

Summary
The filamentous fungus Ashbya gossypii is currently used for the industrial production of vitamin B2. Furthermore, the ability of A. gossypii to grow using low‐cost substrates together with the inexpensive downstream processing makes this fungus an attractive biotechnological chassis. Indeed, the production in A. gossypii of other high‐added value compounds such as folic acid, nucleosides and biolipids has been described. Hence, the development of new methods to expand the molecular toolkit for A. gossypii genomic manipulation constitutes an important issue for the biotechnology of this fungus. In this work, we present a one‐vector CRISPR/Cas9 system for genomic engineering of A. gossypii. We demonstrate the efficiency of the system as a marker‐less approach for nucleotide deletions and substitutions both with visible and invisible phenotypes. Particularly, the system has been validated for three types of genomic editions: gene inactivation, the genomic erasure of loxP scars and the introduction of point mutations. We anticipate that the use of the CRISPR/Cas9 system for A. gossypii will largely contribute to facilitate the genomic manipulations of this industrial fungus in a marker‐less manner.
Introduction
Ashbya gossypii is an industrial filamentous fungus that is currently used for the microbial production of vitamin B2 (Revuelta et al., 2017). Indeed, the worldwide riboflavin market mostly relies on A. gossypii bioprocessing, which provides the vitamin as a safe additive for human foodstuffs and animal feeding (Schwechheimer et al., 2016; Rychen et al., 2018). In addition, A. gossypii has been also shown as an appropriate biotechnological chassis for other applications such as the production of folic acid (Serrano‐Amatriain et al., 2016), microbial oils (Ledesma‐Amaro et al., 2015b, 2018; Lozano‐Martínez et al., 2016; Díaz‐Fernández et al., 2017), nucleosides (Ledesma‐Amaro et al., 2015a) and recombinant proteins (Aguiar et al., 2015, 2017). The main advantages of A. gossypii as a microbial factory are based on the ability to grow using low‐cost substrates and an inexpensive downstream processing (Schwechheimer et al., 2016). Furthermore, a large number of genomic and biotechnological tools for strain engineering are available for A. gossypii (Dietrich et al., 2004; Aguiar et al., 2015; Ledesma‐Amaro et al., 2018). The genomic manipulations of A. gossypii are mainly achieved using integrative cassettes (Ledesma‐Amaro et al., 2018). Consequently, the extensive presence of selection markers and loxP scars in the genome, derived from Cre‐loxP marker excision events, usually decrease both the fitness and the sporulation ability.
Within the last few years, the Clustered Regularly Interspaced Short Palindromic Repeats‐associated Cas system (CRISPR/Cas9) has become an efficient and marker‐less approach for genome editing in yeast and filamentous fungi (Shi et al., 2017; Raschmanová et al., 2018). In the model yeast S. cerevisiae, an extensive toolbox of CRISPR‐related applications has been developed since the yeast system was first described (Dicarlo et al., 2013; Raschmanová et al., 2018). Therefore, the development of a marker‐less CRISPR/Cas9 method for genomic engineering of A. gossypii represents an important challenge willing to expand the molecular toolbox of this industrial fungus.
In a CRISPR/Cas9 system, a guide RNA (gRNA) directs the Cas9 endonuclease at specific genomic loci to produce double‐strand breaks (DSBs) (Brouns et al., 2008). There are two pathways to repair DSBs in DNA: homologous recombination (HR) and non‐homologous end joining (NHEJ). The CRISPR/Cas9 system relies on the ability to repair a targeted DSB with a synthetic donor DNA by HR (Raschmanová et al., 2018). Hence, the preference of the DSB repair pathway among different species largely affects the efficiency of the CRISPR/Cas9 systems. Particularly, in S. cerevisiae HR is the predominant pathway, which is confirmed by the correct integration of virtually all the exogenous DNA with homology flanks (Manivasakam et al., 1995). In this regard, in contrast to other filamentous fungi, integration of homologous exogenous DNA by HR is also highly frequent in A. gossypii (Steiner et al., 1995).
As mentioned above, the CRISPR/Cas9 system comprises an RNA guided DNA endonuclease (Cas9) and a guide RNA (gRNA), which is complementary to the genomic target region (Jinek et al., 2012). The Cas9 nuclease requires a 5′‐NGG‐3′ protospacer adjacent motif (PAM) to generate a DSB in the genomic target (Fig. 1). A CRISPR targeting RNA (crRNA) and a trans‐activating crRNA (tracrRNA) are required to form the catalytic active complex with Cas9 (Jinek et al., 2012). The crRNA and tracrRNA can be joined in a synthetic guide RNA (sgRNA) which is able to bind and target the Cas9 (Jinek et al., 2012). Therefore, the sgRNA comprises a structural sequence for Cas9 binding and a variable 20 bp sequence to match the genomic target sequence. In addition, a donor DNA (dDNA) containing the engineered genomic target with homology flanks must be provided for the DSB repair by HR (Jinek et al., 2012; Raschmanová et al., 2018).
Figure 1.
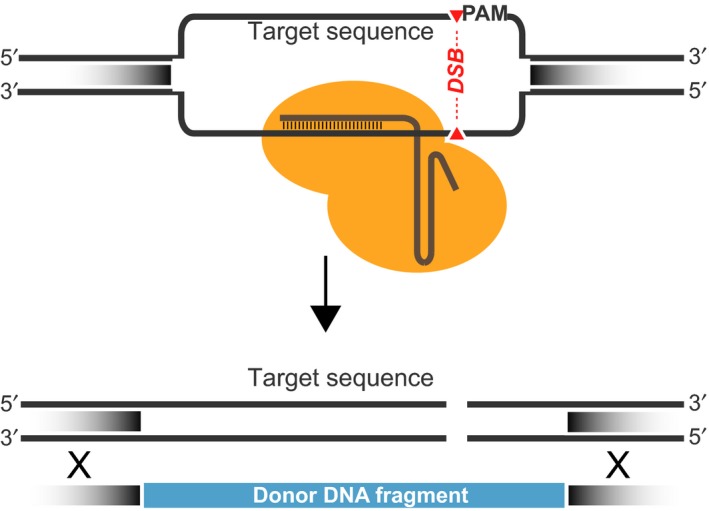
Schematic representation of a general CRISPR/Cas9 system. The ribonucleoprotein Cas9 (orange)/sgRNA is targeted to a selected genomic region. The Cas9 introduces a DSB in a sequence adjacent to the PAM (5′‐NGG‐3′). The DSB can be repaired with a homologous donor DNA fragment.
Genomic engineering in A. gossypii is essential for the development of new strains with the ability to produce metabolites of industrial interest. However, the introduction of a high number of modifications can drastically reduce both the fitness and the sporulation capacity. In the case of overexpression approaches, the number of manipulations can be minimized using a multiple gene overexpression cassette (Ledesma‐Amaro et al., 2018); however, both gene inactivation and gene mutation strategies require the use of a selection marker for each manipulation, thus producing a large number of loxP scars after many rounds of manipulations (Ledesma‐Amaro et al., 2018).
In this work, we have developed an efficient CRISPR/Cas9 system for A. gossypii. This system contains all the components in a single vector, which can be assembled in a one‐step protocol. The efficiency of the system has been confirmed and validated both with a colour‐associated mutant phenotype and non‐visible phenotypes. Future applications of the system in A. gossypii are also discussed.
Results and discussion
Assembly of a one‐vector CRISPR/Cas9 system for A. gossypii
A CRISPR/Cas9 system was reported in S. cerevisiae comprising two vectors for the expression of CAS9 and sgRNA that were co‐transformed with the dDNA (Dicarlo et al., 2013). Alternative strategies from the original approach included different expression modules for CAS9 and sgRNA, CAS9 variants and different strategies for the introduction of the system into the cells (i.e. episomal modules vs. genomic integration) (Raschmanová et al., 2018).
Ashbya gossypii and Saccharomyces cerevisiae are closely related species, with more than 90% of A. gossypii genes showing both homology and synteny with S. cerevisiae (Dietrich et al., 2004). In addition, some genetic elements from S. cerevisiae are fully functional in A. gossypii (Aguiar et al., 2015). Hence, we selected the CAS9 expression module that was previously reported in S. cerevisiae, where the human codon‐optimized Streptococcus pyogenes CAS9 gene is under the control of the yeast TEF1 promoter and CYC1 terminator sequences (Dicarlo et al., 2013).
The mycelium of A. gossypii is organized as multinucleated cells that are separated by septa along the hyphae. Hence, after transformation of A. gossypii germlings, only a limited number of nuclei within each syncytium are recipients of the transforming DNA. Therefore, the sporulation of the primary heterokaryotic transformants is a required step for the isolation of homokaryotic clones, which are derived from uninucleated spores. In this context, the probability that a single nucleus in the multinucleate germling is co‐transformed by two different transforming DNAs would be extremely low. Thus, we decided to follow a one‐vector strategy for the expression of CAS9 and sgRNA. Moreover, we also included the dDNA as a part of the CRISPR/Cas9 vector (Fig. 2).
Figure 2.
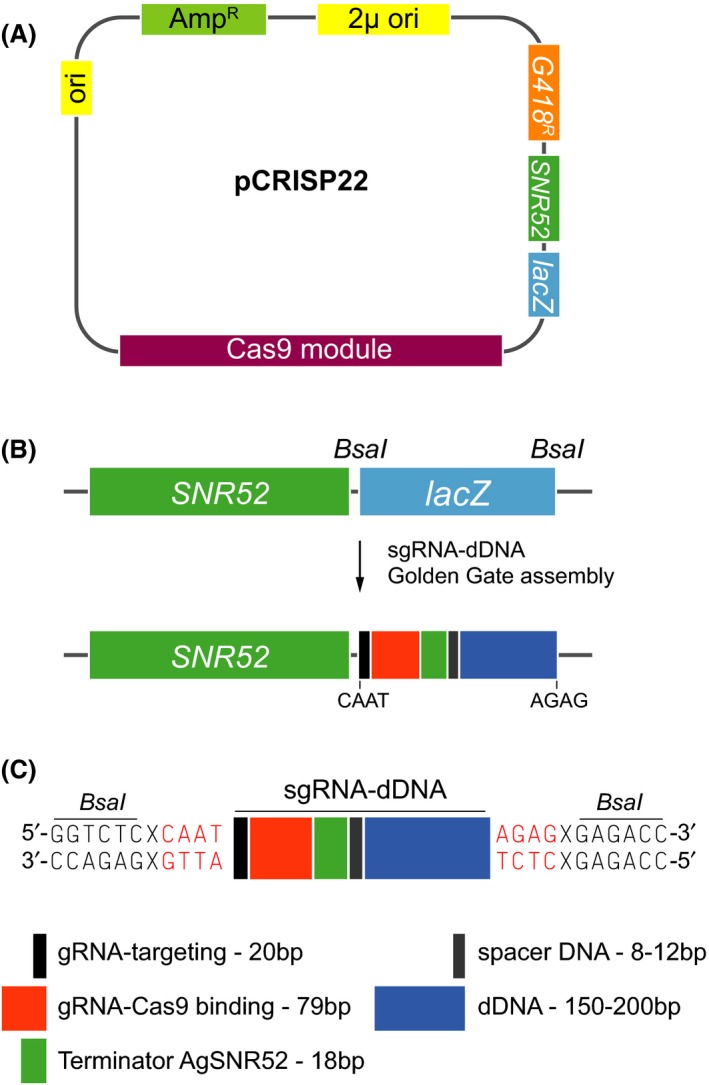
Design of a CRISPR/Cas9 vector for A. gossypii.A. Schematic map of the vector pCRISP22, which comprise the CRISPR/Cas9 system for A. gossypii.B. Representation of the directional cloning of the sgRNA‐dDNA module into the pCRISP22. The BsaI restriction sites and the 4‐nt sticky ends are indicated.C. Genetic organization of the sgRNA‐dDNA sequence.
Next, we combined both the targeting crRNA and tracrRNA in a merged sgRNA comprising two sequences: a 20 bp sequence that match the genomic target and a 79 bp sequence for Cas9 binding (Fig. 2). The sgRNA was expressed under the control of the promoter and terminator sequences from the A. gossypii SNR52 gene, which is transcribed by RNA Polymerase III (see Appendix S1 for sequences).
In addition to the CAS9 and sgRNA expression modules, the vector for the CRISPR/Cas9 system in A. gossypii also contained the dDNA for DSB repair by HR. The dDNA is located in the vector next to an 8–12 bp spacer DNA, following the AgSNR52 terminator and expands for 150–200 bp (Fig. 2). The dDNA included the designed mutation (see below) and a disrupted PAM sequence in order to avoid repeated Cas9 cleavage of both the dDNA and the edited genomic targets.
Other genetic elements in the CRISPR/Cas9 vector were as follows: (i) a bacterial origin of replication (pUC19 origin), an ampicillin resistance marker and a lacZ module for bacterial cloning and (ii) a 2‐micron replicating sequence and a loxP‐KanMX‐loxP marker (G418R) for plasmid selection in A. gossypii. In this regard, although episomic plasmids containing S. cerevisiae replicating sequences are not stable in A. gossypii, they are fully functional for transient gene expression as far as the selection conditions are maintained. Therefore, the CRISPR/Cas9 vector adapted for A. gossypii is readily lost in media lacking G418.
The assembly of the CRISPR/Cas9 vector with the synthetic sgRNA‐dDNA is carried out using a directional cloning strategy, by introducing two BsaI sites in the plasmid, flanking the lacZ module, next to the AgSNR52 promoter. These BsaI sites are flanked by sequences of 4‐nucleotide (nt) sticky ends. Hence, the sgRNA‐dDNA cassette should contain two BsaI sites and be compatible with the 4‐nt sticky ends for a single‐step assembly of the CRISPR/Cas9 vector (Fig. 2).
ADE2 gene inactivation using the CRISPR/Cas9 system for A. gossypii
For the validation of the CRISPR/Cas9 system for introducing deletions, insertions and point mutations into A. gossypii, the coding sequence of the ADE2 gene was chosen as the genomic target. The ade2 − defective mutants show red colour due to the accumulation of intermediates of the purine pathway, thereby leaving the ADE2 gene as a suitable reporter for gene inactivation.
To introduce a deletion, a synthetic sgRNA‐dDNA for ADE2 targeting was designed (ade2_INT1) to contain a dDNA with an 8‐bp deletion that generated an ade2 non‐functional protein (see Appendix S1 for sequences). The 8‐bp deletion produced a frameshift after the nucleotide 629 of the coding region (Val209) and a premature stop codon at nucleotides 676–678 (Fig. 3), thus generating a C‐terminal truncated protein lacking 340 amino acids.
Figure 3.
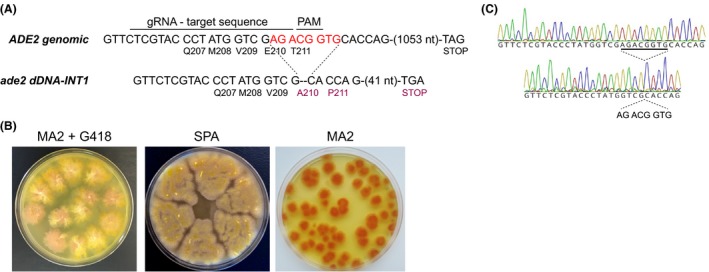
Inactivation of the ADE2 gene using the CRISPR/Cas9 system.A. Sequence detail of the wild‐type ADE2 locus and the mutant ade2‐INT1. The target sequence for the gRNA and the PAM sequence are indicated. The sequence of the 8‐bp deletion is highlighted in red. The corresponding amino acids of the coding sequence are shown below the DNA sequence.B. Culture plates showing the heterokaryotic primary transformants in MA2 + G418 (left) and SPA sporulation media (centre). Homokaryotic ade2 − clones are shown in MA2 media (right).C. Chromatograms of the sequences of the wild‐type ADE2 locus and the mutant ade2‐INT1. The sequence of the 8‐bp deletion is indicated.
The CRISPR/Cas9‐ade2_INT1 plasmid was used to transform spores of the wild‐type strain of A. gossypii and positive clones were selected in G418‐containing medium, thus confirming the functionality of the plasmid. Next, sporulation of the primary heterokaryotic transformants in G418‐free medium allowed at isolating homokaryotic clones. Our results showed that 85% of the homokaryotic clones exhibited a pink‐red colour in MA2 rich media, indicating the inactivation of the ADE2 gene (Fig. 3). Furthermore, all the isolated ade2 − mutants were unable to grow on G418‐containing medium (not shown), confirming that the CRISPR/Cas9‐ADE2 plasmid was lost. The loss of the plasmid likely occurs during the growth of the heterokaryotic transformants to allow sporulation in SPA G418‐free medium. In contrast, the sporulation of the primary transformants in G418‐containing medium led to random integration of the plasmid, pointing out the importance of using G418‐free medium for sporulation of the heterokaryotic clones.
The presence of the 8‐bp deletion in the ade2 − mutants was verified by DNA sequencing (Fig. 3). The stability of the mutation was then confirmed, since no phenotypic variations were observed after five rounds of sporulation (not shown).
Next, we further investigated the ability of the CRISPR/Cas9 system to introduce non‐sense mutations and frameshift insertions. Hence, two additional sgRNA‐dDNAs for ADE2 targeting at two different target sequences within the ADE2 coding region were used. For the introduction of a non‐sense mutation, a sgRNA‐dDNA (ade2_INT2) was designed to contain a single‐point mutation (C‐G substitution at nucleotide 618 of the coding region; Y206*). For the generation of a frameshift insertion, a sgRNA‐dDNA (ade2_INT3) was designed to include a TA‐insertion after the nucleotide 150 of the coding region. This insertion generates a frameshift that results in a premature stop codon after nucleotide 150 (Fig. 4 and Appendix S1).
Figure 4.
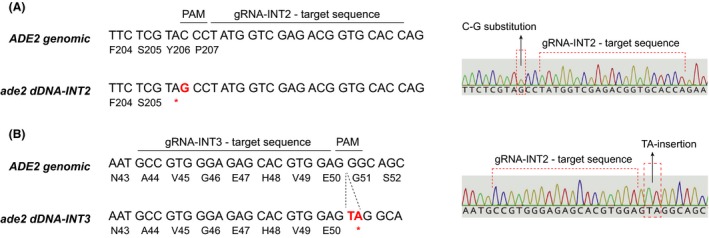
Single‐point mutations and insertions of the ADE2 gene using the CRISPR/Cas9 system.A. Genomic sequence and ade2 dDNA‐INT2 sequence for the introduction of a C‐G substitution. Right, chromatogram of the sequence of an ade2 Y206* mutant.B. Genomic sequence and ade2 dDNA‐INT3 sequence for a TA‐insertion. Right, chromatogram of the sequence of a homokaryotic clone harbouring the TA‐insertion.
The CRISPR/Cas9‐ade2_INT2 and CRISPR/Cas9‐ade2_INT3 plasmids were used to transform the wild‐type strain of A. gossypii. Heterokaryotic primary transformants were obtained and homokaryotic ade − clones were isolated. Sequencing of homokaryotic clones that exhibited a pink‐red colour confirmed the presence of the non‐sense mutation from the ade2_INT2 dDNA and the TA‐insertion from the ade2_INT3 dDNA (Fig. 4).
Editing efficiency of the ADE2 manipulations was estimated as the frequency of primary heterokaryotic transformants containing edited nuclei. The presence of edited nuclei in the heterokaryotic transformants was ascertained by the presence of at least one homokaryotic, pink‐red colony, among 100 homokaryotic colonies derived from sporulation of the primary heterokaryotic clones. Our results revealed a high editing efficiency of the ade2_INT1 sgRNA‐dDNA, which showed a 76% of heterokaryotic clones containing edited ade2 − nuclei. Nevertheless, some variability of editing efficiency (44–76%) between the three ADE2 sgRNA‐dDNAs was found (Table 1).
Table 1.
Editing efficiency of the sgRNA‐dDNA fragments used in this work
| sgRNA‐dDNA | Editing efficiency (%)a |
|---|---|
| ade2_INT1 | 76 |
| ade2_INT2 | 64 |
| ade2_INT3 | 44 |
| A754 | 36 |
| fmp27‐F70Y | 80 |
a. Twenty‐five primary heterokaryotic colonies were analysed by analytical PCR to calculate the editing efficiency.
Removal of genomic loxP scars using the CRISPR/Cas9 system for A. gossypii
Whereas the use of the ade2 − mutations permits the direct visualization of genome editing events, in most instances the phenotypic effects of an intended mutation were not visible. For example, this is the case of the removal of the loxP sequences (loxP scars) that remain in the genome after Cre‐mediated excision of the frequently used loxP‐KanMX‐loxP (loxPMK) marker.
To check the efficiency of the CRISPR/Cas9 system to eliminate a loxP scar in A. gossypii, we used as a model the strain A754 (ACL074WL‐PCK1‐loxP‐ACL074WR; a derivative of the strain A705 after the excision of the loxPMK marker) (Fig. 5). We designed a sgRNA‐dDNA (sgA754‐dA754) to precisely eliminate the loxP sequence from the genome of the A754 strain (Fig. 5 and Appendix S1).
Figure 5.
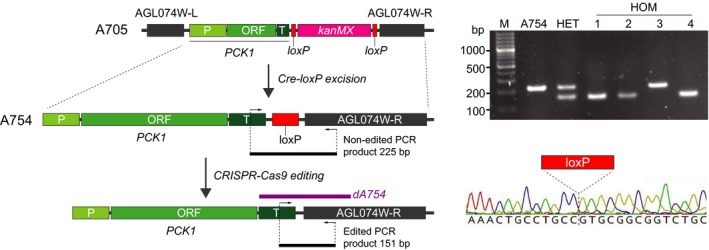
Removal of genomic loxP scars. Left panel, genomic organization of the agl074w locus in the A705 strain. Removal of the loxP‐kanMX marker by Cre recombinase generates a loxP scar. Genomic edition of the loxP sequence using the CRISPR/Cas9 system erased the loxP scar. The location of the primers for analytical PCR and the sequence of the dA754 donor DNA are indicated. Right upper panel, analytical PCR of the A754 strain, one heterokaryotic transformant (HET) and four homokaryotic clones (HOM). Both the edited (151 bp) and non‐edited (225 bp) PCR fragments are shown. Right lower panel, chromatogram of the genomic sequence of the homokaryotic clone 1. The absence of the loxP sequence is indicated.
After cloning the synthetic sgA754‐dA754 fragment into the CRISPR/Cas9 vector, the recombinant plasmid was used to transform spores of the A754 strain. Since the elimination of the loxP scar does not have a visible phenotypic effect, the presence of edited nuclei lacking the loxP sequence in the heterokaryotic transformants was analysed by PCR (Fig. 5). Hence, PCR products of 151 and 225 bp corresponded to edited and non‐edited nuclei, respectively (Fig. 5). An editing efficiency of 35% was calculated for the sgA754‐dA754 fragment (Table 1), thus confirming the ability of the CRISPR/Cas9 system for the removal of loxP scars in A. gossypii. Subsequently, the G418R primary transformants containing edited nuclei were transferred to G418‐free sporulation medium and homokaryotic edited clones were identified by analytical PCR and further corroborated by DNA sequencing (Fig. 5).
Introduction of point mutations using the CRISPR/Cas9 system for A. gossypii
The CRISPR/Cas9 system was also tested for the generation of precise single‐point mutations in A. gossypii that results in not visible phenotypes. As an example, we used the FMP27 (ABR180W) gene of A. gossypii, which is homologous to a yeast gene encoding a mitochondrial protein of unknown function (Reinders et al., 2006). Genome sequencing of a riboflavin overproducing strain (A81) obtained by UV mutagenesis revealed the presence of many point mutations (unpublished results), one of which affects to the FMP27 ORF of A. gossypii, generating an amino acid substitution in the Fmp27 protein (F70Y). To further investigate the effect of this amino acid change in the biosynthesis of riboflavin in A. gossypii, we decided to individually introduce that mutation on a wild‐type genetic background using the CRISPR‐Cas9 system.
A sgRNA‐dDNA for FMP27 targeting was designed to introduce a non‐synonymous mutation (T‐A substitution at nucleotide 209 of the coding region) that produce the intended amino acid change (Phe to Tyr) at position 70 (Fig. 6 and Supporting Information File 1). After the assembly of the sgRNA‐dDNA for fmp27‐F70Y into the CRISP22 vector, the plasmid was used to transform spores of the wild‐type strain of A. gossypii. Heterokaryotic clones were analysed by three primer PCR (TP‐PCR) in such a way that the pair of primers gFMP27‐ver‐fw and gFMP27‐ver‐rv are specific for the amplification of a 200 bp region encompassing position 70 of FMP27 in both edited and non‐edited nuclei/genomes/DNAs, and the pair of primers gFMP27‐mut‐fw and gFMP27‐ver‐rv are exclusively specific for the amplification of a 113 bp fragment only in edited genome templates. The presence of the nucleotide substitutions was confirmed by TP‐PCR and DNA sequencing of three positive homokaryotic clones derived from the three different primary heterokaryotic transformants (Fig. 6). The phenotype of the fmp27‐F70Y strain was undistinguishable from the wild‐type strain with regard to the growth rate and the production of riboflavin (not shown), thus indicating that the only presence of the F70Y mutation in the FMN27 gene is not enough to increase the biosynthesis of the vitamin in A. gossypii. However, our results showed that the CRISPR/Cas9 system was able to introduce specific designed single‐point mutations in the FMN27 gene with high efficiency (Fig. 6 and Table 1).
Figure 6.
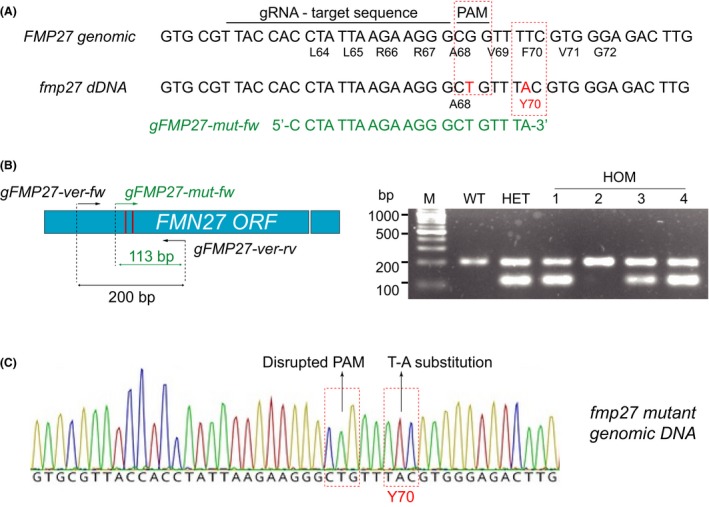
Point mutations using the CRISPR/Cas9 system for A. gossypii.A. Genomic sequence of the FMP27 gene and sequence of the fmp27 dDNA. The target sequence for the gRNA is indicated. The mutations introduced by the fmp27 dDNA are highlighted in red. Amino acid residues are indicated below the DNA sequence. The sequence of the gFMP27‐mut‐fw primer for the analytical PCR is shown in green.B. Locations of the primers used for analytical TP‐PCR in the FMP27 ORF (location of the base substitutions are indicated with red lines) and gel electrophoresis of the PCR products in the wild‐type strain, one heterokaryotic transformant (HET) and four homokaryotic clones (HOM).C. Chromatogram of the sequence of the edited genomic DNA of the fmp27 F70Y homokaryotic clone 1.
In the scope of our results, the introduction of all the modules for the CRISPR/Cas9 system in a single vector provides high efficiency to the system. Indeed, the lowest efficiency that we have obtained with this marker‐less system was 35%. This percentage still allows the convenient use of the CRISPR/Cas9 in A. gossypii with not visible phenotypes, by using analytical PCR and DNA sequencing to identify the edited sequence, as demonstrated for the A754 loxP edition and the fmp27 mutants (Figs 5 and 6).
Thereby, the CRISPR/Cas9 system for A. gossypii represents a novel methodology for marker‐less gene deletions/disruptions and single‐point mutations. This methodology combined with the multigene overexpression system for heterologous genes (Ledesma‐Amaro et al., 2018) will allow to avoid the use of selectable markers for metabolic engineering applications in A. gossypii. Similar toolkits have been described in other biotechnological microorganisms such as Yarrowia lypolytica, thus enabling the rapid engineering of metabolic pathways in that oleaginous yeast (Wong et al., 2017; Holkenbrink et al., 2018; Larroude et al., 2018).
The CRISPR/Cas9 system of A. gossypii described here can be largely expanded using novel RNA guided endonucleases such as FnCpf1, which shows preference for alternative PAM sequences (Swiat et al., 2017). Also, multiplexing CRISPR/Cas9 engineering can be achieved for the simultaneous edition of different targets and metabolic pathways, as previously demonstrated for S. cerevisiae (Bao et al., 2015; Jakočinas et al., 2015; Jakočiunas et al., 2015). In addition, the CRISPR‐Cas9 system can be adapted for transcriptional modulation using an endonuclease‐deficient Cas9 (dCas9) alone (Qi et al., 2013) or fusion proteins consisting of dCas9 and different transcriptional effector domains (Farzadfard et al., 2013). In summary, the CRISPR/Cas9 system for A. gossypii will contribute to the rapid expansion of precise and efficient genomic engineering strategies for this filamentous fungus with industrial interest.
Experimental procedures
A. gossypii strains and growth conditions
The A. gossypii ATCC 10895 strain was used and considered a wild‐type strain. Other A. gossypii strains used in this work are described in Appendix S1. A. gossypii cultures were carried out at 28°C in MA2 rich medium (Jiménez et al., 2005). A. gossypii transformation, sporulation and spore isolation were as described previously (Jiménez et al., 2005). Geneticin (G418) (Gibco‐BRL) was used where indicated at concentrations of 250 mg l−1.
Assembly of the CRISPR/Cas9 vector
The CRISPR/Cas9 vector has been assembled using functional modules from the plasmids p414 and pCRCT from George Church and Huimin Zhao's laboratories, respectively (Dicarlo et al., 2013; Bao et al., 2015). In addition, regulatory elements for sgRNA expression were PCR‐amplified from A. gossypii genomic DNA.
The plasmid pCRCT (Bao et al., 2015), containing the yeast 2μ ori, the Amp R and lacZ markers and the bacterial ColE1 ori, was used as a backbone (see Fig. S1). First, the yeast URA3 marker was replaced with the A. gossypii loxP‐KanMX‐loxP marker (G418 R) by Gibson assembly. Second, the promoter sequence from AgSNR52 was PCR‐amplified from A. gossypii genomic DNA and inserted next to the G418 R marker by Gibson assembly (see Appendix S1 for sequences). Third, the human codon‐optimized Cas9 module (P TEF1‐CAS9‐T CYC1) from p414 (Dicarlo et al., 2013) was PCR‐amplified and used to replace the Cas9 module of pCRCT by Gibson assembly. The final construct (pCRISP22) contained the lacZ marker flanked by two BsaI sites to facilitate the Golden Gate assembly of the sgRNA‐dDNA (Fig. 2). Hence, the sgRNA‐dDNA sequences were designed to contain two BsaI sites flanking the 4‐nt sticky ends for the Golden Gate assembly. The sequences of the primers used for the amplification of each module and Gibson assembly are listed in the Appendix S1. CRISPR target sites (i.e. sgRNA sequences) with an activity score higher than 0.5 and a maximum of three mismatches against off‐targets in the A. gossypii genome were selected according to Doench et al. (2014). The sequences of all the sgRNA‐dDNA fragments are described in Appendix S1.
The assembly of the final CRISPR/Cas9 plasmids was carried out in a one‐step reaction using equimolar amounts of the pCRISP22 vector and the corresponding sgRNA‐dDNA fragment. The Golden Gate assembly reaction included BsaI and T4 DNA ligase (NEB) with the following conditions on a thermocycler: 30 cycles of 37°C for 3 min and 16°C for 4 min; 50°C for 5 min and 80°C for 5 min. The recombinant CRISPR/Cas9 plasmids harbouring sgRNA‐dDNA inserts were selected as lacZ −, kanamycin resistant clones. All the CRISPR/Cas9 plasmids were verified by DNA sequencing.
CRISPR/Cas9 genome engineering in A. gossypii
Spores of indicated strains of A. gossypii were transformed with 5–10 μg of the corresponding CRISPR/Cas9 vector. Selection of heterokaryotic transformants was carried out in G418‐containing MA2 media. Positive G418 resistant clones were isolated and grown in G418‐MA2 media during 2 days. Next, loss of the CRISPR/Cas9 vector was achieved during sporulation of the heterokaryotic clones in SPA media lacking G418. Homokaryotic clones were isolated in MA2 media lacking G418 (see the workflow for the CRISPR‐Cas9 system in Fig. S2). Loss of the CRISPR/Cas9 plasmid was confirmed in G418‐MA2 media. Screening of the CRISPR/Cas9 mutations was carried out either by phenotypic analysis or by analytical PCR. For the ade2 − mutations, homokaryotic clones showing a pink‐red colour were isolated. For the removal of loxP scars, the homokaryotic clones were analysed by PCR using the primers gA754‐CRSP‐ver‐fw and gA754‐CRSP‐ver‐rv. For the AgFMP27 genomic edition, the primers gFMN27‐ver‐fw, gFMN27‐mut‐fw and gFMN27‐ver‐rv were used for analytical TP‐PCR. All the genomic editions were further confirmed by DNA sequencing.
Calculation of the editing efficiency of CRISPR/Cas9 vectors in A. gossypii
The editing efficiency of the CRISPR/Cas9 system was estimated as the frequency of primary heterokaryotic transformants containing edited nuclei. The presence of edited nuclei in the heterokaryotic transformants was ascertained by the presence of at least one homokaryotic edited clone among 100 homokaryotic colonies derived from sporulation of the primary heterokaryotic clones. The presence of edited clones for the ADE2 gene inactivation was confirmed by phenotypic observation (pink‐red colour of the homokaryotic clones). Instead, the presence of edited clones for the loxP elimination and fmp27 mutations was confirmed by analytical PCR.
Conflict of interest
The authors declare that they have no competing interests.
Supporting information
Fig. S1. Assembly of the pCRISP22 vector.
Fig. S2. Workflow of the CRISPR‐Cas9 system for A. gossypii.
Appendix S1. Sequences of genetic elements of CRISPR/Cas9 vectors for A. gossypii, A. gossypii strains and list of primers used in this work.
Acknowledgements
This work was financed by grants from the Spanish Ministerio de Economía y Competitividad (BIO2014‐56930‐P and BIO2017‐88435‐R) and Junta de Castilla y León (SA016P17) to JLR and AJ. We thank María Dolores Sánchez and Silvia Domínguez for excellent technical help and Kyle Brandon del Valle for correcting the manuscript.
Microbial Biotechnology (2019) 12(6), 1293–1301
Funding Information
This work was financed by grants from the Spanish Ministerio de Economía y Competitividad (BIO2014‐56930‐P and BIO2017‐88435‐R) and Junta de Castilla y León (SA016P17) to JLR and AJ.
References
- Aguiar, T.Q. , Silva, R. , and Domingues, L. (2015) Ashbya gossypii beyond industrial riboflavin production: a historical perspective and emerging biotechnological applications. Biotechnol Adv 33: 1774–1786. [DOI] [PubMed] [Google Scholar]
- Aguiar, T.Q. , Silva, R. , and Domingues, L. (2017) New biotechnological applications for Ashbya gossypii: challenges and perspectives. Bioengineered 8: 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, Z. , Xiao, H. , Liang, J. , Zhang, L. , Xiong, X. , Sun, N. , et al (2015) Homology‐integrated CRISPR‐Cas (HI‐CRISPR) system for one‐step multigene disruption in Saccharomyces cerevisiae . ACS Synth Biol 4: 585–594. [DOI] [PubMed] [Google Scholar]
- Brouns, S.J.J. , Jore, M.M. , Lundgren, M. , Westra, E.R. , Slijkhuis, R.J.H. , Snijders, A.P.L. , et al (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz‐Fernández, D. , Lozano‐Martínez, P. , Buey, R.M. , Revuelta, J.L. , and Jiménez, A. (2017) Utilization of xylose by engineered strains of Ashbya gossypii for the production of microbial oils. Biotechnol Biofuels 10: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicarlo, J.E. , Norville, J.E. , Mali, P. , Rios, X. , Aach, J. , and Church, G.M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR‐Cas systems. Nucleic Acids Res 41: 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, F.S. , Voegeli, S. , Brachat, S. , Lerch, A. , Gates, K. , Steiner, S. , et al (2004) The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304: 304–307. [DOI] [PubMed] [Google Scholar]
- Doench, J.G. , Hartenian, E. , Graham, D.B. , Tothova, Z. , Hegde, M. , Smith, I. , et al (2014) Rational design of highly active sgRNAs for CRISPR‐Cas9‐mediated gene inactivation. Nat Biotechnol 32: 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard, F. , Perli, S.D. , and Lu, T.K. (2013) Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol 2: 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkenbrink, C. , Dam, M.I. , Kildegaard, K.R. , Beder, J. , Dahlin, J. , Doménech Belda, D. , and Borodina, I. (2018) EasyCloneYALI: CRISPR/Cas9‐based synthetic toolbox for engineering of the Yeast Yarrowia lipolytica. Biotechnol J 13: e1700543. [DOI] [PubMed] [Google Scholar]
- Jakočinas, T. , Bonde, I. , Herrgård, M. , Harrison, S.J. , Kristensen, M. , Pedersen, L.E. , et al (2015) Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae . Metab Eng 28: 213–222. [DOI] [PubMed] [Google Scholar]
- Jakočiunas, T. , Rajkumar, A.S. , Zhang, J. , Arsovska, D. , Rodriguez, A. , Jendresen, C.B. , et al (2015) CasEMBLR: Cas9‐facilitated multiloci genomic integration of in vivo assembled DNA parts in Saccharomyces cerevisiae . ACS Synth Biol 4: 1126–1134. [DOI] [PubMed] [Google Scholar]
- Jiménez, A. , Santos, M.A. , Pompejus, M. , and Revuelta, J.L. (2005) Metabolic engineering of the purine pathway for riboflavin production in Ashbya gossypii . Appl Environ Microbiol 71: 5743–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J.A. , and Charpentier, E. (2012) A programmable dual‐RNA – guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larroude, M. , Rossignol, T. , Nicaud, J.‐M. , and Ledesma‐Amaro, R. (2018) Synthetic biology tools for engineering Yarrowia lipolytica. Biotechnol Adv 36: 2150–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma‐Amaro, R. , Buey, R.M. , and Revuelta, J.L. (2015a) Increased production of inosine and guanosine by means of metabolic engineering of the purine pathway in Ashbya gossypii . Microb Cell Fact 14: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma‐Amaro, R. , Lozano‐Martínez, P. , Jiménez, A. , and Revuelta, J.L. (2015b) Engineering Ashbya gossypii for efficient biolipid production. Bioengineered 6: 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma‐Amaro, R. , Jiménez, A. , and Revuelta, J.L. (2018) Pathway grafting for polyunsaturated fatty acids production in Ashbya gossypii through Golden Gate Rapid Assembly. ACS Synth Biol 7: 2340–2347. [DOI] [PubMed] [Google Scholar]
- Lozano‐Martínez, P. , Buey, R.M. , Ledesma‐Amaro, R. , Jiménez, A. , and Revuelta, J.L. (2016) Engineering Ashbya gossypii strains for de novo lipid production using industrial by‐products. Microb Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manivasakam, P. , Weber, S.C. , McElver, J. , and Schiestl, R.H. (1995) Micro‐homology mediated PCR targeting in Saccharomyces cerevisiae . Nucleic Acids Res 23: 2799–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, L.S. , Larson, M.H. , Gilbert, L.A. , Doudna, J.A. , Weissman, J.S. , Arkin, A.P. , and Lim, W.A. (2013) Repurposing CRISPR as an RNA‐γuided platform for sequence‐specific control of gene expression. Cell 152: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschmanová, H. , Weninger, A. , Glieder, A. , Kovar, K. , and Vogl, T. (2018) Implementing CRISPR‐Cas technologies in conventional and non‐conventional yeasts: current state and future prospects. Biotechnol Adv 36: 641–665. [DOI] [PubMed] [Google Scholar]
- Reinders, J. , Zahedi, R.P. , Pfanner, N. , Meisinger, C. , and Sickmann, A. (2006) Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res 5: 1543–1554. [DOI] [PubMed] [Google Scholar]
- Revuelta, J.L. , Ledesma‐Amaro, R. , Lozano‐Martinez, P. , Díaz‐Fernández, D. , Buey, R.M. , and Jiménez, A. (2017) Bioproduction of riboflavin: a bright yellow history. J Ind Microbiol Biotechnol: 44. [DOI] [PubMed] [Google Scholar]
- Rychen, G. , Aquilina, G. , Azimonti, G. , Bampidis, V. , de Bastos, M.L. , Bories, G. , et al (2018) Safety and efficacy of vitamin B2 (riboflavin) produced by Ashbya gossypii for all animal species based on a dossier submitted by BASF SE. EFSA J 16: e05337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwechheimer, S.K. , Park, E.Y. , Revuelta, J.L. , Becker, J. , and Wittmann, C. (2016) Biotechnology of riboflavin. Appl Microbiol Biotechnol: 100. [DOI] [PubMed] [Google Scholar]
- Serrano‐Amatriain, C. , Ledesma‐Amaro, R. , López‐Nicolás, R. , Ros, G. , Jiménez, A. , and Revuelta, J.L. (2016) Folic acid production by engineered Ashbya gossypii . Metab Eng 38: 473–482. [DOI] [PubMed] [Google Scholar]
- Shi, T.Q. , Liu, G.N. , Ji, R.Y. , Shi, K. , Song, P. , Ren, L.J. , et al (2017) CRISPR/Cas9‐based genome editing of the filamentous fungi: the state of the art. Appl Microbiol Biotechnol 101: 7435–7443. [DOI] [PubMed] [Google Scholar]
- Steiner, S. , Wendland, J. , Wright, M.C. , and Philippsen, P. (1995) Homologous recombination as the main mechanism for DNA integration and cause of rearrangements in the filamentous ascomycete Ashbya gossypii . Genetics 140: 973–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiat, M.A. , Dashko, S. , Den Ridder, M. , Wijsman, M. , Van Der Oost, J. , Daran, J.M. , and Daran‐Lapujade, P. (2017) FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae . Nucleic Acids Res 45: 12585–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, L. , Engel, J. , Jin, E. , Holdridge, B. , and Xu, P. (2017) YaliBricks, a versatile genetic toolkit for streamlined and rapid pathway engineering in Yarrowia lipolytica. Metab Eng Commun 5: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Assembly of the pCRISP22 vector.
Fig. S2. Workflow of the CRISPR‐Cas9 system for A. gossypii.
Appendix S1. Sequences of genetic elements of CRISPR/Cas9 vectors for A. gossypii, A. gossypii strains and list of primers used in this work.