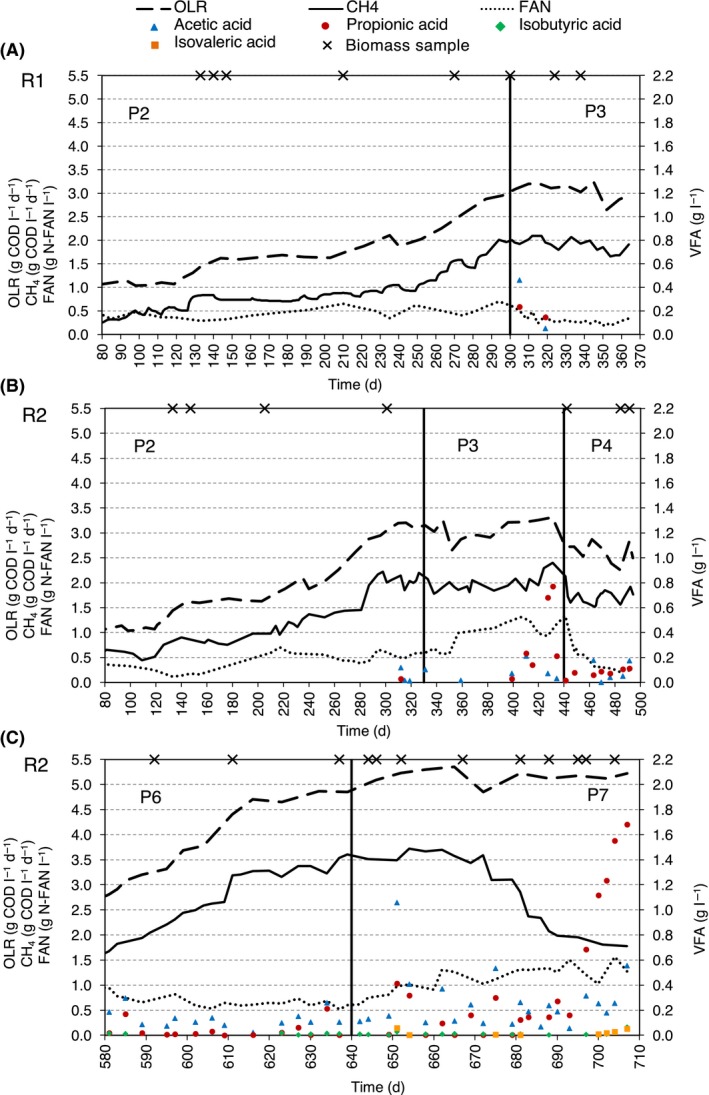
Organic loading rate (OLR), methane production, free ammonia nitrogen (FAN) and main volatile fatty acid (VFA) concentrations in the reactors during the experiment. (A) Periods 2 and 3 of R1; (B) Periods 2, 3 and 4 of R2; and (C) Periods 6 and 7 of R2. Vertical lines separate operational periods. Crosses indicate biomass sampling days.
Overview of community diversity
A total of 787 053 bacterial and 1 295 823 archaeal high‐quality 16S rRNA gene partial sequences were obtained for the 28 biomass samples (Table S1). Community richness (number of species) and evenness (equality of abundance of species) estimations showed a less diverse and complex archaeal community compared to bacterial microbiome (Fig. 2), in concordance with previous observations of biogas‐producing communities (Weiss et al., 2008; Wirth et al., 2012). Bacterial richness and evenness tended to increase during P2, the phase in which the OLR and FAN increased from 1.0 to 3.0 g COD l−1 day−1 and from 0.2 to 0.6 g N‐FAN L−1 respectively (Fig. 1), especially in R2. Later, while richness become constant, bacterial evenness oscillated with time to notably decrease in P7, the phase in which R1 was operated at high OLR (5.1 g COD l−1 day−1) but stripping was not applied with the consequent FAN increment to 1.5 g N‐FAN l−1 and VFA accumulation (Fig. 1). Archaeal richness and evenness were constant to some extent in R1, whereas in R2, they fluctuated with time, including a remarkably increasing trend in P7 in both measurements. The trends observed in R2 during P7 seem to be related to the FAN levels in the reactor (Fig. 1C). Lower levels of bacterial uniformity related to high ammonia levels in AD have been previously observed (Werner et al., 2014) and might be related to ammonia inhibition of many species of bacteria. By contrast, the increase in archaeal community richness and evenness with FAN levels is probably related to the promotion of new species of methanogens that are probably incoming with the feeding and that can face the high‐FAN conditions.
Figure 2.
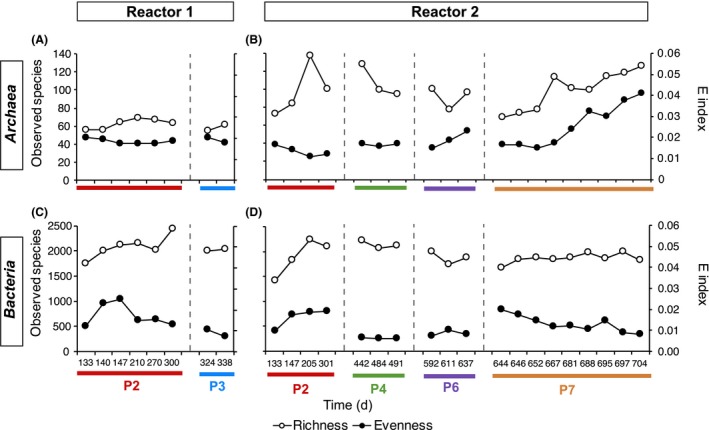
Temporal changes in archaeal and bacterial estimated richness and evenness for R1 (A, C) and R2 (B, D). The different operational periods are indicated in the horizontal axis. The Simpson E index ranges from 0 to 1, where 1 implies total community uniformity.
Linkage of community dynamics and operational conditions
Overall, bacterial and archaeal community structure was dynamic through time and the changes were mainly correlated with alterations in the OLR and FAN levels (Fig. 3, S2–S5). In P2 of both reactors, bacterial but not archaeal community structure shifted in parallel to a moderate OLR increase from 1 to 3 g COD l−1 day−1, which also caused FAN to rise from 0.2 to 0.6 g N‐FAN l−1 due to the introduction of co‐substrates.
Figure 3.
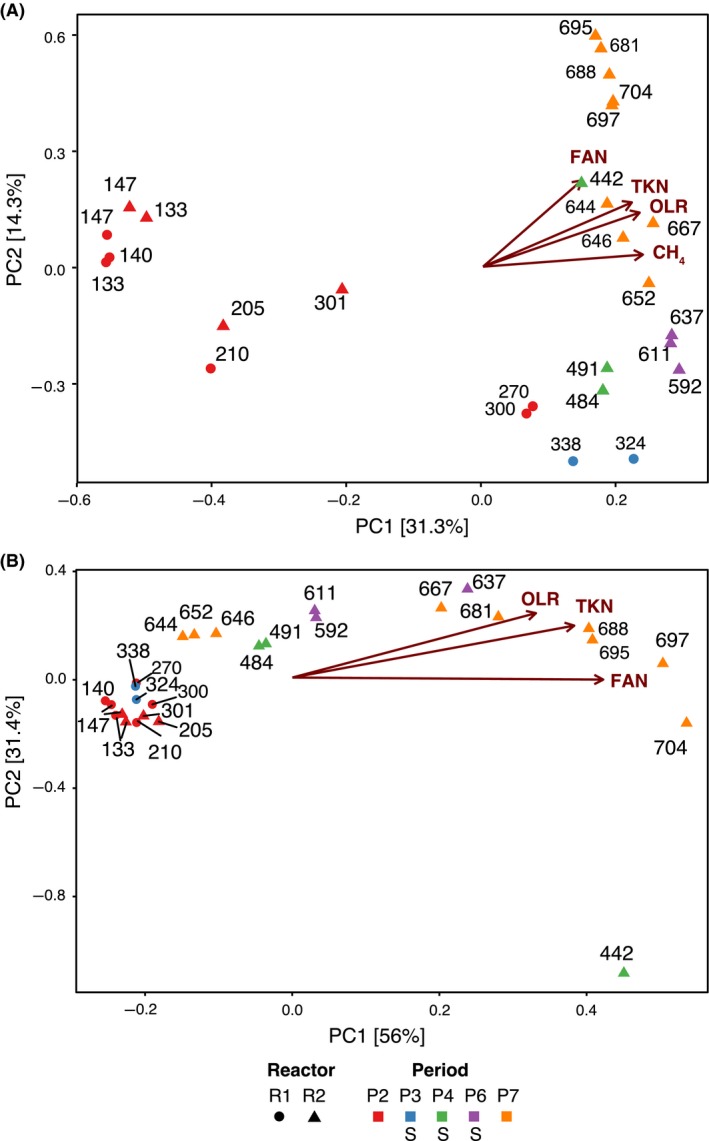
tb‐PCA showing community structure changes and its correlation with operational parameters for bacteria (A) and archaea (B). Each point represents the community composition of a reactor (indicated by the form shape) at a given time point (sample labels indicate operational day; form colour indicates the period). Vectors indicate the increasing values of operational variables (only the statistically significant variables (P‐value < 0.05) are shown). Periods in which the biomass was exposed to stripping are indicated with an S in the legend.
To study the decoupled effects of FAN and OLR, FAN was gradually increased from 0.6 to 1.3 g N‐FAN l−1 in R2 between P2 and P4 (days 330–439; Fig. 1) while keeping a constant OLR (Pedizzi et al., 2017). In this case, bacterial and archaeal community structures were different (Fig. 3) before (day 301) and after the FAN increase (day 442). During P4 of R2, when the stripping unit was used to decrease the FAN levels from 1.3 to 0.2 g N‐FAN l−1, while the OLR was still constant, both archaeal and bacterial community structures largely changed again (Fig. 3). These data indicate the strong influence that FAN concentrations impose to microbial communities. Then, the effect of increasing OLR was tested by keeping a low and constant FAN by applying the stripping process during P6 (Fig. 1). In this case, the overall community structure was not very altered, apart from archaeal communities that become more dissimilar when the OLR reached values close to 5 g COD l−1 day−1 (day 637; Fig. 3B). In P7, a high OLR (5.1 g COD l−1 day−1) was maintained without the application of the stripping process, resulting in a raise of FAN concentration from 0.6 to 1.5 g N‐FAN l−1 and increasing concentrations of VFAs (Fig. 1). In this case, the structures of both communities were gradually modified (Fig. 3). These results confirm previous observations on the importance of OLR and FAN levels on community organization of AD reactors under both mesophilic (Werner et al., 2014; Regueiro et al., 2015, 2016) and thermophilic conditions (Yenigün and Demirel, 2013; Kuroda et al., 2015). Interestingly, the largest community shift of this period occurred earlier in archaeal than in bacterial communities. The largest Archaea community modification occurred between days 652 and 667 when the increasing FAN concentrations crossed the boundary of 1 g N‐FAN l−1, when a peak in the concentrations of VFAs was observed and when methane production started a stepwise decrease. However, the largest bacterial community change occurred later, between days 667 and 688. The latter does not agree with Li et al. (2017), who observed that bacterial communities responded earlier during mesophilic solid‐state AD of protein‐rich organic wastes.
During P3, P4 and P6 when stripping process was applied, part of the biomass was temporarily exposed to high pH, temperatures and oxygen concentrations, conditions that individually can impair AD communities (Karakashev et al., 2005; Yuan et al., 2006; Yang and Wang, 2018). However, neither bacterial nor archaeal community structure data clustered by the application of stripping (Fig. 3; S1). These observations were confirmed by the nonparametric multivariate analysis of variance (PERMANOVA) (Anderson, 2001) that was applied to test the influence of stripping and FAN on community composition. One critical assumption of a PERMANOVA is that the variance between the groups of samples compared needs to be similar, or the PERMANOVA can produce in a false‐positive result. In the case of the bacterial communities, the variance homogeneity assumption was not met for the stripping condition (Pr: 0.002). In that case, the PERMANOVA was not reliable as it could lead to a false‐positive result; therefore, the analysis was not performed. In the case of archaeal communities, the variance was homogeneous among the conditions considered: stripping (Pr > 0.7) and FAN (Pr > 0.9). Accordingly one way PERMANOVA analysis indicated the significant separation of bacterial community according to FAN levels (F: 5.4564, Pr: 0.001). For archaeal communities, two‐way PERMANOVA indicated that while they did not differ according to stripping (F: 2.052, Pr > 0.1), communities exposed to different FAN concentrations were different (F: 40.799, Pr: 0.001). The results suggest that the effects of stripping process on thermophilic AD microbial communities are negligible compared with the strong pressures imposed by FAN concentrations and other operational factors.
Taxonomic composition of bacterial community
Bacteria community was mainly composed of Firmicutes organisms, which on average represented 80.2 ± 7.6% of Bacteria, followed by Bacteroidetes (8.6 ± 5.5%) and Thermotogae (6.1 ± 5.7%) (Fig. S6A). Dominance of Firmicutes and Bacteroidetes phyla and the presence of Thermotogae phylum are common under thermophilic AD conditions (Ritari et al., 2012; Werner et al., 2014).
The bacteria of the Firmicutes phylum were very diverse, although two taxa represented together 70% of the bacteria (Table S2): the Clostridiales order (41.1 ± 9.3%) and the uncultured Firmicutes OPB54 group (29.0 ± 8.2%). A high presence of Clostridiales order is also characteristic of AD reactors (Moset et al., 2015; Cardinali‐Rezende et al., 2016). However, the taxonomy and metabolic capacities of the unknown group Firmicutes OPB54, which was the most abundant bacterial genus through the entire experiment (Fig. 4A), are still unclear as only one species has been isolated so far (Liu et al., 2014), and most of the knowledge about the group is limited to environmental clones found in a wide range of habitats including anaerobic reactors (Dunfield et al., 2012). The other Firmicutes organisms were mostly strict or facultative anaerobic bacteria with broad fermentative capabilities (Niu et al., 2009), like the genera Clostridium, Defluvitalea and Tepidimicrobium (Fig. 4A), or more specialized fermentative genus like Ruminiclostridium, whose members degrade complex carbohydrates such as cellulose, xylan and cellobiose (Yutin and Galperin, 2013), and Caldicoprobacter bacteria, which degrade hemicelluloses and mono‐ and oligosaccharides (Yokoyama et al., 2017). Many of these taxonomic groups, such as Ruminiclostridium, Defluvitalea or Caldicoprobacter, contain thermophilic organisms. Among Bacteroidetes, the most abundant genus was Proteiniphilum, which mostly hydrolyses and ferments proteins (Sakamoto, 2014), and within the Thermotogae phylum, Defluviitoga genus, with a single described species that ferments carbohydrates to acetate, H2 and CO2 (Hania et al., 2012).
Figure 4.
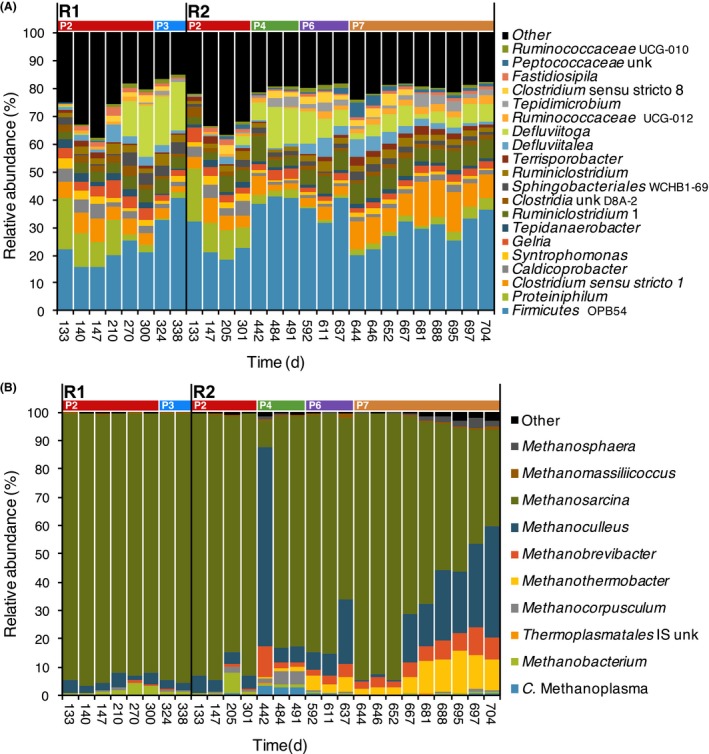
Microbial community composition of AD digesters.
A. Relative abundances of the 20 top most abundant bacteria genera, which individually represented over the 1.5% of the bacterial community in at least one sample.
B. Ten top most abundant archaea genera, which individually represented over the 0.4% of the archaeal community in at least one sample. Operational periods are indicated on top with coloured horizontal bars. IS, incertae sedis; unk, unknown.
Interestingly, the diversity of syntrophic bacteria in the studied reactors was large. It included organisms of the Syntrophomonadaceae family and Thermoanaerobacterales order like Gelria ssp. (Table S2), a group of thermophilic, saccharolytic and fermentative bacteria (Plugge et al., 2002). Moreover, some other bacterial taxa that have been either confirmed or suggested to belong to the syntrophic acetate‐oxidizing (SAO) bacteria were detected as well, like Tepidanaerobacter (Westerholm et al., 2011) and Syntrophaceticus (Westerholm et al., 2010). Other proposed, although not yet confirmed, SAO bacteria present in R1 and R2 were Coprothermobacter, a proteolytic genus commonly found in manure‐based digesters (Tsapekos et al., 2017), Clostridium sp. and Thermotogae organisms (Westerholm et al., 2016).
Taxonomic composition of archaeal community
Most archaea belonged to the Euryarchaeota phylum (99.98 ± 0.04%; Table S3), mainly from the Methanomicrobia (91.3 ± 7.9%) and Methanobacteria (7.7 ± 7.6%) classes (Fig. S6B). Methanosarcina genus dominated archaeal communities for most of the operational period (Fig. 4B). Methanosarcina is an acetoclastic methanogen that also uses the hydrogenotrophic pathway. It is a robust and adaptable methanogen that grows at different temperature ranges and tolerates abrupt changes on it, a capacity probably related to the presence of heat stress‐ and cold stress‐regulating genes in its genome (Vrieze et al., 2012). For that reason, it is not surprising that Methanosarcina members are often reported as the main acetoclastic methanogens at elevated total ammonia, salt or volatile fatty acid concentrations and dominate the methanogenic communities in thermophilic digesters fed with manure (Karakashev et al., 2005; Vavilin et al., 2008; Vrieze et al., 2012, 2015; Moset et al., 2015). Other hydrogenotrophic methanogens, mostly from the genera Methanoculleus, Methanothermobacter and Methanobrevibacter, also represented an important fraction of the archaeal community (Fig. 4), especially during day 442 and P7 of R2, periods in which FAN levels were high (Fig. 1). Methanoculleus , which can dominate pig manure reactors (Barret et al., 2013), comprises robust methanogens that thrive under thermophilic or ammonia stress (Pap et al., 2015; Poirier et al., 2016), and its members are often reported as partners of SAO bacteria (Westerholm et al., 2016). The rise of Methanoculleus genus during P7 of R2 occurred in parallel to the drop in methane production (Figs 1 and 4), which could lead to consider this genus as an early indicator of AD process failure. However, there are examples of well‐functioning AD reactors under high ammonia concentrations with communities dominated by this genus (Pap et al., 2015; Poirier et al., 2016).
Dominance and dynamics of co‐occurring groups of microorganisms
To find potential associations among microorganisms, a co‐occurring network was calculated for the OTUs with relative abundances higher than 0.1%. Twenty‐five groups of co‐occurring OTUs called clusters (CL) were found (Figs S7 and S8), each of them showing a unique temporal trend and taxonomic profile (Figs 5A and S8). The clusters that contained more than 10 OTUs (CL2, CL3, CL4, CL6 and CL7) were selected for further analysis. The two largest clusters, CL2 and CL3, seemed to be antagonist because they showed opposite temporal trends and relationships with OLR, FAN and VFA concentrations (Figs 5A, S9). CL3 dropped during Period 2, when the OLR was moderately increased by the addition of the co‐substrates. Indeed, relative abundance of CL3 was negatively correlated with increasing levels of OLR, FAN and VFAs (Fig. S9). Most microorganisms in CL3 are specialized in the degradation of proteinaceous substrates: mostly the proteolytic bacteria of the genus Proteiniphilum (on average, 40.8% of bacteria within the cluster; Fig. 5B), accompanied by syntrophic bacteria such as those from the proteolytic genus Coprothermobacter (7.7% of CL3), consisting of acetate‐degraders in syntrophic association with hydrogenotrophic methanogens (Ho et al., 2014), and Pelotomaculum (4.9% of CL3), a genus of syntrophic propionate‐oxidizing bacteria (Bok et al., 2005). Changes in microbial communities linked to transitions from mono‐ to co‐digestion have been previously observed (Fitamo et al., 2017) and can be expected given the strong influence of substrates on AD microbial communities (Regueiro et al., 2012).
Figure 5.
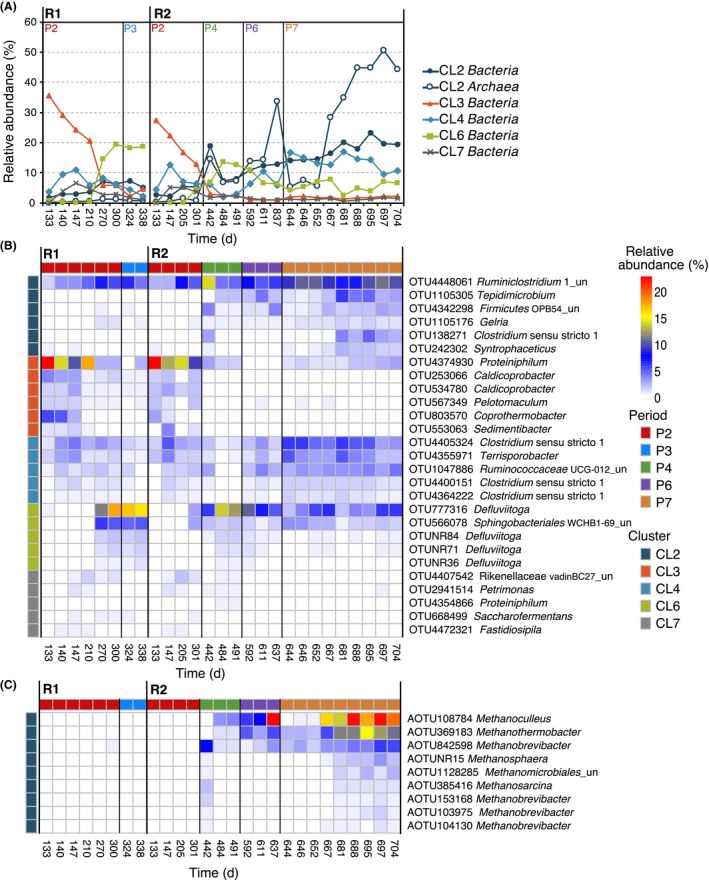
Temporal trends of main clusters of co‐occurring microorganisms and their most relatively abundant OTUs. A: Accumulated relative abundance of OTUs in the clusters by domain. Heatmaps of the most abundant OTUs within each cluster (relative abundances over 0.25% in at least one observation) for bacteria (B) and archaea (C). Coloured cells on the left indicate the cluster, and cells on the top designate the periods. Only data from CL2, CL3, CL4, CL6 and CL7 are shown.
In contrast, the relative abundances of CL2 microorganisms increased under high levels of OLR and FAN, especially during P6 and P7 (Fig. 5). Indeed, CL2 was positively correlated with OLR, FAN and VFAs (Fig. S9). CL2 was dominated by Ruminiclostridium bacteria (42% of bacteria within CL2), which hydrolyse complex carbohydrates. Although high ammonium content inhibits the glycolysis pathway during AD, thus impeding carbohydrate degradation (Mata‐Alvarez et al., 2000), the microbial consortia of these reactors seem to become adapted. Cardinali‐Rezende et al. (2016) already found carbohydrate‐degrading communities adapted to high ammonium concentrations in AD. Syntrophic bacteria composed a significant fraction of CL2, although in this case, the SAO Tepidimicrobium (10% of bacteria within CL2) and Gelria (7.2%) bacteria were the most abundant. CL2 also contained a diverse group of hydrogenotrophic methanogens (Methanoculleus (43% of Archaea within the cluster), Methanothermobacter (24%) and Methanobrevibacter (20%); Fig. 5C).
CL6 was mainly formed by Defluviitoga OTUs (79.3% of cluster microorganisms), which reaches its highest abundance under moderate OLR levels (P2, P3, P4; Fig. 5B). Bacteria of both Defluviitoga and Ruminiclostridium genera hydrolyse complex carbohydrates, and it is plausible that these organisms compete at moderate organic loads but Ruminiclostridium likely displaces Defluviitoga when OLR and FAN increase.
The other two most abundant clusters (CL4 and CL7) mainly contained organisms that use a wider range of substrates. CL4 was composed of OTUs, such as Clostridium (51.9% of CL4) or Terrisporobacter sp. (15.7% of CL4) species that degrade a broad array of simple substrates like amino acids or mono‐ and disaccharides. Most microorganisms in CL4 are likely to be involved in the acidogenesis step of the AD, since they are present throughout the experiment, although they tend to become more abundant during periods with high OLR (Fig. 5B, S9) and less abundant in P7, indicating a lower overall resistance to high FAN and VFA concentrations. Likewise, CL7 contained OTUs with diverse metabolic capabilities, although their abundances are quite low (Fig. 6B), for instance Petrimonas (17.8% of the cluster), whose members degrade carbohydrates and some organic acids, or Fastidiosipila (16.0%), which is a genus of proteolytic bacteria (Falsen et al., 2005). CL7 is probably a group of transient microorganisms with almost no correlation with any operational parameter (Fig. S9).
To conclude, this work shows that even though part of the thermophilic biomass was periodically exposed to the stripping conditions (65°C, high pH and oxygen at saturation levels), overall community structure was not predominantly affected by those harsh conditions. Instead, the thermophilic communities were mainly shaped by the organic loading rate, FAN and VFA concentrations, variables that are interrelated. Hence, air‐side stripping indirectly modulated microbial communities by controlling FAN concentrations in the digester allowing to optimize the treatment of N‐rich wastes.
Experimental procedures
Experimental set‐up and sampling
Two (R1 and R2) continuous stirred tank reactors (CSTR) with 14L working volume were operated in thermophilic range (55 ± 1°C) during 365 and 707 days respectively. The feeding consisted of a mixture of pig manure, maize silage and Ecofrit®, and the experiment was divided into seven operating periods. During Period 3 (P3) of R1 and Periods 4 (P4) and 6 (P6) of R2, the reactors were connected to a stripping column, which worked at 65°C for 3 h with an airflow of 1 Lair Ldigestate −1 min−1 without pH control in batch mode, treating 21% of the reactor volume in each batch. The stripping column was operated three times per week in P3 (R1) and P6 (R2) and five times per week in P4 (R2) to investigate the effects of higher recirculating ratio. A detailed description of the operation of reactors and the stripping column can be found in Pedizzi et al. (2017). Five 1‐ml aliquots of well‐homogenized biomass samples were taken on days 133, 140, 147, 210, 270, 300, 324 and 338 of R1 operation and days 133, 147, 205, 301, 442, 484, 491, 592, 611, 637, 644, 646, 652, 667, 681, 688, 695, 697 and 704 for R2 and immediately frozen at −20°C until further analysis. During the periods when the stripping was applied, biomass samples were always taken before feeding and 30 min after recirculating the stripped digestate into the anaerobic reactor.
DNA extraction and 16S rRNA gene amplicon sequencing
DNA was extracted using the PowerSoil DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, NM, USA) following the manufacturer's instructions. DNA concentrations were quantified by fluorometry (Qubit fluorometer; Thermo Fisher Scientific, Waltham, MA, USA), and standard electrophoresis was used to assess DNA size and integrity. Amplicon libraries were prepared for fragments of the 16S rRNA gene of both bacteria and archaea domains with primers including Illumina adaptors and barcodes. The V3V4 region of the bacterial 16S rRNA gene was amplified with the primer pair S‐D‐Bact‐0341‐b‐S‐17 and S‐D‐Bact‐0785‐a‐A (Klindworth et al., 2013). In the case of Archaea, the V2V3 region was amplified with the primer set Arch1F and Arch1R as previously described (Cruaud et al., 2014). Prepared DNA libraries were analysed for quality in a Bioanalyzer (Bioanalyzer; Agilent Technologies, Santa Clara, CA, USA) and subsequently quantified by qPCR. The multiplexed libraries were pooled in equimolar amounts and sequenced at the genomics unit of the Parque Científico de Madrid (Spain) on an Illumina MiSeq System (Illumina, San Diego, CA, USA) using MiSeq Reagent Kit v3 (Illumina).
Sequence processing
The paired‐end sequence reads were demultiplexed and trimmed to remove Illumina adapters, barcodes and primers. After truncating read length to 250 bp to remove low‐quality ends, paired reads were merged as previously described (Eren et al., 2013), discarding any sequence with quality scores below 30 and any indetermination. The obtained high‐quality sequences were clustered into operational taxonomic units (OTUs) at a 97% cut‐off for 16S rRNA gene identity with the open‐reference OTU‐picking method using UCLUST (Edgar, 2010) and SILVA v123 database (Quast et al., 2013) in QIIME v.1.9.1 (Caporaso et al., 2010b; Rideout et al., 2014). Representative sequences from each OTU were aligned using PyNAST (Caporaso et al., 2010a). Any unaligned sequence, singletons, OTUs classified as unknown or to the incorrect domain and chimeric sequences analysed with VSEARCH (Rognes et al., 2016) were removed from further analysis. Raw reads are available at the Short Read Archive (SRA) of the NCBI under the accession number SRP133112.
Data analysis
To calculate diversity estimations data was rarefied to 20 000 and 20 700 sequences for archaea and bacteria data sets respectively. Richness was determined as the estimated number of species, and evenness was measured with the Simpson evenness (E index):
where S is the number of observed species (richness) and p i is the proportion of species i relative to the total number of species. E takes values from 0 to 1, with 1 being complete evenness or uniformity. Beta diversity, the degree of community differentiation between samples, was measured with Bray–Curtis dissimilarities using Hellinger‐transformed OTU data (Legendre and Gallagher, 2001) and visualized using transformation‐based principal component analysis (tb‐PCA). Additionally, Bray–Curtis dissimilarities were analysed by average linkage clustering. PERMANOVAs were performed using Bray–Curtis dissimilarity matrices. First, the analysis of multivariate homogeneity of group dispersions was performed using the Anderson procedure (Anderson, 2006) through the betadisper and anova functions for each factor tested. Next, PERMANOVA was performed with the adonis function including 999 permutations. Co‐occurring microbial networks were constructed with CoNet v.1.1.1.beta (Faust et al., 2012) in Cytoscape v.3.4.0 (Shannon et al., 2003). The input data matrix included only OTUs with relative abundances over 0.1% (522 OTUs). Compositional effects were avoided using the bootstrap‐renormalization procedure (Faust et al., 2012). The co‐occurrence measures employed were as follows: Spearman and Pearson correlations, Bray–Curtis and Kullback–Leibler dissimilarities and their P‐values merged. The Benjamini–Hochberg procedure was used to correct for multiple comparisons, discarding edges that were not significant (q ≤ 0.05). Only edges supported by at least two measurements were kept. The resulting network was then analysed to detect highly interacting groups of nodes (clusters) using the GLay algorithm (Su et al., 2010). Pearson's correlations were calculated between total relative abundance of clusters and OLR, FAN, CH4 production and VFA concentrations. All statistical analyses were performed in R (R Core Team, 2016) using the R packages vegan (Oksanen et al., 2018), phyloseq (McMurdie and Holmes, 2013) and Rhea (Lagkouvardos et al., 2017).
Conflicts of interest
None declred.
Supporting information
Fig. S1. Clustering of Bray Curtis dissimilarities of bacteria (A) and archaea (B) communities. Samples exposed to stripping are in red and bold.
Fig. S2. tb‐PCA of archaeal community structure changes and their correlations with operational parameters for R1 (A) and R2 (B). Each point represents the community composition of a reactor at a given time point (sample labels indicate operational day; form color indicates the period). Vectors indicate the increasing values of operational variables (only the statistically significant variables (P‐value < 0.05) are shown.
Fig. S3. tb‐PCA of bacterial community structure changes and their correlations with operational parameters for R1 (A) and R2 (B). Each point represents the community composition of a reactor at a given time point (sample labels indicate operational day; form color indicates the period). Vectors indicate the increasing values of operational variables (only the statistically significant variables (P‐value < 0.05) are shown.
Fig. S4. tb‐PCA showing archaeal community structure changes and N‐FAN concentrations. Each point represents the community composition of a reactor (indicated by the form shape) at a given time point (sample labels indicate operational day). The color scale indicates the N‐FAN concentrations on each time point.
Fig. S5. tb‐PCA showing bacterial community structure changes and N‐FAN concentrations. Each point represents the community composition of a reactor (indicated by the form shape) at a given time point (sample labels indicate operational day). The color scale indicates the N‐FAN concentrations on each time point.
Fig. S6. The relative abundances of the most abundant taxa are shown for: A: 10 top bacterial phyla and B: 3 top archaeal classes. Operational periods are indicated on top with colored horizontal bars.
Fig. S7. Co‐occurring network of the abundant OTUs (relative abundance > 0.1%). Each node represents an individual OTU that is linked with other OTUs with similar temporal patters. Highly interconnected nodes have been separated into 25 clusters. Cluster number is indicated on top. For major clusters the number of OTUs within the cluster is shown in parenthesis. Node color indicates the phylum of the OTU.
Fig. S8. Normalized relative abundances of OTUs belonging to the clusters with more than 10 OTUs (CL2, CL3, CL4, CL6 and CL7). Each row shows the OTUs within one of the clusters. Left column correspond to values from R1 and right column to values from R2.
Fig. S9. Pearson correlations between bacterial relative abundances of clusters and volatile fatty acids, free ammonia nitrogen (FAN, g NH3‐N l−1), organic loading rate (OLR, g COD l−1 day−1) and methane production (g COD l−1 day−1). Color corresponds to Pearson´s r values (blue for positive and red for negative). Circle diameter indicates the P‐value.
Table S1. Number of high quality sequences obtained per sample. Sample name indicates: reactor, period and sampling day.
Table S2. Relative abundances of the 10 more abundant Bacteria classes, orders and families. Values are in percentages of total Bacteria.
Table S3. Relative abundances of the Archaea phylum and 10 more orders and families. Values are in percentages of total Archaea.
Acknowledgements
This research was supported by the European Community Seventh Framework Programme (ManureEcoMine project – 603744); and by the Spanish Government (AEI) through CDTI (SmartGreenGas project – 2014‐CE224). The authors belong to the Galician Competitive Research Group GRC 2013‐032 and to the CRETUS Strategic Partnership (AGRUP2015/02), co‐funded by FEDER (UE). Computational resources were kindly provided and supported by Fundacion Pública Galega Centro Tecnolóxico de Supercomputación de Galicia (CESGA).
Microbial Biotechnology (2019) 12(6), 1403–1416
Funding Information
This research was supported by the European Community Seventh Framework Programme (ManureEcoMine project – 603744); and by the Spanish Government (AEI) through CDTI (SmartGreenGas project – 2014‐CE224). The authors belong to the Galician Competitive Research Group ED431C 2017/029 and to the CRETUS Strategic Partnership (ED431E 2018/01), co‐funded by FEDER (UE). Computational resources were kindly provided and supported by Fundacion Pública Galega Centro Tecnolóxico de Supercomputación de Galicia (CESGA).
References
- Amha, Y.M. , Anwar, M.Z. , Brower, A. , Jacobsen, C.S. , Stadler, L.B. , Webster, T.M. , and Smith, A.L. (2018) Inhibition of anaerobic digestion processes: applications of molecular tools. Bioresour Technol 247: 999–1014. [DOI] [PubMed] [Google Scholar]
- Anderson, M.J. (2001) A new method for non‐parametric multivariate analysis of variance. Austral Ecol 26: 32–46. [Google Scholar]
- Anderson, M.J. (2006) Distance‐based tests for homogeneity of multivariate dispersions. Biometrics 62: 245–253. [DOI] [PubMed] [Google Scholar]
- Angelidaki, I. , and Ahring, B.K. (1993) Thermophilic anaerobic digestion of livestock waste: the effect of ammonia. Appl Microbiol Biotechnol 38: 560–564. [DOI] [PubMed] [Google Scholar]
- Barret, M. , Gagnon, N. , Kalmokoff, M.L. , Topp, E. , Verastegui, Y. , Brooks, S.P.J. , et al (2013) Identification of Methanoculleus spp. as active methanogens during anoxic incubations of swine manure storage tank samples. Appl Environ Microbiol 79: 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok, F.A.M.D. , Harmsen, H.J.M. , Plugge, C.M. , Vries, M.C.D. , Akkermans, A.D.L. , Vos, W.M.D. , and Stams, A.J.M. (2005) The first true obligately syntrophic propionate‐oxidizing bacterium Pelotomaculum schinkii sp. nov., co‐cultured with Methanospirillum hungatei, and emended description of the genus Pelotomaculum . Int J Syst Evol Microbiol 55: 1697–1703. [DOI] [PubMed] [Google Scholar]
- Bousek, J. , Scroccaro, D. , Sima, J. , Weissenbacher, N. , and Fuchs, W. (2016) Influence of the gas composition on the efficiency of ammonia stripping of biogas digestate. Bioresour Technol 203: 259–266. [DOI] [PubMed] [Google Scholar]
- Caporaso, J.G. , Bittinger, K. , Bushman, F.D. , Desantis, T.Z. , Andersen, G.L. , and Knight, R. (2010a) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J.G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F.D. , Costello, E.K. , et al (2010b) QIIME allows analysis of high‐ throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinali‐Rezende, J. , Rojas‐Ojeda, P. , Nascimiento, A.M.A. , and Sanz, J.L. (2016) Proteolytic bacterial dominance in a full‐scale municipal solid waste anaerobic reactor assessed by 454 pyrosequencing technology. Chemosphere 146: 519–525. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Jiang, X. , Xiao, K. , Shen, N. , Zeng, R.J. , and Zhou, Y. (2017) Enhanced volatile fatty acids (VFAs) production in a thermophilic fermenter with stepwise pH increase. Investigation on dissolved organic matter transformation and microbial community shift. Water Res 112: 261–268. [DOI] [PubMed] [Google Scholar]
- Cruaud, P. , Vigneron, A. , Lucchetti‐Miganeh, C. , Ciron, P.E. , Godfroy, A. , and Cambon‐Bonavita, M.A. (2014) Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl Environ Microbiol 80: 4626–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francisci, D. , Kougias, P.G. , Treu, L. , Campanaro, S. , and Angelidaki, I. (2015) Microbial diversity and dynamicity of biogas reactors due to radical changes of feedstock composition. Bioresour Technol 176: 56–64. [DOI] [PubMed] [Google Scholar]
- Dunfield, P.F. , Tamas, I. , Lee, K.C. , Morgan, X.C. , Mcdonald, I.R. , and Stott, M.B. (2012) Electing a candidate: a speculative history of the bacterial phylum OP10. Environ Microbiol 14: 3069–3080. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Eren, A.M. , Vineis, J.H. , Morrison, H.G. , and Sogin, M.L. (2013) A filtering method to generate high quality short reads using illumina paired‐end technology. PLoS ONE 8: e66643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsen, E. , Collins, M.D. , Welinder‐olsson, C. , Song, Y. , Finegold, S.M. , and Lawson, P.A. (2005) Fastidiosipila sanguinis gen. nov., sp. nov., a new Gram‐positive, coccus‐shaped organism from human blood. Int J Syst Evol Microbiol 55: 853–858. [DOI] [PubMed] [Google Scholar]
- Faust, K. , Sathirapongsasuti, J.F. , Izard, J. , Segata, N. , Gevers, D. , Raes, J. , and Huttenhower, C. (2012) Microbial co‐occurrence relationships in the human microbiome. PLoS Comput Biol 8: e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitamo, T. , Treu, L. , Boldrin, A. , Sartori, C. , Angelidaki, I. , and Scheutz, C. (2017) Microbial population dynamics in urban organic waste anaerobic co‐digestion with mixed sludge during a change in feedstock composition and different hydraulic retention times. Water Res 118: 261–271. [DOI] [PubMed] [Google Scholar]
- Gao, S. , Zhao, M. , Chen, Y. , Yu, M. , and Ruan, W. (2015) Tolerance response to in situ ammonia stress in a pilot‐scale anaerobic digestion reactor for alleviating ammonia inhibition. Bioresour Tehcnol 198: 372–379. [DOI] [PubMed] [Google Scholar]
- Guo, X. , Wang, C. , Sun, F. , Zhu, W. , and Wu, W. (2014) A comparison of microbial characteristics between the thermophilic and mesophilic anaerobic digesters exposed to elevated food waste loadings. Bioresour Technol 152: 420–428. [DOI] [PubMed] [Google Scholar]
- Hania, W.B. , Godbane, R. , Postec, A. , Hamdi, M. , Ollivier, B. , and Fardeau, M. (2012) Defluviitoga tunisiensis gen. nov., s p., nov., a thermophilic bacterium isolated from a mesothermic and anaerobic whey digester. Int J Syst Evol Microbiol 62: 1377–1382. [DOI] [PubMed] [Google Scholar]
- Ho, D. , Jensen, P. , and Batstone, D. (2014) Effects of temperature and hydraulic retention time on acetotrophic pathways and performance in high‐rate sludge digestion. Environ Sci Technol 48: 6468–6476. [DOI] [PubMed] [Google Scholar]
- Karakashev, D. , Batstone, D.J. , Karakashev, D. , Batstone, D.J. , and Angelidaki, I. (2005) Influence of environmental conditions on methanogenic compositions in anaerobic biogas reactors. Appl Environ Microbiol 71: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , and Glöckner, F.O. (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Res 41: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakat, N. , Demirel, B. , Anjum, R. , and Dietz, D. (2017) Methods of ammonia removal in anaerobic digestion: a review. Water Sci Technol 76: 1925–1938. [DOI] [PubMed] [Google Scholar]
- Kuroda, K. , Chosei, T. , Nakahara, N. , Hatamoto, M. , Wakabayashi, T. , Kawai, T. , et al (2015) High organic loading treatment for industrial molasses wastewater and microbial community shifts corresponding to system development. Bioresour Technol 196: 225–234. [DOI] [PubMed] [Google Scholar]
- Lagkouvardos, I. , Fischer, S. , Kumar, N. , and Clavel, T. (2017) Rhea: a transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 5: e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre, P. , and Gallagher, E.D. (2001) Ecologically meaningful transformations for ordination of species data. Oecologia 129: 271–280. [DOI] [PubMed] [Google Scholar]
- Li, N. , Xue, Y. , Chen, S. , Takahashi, J. , Dai, L. , and Dai, X. (2017) Methanogenic population dynamics regulated by bacterial community responses to protein‐rich organic wastes in a high solid anaerobic digester. Chem Eng J 317: 444–453. [Google Scholar]
- Lin, Q. , Vrieze, J.D. , Li, J. , and Li, X. (2016) Temperature affects microbial abundance, activity and interactions in anaerobic digestion. Bioresour Technol 209: 228–236. [DOI] [PubMed] [Google Scholar]
- Liu, T. , and Sung, S. (2002) Ammonia inhibition on thermophilic acetoclastic methanogens. Water Sci Technol 45: 113–120. [PubMed] [Google Scholar]
- Liu, Y. , Qiao, J. , Yuan, X. , Guo, R. , and Qiu, Y. (2014) Hydrogenispora ethanolica gen. nov., sp. nov., an anaerobic carbohydrate‐fermenting bacterium from anaerobic sludge. Int J Syst Evol Microbiol 64: 1756–1762. [DOI] [PubMed] [Google Scholar]
- Lü, F. , Hao, L. , Guan, D. , Qi, Y. , Shao, L. , and He, P. (2013) Synergetic stress of acids and ammonium on the shift in the methanogenic pathways during thermophilic anaerobic digestion of organics. Water Res 47: 2297–2306. [DOI] [PubMed] [Google Scholar]
- Mata‐Alvarez, J. , Macé, S. , and Llabrés, P. (2000) Anaerobic digestion of organic solid wastes. An overview of research achievements and perspectives. Bioresour Technol 74: 3–16. [Google Scholar]
- McMurdie, P.J. , and Holmes, S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moset, V. , Poulsen, M. , Højberg, O. , and Møller, H.B. (2015) Mesophilic versus thermophilic anaerobic digestion of cattle manure: methane productivity and microbial ecology. Microb Biotechnol 8: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, L. , Song, L. , Liu, X. , and Dong, X. (2009) Tepidimicrobium xylanilyticum sp. nov., and anaerobic xylanolytic bacterium, and emended description of the genus Tepidimicrobium . Int J Syst Evol Microbiol 59: 2698–2701. [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , et al (2018) Vegan: community ecology package. R package version 2.4‐6.
- Pap, B. , Györkei, Á. , Zoltan, I. , Nagy, I.K. , Bíró, T. , Kondorosi, É. , and Maróti, G. (2015) Temperature‐dependent transformation of biogas‐producing microbial communities points to the increased importance of hydrogenotrophic methanogenesis under thermophilic operation. Bioresour Technol 177: 375–380. [DOI] [PubMed] [Google Scholar]
- Pedizzi, C. , Lema, J.M. , and Carballa, M. (2017) Enhancing thermophilic co‐digestion of nitrogen‐rich substrates by air side‐stream stripping. Bioresour Technol 241: 397–405. [DOI] [PubMed] [Google Scholar]
- Plugge, C.M. , Balk, M. , Zoetendal, E.G. , and Stams, A.J.M. (2002) Gelria glutamica gen. nov., sp. nov., a thermophilic, obligately syntrophic, glutamate‐degrading anaerobe. Int J Syst Evol Microbiol 52: 401–407. [DOI] [PubMed] [Google Scholar]
- Poirier, S. , Desmond‐Le Quéméner, E. , Madigou, C. , Bouchez, T. , and Chapleur, O. (2016) Anaerobic digestion of biowaste under extreme ammonia concentration: identification of key microbial phylotypes. Bioresour Technol 207: 92–101. [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2016) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Regueiro, L. , Veiga, P. , Figueroa, M. , Alonso‐Gutierrez, J. , Stams, A.J.M. , Lema, J.M. , and Carballa, M. (2012) Relationship between microbial activity and microbial community structure in six full‐scale anaerobic digesters. Microbiol Res 167: 581–589. [DOI] [PubMed] [Google Scholar]
- Regueiro, L. , Carballa, M. , and Lema, J.M. (2014) Outlining microbial community dynamics during temperature drop and subsequent recovery period in anaerobic co‐digestion systems. J Biotechnol 192: 179–186. [DOI] [PubMed] [Google Scholar]
- Regueiro, L. , Lema, J.M. , and Carballa, M. (2015) Key microbial communities steering the functioning of anaerobic digesters during hydraulic and organic overloading shocks. Bioresour Technol 197: 208–216. [DOI] [PubMed] [Google Scholar]
- Regueiro, L. , Carballa, M. , and Lema, J.M. (2016) Microbiome response to controlled shifts in ammonium and LCFA levels in co‐digestion systems. J Biotechnol 220: 35–44. [DOI] [PubMed] [Google Scholar]
- Rideout, J.R. , He, Y. , Navas‐Molina, J.A. , Walters, W.A. , Ursell, L.K. , Gibbons, S.M. , et al (2014) Subsampled open‐reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2: e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritari, J. , Koskinen, K. , Hultman, J. , Kurola, J.M. , Kymäläinen, M. , Romantschuk, M. , et al (2012) Molecular analysis of meso‐ and thermophilic microbiota associated with anaerobic biowaste degradation. BMC Microbiol: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , and Mahé, F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, M. (2014) The family Porphyromonadaceae In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea. Rosenberg E., DeLong E.F., Lory S., Stackebrandt E., and Thompson F. (eds). New York: Springer, pp. 813–824. [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stams, A.J.M. , and Plugge, C.M. (2009) Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol 7: 568–577. [DOI] [PubMed] [Google Scholar]
- Su, G. , Kuchinsky, A. , Morris, J.H. , States, D.J. , and Meng, F. (2010) GLay: community structure analysis of biological networks. Bioinformatics 26: 3135–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C. , Cao, W. , Banks, C.J. , Heaven, S. , and Liu, R. (2016) Biogas production from undiluted chicken manure and maize silage: a study of ammonia inhibition in high solids anaerobic digestion. Bioresour Technol 218: 1215–1223. [DOI] [PubMed] [Google Scholar]
- Sundberg, C. , Al‐Soud, W.A. , Larsson, M. , Alm, E. , Yekta, S.S. , Svensson, B.H. , et al (2013) 454 Pyrosequencing analyses of bacterial and archaeal richness in 21 full‐scale biogas digesters. FEMS Microbiol Ecol 85: 612–626. [DOI] [PubMed] [Google Scholar]
- Theuerl, S. , Klang, J. , and Prochnow, A. (2019) Process disturbances in agricultural biogas production – causes, mechanisms and effects on the biogas microbiome: a review. Energies 12: 365. [Google Scholar]
- Tsapekos, P. , Kougias, P.G. , Treu, L. , Campanaro, S. , and Angelidaki, I. (2017) Process performance and comparative metagenomic analysis during co‐digestion of manure and lignocellulosic biomass for biogas production. Appl Energy 185: 126–135. [Google Scholar]
- Vanwonterghem, I. , Jensen, P.D. , Rabaey, K. , and Tyson, G.W. (2015) Temperature and solids retention time control microbial population dynamics and volatile fatty acid production in replicated anaerobic digesters. Sci Rep 5: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavilin, V.A. , Qu, X. , Mazeas, L. , Lemunier, M. , Duquennoi, C. , He, P.J. , and Bouchez, T. (2008) Methanosarcina as the dominant aceticlastic methanogens during mesophilic anaerobic digestion of putrescible waste. Antonie Van Leeuwenhoek 94: 539–605. [DOI] [PubMed] [Google Scholar]
- Vrieze, J.D. , Hennebel, T. , Boon, N. , and Verstraete, W. (2012) Methanosarcina: the rediscovered methanogen for heavy duty biomethanation. Bioresour Technol 112: 1–9. [DOI] [PubMed] [Google Scholar]
- Vrieze, J.D. , Marc, A. , He, Y. , Fang, J. , Halkjaer, P. , Verstraete, W. , and Boon, N. (2015) Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome. Water Res 75: 312–323. [DOI] [PubMed] [Google Scholar]
- Weiss, A. , Jérôme, V. , Freitag, R. , and Mayer, H.K. (2008) Diversity of the resident microbiota in a thermophilic municipal biogas plant. Appl Microbiol Biotechnol 81: 163–173. [DOI] [PubMed] [Google Scholar]
- Werner, J.J. , Garcia, M.L. , Perkins, S.D. , Yarasheski, K.E. , Smith, S.R. , Muegge, B.D. , et al (2014) Microbial community dynamics and stability during an ammonia‐induced shift to syntrophic acetate oxidation. Appl Environ Microbiol 80: 3375–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerholm, M. , Roos, S. , Schnürer, A. , and Schn, A. (2010) Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate‐oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett 309: 100–104. [DOI] [PubMed] [Google Scholar]
- Westerholm, M. , Roos, S. , and Schnürer, A. (2011) Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate‐oxidizing bacterium isolated from two ammonium‐enriched mesophilic methanogenic processes. Syst Appl Microbiol 34: 260–266. [DOI] [PubMed] [Google Scholar]
- Westerholm, M. , Levén, L. , and Schnürer, A. (2012) Bioaugmentation of syntrophic acetate‐oxidizing culture in biogas reactors exposed to increasing levels of ammonia. Appl Environ Microbiol 78: 7619–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerholm, M. , Moestedt, J. , and Schnürer, A. (2016) Biogas production through syntrophic acetate oxidation and deliberate operating strategies for improved digester performance. Appl Energy 179: 124–135. [Google Scholar]
- Westerholm, M. , Isaksson, S. , Karlsson Lindsjö, O. , and Schnürer, A. (2018) Microbial community adaptability to altered temperature conditions determines the potential for process optimisation in biogas production. Appl Energy 226: 838–848. [Google Scholar]
- Wirth, R. , Kovács, E. , Maróti, G. , Bagi, Z. , Rákhely, G. , and Kovács, K.L. (2012) Characterization of a biogas‐producing microbial community by short‐read next generation DNA sequencing. Biotechnol Biofuels 5: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, M. , Fotidis, I.A. , Tian, H. , Khoshnevisan, B. , Treu, L. , Tsapekos, P. , and Angelidaki, I. (2019) Acclimatization contributes to stable anaerobic digestion of organic fraction of municipal solid waste under extreme ammonia levels: focusing on microbial community dynamics. Bioresour Technol 286: 121376. [DOI] [PubMed] [Google Scholar]
- Yang, G. , and Wang, J. (2018) Kinetics and microbial community analysis for hydrogen production using raw grass inoculated with different pretreated mixed culture. Bioresour Technol 247: 954–962. [DOI] [PubMed] [Google Scholar]
- Yenigün, O. , and Demirel, B. (2013) Ammonia inhibition in anaerobic digestion: a review. Process Biochem 48: 901–911. [Google Scholar]
- Yokoyama, H. , Wagner, I.D. , and Wiegel, J. (2017) Caldicoprobacter oshimai gen. nov., sp. nov., an anaerobic, xylanolytic, extremely thermophilic bacterium isolated from sheep faeces, and proposal of Caldicoprobacteraceae fam. nov. Int J Syst Evol Microbiol 60: 67–71. [DOI] [PubMed] [Google Scholar]
- Yuan, H. , Chen, Y. , Zhang, H. , Jiang, S. , Zhou, Q. , and Gu, G. (2006) Improved bioproduction of short‐chain ratty acids (SCFAs) from excess sludge under alkaline conditions. Environ Sci Technol 40: 2025–2029. [DOI] [PubMed] [Google Scholar]
- Yun, Y.M. , Kim, D.H. , Cho, S.K. , Shin, H.S. , Jung, K.W. , and Kim, H.W. (2016) Mitigation of ammonia inhibition by internal dilution in high‐rate anaerobic digestion of food waste leachate and evidences of microbial community response. Biotechnol Bioenge 113: 1892–1901. [DOI] [PubMed] [Google Scholar]
- Yutin, N. , and Galperin, M.Y. (2013) A genomic update on clostridial phylogeny: gram‐negative spore formers and other misplaced clostridia. Environ Microbiol 15: 2631–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Lee, Y.‐W. , and Jahng, D. (2012) Ammonia stripping for enhanced biomethanization of piggery wastewater. J Hazard Mater 199–200: 36–42. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Werner, J.J. , Agler, M.T. , and Angenent, L.T. (2014) Substrate type drives variation in reactor microbiomes of anaerobic digesters. Bioresour Technol 151: 397–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Clustering of Bray Curtis dissimilarities of bacteria (A) and archaea (B) communities. Samples exposed to stripping are in red and bold.
Fig. S2. tb‐PCA of archaeal community structure changes and their correlations with operational parameters for R1 (A) and R2 (B). Each point represents the community composition of a reactor at a given time point (sample labels indicate operational day; form color indicates the period). Vectors indicate the increasing values of operational variables (only the statistically significant variables (P‐value < 0.05) are shown.
Fig. S3. tb‐PCA of bacterial community structure changes and their correlations with operational parameters for R1 (A) and R2 (B). Each point represents the community composition of a reactor at a given time point (sample labels indicate operational day; form color indicates the period). Vectors indicate the increasing values of operational variables (only the statistically significant variables (P‐value < 0.05) are shown.
Fig. S4. tb‐PCA showing archaeal community structure changes and N‐FAN concentrations. Each point represents the community composition of a reactor (indicated by the form shape) at a given time point (sample labels indicate operational day). The color scale indicates the N‐FAN concentrations on each time point.
Fig. S5. tb‐PCA showing bacterial community structure changes and N‐FAN concentrations. Each point represents the community composition of a reactor (indicated by the form shape) at a given time point (sample labels indicate operational day). The color scale indicates the N‐FAN concentrations on each time point.
Fig. S6. The relative abundances of the most abundant taxa are shown for: A: 10 top bacterial phyla and B: 3 top archaeal classes. Operational periods are indicated on top with colored horizontal bars.
Fig. S7. Co‐occurring network of the abundant OTUs (relative abundance > 0.1%). Each node represents an individual OTU that is linked with other OTUs with similar temporal patters. Highly interconnected nodes have been separated into 25 clusters. Cluster number is indicated on top. For major clusters the number of OTUs within the cluster is shown in parenthesis. Node color indicates the phylum of the OTU.
Fig. S8. Normalized relative abundances of OTUs belonging to the clusters with more than 10 OTUs (CL2, CL3, CL4, CL6 and CL7). Each row shows the OTUs within one of the clusters. Left column correspond to values from R1 and right column to values from R2.
Fig. S9. Pearson correlations between bacterial relative abundances of clusters and volatile fatty acids, free ammonia nitrogen (FAN, g NH3‐N l−1), organic loading rate (OLR, g COD l−1 day−1) and methane production (g COD l−1 day−1). Color corresponds to Pearson´s r values (blue for positive and red for negative). Circle diameter indicates the P‐value.
Table S1. Number of high quality sequences obtained per sample. Sample name indicates: reactor, period and sampling day.
Table S2. Relative abundances of the 10 more abundant Bacteria classes, orders and families. Values are in percentages of total Bacteria.
Table S3. Relative abundances of the Archaea phylum and 10 more orders and families. Values are in percentages of total Archaea.