

Abstract
Objective
Ala97Ser (A97S) is the major transthyretin (TTR) mutation in Taiwanese patients of familial amyloid polyneuropathy (FAP), characterized by a late‐onset but rapidly deteriorated neuropathy. Tafamidis can restore the stability of some mutant TTR tetramers and slow down the progression of TTR‐FAP. However, there is little understanding of the biophysical features of A97S‐TTR mutant and the pharmacological modulation effect of tafamidis on it. This study aims to delineate the biophysical characteristics of A97S‐TTR and the pharmacological modulation effect of tafamidis on this mutant.
Method
The stability of TTR tetramers was assessed by urea denaturation and differential scanning calorimetry. Isothermal titration calorimetry (ITC) was used to measure the binding constant of tafamidis to TTR. Nuclear magnetic resonance spectroscopy (NMR) titration experiment was used to map out the tafamidis binding site.
Results
Chemical and thermal denaturation confirmed the destabilization effect of A97S. Consistent with other the amyloidogenic mutant, A97S‐TTR has slightly lower conformational stability. NMR revealed the binding site of A97S‐TTR with tafamidis is at the thyroxine binding pocket. The ITC experiments documented the high affinity of the binding which can effectively stabilize the A97S‐TTR tetramer.
Interpretation
This study confirmed the structural modulation effect of tafamidis on A97S‐TTR and implied the potential therapeutic benefit of tafamidis for A97S TTR‐FAP. This approach can be applied to investigate the modulation effect of tafamidis on other rare TTR variants and help to make individualized choices of available treatments for FAP patients.
Introduction
Familial amyloid polyneuropathy (FAP) is the most common hereditary amyloid diseases. Transthyretin‐related FAP (TTR‐FAP) is the most prevalent genetic subtype characterized by debilitating polyneuropathy and life‐threatening cardiomyopathy.1, 2 The disease‐causing gene is TTR (NM_000371). More than 150 disease‐causing TTR mutations associated with highly variable clinical manifestations have been identified.2 Val30Met (V30M) is the most frequent mutation worldwide.2, 3 V30M TTR‐FAP typically presents by a slowly progressive polyneuropathy with the onset at around 30 years and most patients could still walk independently even after 20 years of disease onset. Cardiac manifestations usually occur in the later stage of the disease.4, 5, 6, 7 The overall survival rate could stay around 80% after 10 years of follow‐up.4 It has been reported that Ala97Ser (A97S) is the major TTR mutation, accounting for 91.2% of the hereditary transthyretin amyloidosis (ATTR) pedigrees.8, 9, 10 Contrary to the classical early‐onset V30M TTR‐FAP, the patients with A97S TTR‐FAP have a much later onset with the mean age of onset of 58.2 years but would become bed ridden or wheel‐chair dependent within 8.4 years of the onset on average.8, 9, 10 The rapidly deteriorative course may be partially due to severe cardiac dysfunction. Life‐threatening cardiac arrhythmia (15.1%) and heart failure (16.4%) are prevalent in the A97S patients and might become severe before the full brown of peripheral neuropathy.8, 10 Although only a limited number of cases were available to provide a longitudinal follow‐up information, unfavorable events like syncope/cardiac arrest/ cardiopulmonary‐cerebral resuscitation tended to occur approximately 4 years after the appearance of the initial neurological symptoms and up to half died within 11 years after the first symptom or event.9
The encoding protein of TTR is transthyretin (TTR), a 55‐kDa homo‐tetrameric plasma protein circulating in the human serum and cerebrospinal fluid. The toxic species of most pathogenic TTR mutations is comprised of mutant monomers with a lower conformational stability and a higher propensity to dissociation from the tetramer then leading to amyloid fibril formation.11, 12 Tafamidis, one of the TTR stabilizers, has shown the potency to slow down the progression of neuropathy with a long‐term safety and to reduce the mortality and hospitalizations due to TTR amyloid cardiomyopathy (TAC).13, 14, 15, 16, 17, 18, 19, 20 However, only very limited patients of A97S TTR‐FAP were recruited in previous clinical trials because of the late‐onset nature and the moderate to severe disability at diagnosis. So far there is little information of the effect of tafamidis on A97S TTR‐FAP.
Understanding the biophysical characteristics of A97S‐TTR mutant and the modulation effects by tafamidis will help understand the pathogenicity of this mutation and the potential benefits from the pharmacotherapy. In this study, we assessed the stability of the A97S‐TTR tetramer by monitoring urea denaturation and measuring the melting temperature using differential scanning calorimetry (DSC). Isothermal titration calorimetry (ITC) was used to measure the binding constants of tafamidis to A97S‐TTR. Solution nuclear magnetic resonance (NMR) spectroscopy was used to map out the structure and the tafamidis binding site of A97S‐TTR. Our results will generate significant impacts on the clinical management and influence the healthy policy for patients of A97S TTR‐FAP. The approach can be applied to evaluate the potential benefits of pharmacological modulation for other TTR variants.
Materials and Methods
TTR unfolding assay using urea
Proteins unfolding of WT‐TTR, A97S‐TTR, T119M‐TTR, V30M‐TTR, and L55P‐TTR were assessed using urea denaturation assay.21 TTR sample solutions (0.1 mg/mL) were prepared by diluting 200 μL of 1.0 mg/mL TTR stock into 1.8 mL buffer, containing 10 mmol/L Na phosphate (pH7.5) with 100 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L DTT, 0.02 % sodium azide, and various concentrations of urea. The fluorescence emission ratio at 355 and 335 nm (F355/F335) of each sample was then monitored using a fluorescence spectrometer (Perkin Elmer LS‐55) after 96 h of incubation at room temperature. The excitation slits were set at 2.5 nm, while the emission slits were set at 3.5 nm. Excitation wavelength was fixed at 295 nm and the emission spectra were collected from 310 to 450 nm.
Differential scanning calorimetry (DSC)
DSC measurements were performed on a Nano DSC differential scanning microcalorimeter (TA Instruments). By measuring the critical concentration for different TTR variants to re‐associate to be TTR tetramer, we can provide a parameter to assess the tendency of different TTR proteins to be in tetrameric state. In addition to WT‐TTR, A97S‐TTR, V30M‐TTR, and L55P‐TTR, the TTR protein with the protective variants R104H and T119M were also studies. Experiments were carried out at the concentrations of the TTR protein ranging from 1.5 to 2 mg/mL at the scan rate of 1.0°C/min. Protein samples were prepared in PBS buffer, as well as in PBS buffer containing 5% DMSO. To examine the effect of tafamidis‐binding on the Tm of TTR proteins, we needed to ensure that tafamidis can be completely solubilized. Thus, PBS buffer containing 5% DMSO was also used in the experiment. For DSC experiments of tafamidis bound TTR, the ratio of tafamidis to TTR tetramer equaled to 1.6 and only PBS buffer containing 5% DMSO was used. Before the measurements, the sample and reference solutions were properly degassed. An overpressure of 3 atm was always kept over the liquid in the cells throughout the scans to prevent any degassing during heating. The buffer scan (i.e., instrumental baseline) was determined before each sample scan, by filling both the sample and reference cells with the buffer used for the protein sample and using the same scanning parameters.
Isothermal titration calorimetry (ITC)
ITC experiments were performed for WT‐TTR, the TTR mutants of A97S, V30M, and L55P based on Bulawa et al 2012.14 The detailed experimental conditions were documented in Table S1. Dissociation constants for tafamidis and each TTR protein were determined using a Microcal VP‐ITC isothermal titration calorimeter (Microcal Inc.). The concentrations of proteins and tafamidis used in all ITC experiments are listed in Table S1. The initial injection of 2.5 µL of tafamidis solution was followed by 49 injections of 5 µL each (25°C). The integrated heat was plotted against the molar ratio of tafamidis added to protein in the cell, yielding complete binding isotherms.
Nuclear magnetic resonance spectroscopy
NMR spectroscopic characterization of the structural changes by tafamidis binding to A97S‐TTR and WT‐TTR was performed. NMR experiments were carried out at 37°C on Bruker spectrometers, operating at 600 MHz, 800 MHz and 850 MHz (proton frequencies.) Each NMR spectrometer was equipped with a Bruker TXI probes. Transverse relaxation‐optimized spectroscopy (TROSY) based triple‐resonance NMR experiments, including TROSY‐HNCA, TROSY‐HN(co)CA, TROSY‐HNCACB and NOESY‐TROSY‐HSQC were recorded for sequence‐specific resonance assignment.22 A typical NMR sample for this purpose contained 0.6–0.9 mmol/L TTR in a buffer containing 25 mmol/L Na phosphate, 100 mmol/L NaCl, 0.5 mmol/L EDTA, 1 mmol/L TCEP at pH 6.5. The TROSY‐based 1H‐15N heteronuclear single quantum coherence spectroscopy (TROSY‐HSQC) spectra of TTR with and without the addition of tafamidis were recorded to analyze the chemical shift changes due to drug binding. To reliably assign the peaks shift due to the binding with tafamidis, TROSY‐HNCA spectra of both samples were also recorded. The NMR sample buffer for this purpose contained an additional 5% of DMSO in order to completely dissolve tafamidis at 2.5:1 molar ratio (tafamidis to TTR) to saturate the tafamidis binding sites of TTR. The NMR data were processed with NMRPipe and the assignments were made using the program CARA.23
Results
Destabilization effect of A97S‐TTR mutant in chemical denaturation
By monitoring urea denaturation using F355/F335,21 we compared the stability of A97S‐TTR with WT‐TTR. Two pathogenic mutants V30M‐TTR and L55P‐TTR, as well as one clinically protective mutant T119M,11 were also assessed. V30M‐TTR is the most prevalent disease‐causing variant worldwide2, 3 and L55P‐TTR is well recognized for its high amyloidogenic potential.24, 25, 26 The dissociation tendency revealed that the highest unfolding change in the L55P‐TTR tetramer, compatible with its notorious amyloidogenic potential (blue open circles in Fig. 1). On the other hand, T119M‐TTR possesses the greatest the tetramer stability against urea‐induced unfolding (pink filled squares in Fig. 1), consistent with the clinical observation of T119M as a protective mutant.27, 28 V30M‐TTR bears a tetramer stability slightly worse than WT‐TTR (green open circles in Fig. 1), which probably reflects the early‐onset but insidiously progressive disease course. For A97S‐TTR (red open circles in Fig. 1), the result indicates that the tetramer stability of A97S‐TTR is worse than WT‐TTR, confirming the pathogenicity of the mutant. Compared with V30M‐TTR and L55P‐TTR mutants, A97S‐TTR is shown to have slightly better resistivity against urea denaturation.
Figure 1.
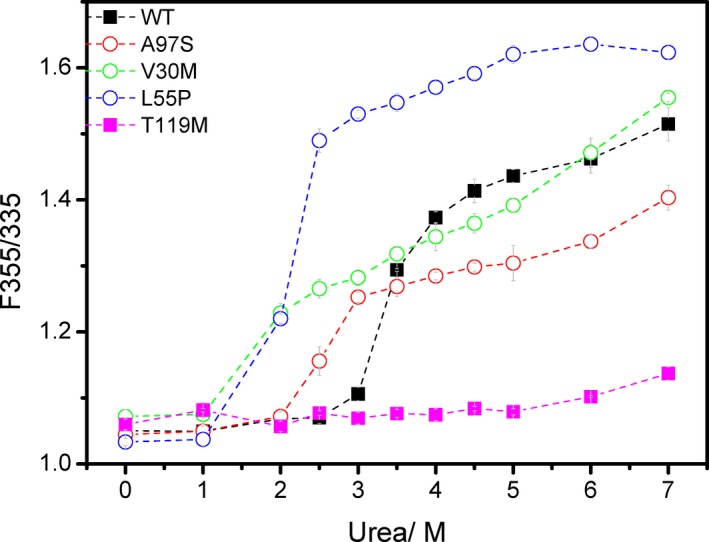
Urea denaturation monitored by tryptophan fluorescence (F355/F335). WT, wild type TTR, black filled squares; A97S‐TTR, red open circles; V30M TTR, green open circles; L55P TTR, blue open circles; T119M TTR, pink filled squares. Dashed lines are drawn to guide the eye.
Destabilization effect of A97S‐TTR mutant in thermal denaturation
The denaturing temperatures (Tm°C) of WT‐TTR and several mutant proteins were measured by DSC. In addition to the three pathogenic mutants A97S‐, V30M‐, and L55P‐ TTR, the proteins carrying R104H and T119M were also studied. The two variants are believed to be protective because TTR M30/H104 and TTR M30/M119 compound heterozygotes can induce structural alterations that increase the stability of the tetramer and protect from amyloid fibril formation.27, 28 The results are listed in Table 1. In both the conditions of PBS buffer only and PBS buffer containing 5% DMSO, the three amyloidogenic mutants had lower Tm than WT‐TTR while the Tm of two protective variants were similar to WT‐TTR. Shnyrov VL et al. clearly showed the tetrameric forms of non‐amyloidogenic variants had a slightly higher conformational stability than amyloidogenic variants, even though the stability among the amyloidogenic variants did not correlate with the amyloidogenic potential of these proteins.29 Our DSC data showed that the stability of A97S‐TTR falls below WT‐TTR and the two protective mutants R104H and T119M. In parallel with the results of chemical denaturation, this finding suggests that A97S‐TTR is an amyloidogenic mutant since it has slightly lower conformational stability. By comparing the Tm of TTR proteins measured with and without the presence of tafamidis, we observed an increase in Tm of all TTR variants upon tafamidis binding. This observation could serve as evidence of a direct interaction between tafamidis and TTR proteins. Among the three pathogenic mutants, only the Tm of A97S‐TTR reached the levels of WT‐TTR and the two protective R104H and T119M variants, implying tafamidis could help A97S‐TTR resume the stability.
Table 1.
The denaturing temperatures (Tm °C) of TTR proteins measured with differential scanning calorimetry (DSC) (60°C/h).
| TTR protein | Buffer condition | ||
|---|---|---|---|
| PBS | PBS containing 5% DMSO | PBS containing 5% DMSO and tafamidis | |
| WT‐TTR | 101.1 ± 0.3 | 97.2 ± 0.1 | 103.8 ± 0.5 |
| V30M‐TTR | 92.8 ± 0.1 | 89.2 ± 0.1 | 94.8 ± 0.1 |
| L55P‐TTR | 97.0 ± 0.2 | 92.3 ± 0.1 | 97.0 ± 0.1 |
| A97S‐TTR | 98.5 ± 0.1 | 93.9 ± 0.3 | 100.0 ± 0.3 |
| R104H‐TTR | 101.3 ± 0.1 | 97.6 ± 0.1 | 103.5 ± 0.1 |
| T119M‐TTR | 102.3 ± 0.1 | 98.2 ± 0.1 | 102.2 ± 0.2 |
Binding affinity of tafamidis with A97S‐TTR mutant
We then assessed the binding constants of tafamidis to WT‐TTR, A97S‐TTR, V30M‐TTR and L55P‐TTR by ITC. The integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to TTR proteins are listed in Table S2, while the experimental and fitted binding thermograms of ITC experiments are shown in Figure S2. The binding constants of tafamidis to various TTR mutants are listed in Table 2. The binding constants that were obtained consistent with the values previously reported by Bulawa et al.14 Our results revealed that the binding affinity of tafamidis with A97S‐TTR is comparable to that with WT‐TTR. In contrast, the binding affinities of tafamidis with V30M‐TTR and with L55P‐TTR are significantly weaker.
Table 2.
The characteristics of isothermal titration calorimetry (ITC) experiments for tafamidis binding to different TTR mutants.
| TTR protein | Ka1 (M−1) | ΔH1 (kcal/mol) | Ka2 (M−1) | ΔH2 (kcal/mol) | Kd1 (nmol/L) | Kd2 (nmol/L) |
|---|---|---|---|---|---|---|
| WT | 4.2 × 108 | −6.48 | 3.0 × 106 | −6.8 | 2.4 | 333.3 |
| A97S | 3.5 × 108 | −5.95 | 3.8 × 106 | −6.6 | 2.9 | 263.2 |
| V30M | 6.5 × 106 | −7.2 | 1.6 × 106 | −6.4 | 153.8 | 625 |
| L55P | 9 × 106 | −8.584 | 1.2 × 106 | −5.12 | 111 | 833 |
The binding isotherm were fitted with a model of two interacting sites exhibiting negative cooperativity to obtain the binding parameters, including dissociation constants, Kd1 and Kd2, and enthalpy, ΔH1 and ΔH2. Dissociation constants of tafamidis:WT‐TTR, tafamidis:A97S‐TTR, tafamidis:V30M‐TTR and tafamidis:L55P‐TTR were determined by finding the best fitted parameters to the experimental thermograms. The binding constants of tafamidis binding Kd1 and Kd2 were determined as the inverse of the association constants. All ITC experiments were performed at least three times in order to obtain the statistical results.
NMR spectroscopic characterization of tafamidis binding to A97S‐TTR mutant
The 1H‐15N TROSY‐HSQC spectra of both WT‐TTR and A97S‐TTR samples, with and without the addition of tafamidis, are shown in Figure 2A and B. The resonance peaks showed in the TROSY HSQC spectra are associated mainly with backbone amide groups. The chemical shift perturbation (CSP) of tafamidis binding, to both WT‐TTR and A97S‐TTR are plotted in Figure 3A and B. Those resonance peaks significant shifted as a result of drug binding are mainly located in the dimer‐dimer interface, previously reported as the thyroxine‐binding site by X‐ray crystallography studies.30 Thus, the tafamidis binding site of WT‐TTR is well associated with NMR CSP mapping results. Based on the CSP mapping results, tafamidis also affects the chemical shifts of the residues around thyroxine‐binding pocket of A97S‐TTR. To visualize the NMR results, we mapped the significantly perturbed residues on the cartoon presentation of WT‐TTR proteins structure generated with Pymol based on the X‐ray crystal structure (PDB ID: 4pvl)31, as shown in Figure 3C.
Figure 2.
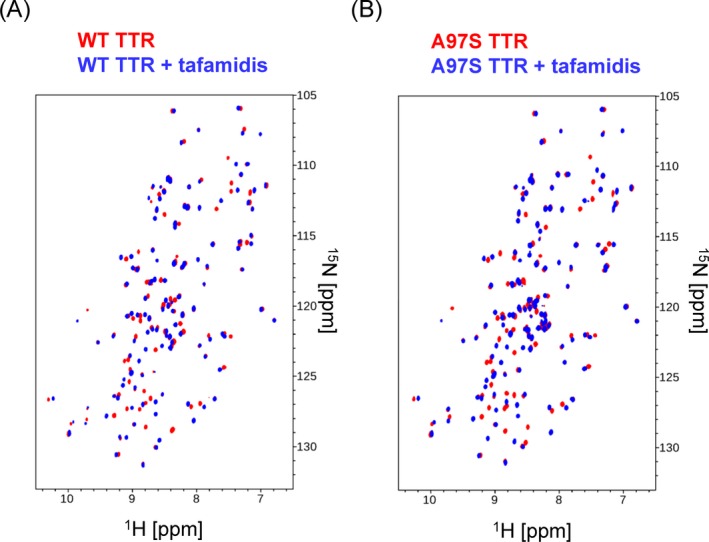
NMR spectroscopic characterization of Tafamidis binding to wildtype transthyretin and A97S transthyretin. (A) 2D TROSY HSQC spectra of 15N labeling WT‐TTR with (blue) and without (red) the presence of tafamidis at 2.5:1 molar ratio (tafamidis to protein). (B) 2D TROSY HSQC spectra of 15N labeling A97S‐TTR with (blue) and without (red) the presence of tafamidis at 2.5:1 molar ratio (tafamidis to protein).
Figure 3.
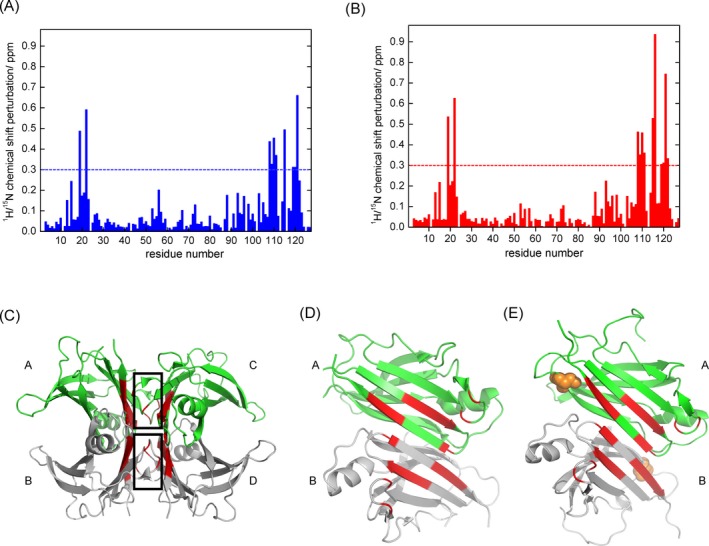
Characterizations of tafamidis binding sites of wildtype transthyretin and A97S transthyretin. (A) Normalized chemical shift changes for each residue of wildtype TTR due to tafamidis binding; (B) normalized chemical shift changes for each residue of A97S‐TTR due to tafamidis binding; (C) mapping of significantly perturbed (>0.3 ppm) residues in red color on the ribbon representation of the wildtype transthyretin using crystal structure (PBD ID: 4pvl), where the thyroxine binding sites are indicated with black box; (D) mapping of significantly perturbed (>0.3 ppm) residues on the ribbon representation of the dimer‐dimer interface of the wildtype transthyretin using crystal structure (PBD ID: 4pvl); (E) mapping of significantly perturbed (>0.3 ppm) residues on the ribbon representation of the dimer‐dimer interface of A97S transthyretin using a homology model generated based on the wildtype transthyretin crystal structure (PBD ID: 4pvl), where Ser‐97 residues are highlighted as orange spheres. In Figure 3D and E, the structural models were turned 90 degrees, with subunits C and B removed, to clearly show the TTR dimer‐dimer interface of subunit A and B.
The four TTR monomer subunits are labeled with A, B, C, and D and the thyroxine binding pocket highlighted with black box. The red‐color highlighted residues are associated with the resonance peaks significantly perturbed upon tafamidis binding. The cartoon presentation of A97S‐TTR structure was generated with Pymol using the homology model created with modeller 9.2 based on the X‐ray crystal structure (PDB ID: 4pvl). By comparing CSP mapping of tafamidis to both WT‐TTR and A97S‐TTR, we show that tafamidis binds to thyroxine binding sites of A97S TTR as well. Together with our ITC results, we concluded that tafamidis also binds strongly and specifically to the pathogenic mutant A97S‐TTR. The chemical shifts of backbone proton, backbone nitrogen and alpha carbon of both WT‐TTR and A97S‐TTR have been documented and uploaded to Biological Magnetic Resonance Data Bank (BMRB). The BMRB entry assigned accession numbers are 27575 and 27576 for WT‐TTR and A97S‐TTR, respectively.
Discussion
Over the past decade, the advancement of genetic diagnosis has greatly expanded our knowledge of TTR‐FAP and facilitated the development of mechanism‐driven therapies targeting to different stages of TTR amyloid formation and deposition, including liver transplantation, TTR stabilizers, gene modifiers like silencing RNA and antisense oligonucleotide therapies.32, 33, 34 Among them, disease‐modifying pharmacotherapy by the TTR stabilizer tafamidis has been proved to be able to significantly slow down the progression of polyneuropathy and deterioration of life quality with a long‐term safety and efficacy.13, 14, 15, 16, 17, 18 Very recently, a multicenter, international, double‐blind phase 3 trial further revealed that tafamidis was associated with reductions in all‐cause mortality and cardiovascular‐related hospitalizations in patients with TTR amyloid cardiomyopathy (TAC).19, 20
All previous clinical trials were targeted to the patient at the early stage and V30M TTR‐FAP was the main recruited genotype. Nevertheless, numerous TTR variants other than V30M have been identified and associated with highly variable age of onset, severity and progression rates of the neuropathy and cardiopathy. The wide range of clinical manifestations reflects the diverse pathological effects caused by different TTR variants, which further raises the issue that the best treatment may vary with the genotypes. One of the key factors in determining the pathogenicity of a TTR variant is its destabilization effect on the tetramer which in turn accelerates amyloid fibril formation. For example, the highly amyloidogenic L55P variant is associated with early‐onset aggressive diffuse amyloidosis and severe cardiac and neurologic dysfunctions, whereas the compound heterozygous carriers with V30M mutation and the protective T119M variant present by a late‐onset and mild phenotype.35 This implies that the tetramer stability would influence the treatment response to TTR stabilizers and bring the clinical importance to elucidate the structural changes and related pathogenesis of specific TTR variants.
A97S is the predominant genotype of Taiwanese TTR‐FAP patients, presenting by a unique phenotype composed of a late‐onset but rapid‐progressive neuropathy and life‐threatening cardiopathy. Nevertheless, there is little information of the impacts of the mutation on the TTR tetramer stabilization and the amyloidogenic potential of the mutant protein. To fill the knowledge gap, we assessed A97S‐TTR tetramer stability against external stresses using both chemical denaturation assay and thermal denaturation assays and compared with the stability of WT‐TTR. Our results revealed that A97S mutation did cause destabilization of the TTR tetramer as the mutated proteins had a higher dissociation tendency and a lower denaturing temperature. Of note, the stability of the late‐onset A97S‐TTR tetramer is still better than that of the V30M‐TTR and L55P‐TTR mutants, both are clinically associated with an early‐onset polyneuropathy. These findings supported that the propensity of destabilization of the TTR tetramer could be one of the determinants of the onset and progression of the disease, indirectly explaining the wide clinical spectrum of TTR‐FAP.
In the DSC assay, adding of tafamidis was found to increase the denaturing temperatures of the three pathogenic mutants, indicating that the drug is able to enhance the structural stability. Particularly for the A97S‐TTR, the Tm raised up to the level very close to those of WT‐TTR and the protective R104H and T119M variants. The strong affinity between tafamidis and A97S‐TTR was also confirmed by the ITC experiment. As listed in Table 2, our data provided strong evidence that tafamidis can bind to A97S‐TTR with the affinity comparable to WT‐TTR and the binding is much stronger than V30M‐TTR and L55P‐TTR. Together with the DSC result, our ITC result suggested that tafamidis can effectively bind to A97S‐TTR tetramer to provide stabilization effect.
Considering the stability of TTR tetramers against denaturing agents may not reflect their physiologically relevant condition, we then used solution NMR to characterize the structural changes associated with the A97S mutation and the binding effects of tafamidis. By plotting the chemical shift pertubation of tafamidis binding, we documented that tafamidis binds to thyroxine‐binding pocket of A97S‐TTR tetramer, like it behaves to WT‐TTR (Fig. 3). NMR measurements can reveal the spatial and dynamic alternations of the tetramer structures of different pathogenic mutations which are not readily observed in the crystal structures. Solution NMR revealed that AB loop regions interacting with strand A in the DAGH β‐sheet undergo conformational changes, leading to the destabilized β‐sheet.36 And the DA β‐structure is believed to be the main site undergoing conformational changes leading to amyloid formation in the pathogenic L55P and V30M mutations.37, 38 A97 is on the strand G in the DAGH β‐sheet 39 and spatially close to the thyroxine‐binding pocket on the dimer‐dimer interface.40 Based on the NMR and ITC experiments, we proposed that the A97S mutation may disrupt β‐sheet structure but the influence can be reversed significantly by tafamidis binding.
To sum up the biophysical characters of A97S‐TTR, destabilization effect of this genetic variant has been clearly observed in both chemical and thermal denaturation. It has been well recognized that the amyloidogenic potential of human pathogenic TTR variants is determined by the destabilization of their native structures. Structural alterations of TTR tetramer induced by genetic variation are essential to induce protein destabilization, the representative early events leading to amyloid fibril dissociations and depositions.11 Furthermore, via solution NMR and ITC assessment, this study documented at the conformational destabilization can be reversed though the strong affinity of A97S‐TTR and tafamidis. These findings have several important clinical implications on TTR‐FAP. First, our results suggested the therapeutic benefit of tafamidis for the majority of the TTR‐FAP patients in Taiwan as A97S is the hot‐spot TTR mutation in our population. By providing evidence of the stabilization effect from tafamidis, we hope to raise clinical awareness and to encourage early diagnosis of A97S TTR‐FAP. This is also an endeavor in order to strike for more public resources in treatment for this disease. Furthermore, these in‐vitro assessments can be applied to address the biophysical features of other rare TTR variants. This study provides a feasible approach to evaluate the modulational effects of tafamidis on other rare TTR variants.
Although gene modifiers like the RNA interference therapeutic agent and the antisense oligonucleotide are effective in the upstream process of mutant TTR protein production, the safety under a long‐term profound reduction of TTR remains unknown. Although not frequent, two recent large‐scale trials showed severe adverse events under gene modifier therapy could be fatal, including cardiac failure, acute kidney injury and thrombocytopenia.33, 34 This makes safety of gene modifier therapy a concern for patients with a late onset, rapid progression, or marked cardiac dysfunction, just like the condition of A97S TTR‐FAP. On the other hand, a recent meta‐analysis has demonstrated that tafamidis can preserve a better life quality for patients with a rate of adverse events similar to the placebo.41 The safety and long‐term efficacy of tafamidis provide the advantage when treating patients of TTR‐FAP, therefore, it is important to understand which genetic subtype may benefit more from the drug.
Treatment of TTR‐FAP is stepping into a new era as several therapeutic options are now available. Specific genotypes, in association with disease onset and progression, will be important concerns for tailored pharmacotherapy. Our results delineated the biophysical characters of A97S‐TTR tetramer and provided multiple lines of evidence of the modulation effects of tafamidis on its stability. These deepened our understanding of the pathogenesis of A97S TTR‐FAP. We hope this study pave a road with stone leading to precision medicine of TTR amyloidosis.
Conflict of Interest
None declared.
Supporting information
Figure S1. Recombinant transthyretin (TTR) expression, purification, and characterization.
Figure S2. The experimental and fitted binding thermograms of ITC experiments.
Table S1 . Concentrations of proteins and tafamidis used in isothermal titration calorimetry experiments.
Table S2 . (A) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to WT TTR. (B) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to A97S TTR. (C) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to V30M TTR. (D) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to L55P TTR.
Acknowledgments
This work was supported by Ministry of Science and Technology (MOST 105‐2314‐B‐075 ‐066 and 105‐2113‐M‐001 ‐021 ‐MY2); Academia Sinica (AS‐iMATE‐107‐31); National Health Research Institutes (NHRI‐EX105‐10507EC, NHRI‐EX106‐10507EC, NHRI‐EX107‐10507EC); Yen Tjing Ling Medical Foundation (CI‐106‐6); Taipei Veterans General Hospital (V104‐B015, V105C‐166, V106C‐153, V107C‐152, VGHUST106‐G7‐5‐1, VGHUST107‐G7‐1‐3); 2017 Global ASPIRE Transthyretin (TTR) Amyloidosis Competitive Research Grant Awards (WI229986). All above funding sources had no involvement in study design, collection, analysis/interpretation of data; the writing of the report; or the decision to submit the article for publication.
Funding Information
This work was supported by Ministry of Science and Technology (MOST 105‐2314‐B‐075 ‐066 and 105‐2113‐M‐001 ‐021 ‐MY2); Academia Sinica (AS‐iMATE‐107‐31); National Health Research Institutes (NHRI‐EX105‐10507EC, NHRI‐EX106‐10507EC, NHRI‐EX107‐10507EC); Yen Tjing Ling Medical Foundation (CI‐106‐6); Taipei Veterans General Hospital (V104‐B015, V105C‐166, V106C‐153, V107C‐152, VGHUST106‐G7‐5‐1, VGHUST107‐G7‐1‐3); 2017 Global ASPIRE Transthyretin (TTR) Amyloidosis Competitive Research Grant Awards (WI229986) .
Funding Statement
This work was funded by Academia Sinica, Taipei, Taiwan grant AS‐iMATE‐107‐31; National Health Research Institutes grant NHRI‐EX105‐10507EC, NHRI‐EX106‐10507EC, NHRI‐EX107‐10507EC; Taipei Veterans General Hospital, Taiwan grant V104‐B015, V105C‐166, V106C‐153, V107C‐152, VGHUST106‐G7‐5‐1, VGHUST107‐G7‐1‐3; 2017 Global ASPIRE Transthyretin (TTR) Amyloidosis Competitive Research Grant Awards grant WI229986; Ministry of Science and Technology, Taiwan grants MOST 105‐2314‐B‐075 ‐066 and 105‐2113‐M‐001 ‐021 ‐MY2; Yen Tjing Ling Medical Foundation grant CI‐106‐6.
Contributor Information
Kon‐Ping Lin, Email: kplin@vghtpe.gov.tw.
Tsyr‐Yan Yu, Email: tyyu@pub.iams.sinica.edu.tw.
References
- 1. Plante‐Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol 2014;261:1227–1233. [DOI] [PubMed] [Google Scholar]
- 2. Plante‐Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol 2018;265:976–983. [DOI] [PubMed] [Google Scholar]
- 3. Plante‐Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol 2011;10:1086–1097. [DOI] [PubMed] [Google Scholar]
- 4. Mariani LL, Lozeron P, Theaudin M, et al. Genotype‐phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol 2015;78:901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late‐onset cases from non‐endemic areas. J Neurol Neurosurg Psychiatry 2012;83:152–8. [DOI] [PubMed] [Google Scholar]
- 6. Conceicao I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late‐ and early‐onset cases in Portugal. Muscle Nerve 2007;35:116–8. [DOI] [PubMed] [Google Scholar]
- 7. Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early‐ vs late‐onset form. Arch Neurol 2002;59:1771–1776. [DOI] [PubMed] [Google Scholar]
- 8. Chao HC, Liao YC, Liu YT, et al. Clinical and genetic profiles of hereditary transthyretin amyloidosis in Taiwan. Ann Clin Transl Neurol 2019;6:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsu HC, Liao MF, Hsu JL, et al. Phenotypic expressions of hereditary Transthyretin Ala97Ser related Amyloidosis (ATTR) in Taiwanese. BMC Neurol 2017;17:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu YT, Lee YC, Yang CC, et al. Transthyretin Ala97Ser in Chinese‐Taiwanese patients with familial amyloid polyneuropathy: genetic studies and phenotype expression. J Neurol Sci 2008;267:91–99. [DOI] [PubMed] [Google Scholar]
- 11. Cendron L, Trovato A, Seno F, et al. Amyloidogenic potential of transthyretin variants: insights from structural and computational analyses. J Biol Chem 2009;284:25832–25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sousa MM, Cardoso I, Fernandes R, et al. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol 2001;159:1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams D, Cauquil C, Theaudin M, et al. Current and future treatment of amyloid neuropathies. Expert Rev Neurother 2014;14:1437–1451. [DOI] [PubMed] [Google Scholar]
- 14. Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA 2012;109:9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merkies IS. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2013;80:1444–1445. [DOI] [PubMed] [Google Scholar]
- 17. Plante‐Bordeneuve V, Carayol J, Ferreira A, et al. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet 2003;40:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barroso FA, Judge DP, Ebede B, et al. Long‐term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid 2017;24:194–204. [DOI] [PubMed] [Google Scholar]
- 19. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–1016. [DOI] [PubMed] [Google Scholar]
- 20. Le Bras A. Tafamidis: a new treatment for ATTR cardiomyopathy. Nat Rev Cardiol 2018;15:652. [DOI] [PubMed] [Google Scholar]
- 21. Groenning M, Campos RI, Fagerberg C, et al. Thermodynamic stability and denaturation kinetics of a benign natural transthyretin mutant identified in a Danish kindred. Amyloid 2011;18:35–46. [DOI] [PubMed] [Google Scholar]
- 22. Salzmann M, Pervushin K, Wider G, et al. TROSY in triple‐resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA 1998;95:13585–13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delaglio F, Grzesiek S, Vuister GW, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 24. Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early‐onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet 1992;89:353–356. [DOI] [PubMed] [Google Scholar]
- 25. Lashuel HA, Wurth C, Woo L, Kelly JW. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry 1999;38:13560–13573. [DOI] [PubMed] [Google Scholar]
- 26. Rodrigues JR, Simoes CJ, Silva CG, Brito RM. Potentially amyloidogenic conformational intermediates populate the unfolding landscape of transthyretin: insights from molecular dynamics simulations. Protein Sci 2010;19:202–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Batista AR, Gianni D, Ventosa M, et al. Gene therapy approach to FAP: in vivo influence of T119M in TTR deposition in a transgenic V30M mouse model. Gene Ther 2014;21:1041–1050. [DOI] [PubMed] [Google Scholar]
- 28. Almeida MR, Alves IL, Terazaki H, et al. Comparative studies of two transthyretin variants with protective effects on familial amyloidotic polyneuropathy: TTR R104H and TTR T119M. Biochem Biophys Res Commun. 2000;270:1024–1028. [DOI] [PubMed] [Google Scholar]
- 29. Shnyrov VL, Villar E, Zhadan GG, et al. Comparative calorimetric study of non‐amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin. Biophys Chem 2000;88:61–67. [DOI] [PubMed] [Google Scholar]
- 30. Blake CC, Geisow MJ, Oatley SJ, et al. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol 1978;121:339–356. [DOI] [PubMed] [Google Scholar]
- 31. Haupt M, Blakeley MP, Fisher SJ, et al. Binding site asymmetry in human transthyretin: insights from a joint neutron and X‐ray crystallographic analysis using perdeuterated protein. IUCrJ 2014;1(Pt 6):429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mundayat R, Stewart M, Alvir J, et al. Positive Effectiveness of tafamidis in delaying disease progression in transthyretin familial amyloid polyneuropathy up to 2 years: an analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther 2018;7:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benson MD, Waddington‐Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018;379:22–31. [DOI] [PubMed] [Google Scholar]
- 34. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018;379:11–21. [DOI] [PubMed] [Google Scholar]
- 35. Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003;299:713–716. [DOI] [PubMed] [Google Scholar]
- 36. Lim KH, Dasari AK, Hung I, et al. Structural changes associated with transthyretin misfolding and amyloid formation revealed by solution and solid‐state NMR. Biochemistry 2016;55:1941–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leach BI, Zhang X, Kelly JW, et al. NMR measurements reveal the structural basis of transthyretin destabilization by pathogenic mutations. Biochemistry 2018;57:4421–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim KH, Dasari AKR, Ma R, et al. Pathogenic mutations induce partial structural changes in the native beta‐sheet structure of transthyretin and accelerate aggregation. Biochemistry 2017;56:4808–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Groenning M, Campos RI, Hirschberg D, et al. Considerably unfolded transthyretin monomers preceed and exchange with dynamically structured amyloid protofibrils. Sci Rep 2015;5:11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Penchala SC, Connelly S, Wang Y, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy‐associated V122I transthyretin. Proc Natl Acad Sci USA 2013;110:9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao Y, Xin Y, Song Z, et al. Tafamidis, a noninvasive therapy for delaying transthyretin familial amyloid polyneuropathy: systematic review and meta‐analysis. J Clin Neurol 2019;15:108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Recombinant transthyretin (TTR) expression, purification, and characterization.
Figure S2. The experimental and fitted binding thermograms of ITC experiments.
Table S1 . Concentrations of proteins and tafamidis used in isothermal titration calorimetry experiments.
Table S2 . (A) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to WT TTR. (B) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to A97S TTR. (C) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to V30M TTR. (D) Integrated heat release (kcal/mol) vs. molar ratio of tafamidis added to L55P TTR.