

Abstract
Background: During the previous decade a new class of benzamide-based inhibitors of 2-trans enoyl-acyl carrier protein reductase (InhA) of Mycobacterium tuberculosis (Mt) with unusual binding mode have emerged. Here we report in silico design and evaluation of novel benzamide InhA-Mt inhibitors with favorable predicted pharmacokinetic profiles. Methods: By using in situ modifications of the crystal structure of N-benzyl-4-((heteroaryl)methyl) benzamide (BHMB)-InhA complex (PDB entry 4QXM), 3D models of InhA-BHMBx complexes were prepared for a training set of 19 BHMBs with experimentally determined inhibitory potencies (half-maximal inhibitory concentrations IC50exp). In the search for active conformation of the BHMB1-19, linear QSAR model was prepared, which correlated computed gas phase enthalpies of formation (∆∆HMM) of InhA-BHMBx complexes with the IC50exp. Further, taking into account the solvent effect and entropy changes upon ligand, binding resulted in a superior QSAR model correlating computed complexation Gibbs free energies (∆∆Gcom). The successive pharmacophore model (PH4) generated from the active conformations of BHMBs served as a virtual screening tool of novel analogs included in a virtual combinatorial library (VCL) of compounds containing benzamide scaffolds. The VCL filtered by Lipinski’s rule-of-five was screened by the PH4 model to identify new BHMB analogs. Results: Gas phase QSAR model: −log10(IC50exp) = pIC50exp = −0.2465 × ∆∆HMM + 7.95503, R2 = 0.94; superior aqueous phase QSAR model: pIC50exp = −0.2370 × ∆∆Gcom + 7.8783, R2 = 0.97 and PH4 pharmacophore model: = 1.0013 × p − 0.0085, R2 = 0.95. The VCL of more than 114 thousand BHMBs was filtered down to 73,565 analogs Lipinski’s rule. The five-point PH4 screening retained 90 new and potent BHMBs with predicted inhibitory potencies IC50pre up to 65 times lower than that of BHMB1 (IC50exp = 20 nM). Predicted pharmacokinetic profile of the new analogs showed enhanced cell membrane permeability and high human oral absorption compared to current anti-tuberculotics. Conclusions: Combined use of QSAR models that considered binding of the BHMBs to InhA, pharmacophore model, and ADME properties helped to recognize bound active conformation of the benzamide inhibitors, permitted in silico screening of VCL of compounds sharing benzamide scaffold and identification of new analogs with predicted high inhibitory potencies and favorable pharmacokinetic profiles.
Keywords: Tuberculosis, N-benzyl-4-((heteroaryl)methyl)benzamides, 2-trans enoyl-acyl carrier protein reductase, molecular modeling, QSAR models, pharmacophore, combinatorial library, in silico screening, ADME properties prediction
1. Introduction
“United to end tuberculosis: an urgent global response to a global epidemic” is the title of the declaration adopted by the UN General Assembly on the fight against tuberculosis (TB), which reaffirmed the commitment to end the tuberculosis epidemic globally by 2030 [1]. The General Assembly acknowledged that the Millennium Development Goals [2] and associated strategies helped to reverse the trend of the tuberculosis epidemic and until 2016 reduced the tuberculosis mortality by 37%. Nonetheless, the current WHO year report [3,4] revealed alarming statistics showing that throughout the world about 3 TB deaths occurred and about 19 persons developed TB every minute. On the other hand, drug development against Mycobacterium tuberculosis (Mt), despite the increased occurrence of multiple drug resistant (MDR-TB) and extensively drug resistant (XDR-TB) strains, has reached a noticeable progress on inhibitor design against 2-trans enoyl-acyl carrier protein reductase (InhA), the most frequently addressed validated mycobacterial drug target. The division of the InhA substrate binding site into three structural subsites (site I, catalytic; site II, hydrophobic; site III, hydrophilic) and the determination of the catalytic residue Tyr158 conformation (‘in’ or ‘out’), have paved the way for the improvement in structure-based design and development of novel InhA inhibitors [5]. Another important observation is connected with the presence of a previously unnoticed interaction pocket formed by the side chains of Phe41 and Arg43 at the InhA active site displaying Tyr158 ‘out’ conformation [6]. Promising drug candidates, which target the isoniazid-resistant Mt, are direct InhA inhibitors that do not require KatG (Mycobacterium tuberculosis catalase-peroxidase) activation [7]. Recent useful structural information involving key binding site residues identified by site-directed mutations of the InhA gene revealed that these residues (except Ser94 and Tyr158) interact with the ligand mostly through hydrophobic contacts [8]. The long list of known InhA inhibitors may be divided into, on the one hand, class 1 scaffolds: triclosan derivatives (TCL) [9], diphenyl ether [10,11], pyrrolidine carboxamide (PCAM) [12], and aryl amide derivatives [13] with Tyr158 ‘in’ conformation and typical stacking interaction with the Phe97 residue. On the other hand, class 2 scaffolds include methyl-thiazole derivatives [5], pyrazoles [14], benzamides [15] with Tyr158 ‘out’ conformation and interaction with the Phe41 and Arg43 pocket instead of the stacking with Phe97. The 3D-QSAR pharmacophores (PH4) for InhA inhibition are available for class 1 TCL and PCAM inhibitors only [16,17] but not for the class 2 compounds. Figure 1A,B show various numbers of hydrophobic features (HYD) for the PH4 of TCL and PCAM. The third HYD feature of TCL PH4 suggests that a bulky group can fill large hydrophobic pocket (LHP, site II) delimited by residues Met155, Pro193, Ile215, Leu217, Leu218, and Trp222 as a major structural requirement for efficient InhA inhibition [18]. Indeed, the best substitutions on candidates with the Triclosan scaffold direct a nonpolar group containing an ethyl linker capped by phenyl (IC50exp = 21 nM) or pentyl group (IC50exp = 11 nM with removal of all Cl atoms) to this LHP. The preliminary ‘interaction generation’ analysis of the InhA active site with no ligand bound (PDB: 4DRE, Figure 1C) revealed at least four HYD features, two of them located in the LHP. Structure–activity relationships involving interactions of 3D pharmacophore have been previously reported for HIV-1 inhibition, genetic disorders treatment, or proton pump inhibition [19,20,21].
Figure 1.

(A) 3D-QSAR pharmacophore model (PH4) for triclosan (TCL) derivatives displaying 3 HYD (cyan) features and the mapping of the most active analog synthesized (IC50 = 21 nM [16], PDB: 3FNH [21], five key interactions with InhA: HB—Tyr158, π–π—NAD and hydrophobic contacts). (B) PH4 for pyrrolidine carboxamide (PCAM) derivatives displaying 2 HYD (light blue) and the mapping of the most active derivative synthesized (IC50 = 390 nM [17], PDB: 4U0J [12], main interactions with InhA: HB—Tyr158, HB—NAD). (C) PH4 for the active site of InhA, depicted in ribbon, (PDB: 4DRE [22]) with one acceptor in green color, one donor in purple color, and 4 HYD features (cyan), for clarity the acceptor and donor spheres were removed. The LHP is enclosed by 2 residues in yellow color and labeled in green surrounding two HYDs.
The main objective of this work was to design novel potent N-benzyl-4-((heteroaryl)methyl) benzamides (BHMBs) based on a series of 19 (training set) plus 6 (validation set) nanomolar inhibitors with observed inhibitory potencies as low as IC50exp = 20 nM [23]. Starting with in situ modification of the crystal structure of InhA-BHMB2 complex (PDB: 4QXM), we have elaborated a QSAR model which correlated Gibbs free energies of InhA-BHMBx complex formation with the potencies IC50exp and determined the active conformation of BHMBs bound at the active site of InhA of Mt (MM-PB complexation approach). Based on this active conformation we have formulated 3D QSAR pharmacophore of InhA inhibition (PH4). Large virtual library of compounds sharing the BHMB scaffold has been generated and in silico screened with the PH4. The screening yielded virtual hits that exhibited predicted inhibitory potencies IC50pre more than 60 times lower than the most active training set compound BHMB1. Several of the identified putative inhibitors displayed favorable ADME profiles. Moreover, a series of drugs currently used in clinical practice, which include the benzamide scaffold in their molecular structure, were assessed with our new PH4 for InhA inhibition. Top five approved drugs identified by the PH4 screening exhibited predicted potencies IC50pre ranging from 1.7 to 60 nM.
2. Results
2.1. Training and Validation Sets
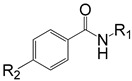
The training set of 19 BHMBs and validation set of another 6 analogs (Table 1) were selected from a homogeneous series of InhA inhibitors with known experimentally determined inhibitory activities originating from a single laboratory [23]. The whole series was obtained by variations at two positions R1 and R2 of the phenyl ring and amide group as shown in Table 1. The experimental half-maximal inhibitory concentrations (20 ≤ IC50exp ≤ 5930 nM) [23] cover a sufficiently wide concentration range for building of a reliable QSAR model. The ratio between the sizes of training and validation sets remains a critical point of correct classification but is limited by the count of the set of homologous compounds available from the literature [24].
Table 1.
Set (BHBM1-19) and validation set (BHBV1-6) of InhA inhibitors [23] used in the preparation of QSAR models of inhibitor binding. The R1 and R2 groups are numbered in the first part of the Table as #R ≡ group index.

| #R | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| R group |

|

|

|

|

|

|

|

|
| #R | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| R group |

|

|

|
|
|

|
|

|
| #R | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| R group |

|

|

|

|
|

|

|

|
| #R | 25 | 26 | ||||||
| R group |

|

|
||||||
| Training Set | BHMB1 | BHMB2 | BHMB3 | BHMB4 | BHMB5 | BHMB6 | BHMB7 | BHMB8 |
| #R1–#R2 | 8–17 | 8–19 | 8–14 | 8–15 | 12–19 | 13–19 | 8–26 | 18–19 |
| IC50exp (nM) | 20 | 50 | 50 | 60 | 80 | 90 | 90 | 120 |
| Training Set | BHMB9 | BHMB10 | BHMB11 | BHMB12 | BHMB13 | BHMB14 | BHMB15 | BHMB16 |
| #R1–#R2 | 10–19 | 8–16 | 9–19 | 8–22 | 3–19 | 8–25 | 6–19 | 5–19 |
| IC50exp (nM) | 250 | 260 | 350 | 500 | 540 | 1400 | 1580 | 1580 |
| Training Set | BHMB17 | BHMB18 | BHMB19 | |||||
| #R1–#R2 | 4–19 | 7–19 | 1–19 | |||||
| IC50exp (nM) | 2510 | 3100 | 5930 | |||||
| Validation Set | BHMV1 | BHMV2 | BHMV3 | BHMV4 | BHMV5 | BHMV6 | ||
| #R1–#R2 | 8–24 | 21–19 | 8–20 | 2–19 | 11–19 | 8–23 | ||
| IC50exp (nM) | 60 | 90 | 320 | 1260 | 1580 | 3250 |
2.2. QSAR Model
2.2.1. One Descriptor QSAR Models
Each of the 19 training sets (TS) and 6 validation sets (VS) InhA-BHMBx complexes (Table 1), was prepared by in situ modification of the refined template crystal structure (PDB entry code 4QXM [23]) of the complex InhA-BHMB2 as described in the Methods section. Further, the relative Gibbs free energy of the InhA-BHMBx complex formation (∆∆Gcom) was computed for each of the 25 optimized enzyme–inhibitor complexes. Table 2 lists computed values of ∆∆Gcom and its components as defined in Equation (7), for the TS and VS of benzamides [23]. The QSAR model explained variation in the BHMBs experimental inhibitory potencies (pIC50exp = −log10(IC50exp) [23]) by correlating it with computed GFE ∆∆Gcom through linear regression (Equation (8), Table 2). In addition, significant correlation obtained in this QSAR relationship permitted to determine the active bound conformation of the BHMBs at the InhA binding site and enabled definition of the PH4 pharmacophore. In search for a better insight into the binding affinity of BHBMs towards MtInhA, we have analyzed the enthalpy of complexation in gas phase ∆∆HMM by correlating it with the pIC50exp. The validity of this linear correlation (for statistical data of the regression see Table 3, Equation A) allowed assessment of the significance of inhibitor-enzyme interactions (∆∆HMM) when solvent effect and loss of entropy of the inhibitor upon binding to the enzyme were neglected. This correlation explained about 94% of the pIC50exp data variation and underlined the role of the enthalpic contribution to the binding affinity of the ligand. Similarly, the more advanced descriptor, namely the GFE of the InhA-BHMBx complex formation including all components: ∆∆HMM, ∆∆TSvib and ∆∆Gsol, has been assessed (for statistical data see Table 3, Equation B). Relatively high values of the regression coefficient R2, leave-one-out cross-validated regression coefficient R2xv and Fischer F-test of the correlation suggest strong relationship between the 3D model of inhibitor binding and the observed inhibitory potencies of the BHBMs [23]. Therefore, structural information derived from the 3D models of InhA-BHMBx complexes can be expected to lead to reliable prediction of InhA inhibitory potencies for new BHMBs analogs based on the QSAR model B, Table 3.
Table 2.
Gibbs free energy (binding affinity) and its components for the training set of InhA inhibitors BHMB1-19 and validation set inhibitors BHMV1-6 [23].
| Training Set a | Mwb | ∆∆HMMc | ∆∆Gsold | ΔΔTSvibe | ∆∆Gcomf | IC50exp g |
| [g⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | [nM] | |
| BHMB1 | 385 | 0 | 0 | 0 | 0 | 20 |
| BHMB2 | 372 | 3.14 | −1.75 | −0.51 | 1.90 | 50 |
| BHMB3 | 369 | 2.99 | −0.56 | −0.33 | 2.76 | 50 |
| BHMB4 | 387 | 3.01 | −3.09 | −2.50 | 2.41 | 60 |
| BHMB5 | 487 | 4.31 | −5.38 | −3.96 | 2.89 | 80 |
| BHMB6 | 488 | 4.70 | −1.02 | −0.30 | 3.98 | 90 |
| BHMB7 | 377 | 4.24 | −3.22 | −3.45 | 4.47 | 90 |
| BHMB8 | 391 | 4.66 | −0.01 | −0.46 | 5.11 | 120 |
| BHMB9 | 405 | 4.77 | 0.64 | 0.18 | 5.23 | 250 |
| BHMB10 | 401 | 4.63 | 0.33 | −0.62 | 5.58 | 260 |
| BHMB11 | 369 | 6.31 | 1.59 | 1.58 | 6.32 | 350 |
| BHMB12 | 358 | 7.61 | −1.80 | −1.63 | 7.45 | 500 |
| BHMB13 | 398 | 7.06 | 0.29 | 0.65 | 6.70 | 540 |
| BHMB14 | 389 | 7.62 | −1.31 | −2.20 | 8.51 | 1400 |
| BHMB15 | 398 | 8.81 | 0.77 | 1.00 | 8.57 | 1580 |
| BHMB16 | 354 | 8.26 | 1.50 | 1.04 | 8.72 | 1580 |
| BHMB17 | 364 | 8.54 | 2.95 | 1.94 | 9.55 | 2510 |
| BHMB18 | 337 | 9.26 | 1.80 | 2.01 | 9.05 | 3100 |
| BHMB19 | 319 | 11.72 | 1.81 | 2.84 | 10.69 | 5930 |
| Validation Set | Mwb | ∆∆HMMc | ∆∆Gsold | ΔΔTSvibe | ∆∆Gcomf | pIC50pre/pIC50exp h |
| [g⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | [kcal⋅mol−1] | ||
| BHMV1 | 375 | 2.41 | −2.49 | −2.95 | 2.87 | 0.99 |
| BHMV2 | 416 | 4.58 | 0.32 | 1.45 | 3.44 | 1.00 |
| BHMV3 | 386 | 4.61 | 4.53 | 2.47 | 6.67 | 0.97 |
| BHMV4 | 354 | 8.63 | 1.49 | 2.12 | 8.01 | 1.01 |
| BHMV5 | 377 | 8.47 | 0.47 | 0.77 | 8.17 | 1.02 |
| BHMV6 | 361 | 8.20 | 0.48 | −2.62 | 11.29 | 0.95 |
a for the chemical structures of the training set of inhibitors see Table 1; b Mw is the molar mass of inhibitors; c ΔΔHMM is the relative enthalpic contribution to the GFE change related to E-I complex formation derived by MM; ΔΔHMM ≈ [EMM{E-Ix} − EMM{Ix}] − [EMM{E-Iref} − EMM{Iref}], Iref is the reference inhibitor BHMB1; d ΔΔGsol is the relative solvent effect contribution to the GFE change of E-I complex formation: ΔΔGsol = [Gsol{E-Ix} − Gsol{Ix}] − [Gsol{E-Iref} − Gsol{Iref}]; e −ΔΔTSvib is the relative entropic contribution of inhibitor Ix to the GFE of E-Ix complex formation: ΔΔTSvib = [TSvib{Ix}E − TSvib{Ix}] − [TSvib{Iref}E − TSvib{Iref}]; f ΔΔGcom is the overall relative GFE change of E-Ix complex formation: ΔΔGcom ≈ ΔΔHMM + ΔΔGsol − ΔΔTSvib; g IC50exp is the experimental half-maximal inhibition concentration of InhA obtained from ref. [23]; h ratio of predicted and experimental half-maximal inhibition concentrations pIC50pre/pIC50exp (pIC50pre = −log10IC50pre) was predicted from computed ΔΔGcom using the regression equation for InhA shown in Table 3, B.
Table 3.
Analysis of computed binding affinities ∆∆Gcom, its enthalpic component ∆∆HMM, and experimental half-maximal inhibitory concentrations pIC50exp = −log10IC50exp of BHMBs towards MtInhA [23].
| Statistical Data of Linear Regression | (A) | (B) |
|---|---|---|
| pIC50exp = −0.2465 × ∆∆HMM + 7.9554 (A) | ||
| pIC50exp = −0.2370 × ∆∆Gcom + 7.8783 (B) | ||
| Number of compounds n | 19 | 19 |
| Squared correlation coefficient of regression R2 | 0.94 | 0.97 |
| LOO cross-validated squared correlation coefficient R2xv | 0.92 | 0.95 |
| Standard error of regression σ | 0.178 | 0.135 |
| Statistical significance of regression, Fisher F-test | 274.92 | 493.24 |
| Level of statistical significance α | >95 % | >95 % |
| Range of activities IC50exp [nM] | 20–5930 | |
The statistical data confirmed validity of the correlation Equations (A) and (B) plotted on Figure 2. The ratio pIC50pre/pIC50exp ≅ 1 (the pIC50pre values were estimated using correlation Equation B, Table 3) calculated for the validation set BHMV1-6 documents the substantial predictive power of the complexation QSAR model from Table 2. Thus, the regression Equation B (Table 3) and computed ∆∆Gcom GFEs can be used for prediction of inhibitory potencies IC50pre against MtInhA for novel BHMB analogs, provided they share the same binding mode as the training set benzamides BHMB1-19.
Figure 2.

(Left) plot of correlation equation between pIC50exp and relative enthalpic contribution to the GFE (Equation (6)) ∆∆HMM [kcal⋅mol−1]. (Right) similar plot for relative complexation Gibbs free energies of the InhA-BHMBx complex formation ∆∆Gcom [kcal⋅mol−1] of the training set [23]. The validation set data points are shown in red color.
2.2.2. Binding Mode of BHMBs
Structural information enzyme–inhibitor interactions retrieved from the crystal structure of InhA-BHMB1 complex [23] showed that BHMBs are InhA class 2 specific inhibitors. As indicated in Figure 3, in the catalytic site I residue Tyr158 adopted the ‘out’ conformation [5]; the pyridine moiety of the ligand (referred to as the “warhead” [6]) is in π–alkyl contact with Tyr158 and nicotinamide ring and π–donor with the hydroxyl group adjacent to the ribose of NADH. The central benzene ring of the inhibitor forms a recently observed π–π stacking interaction with Phe97 [6]. In the hydrophobic site II, the dihalide-substituted benzene ring sits in the hydrophobic substrate cavity, surrounded by side chains of predominantly nonpolar residues: Met103, Met98, Ile202, Leu207, Gly104, Phe149, Ala157, Tyr158, and Ile215 [6,23]. In the hydrophilic site III, the inhibitor makes hydrogen bonds with the sidechain of catalytic Met98, a key interaction reported recently [6]. These specific contacts were observed for the novel class of InhA inhibitors except the interaction with the “Phe41–Arg43” pocket previously unreported before Soutter et al. [6] and which will be discussed later.
Figure 3.

(Left) 2D schematic interaction diagram of the most potent inhibitor BHMB1 [23] at the active site of InhA of Mt. (Right) 3D structure of the InhA active site with bound inhibitor BHMB1. (Bottom) Hydrophobic surface of the active site of InhA showing conventional hydrogen bonds (green) and alkyl group hydrophobic contacts (pink). Surface coloring legend: red = hydrophobic, blue = hydrophilic and white = intermediate.
2.3. Interaction Energy
Other key structural information was provided by the interaction energy (IE, ΔEint) diagram obtained for each training set inhibitor. IE breakdown to contributions from InhA active site residue is helpful for the choice of relevant R1-groups (site I) and R2-groups (site II) which could improve the binding affinity of BHMB analogs to the MtInhA and subsequently enhance the inhibitory potency. A comparative analysis of computed IE for training set BHMBs (Figure 4) divided into three classes (highest, moderate, and lowest activity) has been carried out to identify the residues for which the contribution to binding affinity could be increased. However, the comparative analysis showed about the same level of IE contributions from active site residues for all three classes of inhibitors. Therefore, no suggestions of suitable substitutions able to improve the binding affinity as we previously reported for thymine-like inhibitors of Mt thymidine monophosphate kinase design could be proposed [25]. Since specific substitutions could not be proposed, we have adopted a combinatorial approach to novel BHMB analogs design and in silico screened a virtual library of 114921 BHMB analogs with help of the PH4 pharmacophore of InhA inhibition derived from the complexation QSAR model. As we can see from the IE analysis (Figure 4), the TS and VS benzamides [23] do not show significant interaction energies with the “Phe41–Arg43” pocket.
Figure 4.

Mechanics intermolecular interaction energy Eint breakdown to residue contributions in [kcal.mol−1]: (A) the most active inhibitors BHMB1-7, (B) moderately active inhibitors BHMB8-13, (C) less active inhibitors BHMB14-19, Table 2 [23].
2.4. 3D-QSAR Pharmacophore Model
2.4.1. InhA Active Site Pharmacophore
The interaction generation protocol in Discovery Studio molecular modeling program [26] provides the pharmacophore features of the active site of a protein. InhA predominantly displays hydrophobic features at the active site (Figure 1C) as confirmed by previously reported works [16,17], the larger being the site II hydrophobic pocket that accommodates the substrate long alkyl chains. InhA substrate-competitive inhibitors design often exploits the pocket flexibility because of the high mobility of the Tyr158, Phe149 side chains and the substrate-binding loop (Thr196–Gly208) [27].
2.4.2. Generation and Validation of 3D-QSAR Pharmacophore
InhA inhibition 3D-QSAR pharmacophore was generated from the active conformation of 19 TS BHMB1-19 and evaluated by 6 VS BHMV1-6 covering a large range of experimental activity (20–5930 nM) spanning more than two orders of magnitude. The generation process is divided into three main steps: (i) the constructive step, (ii) the subtractive step, and (iii) the optimization step [26] as described earlier [17]. During the constructive phase, BHMB1 alone was retained as the lead (since only the activity of BHMB1 fulfilled the threshold criterion, IC50exp ≤ 2 × 20 nM), and used to generate the starting PH4 features. In the subtractive phase, compounds for which IC50exp > 20 × 103.5 nM = 63246 nM were considered inactive. Accordingly, none of the training set BHMBx was inactive and no starting PH4 features were removed. Finally, during the optimization phase, the score of the pharmacophoric hypotheses was improved. Hypotheses were scored according to errors in activity estimates from regression and complexity via a simulated annealing approach. At the end of the optimization, the top scoring 10 unique pharmacophore hypotheses were kept, all displaying five-point features. The cost values, correlation coefficients, root-mean square deviation (RMSD) values, the pharmacophore features, and the max-fit value of the top 10 ranked hypotheses (Hypo1−Hypo10) are listed in Table 4. They were selected based on significant statistical parameters, such as high correlation coefficient, low total cost, and low RMSD.
Table 4.
Parameters of 10 generated PH4 pharmacophoric hypotheses for InhA inhibitor after Cat-Scramble validation procedure (49 scrambled runs for each hypothesis at the selected level of confidence of 98%).
| Hypothesis | RMSD a | R2 b | Total Costs c | Costs Difference d | Closest Random e |
|---|---|---|---|---|---|
| Hypo1 | 1.610 | 0.97 | 70.1 | 458.1 | 147.15 |
| Hypo2 | 1.973 | 0.96 | 82.6 | 445.6 | 155.68 |
| Hypo3 | 2.281 | 0.95 | 94.8 | 433.4 | 183.64 |
| Hypo4 | 2.673 | 0.93 | 114.3 | 413.9 | 201.42 |
| Hypo5 | 2.751 | 0.92 | 118.9 | 409.3 | 205.69 |
| Hypo6 | 2.916 | 0.91 | 126.3 | 401.9 | 213.65 |
| Hypo7 | 3.396 | 0.88 | 155.5 | 372.8 | 214.63 |
| Hypo8 | 3.586 | 0.87 | 169.1 | 359.2 | 241.58 |
| Hypo9 | 3.709 | 0.86 | 177.2 | 351.1 | 247.34 |
| Hypo10 | 3.809 | 0.85 | 184.7 | 343.5 | 260.41 |
a root mean square deviation; b squared correlation coefficient; c overall cost parameter of the PH4 pharmacophore; d cost difference between Null cost and hypothesis total cost; e lowest cost from 49 scrambled runs at a selected level of confidence of 98%. The Fixed Cost = 45.4 with RMSD = 0, the Null Cost = 528.2 with RMSD = 7.215 and the Configuration cost = 10.63.
The generated pharmacophore models were then assessed for their reliability based on the calculated cost parameters ranging from 70.1 (Hypo1) to 184.7 (Hypo10). The relatively small gap between the highest and lowest cost parameter corresponds well with the homogeneity of the generated hypotheses and consistency of the TS of BHMBx. For this PH4 model, the fixed cost (45.4) is lower than the null cost (528.2) by a difference ∆ = 482.8. This difference is a major quality indicator of the PH4 predictability (∆ > 70 corresponds to an excellent chance or a probability higher than 90% that the model represents a true correlation [26]). To be statistically significant, a hypothesis has to be as close as possible to the fixed cost and as far as possible from the null cost. For the set of 10 hypotheses, the difference ∆ ≥ 343.5, which attests to the high quality of the pharmacophore model. The standard indicators such as the RMSD between the hypotheses ranged from 1.610 to 3.809, and the squared correlation coefficient (R2) falls to an interval from 0.97 to 0.84. The first PH4 hypothesis with the closest cost (70.1) to the fixed one (45.4) and best RMSD and R2 was retained for further analysis. The statistical data for the set of hypotheses (costs, RMSD, R2) are listed in Table 4. The configuration cost (10.63 for all hypotheses) far below 17 confirms this pharmacophore as a reasonable one.
The link between the 98% significance and the number 49 scrambled runs of each hypothesis is based on the formula S = [1 − (1 + X)/Y] × 100, with X the total number of hypotheses having a total cost lower than the original hypothesis (Hypo 1) and Y the total number of HypoGen runs (initial + random runs): X = 0 and Y = (1 + 49), hence 98% = {1 − [(1 + 0)/(49 + 1)]} × 100.
The evaluation of Hypo 1 was performed first through Fischer’s randomization cross-validation test. The CatScramble program was used to randomize the experimental activities of the training set. At 98% confidence level, each of the 49 scramble runs created ten valid hypotheses, using the same features and parameters as in the generation of the original 10 pharmacophore hypotheses. Among them, the cost value of Hypo1 is the lowest compared with those of the 49 randomly generated hypotheses, as we can see in Table 4 where the lowest cost of the 49 random runs is listed for each original hypothesis, and none of them was as predictive as the original hypotheses generated shown in Table 4. Thus, there is a 98% probability that the best selected hypothesis Hypo1 represents a pharmacophore model for inhibitory activity of BHMBs with a similar level of predictive power as the complexation QSAR model, which relies on the BHMBx active conformation from 3D structures of the InhA-BHMBx complexes and computed GFE of enzyme–inhibitor binding ΔΔGcom. Another evaluation of Hypo 1 is the mapping of the best active training set BHMB1 (Figure 5) displaying the geometry of the Hypo1 pharmacophore of InhA inhibition. The regression equation for pIC50exp vs. pIC50pre estimated from Hypo1: pIC50exp = 1.0013 × pIC50pre − 0.0085 (n = 19, R2 = 0.95, Rxv2 = 0.93, F-test = 324.25, σ = 0.165, α > 98 %) is also plotted on Figure 5.
Figure 5.


Features (A) coordinates of centers, (B) angles between centers of pharmacophoric features, (C) distances between centers, (D) mapping of pharmacophore of InhA inhibitor with the most potent molecule BHMB1. The R1 position in the BHMBs of TS compounds is occupied by benzene ring (except BHMB8), therefore the second HYD expected from the InhA active site PH4 (Figure 1C) is replaced by Ar feature in BHMBs PH4. Feature legend: HYDA = Hydrophobic Aliphatic (blue), HYD = Hydrophobic (cyan), Ar = Ring aromatic (orange), HBA = Hydrogen bond Acceptor (green). (E) correlation plot of experimental vs. predicted inhibitory activity (open circles correspond to TS, red dots to VS).
We can carry out computational design and selection of new BHMB analogs with elevated inhibitory potencies against MtInhA, based on a strategy using the noticeable presence of the hydrophobic features included in the best pharmacophore model at the position of R2 coupled with mapping of R1 to the aromatic ring feature and the appropriate ring substitution to the hydrophobic aliphatic feature in Hypo1 (Figure 5).
2.5. Virtual Screnning
In silico screening of a virtual (combinatorial) library can lead to hit identification as it was shown in our previous works on inhibitors design [17,28,29].
2.5.1. Virtual Library
An initial virtual library (VL) was generated by substitutions at positions for R1 and R2 (see Table 5) on the benzamide scaffold. During the virtual library enumeration, all 339 R-groups listed in Table 5 were attached to in positions R1 and R2 of the BHMB scaffold to form a combinatorial library of the size: R1 × R2 = 339 × 339 = 114,921 analogs. This initial diversity library was generated from building blocks (chemicals) listed in the databases of available chemicals [30]. To design a more focused library of a reduced size and increased content of drug-like molecules, we have introduced a set of filters and penalties such as the Lipinski rule-of-five [31], which helped to select a smaller number of suitable BHMBs that could be submitted to in silico screening. This focusing has reduced the size of the initial library to 73,565 analogs, 64% of its original number size.
Table 5.
R1- and R2-groups (fragments, building blocks, substituents) used in the design of the initial diversity virtual combinatorial library of benzamides.

| R-groups a | |||||
|---|---|---|---|---|---|
| 1 | Me | 2 | 1-ClMe | 3 | Et |
| 4 | 1-F-Et | 5 | propyl | 6 | 1-Brpropyl |
| 7 | Me-thiol | 8 | 1-BrMe | 9 | 1-ClEt |
| 10 | 1-F-propyl | 11 | butyl | 12 | 1-F-Me |
| 13 | 1-BrEt | 14 | 1-Clpropyl | 15 | 1-F-butyl |
| 16 | 1-Clbutyl | 17 | 1-F-pentyl | 18 | hexyl |
| 19 | 1-Brhexyl | 20 | isopentyl | 21 | 6,6-diMeheptyl |
| 22 | 6-diMe-heptyl | 23 | 3,3-diMe-butyl | 24 | 3-Mepentyl |
| 25 | 6-Meoctyl | 26 | 3-Etpentyl | 27 | Me-C3H5 |
| 28 | Butyl-C3H5 | 29 | Me-C4H7 | 30 | Butyl-C4H7 |
| 31 | Me-C5H9 | 32 | Butyl-C5H9 | 33 | Me-C6H11 |
| 34 | Butyl-C6H11 | 35 | cycloprop-2-en-1-yl | 36 | Thiophen-2-yl |
| 37 | Thiophen-3-yl | 38 | Furan-2-yl | 39 | 5-Me-thiophen-2-yl |
| 40 | 3,4,5-triMe-thiophen-2-yl | 41 | Thiophen-2-ylMe | 42 | furan-3-ylMe |
| 43 | 3-Me-thiophen-2-yl | 44 | 3,5-diMe-thiophen-2-yl | 45 | Furan-2-ylMe |
| 46 | 2-(thiophen-2-yl)Et | 47 | 4-Me-thiophen-2-yl | 48 | 4,5-diMe-thiophen-2-yl |
| 49 | 2-(furan-2-yl)Et | 50 | Thiophen-3-ylMe | 51 | Ph |
| 52 | 4-ClPh | 53 | 4-F-Ph | 54 | 4-BrPh |
| 55 | p-tolyl | 56 | 3-F-Ph | 57 | 3-ClPh |
| 58 | 3-BrPh | 59 | m-tolyl | 60 | 2-F-Ph |
| 61 | 2-ClPh | 62 | 2-BrPh | 63 | o-tolyl |
| 64 | 4-OHPh | 65 | 4-MeOPh | 66 | 4-OH-Bn |
| 67 | 4-MeO-Bn | 68 | -Bn | 69 | 4-F-Bn |
| 70 | 4-Cl-Bn | 71 | 4-Br-Bn | 72 | 4-Me-Bn |
| 73 | 3,5-diMe-Bn | 74 | 4-((1H-imidazol-2-yl)Me)-Bn | 75 | aminoMe |
| 76 | diClMe | 77 | 2-(1H-imidazol-2-yl)Et | 78 | 2-Clpropyl |
| 79 | 3-Br-2-(thiazol-2-yl)propyl | 80 | (Furan-3-ylMe)thio | 81 | diBrMe |
| 82 | 2-amino-2-ClEt | 83 | 3,3-diBr-3-F-propyl | 84 | 4-(pyridin-3-yl)butyl |
| 85 | F-Me-Cl | 86 | 2-(1,3,4-thiadiazol-2-yl)Et | 87 | 2-Br-2-(1,3,4-thiadiazol-2-yl)Et |
| 88 | 1-Br-3-Clpropyl | 89 | 4-(1H-imidazol-2-yl)butyl | 90 | 4-Cl-3-OHbutyl |
| 91 | 3-((F-Me)amino)propyl | 92 | 4-OHhexyl | 93 | 5-amino-6-Brhexyl |
| 94 | 4-iodo-3-Mebutyl | 95 | 3-(neopentylamino)propyl | 96 | 5-(Meamino)pentyl |
| 97 | 2-Me-3,3-diMebutyl | 98 | 4-Cl-3-Mepentyl | 99 | 6-aminooctyl |
| 100 | 6-amino-3-Et-4-Meoctyl | 101 | cycloprop-2-en-1-ylMe | 102 | (4-Mecyclohexyl)Me |
| 103 | Me-cyclohexyl | 104 | Me-cyclopentyl | 105 | -Bn |
| 106 | 4-Me-Bn | 107 | 4-MeO-Bn | 108 | 4-Et-Bn |
| 109 | 4-Cl-Bn | 110 | 4-F-Bn | 111 | 4-(F-Me)-Bn |
| 112 | 3,4-diF-Bn | 113 | 3,5-diF-Bn | 114 | 2-Cl-4-F-Bn |
| 115 | 4-Cl-2-F-Bn | 116 | 4-Br-5-Et-2-F-Bn | 117 | 2-F-4-Me-Bn |
| 118 | 2,6-diF-Bn | 119 | 2,4-diF-Bn | 120 | 2,4,6-triF-Bn |
| 121 | 3-(CF3)pyridin-2-yl | 122 | 2-(CF3)pyridin-3-yl | 123 | 3-(CF3)pyridin-4-yl |
| 124 | 3-Br-pyridin-2-yl | 125 | 4-MeO-3-(CF3)pyridin-2-yl | 126 | 4-(CF3)pyridin-3-yl |
| 127 | 2-(CF3)Ph | 128 | Ph | 129 | EtO |
| 130 | 2-(6-Et-3,6-dihydro-2H-pyran-2-yl)EtO | 131 | 4-(5-F-1H-pyrazol-1-yl)Ph | 132 | 4-(4-F-1H-pyrazol-1-yl)Ph |
| 133 | 4-(3-F-1H-pyrazol-1-yl)Ph | 134 | 4-(3,4-diF-1H-pyrazol-1-yl)Ph | 135 | 4-(3,4,5-triF-1H-pyrazol-1-yl)Ph |
| 136 | 4-(4,5-diF-1H-pyrazol-1-yl)Ph | 137 | 4-(3,5-diF-1H-pyrazol-1-yl)Ph | 138 | 4-(3-Br-1H-pyrazol-1-yl)Ph |
| 139 | 4-(4-Br-1H-pyrazol-1-yl)Ph | 140 | 4-(5-Br-1H-pyrazol-1-yl)Ph | 141 | 4-(4,5-diBr-1H-pyrazol-1-yl)Ph |
| 142 | 4-(3,4-diBr-1H-pyrazol-1-yl)Ph | 143 | 4-(3,5-diBr-1H-pyrazol-1-yl)Ph | 144 | 4-(3,4,5-triBr-1H-pyrazol-1-yl)Ph |
| 145 | 4-(5-Me-1H-pyrazol-1-yl)Ph | 146 | 4-(4-Me-1H-pyrazol-1-yl)Ph | 147 | 4-(3-Me-1H-pyrazol-1-yl)Ph |
| 148 | 4-(3,4-diMe-1H-pyrazol-1-yl)Ph | 149 | 4-(4,5-diMe-1H-pyrazol-1-yl)Ph | 150 | 4-(3,5-diMe-1H-pyrazol-1-yl)Ph |
| 151 | 4-(3,4,5-triMe-1H-pyrazol-1-yl)Ph | 152 | 4-(3-iodo-1H-pyrazol-1-yl)Ph | 153 | 4-(4-iodo-1H-pyrazol-1-yl)Ph |
| 154 | 4-(5-iodo-1H-pyrazol-1-yl)Ph | 155 | 4-(4,5-diI-1H-pyrazol-1-yl)Ph | 156 | 4-(3,4-diI-1H-pyrazol-1-yl)Ph |
| 157 | 4-(3,4,5-triiodo-1H-pyrazol-1-yl)Ph | 158 | 4-(3,5-diI-1H-pyrazol-1-yl)Ph | 159 | 4-(3-Cl-1H-pyrazol-1-yl)Ph |
| 160 | 4-(4-Cl-1H-pyrazol-1-yl)Ph | 161 | 4-(5-Cl-1H-pyrazol-1-yl)Ph | 162 | 4-(4,5-diCl-1H-pyrazol-1-yl)Ph |
| 163 | 4-(3,5-diCl-1H-pyrazol-1-yl)Ph | 164 | 4-(3,4-diCl-1H-pyrazol-1-yl)Ph | 165 | 4-(3,4,5-triCl-1H-pyrazol-1-yl)Ph |
| 166 | 4-(3-amino-1H-pyrazol-1-yl)Ph | 167 | 4-(4-amino-1H-pyrazol-1-yl)Ph | 168 | 4-(5-amino-1H-pyrazol-1-yl)Ph |
| 169 | 4-(4,5-diamino-1H-pyrazol-1-yl)Ph | 170 | 4-(3,5-diamino-1H-pyrazol-1-yl)Ph | 171 | 4-(3,4-diamino-1H-pyrazol-1-yl)Ph |
| 172 | 4-(3,4,5-triamino-1H-pyrazol-1-yl)Ph | 173 | 4-(3-Me-1H-pyrazol-1-yl)Ph | 174 | 4-(4-Me-1H-pyrazol-1-yl)Ph |
| 175 | 4-(5-Me-1H-pyrazol-1-yl)Ph | 176 | 4-(4,5-diMe-1H-pyrazol-1-yl)Ph | 177 | 4-(3,5-diMe-1H-pyrazol-1-yl)Ph |
| 178 | 4-(3,4-diMe-1H-pyrazol-1-yl)Ph | 179 | 4-(3,4,5-triMe-1H-pyrazol-1-yl)Ph | 180 | 4-(5-Et-1H-pyrazol-1-yl)Ph |
| 181 | 4-(4-Et-1H-pyrazol-1-yl)Ph | 182 | 4-(5-Et-4-Me-1H-pyrazol-1-yl)Ph | 183 | 4-(5-Et-3,4-diMe-1H-pyrazol-1-yl)Ph |
| 184 | 4-(5-(Me-thio)-1H-pyrazol-1-yl)Ph | 185 | 4-(4-Me-5-(Me-thio)-1H-pyrazol-1-yl)Ph | 186 | 4-(4,5-bis(Me-thio)-1H-pyrazol-1-yl)Ph |
| 187 | 4-(3-Me-4,5-bis(Me-thio)-1H-pyrazol-1-yl)Ph | 188 | 4-(5-(aminothio)-1H-pyrazol-1-yl)Ph | 189 | 4-(4-(aminothio)-1H-pyrazol-1-yl)Ph |
| 190 | 4-(4-(aminothio)-5-ME-1H-pyrazol-1-yl)Ph | 191 | 4-(4,5-bis(aminothio)-1H-pyrazol-1-yl)Ph | 192 | [1,1′-biPh]-4-yl |
| 193 | 4-(5H-tetrazol-5-yl)Ph | 194 | 4-(1H-imidazol-1-yl)Ph | 195 | 4-(1H-1,2,4-triazol-1-yl)Ph |
| 196 | 4-(1H-tetrazol-1-yl)Ph | 197 | 4-(thiophen-2-yl)Ph | 198 | 4-(pyridin-2-yl)Ph |
| 199 | 4-(pyrazin-2-yl)Ph | 200 | 4-(pyrimidin-2-yl)Ph | 201 | 4-(pyridazin-3-yl)Ph |
| 202 | 4-(piperazin-1-yl)Ph | 203 | 3H-indol-2-yl | 204 | 7H-purin-8-yl |
| 205 | 1,8a-dihydroindolizin-2-yl | 206 | isoquinolin-6-yl | 207 | quinolin-6-yl |
| 208 | cyclopenta-2,4-dienecarbonyl | 209 | 2-Mecyclopenta-2,4-dienecarbonyl | 210 | 2-F-cyclopenta-2,4-dienecarbonyl |
| 211 | 2-aminocyclopenta-2,4-dienecarbonyl | 212 | 2-Mecyclopenta-2,4-dienecarbonyl | 213 | 3-Mecyclopenta-2,4-dienecarbonyl |
| 214 | 2,3-diMecyclopenta-2,4-dienecarbonyl | 215 | 2-Clcyclopenta-2,4-dienecarbonyl | 216 | 3-Clcyclopenta-2,4-dienecarbonyl |
| 217 | 2,3-diClcyclopenta-2,4-dienecarbonyl | 218 | 3-Brcyclopenta-2,4-dienecarbonyl | 219 | 2-Brcyclopenta-2,4-dienecarbonyl |
| 220 | 2,3-diBrcyclopenta-2,4-dienecarbonyl | 221 | 2-iodocyclopenta-2,4-dienecarbonyl | 222 | 3-iodocyclopenta-2,4-dienecarbonyl |
| 223 | 2,3-diIcyclopenta-2,4-dienecarbonyl | 224 | amino(cyclopenta-2,4-dien-1-yl)Me | 225 | amino(2-F-cyclopenta-2,4-dien-1-yl)Me |
| 226 | NH2(2,3-diF-cyclopenta-2,4-dien-1-yl)Me | 227 | NH2(2-Mecyclopenta-2,4-dien-1-yl)Me | 228 | HN(2,3-diMecyclopenta-2,4-dien-1-yl)Me |
| 229 | (2,3-diMecyclopenta-2,4-dien-1-yl)(CH3NHMe | 230 | (CH3NH)(2-Mecyclo-penta-2,4-dien-1-yl)Me | 231 | (CH3NH)(3-Mecyclo-penta-2,4-dien-1-yl)Me |
| 232 | (3-F-cyclopenta-2,4-dien-1-yl)(CH3NH)Me | 233 | (2-F-cyclopenta-2,4-dien-1-yl)(CH3NH)Me | 234 | (2,3-diF-cyclopenta-2,4-dien-1-yl)(CH3NH)Me |
| 235 | (2,3-diMecyclopenta-2,4-dien-1-yl)(FNH)Me | 236 | FNH(2-Mecyclopenta-2,4-dien-1-yl)Me | 237 | FNH(3-Mecyclopenta-2,4-dien-1-yl)Me |
| 238 | F-amino(3-F-cyclopenta-2,4-dien-1-yl)Me | 239 | (2,3-diF-cyclopenta-2,4-dien-1-yl)(FNH)Me | 240 | (2,3-diClcyclopenta-2,4-dien-1-yl)(FNH)Me |
| 241 | (2-Clcyclopenta-2,4-dien-1-yl)(FNH)Me | 242 | (3-Clcyclopenta-2,4-dien-1-yl)(FNH)Me | 243 | (3-Brcyclopenta-2,4-dien-1-yl)(F-amino)Me |
| 244 | (2,3-diBrcyclopenta-2,4-dien-1-yl)(FNH)Me | 245 | (2-Brcyclopenta-2,4-dien-1-yl)(FNH)Me | 246 | NH2(2-carbamoylcyclopenta-2,4-dien-1-yl)Me |
| 247 | NH2(3-carbamoylcyclopenta-2,4-dien-1-yl)Me | 248 | NH2(2-carbamoyl-3-F-cyclopenta-2,4-dien-1-yl)Me | 249 | NH2(2-carbamoyl-3-Clcyclopenta-2,4-dien-1-yl)Me |
| 250 | NH2(3-NH-2-carbamoylcyclo penta-2,4-dien-1-yl)Me | 251 | 2-carbamoylPh-HCOO– | 252 | 3-carbamoylPh-HCOO– |
| 253 | 4-carbamoylPh-HCOO– | 254 | 2-MePh-HCOO– | 255 | 3-MePh-HCOO– |
| 256 | 3-MePh-HCOO– | 257 | 2,3-diMePh-HCOO– | 258 | (2-carbamoylPh)(imino)Me |
| 259 | imino(Ph)Me | 260 | 3-carbamoylPh(imino)Me | 261 | 4-carbamoylPh(imino)Me |
| 262 | imino(2-MePh)Me | 263 | 2,3-diMePh(imino)Me | 264 | imino(3-MePh)Me |
| 265 | imino(4-MePh)Me | 266 | (F-imino)(2-F-Ph)Me | 267 | F-imino(3-F-Ph)Me |
| 268 | (3-BrPh)(F-imino)Me | 269 | (2-BrPh)(F-imino)Me | 270 | (2-ClPh)(F-imino)Me |
| 271 | (3-ClPh)(F-imino)Me | 272 | Brimino(3-ClPh)Me | 273 | Brimino(3-BrPh)Me |
| 274 | Cl-imino(3-ClPh)Me | 275 | Cl-imino(2-ClPh)Me | 276 | imino(o-tolyl)Me |
| 277 | imino(2-(CF3)Ph)Me | 278 | imino(3-(CF3)Ph)Me | 279 | 3-formylbenzamide |
| 280 | 2-formylbenzamide | 281 | 4-formylbenzamide | 282 | -Bz-2-Me |
| 283 | -Bz-2,3-diMe | 284 | -Bz-3-Me | 285 | -Bz-4-Me |
| 286 | -Bz-2-Me | 287 | -Bz-2-(CF3) | 288 | -Bz-3-(CF3) |
| 289 | -Bz-2-F- | 290 | NH2(3-Br-2-carbamoylcyclopenta-2,4-dien-1-yl)Me | 291 | Carbamoyl |
| 292 | 4-Cl-1H-pyrazol-1-yl | 293 | 4,5-diCl-1H-pyrazol-1-yl | 294 | 5-Cl-1H-pyrazol-1-yl |
| 295 | 3-Cl-1H-pyrazol-1-yl | 296 | 3-Br-1H-pyrazol-1-yl | 297 | 4-Br-1H-pyrazol-1-yl |
| 298 | 5-Br-1H-pyrazol-1-yl | 299 | 4,5-diBr-1H-pyrazol-1-yl | 300 | 3,4,5-triBr-1H-pyrazol-1-yl |
| 301 | 4-Me-1H-pyrazol-1-yl | 302 | 4,5-diMe-1H-pyrazol-1-yl | 303 | 5-Me-1H-pyrazol-1-yl |
| 304 | 5-iodo-1H-pyrazol-1-yl | 305 | 4-iodo-1H-pyrazol-1-yl | 306 | 3-iodo-1H-pyrazol-1-yl |
| 307 | 3,4-diI-1H-pyrazol-1-yl | 308 | 3,4,5-triiodo-1H-pyrazol-1-yl | 309 | 3,4,5-triF-1H-pyrazol-1-yl |
| 310 | 3-F-1H-pyrazol-1-yl | 311 | 3,4-diF-1H-pyrazol-1-yl | 312 | 4-F-1H-pyrazol-1-yl |
| 313 | 5-F-1H-pyrazol-1-yl | 314 | 3-NH2-1H-pyrazol-1-yl | 315 | 4-amino-1H-pyrazol-1-yl |
| 316 | 5-amino-1H-pyrazol-1-yl | 317 | 5-Me-1H-pyrazol-1-yl | 318 | 5-Et-1H-pyrazol-1-yl |
| 319 | 4-Me-1H-pyrazol-1-yl | 320 | 4,5-diMe-1H-pyrazol-1-yl | 321 | 5-(Me-Me)-1H-pyrazol-1-yl |
| 322 | 4-Me-5-(Me-Me)-1H-pyrazol-1-yl | 323 | 5-(H2N-thio)-4-Me-1H-pyrazol-1-yl | 324 | 4,5-bis(aminothio)-1H-pyrazol-1-yl |
| 325 | 4,5-bis(H2N-thio)-3-Me-1H-pyrazol-1-yl | 326 | 5-Et-4-Me-1H-pyrazol-1-yl | 327 | pyridazin-3-yl |
| 328 | pyridazin-4-yl | 329 | pyrimidin-4-yl | 330 | 1,3,5-triazin-2-yl |
| 331 | pyrimidin-2-yl | 332 | pyrazin-2-yl | 333 | Cyclohexyl |
| 334 | piperidin-1-yl | 335 | tetrahydropyridazin-1(2H)-yl | 336 | piperazin-1-yl |
| 337 | 1,2,4-triazinan-1-yl | 338 | 2-(thiazol-2-yl)butyl | 339 | 2-(thiazol-2-yl)pentyl |
a All fragments were used for substitutions in the R1 and R2 positions.
2.5.2. In Silico Screening of Library of BHMBs
The focused library of 73,565 analogs was further screened for molecular structures matching the 3D-QSAR PH4 pharmacophore model Hypo1 of InhA inhibition. 238 BHMBs mapped to at least 2 pharmacophoric features, 90 of which mapped to at least 4 features of the pharmacophore. These best fitting analogs (PH4 hits) then underwent complexation QSAR model screening. The computed GFE of InhA-BHMBx complex formation, their components, and predicted half-maximal inhibitory concentrations IC50pre calculated from the correlation Equation B (Table 3) are listed in Table 6.
Table 6.
GFE and their components for the top scoring 90 virtual BHMB analogs. The analog numbering concatenates the index of each substituent R1 to R2 with the substituent numbers taken from Table 5 except for hydrogen which is directly specified by the letter H.
| Designed Analogs | Mw a [g⋅mol−1] | ΔΔHMM b [kcal⋅mol−1] | ΔΔGsol c [kcal⋅mol−1] | ΔΔTSvib d [kcal⋅mol−1] | ΔΔGcom e [kcal⋅mol−1] | IC50pre f [nM] | |
|---|---|---|---|---|---|---|---|
| N | BHMB1 | 385 | 0 | 0 | 0 | 0 | 20 g |
| 1 | 32–218 | 389 | −21.96 | 14.78 | 0.15 | −7.33 | 0.24 |
| 2 | 39–95 | 359 | 21.7 | 2.26 | 1.98 | 21.99 | 1996 |
| 3 | 40–232 | 439 | −1.72 | 5.01 | −1.65 | 4.94 | 200 |
| 4 | 40–234 | 405 | −10.02 | 2.93 | 0.99 | −8.08 | 0.16 |
| 5 | 41–93 | 409 | −11.28 | 3.31 | 2.16 | −10.14 | 0.052 |
| 6 | 42–207 | 342 | −7.22 | 6.24 | −4.45 | 3.47 | 90 |
| 7 | 47–235 | 421 | −5.66 | 3.16 | −5.73 | 3.24 | 80 |
| 8 | 54–282 | 505 | −3.55 | 6.63 | −3.02 | 6.1 | 370 |
| 9 | 65–235 | 413 | −5.71 | 3.01 | 1.99 | −4.69 | 1 |
| 10 | 67–67 | 375 | −8.72 | 3.21 | 0.32 | −5.83 | 0.55 |
| 11 | 67–212 | 505 | 0.56 | 2.82 | −2.2 | 5.59 | 280 |
| 12 | 67–215 | 599 | 3.45 | 3.22 | −2.52 | 9.18 | 1990 |
| 13 | 70–241 | 430 | −13.98 | 14.15 | −1.02 | 1.18 | 30 |
| 14 | 72–232 | 401 | −10.09 | 4.74 | −0.7 | −4.65 | 1 |
| 15 | 79–9 | 402 | −12.2 | 15.92 | −2.82 | 6.54 | 470 |
| 16 | 79–92 | 439 | −13.99 | 6.17 | 3.15 | −10.97 | 0.033 |
| 17 | 79–215 | 683 | −4.32 | 2.82 | −5.06 | 3.56 | 94 |
| 18 | 79–338 | 375 | −12.46 | 5.35 | 3.19 | −10.3 | 0.047 |
| 19 | 79–339 | 389 | −13.64 | 5 | 2.01 | −10.65 | 0.039 |
| 20 | 86–233 | 393 | −14.09 | 17.65 | −2.46 | 6.02 | 350 |
| 21 | 87–24 | 410 | −8.64 | 4.95 | 1.59 | −5.28 | 0.74 |
| 22 | 87–122 | 480 | −9.46 | 4.77 | 0.39 | −5.07 | 0.83 |
| 23 | 87–205 | 450 | −13.78 | 17.73 | −4.81 | 8.75 | 1570 |
| 24 | 87–208 | 453 | −7.19 | 3.56 | −4.64 | 1.01 | 20 |
| 25 | 92–169 | 406 | −14.2 | 14.73 | 5.02 | −4.48 | 2 |
| 26 | 95–215 | 606 | −6.76 | 1.94 | −2.28 | −2.53 | 4 |
| 27 | 106–215 | 605 | −6.81 | 2.61 | −6.96 | 2.76 | 60 |
| 28 | 106–233 | 407 | −7.12 | 5.75 | −3.77 | 2.4 | 50 |
| 29 | 106–241 | 432 | −2.77 | 6.09 | −2.2 | 5.51 | 270 |
| 30 | 111–95 | 389 | 9.21 | −0.51 | 0.18 | 8.52 | 1380 |
| 31 | 112–208 | 388 | 2.68 | 0.15 | −2.26 | 5.09 | 210 |
| 32 | 114–232 | 460 | −11.36 | 15.18 | −4.36 | 8.18 | 1150 |
| 33 | 118–84 | 443 | −6.36 | 4.46 | −0.67 | −1.23 | 7 |
| 34 | 118–235 | 500 | −5.5 | 1.49 | −3.13 | −0.89 | 9 |
| 35 | 122–57 | 400 | −12.27 | 15.46 | 1.16 | 2.03 | 40 |
| 36 | 122–88 | 445 | −11.79 | 16.54 | −0.93 | 5.68 | 290 |
| 37 | 122–92 | 390 | −18.45 | 16.69 | 7.09 | −8.85 | 0.1 |
| 38 | 122–282 | 504 | −6.19 | 12.29 | 2.79 | 3.31 | 81 |
| 39 | 128–232 | 493 | −19.75 | 17.06 | −6.07 | 3.38 | 83 |
| 40 | 138–65 | 416 | −18.69 | 16.51 | −0.24 | −1.94 | 5 |
| 41 | 147–282 | 744 | −16.78 | 13.56 | −7.11 | 3.89 | 110 |
| 42 | 158–209 | 453 | −20.41 | 19.64 | −2.82 | 2.05 | 41 |
| 43 | 166–92 | 393 | −14.08 | 7.1 | 1.14 | −8.12 | 0.15 |
| 44 | 169–232 | 485 | −18.91 | 17.42 | −5.38 | 3.9 | 113 |
| 45 | 172–235 | 495 | −23.56 | 16.58 | −4.17 | −2.81 | 3 |
| 46 | 174–214 | 537 | −15.39 | 17.46 | −0.99 | 3.06 | 71 |
| 47 | 174–311 | 399 | −20.87 | 18.65 | −2.04 | −0.18 | 12 |
| 48 | 177–282 | 571 | −15.7 | 15.55 | −7.49 | 7.34 | 725 |
| 49 | 178–233 | 515 | −22.75 | 17.89 | −5.82 | 0.96 | 23 |
| 50 | 185–282 | 494 | −10.48 | 15.12 | −4.02 | 8.66 | 1514 |
| 51 | 201–15 | 315 | −15.79 | 15.32 | 3.25 | −3.72 | 2 |
| 52 | 214–233 | 499 | 3.45 | 2.69 | −2.89 | 9.03 | 1820 |
| 53 | 220–99 | 420 | −7.16 | 3.71 | 5.22 | −8.66 | 0.11 |
| 54 | 225–233 | 424 | −7.4 | 15.23 | −1.23 | 9.05 | 1863 |
| 55 | 232–276 | 451 | −19 | 17.57 | −0.81 | −0.63 | 2 |
| 56 | 234–99 | 408 | −8.67 | 3.96 | 4.31 | −9.02 | 0.09 |
| 57 | 234–234 | 426 | −5.74 | 17.27 | 2.21 | 9.32 | 2138 |
| 58 | 238–25 | 398 | −5.51 | 5.21 | 8.81 | −9.11 | 0.09 |
| 59 | 240–84 | 423 | −15.36 | 16.82 | −0.29 | 1.74 | 34 |
| 60 | 241–63 | 396 | −7.77 | 19.23 | 5.04 | 6.42 | 437 |
| 61 | 244–99 | 426 | −9.46 | 6.19 | 0.9 | −4.17 | 2 |
| 62 | 245–43 | 394 | −14.47 | 19.17 | −0.26 | 4.96 | 200 |
| 63 | 245–174 | 483 | −19.31 | 16.99 | −4.81 | 2.48 | 52 |
| 64 | 261–212 | 585 | −4.95 | 4.87 | −8.29 | 8.21 | 1175 |
| 65 | 266–210 | 478 | −3.42 | 4.54 | −5.75 | 6.88 | 563 |
| 66 | 269–215 | 650 | −4.2 | 3.14 | −5.61 | 4.54 | 159 |
| 67 | 269–233 | 452 | −12.9 | 5.61 | −3.64 | −3.65 | 2 |
| 68 | 269–236 | 575 | −6.69 | 4.6 | −5.62 | 3.53 | 92 |
| 69 | 270–232 | 486 | −8.35 | 5.48 | −6.4 | 3.53 | 92 |
| 70 | 279–214 | 525 | −5.1 | −0.63 | −4.71 | −1.02 | 8 |
| 71 | 279–282 | 522 | −2.8 | 5.06 | −4.06 | 6.32 | 417 |
| 72 | 282–26 | 448 | −13.63 | 18.31 | 5.63 | −0.95 | 8 |
| 73 | 282–280 | 522 | −10.39 | 2.59 | −3.51 | −4.3 | 2 |
| 74 | 297–28 | 423 | −5.81 | 3.84 | 2.05 | −4.01 | 2 |
| 75 | 307–99 | 343 | −7.43 | 6.73 | 3.3 | −4.01 | 2 |
| 76 | 308–25 | 342 | −6.7 | 6.12 | 3.74 | −4.32 | 2 |
| 77 | 310–99 | 357 | −10.86 | 4.79 | 2.67 | −8.74 | 0.11 |
| 78 | 310–74 | 525 | 3.9 | 1.65 | −3.08 | 8.63 | 1479 |
| 79 | 310–209 | 390 | 2.99 | −0.1 | −3.93 | 6.82 | 550 |
| 80 | 310–289 | 374 | 2.6 | −0.5 | −1.85 | 4.1 | 124 |
| 81 | 313–278 | 365 | −20.4 | 18.73 | −0.79 | −0.88 | 9 |
| 82 | 316–150 | 689 | −17.61 | 14.38 | −8.08 | 4.85 | 187 |
| 83 | 317–98 | 446 | −11.26 | 17.19 | 1.24 | 4.69 | 170 |
| 84 | 318–79 | 447 | −10.86 | 9.09 | −1.71 | −0.07 | 13 |
| 85 | 318–89 | 365 | −4.96 | 3.62 | 1.15 | −2.48 | 4 |
| 86 | 318–198 | 370 | 0.06 | 4.96 | 1.85 | 3.17 | 75 |
| 87 | 318–207 | 370 | −11.32 | 5.52 | 1.37 | −7.17 | 0.26 |
| 88 | 318–212 | 493 | −9.55 | 3.93 | −1.59 | −4.02 | 2 |
| 89 | 320–19 | 376 | 3.36 | 3.01 | 4.03 | 2.34 | 48 |
| 90 | 321–25 | 339 | −1.01 | 2.16 | 4.2 | −3.05 | 3 |
a Mw is molar mass of inhibitor; b ΔΔHMM is the relative enthalpic contribution to the GFE change of the InhA-BHMB complex formation ΔΔGcom (for details see footnote pf Table 2); c ΔΔGsol is the relative solvation GFE contribution to ΔΔGcom; d ΔΔTSvib is the relative (vibrational) entropic contribution to ΔΔGcom; e ΔΔGcom is the relative Gibbs free energy change related to the enzyme–inhibitor InhA-BHMB complex formation ΔΔGcom ≅ ΔΔHMM + ΔΔGsol − ΔΔTSvib; f IC50pre is the predicted inhibition potency towards MtInhA calculated from ΔΔGcom using correlation Equation B, Table 3; g IC50exp [19] is given for the reference inhibitor BHMB1 instead of the IC50pre.
2.6. Novel BHMB Analogs
The design of virtual library of novel analogs was guided by structural information retrieved from the BHMBx active conformation and was used for the selection of appropriate substituents (R1- and R2-groups). In order to identify which substituents lead to new inhibitor candidates with the highest predicted potencies towards the InhA of Mt, we have prepared histograms of the frequency of occurrence of R1- and R2-groups among the 90 best fit PH4 hits (Figure 6). The histograms show that the R1 groups 79, 310, and 318 were represented with the highest frequency of occurrence (5) among the 90 BHMB hits. The R2-groups most frequently represented in this subset are 282 (10) and 232, 233 (6) and 99, 215 and 235 (5). The top ten scoring virtual hits, namely, analogs are 79-92 (IC50pre = 33 pM), 79-339 (39 pM), 79-338 (47 pM), 41-93 (52 pM), 234-99 (90 pM), 238-25 (90 pM), 122-92 (100 pM), 220-99 (110 pM), 310-99 (110 pM), and 166-92 (150 pM). They include the following substituents at R1 position: 79: 3-Br-2-(thiazol-2-yl)Pr (3), 41: Thiophen-2-ylMe (1), 234: (2,3-diFC5H7- 2,4-dien-1-yl)(mercapto-amino)Me (1), 238: FNH(3-FC5H7-2,4-dien-1-yl)Me (1), 122: 2-(F3C)Pyri- din-3-yl (1), 220: 2,3-diBr C5H7-2,4-diene-carbonyl (1), 310: 3-F-1-H-pyrazol-1-yl (1), 166: 4-(3-NH2-1- H-pyrazol-1-yl)Ph (1), or at R2 position: 92: 4-HO-Hexyl (3), 339: 2-(thiazol-2-yl)butyl (1), 338: 2-(thiazol-2-yl)pentyl (1), 93: 5-NH2-6-Br Hexyl (1), 99: 6-NH2Octyl (3), 25: 6-MeOctyl (1). Due to amino acid composition of the larger hydrophobic pocket, the R2-groups display preferences for larger aliphatic building blocks from butyl to octyl [5].
Figure 6.

Histograms of frequency of occurrence of individual R-groups in the 90 best selected analogs mapping to four features of the PH4 pharmacophore hypothesis Hypo1 (for the structures of the fragments see Table 5); R1 = -3-Br-2-(thiazol-2-yl)propyl (79); -3-F-1H-pyrazol-1-yl (310); -5-Et-1H- pyrazol-1-yl (318) and R2 = -(3-F-cyclopenta-2,4-dien-1-yl)(MeAmino)Me (232); -(2-F-cyclopenta-2, 4-dien-1-yl)(MeAmino)Me (233) and -Bz-2-Me (282).
The substitutions in R1 and R2 positions of BHMBs led to an overall increase of affinity of InhA binding as exemplified by the inhibitory potencies of majority of new designed analogs. The best designed benzamide BHMB 79–92 displays predicted half-minimal inhibitory concentration of IC50pre = 33 pM that is about 61 times lower than that of the most active compound of the TS, namely the BHMB1 with IC50exp = 20 nM, Figure 7 and Figure 8.
Figure 7.

(A) Close up of virtual hit 79–92, the most active designed BHMB analog (IC50pre = 33 pM) at the active site of InhA. Interacting residues are colored yellow, and NADH is not shown for clarity. (B) Mapping of the BHMB 79–92 to InhA inhibition pharmacophore. (C) 2D schematic interaction diagram of the BHMB 79–92 at the active site of MtInhA. (D) 2D schematic interaction diagram of the analog BHMB 79–339 (IC50pre = 39 pM) at the active site of MtInhA. (E) 2D schematic interaction diagram of the ligand ((2S,4S)-N-methyl-4-[[(2S,3R)-3-[(2-methylpropan-2-yl)oxy]-2-[[4-(pyrazol-1- ylmethyl)phenyl]carbonylamino]butanoyl]amino]-1-(phenylcarbonyl)pyrrolidine-2-carboxamide) in complex with MtInhA (PDB: 5G0W) displaying the HB contact with Arg43 as reported in [6].
Figure 8.

Surface of the active site of MtInhA with bound 5 best active designed BHMB analogs. The binding site surface is colored according to residue hydrophobicity: red = hydrophobic, blue = hydrophilic, and white = intermediate.
2.7. Pharmacokinetic Profile of Novel BHMB Analogs
The pharmacokinetic profile of InhA inhibitors remains an important issue [7]. The best designed triclosan derivative with very low oral bioavailability due to its poor water solubility and rapid phase II metabolism has to be optimized for a possible use as antituberculotic and eventually antimalarial in case of high affinity toward PfEACP [32]. Among the ADME-related properties displayed in Table 7, such as octanol-water partitioning coefficient; aqueous solubility; blood-brain partition coefficient; Caco-2 cell permeability; serum protein binding; number of likely metabolic reactions; and another eighteen descriptors related to absorption, distribution, metabolism, and excretion (ADME) of the new analogs, were computed by the QikProp program [33] based on the method of Jorgensen [34,35]. Experimental data from more than 710 compounds including about 500 drugs and related heterocycles were used to produce regression equations correlating experimental and computed descriptors, resulting in an accurate prediction of pharmacokinetic properties of molecules. In line with the observed unfavorable pharmacokinetic exposure for the best active triclosan derivative, the predicted oral bioavailability of novel BHMB analogs ranges from 86% to 100%. Since a value greater than 80% is considered good, ten analogs among the best predicted 14 display a human oral absorption in gastrointestinal tract (HOA) of 100%. Drug likeness (#stars)—the number of property descriptors that fall outside the range of optimal values determined for 95% of known drugs out of 24 selected descriptors computed by the QikProp—was used as an additional ADME-related compound selection criterion. The values for the best active designed BHMBs are compared with those computed for drugs used for treatment of tuberculosis or currently undergoing clinical trials (Table 7). Our best designed analogs all display #stars equal to zero, meaning that the optimal value range of none of the drug-likeness descriptors was violated. Thus the designed BHMBs display a favorable pharmacokinetic profile.
Table 7.
ADME-related properties of the best designed BHMB analogs and known antituberculotic agents either in clinical use or currently undergoing clinical testing computed by QikProp [33].
| BHMBx a | #stars b | Mw c [g.mol−1] | Smol d [Å2] | Smol,hfo e [Å2] | Vmol f [Å3] | RotB g | HBdon h | HBacc i | logPo/w j | logSwat k | logKHSA l | logB/B m | BIPcaco n [nm.s−1] | #meta o | IC50pre [nM] | HOAq | %HOAr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 32–218 | 0 | 389 | 760 | 380.1 | 1340 | 10 | 3 | 3.5 | 5.1 | −5.8 | 0.94 | −0.49 | 320.6 | 4 | 0.24 | 3 | 89 |
| 40–234 | 1 | 405 | 703 | 285.9 | 1253 | 6 | 2 | 3.5 | 5.8 | −7.2 | 1 | −0.11 | 2551.9 | 8 | 0.16 | 1 | 100 |
| 41–93 | 0 | 409 | 691 | 230.2 | 1218 | 11 | 3 | 3.5 | 4.5 | −4.6 | 0.57 | −0.42 | 301.4 | 5 | 0.05 | 3 | 100 |
| 67–67 | 1 | 375 | 716 | 305.9 | 1278 | 8 | 1 | 4 | 5.8 | −6.5 | 0.98 | −0.42 | 3493.1 | 4 | 0.55 | 1 | 100 |
| 79–92 | 0 | 439 | 737 | 323.6 | 1306 | 12 | 2 | 5.7 | 4.8 | −5.9 | 0.48 | −1.01 | 1105.1 | 4 | 0.033 | 3 | 100 |
| 79–338 | 0 | 375 | 680 | 367.1 | 1268 | 12 | 2 | 5.7 | 4.5 | −4.7 | 0.41 | −0.83 | 1688.3 | 4 | 0.047 | 3 | 100 |
| 79–339 | 0 | 389 | 760 | 431.0 | 1368 | 13 | 2 | 5.7 | 5 | −5.9 | 0.6 | −1.15 | 1371.3 | 4 | 0.039 | 3 | 100 |
| 87–24 | 0 | 410 | 701 | 332.5 | 1226 | 9 | 1 | 4.5 | 4.8 | −6.3 | 0.68 | −0.96 | 882.4 | 3 | 0.74 | 1 | 100 |
| 87–122 | 0 | 480 | 762 | 347.2 | 1363 | 10 | 1 | 6.95 | 5 | −6.3 | 0.5 | −0.62 | 1884.2 | 6 | 0.83 | 1 | 100 |
| 122–92 | 0 | 390 | 808 | 522.4 | 1430 | 13 | 2 | 7.6 | 4.5 | −5.9 | 0.39 | −1.28 | 1347.2 | 6 | 0.10 | 3 | 100 |
| 166–92 | 0 | 393 | 739 | 244.7 | 1324 | 10 | 4 | 5.7 | 3.9 | −5.5 | 0.44 | −1.83 | 254.9 | 3 | 0.15 | 3 | 92 |
| 234–99 | 0 | 408 | 730 | 318.4 | 1343 | 13 | 4 | 4.5 | 4.5 | −4.4 | 0.56 | −0.79 | 180.9 | 5 | 0.09 | 3 | 93 |
| 310–74 | 1 | 525 | 797 | 239.5 | 1419 | 8 | 2 | 5 | 6 | −7.8 | 1.07 | −0.81 | 1168.4 | 3 | 0.11 | 1 | 91 |
| 318–207 | 0 | 370 | 683 | 215.9 | 1198 | 6 | 1 | 5.5 | 4.3 | −6 | 0.53 | −0.73 | 1021.2 | 3 | 0.26 | 3 | 100 |
| Rifampin | 1 | 137.1 | 314 | 0.0 | 480 * | 2 | 3 | 4.5 | −0.7 | 0 | −0.8 | −0.8 | 267.5 | 2 | − | 2 | 67 |
| Isoniazid | 4 | 123.1 * | 300 | 0.0 | 443 * | 1 | 2 | 5 | −0.6 | −0.5 | −0.8 | −0.7 | 298.4 | 4 | − | 2 | 67 |
| Ethambutol | 2 | 204.3 | 476 | 395.8 | 806 | 11 | 4 | 6.4 | −0.2 | 0.6 | −0.8 | 0.0 | 107.8 | 4 | − | 2 | 62 |
| Pyrazinamide | 10 | 823.0 * | 1090 * | 850.0 * | 2300 * | 25 * | 6 | 20.3 * | 3.0 | −3.1 | −0.3 | −2.7 | 38.2 | 11 * | − | 1 | 34 |
| Gatifloxacin | 0 | 375.4 | 598 | 355.7 | 1093 | 2 | 1 | 6.8 | 0.5 | −4.0 | 0 | −0.6 | 17.0 | 1 | − | 2 | 52 |
| Moxifloxacin | 0 | 401.4 | 642 | 395.6 | 1168 | 2 | 1 | 6.8 | 1.0 | −4.7 | 0.2 | −0.6 | 20.9 | 1 | − | 2 | 56 |
| Rifapentine | 10 | 877.0 * | 1025 * | 844.9 * | 2333 * | 24 * | 6 | 20.9 * | 3.6 | −2.2 | −0.2 | −1.5 | 224.0 | 13 * | − | 1 | 51 |
| Bedaquiline | 4 | 555.5 | 787 | 213.7 | 1532 | 9 | 1 | 3.8 | 7.6 * | −6.9 | 1.7 | 0.4 | 1562.2 | 5 | − | 1 | 100 |
| Delamanid | 2 | 534.5 | 796 | 284.4 | 1470 | 7 | 0 | 6.0 | 5.8 | −7.6 | 1.0 | −1.0 | 590.9 | 2 | − | 1 | 85 |
| Linezolid | 0 | 337.4 | 555 | 337.2 | 996 | 2 | 1 | 8.7 | 0.6 | −2.0 | −0.7 | −0.5 | 507.0 | 2 | − | 3 | 79 |
| Sutezolid | 1 | 353.4 | 594 | 330.6 | 1047 | 2 | 1 | 7.5 | 1.3 | −3.4 | −0.4 | −0.4 | 449.3 | 0 | − | 3 | 82 |
| Ofloxacin | 1 | 361.4 | 581 | 337.0 | 1044 | 1 | 0 | 7.3 | −0.4 | −2.8 | −0.5 | −0.4 | 25.9 | 1 | − | 2 | 50 |
| Amikacin | 14 | 585.6 | 739 | 350.3 | 1500 | 22 * | 17 * | 26.9 * | −7.9 * | −0.2 | −2.1 | −3.5 | 0 | 14 * | − | 1 | 0 |
| Kanamycin | 10 | 484.5 | 656 | 258.9 | 1291 | 17 * | 15 * | 22.7 * | −6.7 * | 2.0 | −1.4 | −3.1 | 0 | 12 * | − | 1 | 0 |
| Imipenem | 0 | 299.3 | 487 | 259.1 | 880 | 8 | 3 | 7.2 | 1.0 | −1.8 | −0.7 | −1.4 | 35.0 | 3 | − | 3 | 61 |
| Amoxicillin | 2 | 365.4 | 561 | 164.6 | 1033 | 6 | 4.25 | 8.0 | −2.5 | −0.8 | −1.1 | −1.5 | 1.0 | 5 | − | 1 | 12 |
| Clavulanate | 0 | 199.2 | 397 | 184.6 | 630 | 4 | 2 | 6.5 | −0.8 | 0.3 | −1.3 | −1.3 | 13.3 | 2 | − | 2 | 42 |
a designed BHMB analogs and known antituberculotic agents, Table 6; b drug likeness, number of property descriptors (24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release 14) that fall outside of the range of values for 95% of known drugs; c molar mass in [g.mol-1] (range for 95% of drugs: 130–725 g.mol−1) [33]; d total solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2); e hydrophobic portion of the solvent-accessible molecular surface, in [Å2] (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2); f total volume of molecule enclosed by solvent-accessible molecular surface, in [Å3] (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3); g number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds (range for 95% of drugs: 0–15); h estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can assume non-integer values (range for 95% of drugs: 0.0–6.0); i estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can assume non-integer values (range for 95% of drugs: 2.0–20.0); j logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6.5); k logarithm of predicted aqueous solubility, logS. S in [mol·dm–3] is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (range for 95% of drugs: −6.0 to 0.5); l logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5); m logarithm of predicted brain/blood partition coefficient (range for 95% of drugs: −3.0 to 1.2); n predicted apparent Caco-2 cell membrane permeability in Boehringer-Ingelheim scale in [nm s-1] (range for 95% of drugs: < 25 poor, > 500 nm s−1 great); o number of likely metabolic reactions (range for 95% of drugs: 1–8); p predicted inhibition constants IC50pre. IC50pre was predicted from computed ΔΔGcom using the regression Equation B shown in Table 3; q human oral absorption (1 = low, 2 = medium, 3 = high); r percentage of human oral absorption in gastrointestinal tract (<25% = poor, >80% = high); * star in any column indicates that the property descriptor value of the compound falls outside the range of values for 95% of known drugs.
2.8. Predicted InhA Inhibition Potency for Current Drugs Bearing Benzamide Scaffold
Since the benzamide scaffold has been analyzed in this study, drugs currently used in the clinical practice sharing this scaffold are worth evaluating with the help of our 3D-QSAR generated PH4 pharmacophore. The list of 24 compounds given in Table 8 is mostly indicated for treatment of neural disorders and cancers [36]. As we can see on Figure 9, the mapping of the five most potent predicted MtInhA inhibitors to PH4 pharmacophore sheds light on their affinity towards the enzyme and suggests their experimental evaluation as antituberculotics as they predominantly occupy the large hydrophobic pocket. The best predicted, Sultopride (IC50pre = 1.7 nM) is an approved drug, used in Japan, Hong Kong, and Europe for treatment of schizophrenia [36].
Table 8.
GFE, its components, and predicted InhA inhibitory potencies of 24 approved drugs which contain benzamide scaffold in their molecular structure (for 2D representation see Table 9).
| DrugBank Accession Number | Name a | Mw [g⋅mol−1] | ΔΔHMM [kcal⋅mol−1] | ΔΔGsol [kcal⋅mol−1] | ΔΔTSvib [kcal⋅mol−1] | ΔΔGcom [kcal⋅mol−1] | IC50pre [nM] |
|---|---|---|---|---|---|---|---|
| DB00345 | Aminohippuric Acid | 194 | 10.6 | 3.81 | 2.03 | 12.39 | 11,370 |
| DB00391 | Sulpiride | 341 | 4.53 | 3.59 | 3.91 | 4.2 | 132 |
| DB00409 | Remoxipride | 371 | 11.49 | −1.65 | 7.06 | 2.78 | 60 |
| DB00604 | Cisapride | 465 | 8.47 | −0.06 | 2.97 | 5.44 | 258 |
| DB00619 | Imatinib | 493 | −3.54 | 13.79 | 2.56 | 7.69 | 879 |
| DB01035 | Procainamide | 235 | 12.97 | 2.22 | 6.14 | 9.05 | 1 847 |
| DB01168 | Procarbazine | 221 | 16.54 | 1.73 | 2.12 | 16.15 | 88,971 |
| DB01171 | Moclobemide | 268 | −7.79 | 16.78 | 3.22 | 5.76 | 308 |
| DB01233 | Metoclopramide | 299 | −4.54 | 17.82 | 6.47 | 6.81 | 544 |
| DB01393 | Bezafibrate | 361 | −11.95 | 18.14 | 1.32 | 4.87 | 189 |
| DB06288 | Amisulpride | 369 | 4.91 | 5.02 | 6.5 | 3.42 | 86 |
| DB06421 | Declopramide | 269 | −6.29 | 17.29 | 1.34 | 9.66 | 2577 |
| DB06422 | Tricalopride | 313 | −13.62 | 17.61 | 4.43 | −0.44 | 10 |
| DB06626 | Axtinib | 386 | 5.04 | 0.12 | −0.46 | 5.62 | 281.8 |
| DB07069 | 3-Hydroxyhippuric Acid | 195 | 0.99 | 5.78 | 1.41 | 5.36 | 247 |
| DB08950 | Indoramin | 347 | −2.33 | 18.92 | 4.19 | 12.39 | 11,495 |
| DB09018 | Bromopride | 344 | −8.55 | 17.12 | 3.08 | 5.49 | 265 |
| DB11282 | Diethyltoluamide | 191 | 2.37 | 2.47 | 5.48 | −0.64 | 9 |
| DB11480 | Zoalene | 225 | −0.72 | 7.5 | 0 | 6.78 | 535 |
| DB12518 | Raclopride | 347 | −12 | 18.54 | 1.78 | 4.77 | 178 |
| DB13025 | Tiapride | 328 | 5.98 | 2.72 | 4.83 | 3.86 | 109 |
| DB13273 | Sultopride | 354 | −17.19 | 19.7 | 6.23 | −3.42 | 1.7 |
| DB13523 | Veralipride | 384 | 0.93 | 4.96 | 4.84 | 1.05 | 23 |
| DB15445 | Iodohippuric Acid | 305 | 1.09 | 6.93 | −1.76 | 9.78 | 2751 |
a for definition of the individual quantities see the footnote of Table 2.
Figure 9.

Mapping of the benzamide drugs currently used in clinical practice to the InhA inhibition pharmacophore. Feature legend: HYDA = Hydrophobic Aliphatic (blue), HYD = Hydrophobic (cyan), Ar = Ring aromatic (orange), HBA = Hydrogen bond Acceptor (green). Only the top five predicted IC50pre are displayed.
3. Discussions
3.1. Binding Mode of BHMBs
To date, the most comprehensive study on InhA inhibition has been reported by Soutter et al. [6]. An important output of their DNA-encoded screening is the novel binding mode of inhibitors at the MtInhA active site, where the ligand, in addition to the typical stacking contact with Phe97 reaches a pocket surrounded by Phe41 and Arg43 side chains. This results in either a π–cation or a conventional hydrogen-bond contact. According to our analysis of the InhA-BHMBx complexes of most potent inhibitors, this interaction plays a key role in the significant improvement of predicted inhibitory potencies of novel benzamides. The enzyme–inhibitor interaction energy (Eint) breakdown to InhA active site residues contribution of best designed BHMB analogs (Figure 10) shows that in contrast to the TS and VS inhibitors [23] (Figure 4), while the contribution of Phe97 to Eint is almost the same, the impact of interaction with polar Arg43 increased by at least 4 kcal·mol−1. Surprisingly, the novel benzamide inhibitors display noticeable interaction with Phe97 and Arg43, suggesting that their length, which is comparable to that of TS BHMBs, is sufficient for concomitant interactions. Recently, site exclusion was observed for the ligands in complex with InhA (a pyrrolidine core and three substituents: benzoyl and 1-t-butoxy ethyl groups directly connected to the pyrrolidine core and a pyrazole benzaldehyde connected via the t-butoxy ethyl) benzoyl π–π stacks Phe97, methyl amide HB with Arg43), (PDB: 5G0W) [6]. As displayed in Figure 10, distance between the benzamide carbonyl oxygen and the Arg43 sidechain warhead carbon atom is relatively high for the training set compounds (7.5–9.5 Å). Interestingly, the plot of this distance vs. inhibitory activity pIC50exp displays a correlation. This distance seems to be one of the key determinants for binding affinity for the class 2 InhA direct inhibitors instead of the existence of a HB. This structural specificity that emerged during the PH4 screening of the virtual library of benzamide analogs led to identification of new hits, the best of which are capable of forming a HB with the Arg43 residue.
Figure 10.


Molecular mechanics inter-molecular interaction energy Eint break-down to active site residue contributions in [kcal.mol−1]: (A) designed best five novel BHMB analogs (the color coding refers to ligands given in the legend), (B) correlation between pIC50exp and distance from the benzamide carbonyl oxygen to the Arg43 warhead carbon atom (7.5–9.5 Å).
3.2. PH4-Based Screening of Approved Drugs Containing Benzamide Scaffold
A library of 24 benzamide scaffold drugs from DrugBank [36] was screened with the class 2 InhA inhibition PH4. These drugs are mostly dedicated to a variety of neurological and psychiatric disorders or nausea and vomiting treatment. The top five best PH4 hits have been evaluated also with the complexation QSAR model Equation B, Table 3, to predict their antituberculotic IC50pre towards the MtInhA: Sultopride (IC50pre = 1.7 nM), Diethyltoluamide (9 nM), Tricalopride (10 nM), Veralipride (23 nM) and Remoxipride (60 nM). Their mapping to the pharmacophore features is displayed in Figure 9. Therefore, experimental testing of these five approved drugs for MtInhA inhibition and perhaps also for antibacterial effect against the Mt would be worthwhile.
4. Materials and Methods
4.1. Training and Validation Sets
Chemical structures and biological activities (IC50exp) of training and validation sets of N-benzyl-4-((heteroaryl)methyl) benzamides inhibitors of InhA used in this study were taken from literature [23]. The potencies of these compounds cover a sufficiently broad range of half-maximal inhibitory concentrations (20 ≤ IC50exp ≤ 5930 nM) to allow construction of a QSAR model. The training set (TS) containing 19 BHMB inhibitors and the validation set (VS) including 6 BHMBs were taken from the ref. [23].
4.2. Model Building
Three dimensional (3D) molecular models of enzyme–inhibitor (E-I) complexes MtInhA-BHMBx, free enzyme InhA and free inhibitors BHMBx were prepared from high-resolution (2.2 Å) crystal structure of a reference complex containing the training set compound N-(2-chloro-4-fluorobenzyl)-4-((3,5-dimethyl-1-H-pyrazol-1-yl)methyl)benzamide (BHMB2, Table 1) bound to the mycobacterial InhA (Protein Data Bank [37] entry code 4QXM [23]) using Insight-II molecular modeling program [38].
The structures of InhA and the E-I complexes were considered to be at pH of 7 with neutral N- and C-terminal residues and all protonizable and ionizable residues charged. No crystallographic water molecules were included in the model. The inhibitors were built into the reference structure 4QXM [23] by in situ replacing of derivatized groups in the molecular scaffold of the template inhibitor BHMB2. An exhaustive conformational search over all rotatable bonds of the replacing function groups coupled with a careful gradual energy-minimization of the modified inhibitor and active site residues of the InhA located in the vicinity of the inhibitor (within 5 Å distance) was employed to identify low-energy bound conformations of the modified inhibitor. The resulting low-energy structures of the E-I complexes were then carefully refined by minimization of the whole complex. This procedure has been successfully used for model building of viral, bacterial, and protozoal enzyme–inhibitor complexes and design of peptidomimetic, hydroxynaphthoic, thymidine, triclosan, pyrrolidine carboxamide, nitriles, and chalcone-based inhibitors [16,17,29,39,40,41,42,43,44,45,46,47].
4.3. Molecular Mechanics
Modeling of inhibitors, InhA, and E-I complexes was carried out by molecular mechanics using CFF91 force field [48] as described earlier [17].
4.4. Conformational Search
Free inhibitor conformations were derived from their bound conformations in the E-I complexes by gradual relaxation to the nearest local energy minimum as described earlier [17].
4.5. Solvation Gibbs Free Energies
The electrostatic component of solvation Gibbs free energy (GFE) that includes also the effects of ionic strength via solving nonlinear Poisson–Boltzmann equation [49,50] was computed by the DelPhi module in Discovery Studio [26] as described earlier [17].
4.6. Calculation of Binding Affinity and QSAR Model
The calculation of binding affinity expressed as complexation GFE has been described fully earlier [17].
4.7. Interaction Energy
The calculation of MM interaction energy (Eint) between enzyme residues and the inhibitor CFF91 force field [48] was performed as described earlier [17].
4.8. Pharmacophore Generation
Bound conformations of inhibitors taken from the models of E-I complexes were used for constructing of 3D-QSAR pharmacophore (PH4) by means of Catalyst HypoGen algorithm [51] implemented in Discovery Studio [26] as described earlier [17].
4.9. ADME Properties
The pharmacokinetics profile of BHMBs were computed by the QikProp program [33] as described earlier [17].
4.10. Virtual Library Generation
The virtual library generation was performed as described earlier [17].
4.11. ADME-Based Library Searching
The drug-likeness selection criterion served to focus the initial virtual library as described earlier [17].
4.12. Pharmacophore-Based Library Searching
The pharmacophore model (PH4) described in Section 4.8 and derived from the bound conformations of BHMBs at the active site of InhA served as library searching tool as described earlier [17].
4.13. Inhibitory Potency Prediction
The conformer with the best mapping on the PH4 pharmacophore in each cluster of the focused library subset was used for ΔΔGcom calculation and IC50pre estimation (virtual screening) by the complexation QSAR model as described earlier [17].
5. Conclusions
In this work novel class 2 InhA direct inhibitors with benzamide scaffold have been designed to reach the picomolar inhibitory concentration range of the predicted IC50pre (Table 6, Figure 8). Even though these predicted inhibitory potencies may be somewhat too optimistic, they suggest that benzamide-type MtInhA inhibitors more potent than the known TS and VS analogs [23] may exist. Our QSAR model provided bound InhA inhibitor conformation, from which the enzyme–inhibitor interaction energy breakdown to active site residue contribution clearly revealed structural determinants needed for binding improvement involving the favorable contacts in the three active site subsites (I, II, and III) including combination of π–π stacking with Phe97 and the HB contacts with Arg43. The derived 3D QSAR pharmacophores identified during chemical space exploration around R1 and R2 positions novel BHMBs analogs with predicted picomolar MtInhA inhibitory potencies 79–92 (IC50pre = 33 pM), 79–39 (39 pM) and 79–338 (47 pM) all display also favorable pharmacokinetic profiles compared to current antituberculotics. We believe that they are worth synthesizing and evaluating.
Moreover, current drugs sharing the benzamide scaffold have been in silico evaluated and the top five predicted drugs, Sultopride (IC50pre = 1.7 nM), Diethyltoluamide (9 nM), Tricalopride (10 nM), Veralipride (23 nM), and Remoxipride (60 nM) are also suggested for biological evaluation as potential antituberculotics.
Table 9.
Representation for 24 approved drugs containing benzamide scaffold listed in Table 8.
| DB00345 | DB00391 | DB00409 | DB00604 | DB00619 | DB01035 | DB01168 | DB01171 |

|

|

|

|

|

|

|

|
| DB01233 | DB01393 | DB06288 | DB06421 | DB06422 | DB06626 | DB07069 | DB08950 |

|

|

|

|

|

|

|

|
| DB09018 | DB11282 | DB11480 | DB12518 | DB13025 | DB13273 | DB13523 | DB15445 |

|

|

|

|

|

|

|

|
Acknowledgments
Partial financial support by the Slovak Research and Development Agency grants (APVV-15-111 and APVV-17-0239) as well as Granting Agency of Slovak Ministry of Education and Slovak Academy of Sciences (VEGA 1/0228/17) is gratefully acknowledged.
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| ADME | Absorption, distribution, metabolism, and excretion |
| AHO | Hydroxamic acids inhibitors |
| BHMBx | Training set of N-benzyl-4-((heteroaryl)methyl)benzamides |
| BHMVx | Validation set of N-benzyl-4-((heteroaryl)methyl)benzamides |
| CAMD | computer-aided molecular design |
| Eint | MM enzyme–inhibitor interaction energy per residue |
| ΔΔGcom | Relative complexation GFE |
| GFE | Gibbs free energy |
| ΔΔGsol | Relative solvation GFE |
| HBA | Hydrogen bond Acceptor |
| HBD | Hydrogen bond Donor |
| HMM | Enthalpy component of GFE |
| HOA | Human oral absorption |
| HYD | Hydrophobic |
| HYDA | Hydrophobic Aliphatic |
| IC50 | Half-maximal inhibitory concentration |
| IE | Interaction energy |
| InhA | 2-trans enoyl-acyl carrier protein reductase |
| KatG | Mycobacterium tuberculosis catalase–peroxidase |
| LHP | Large hydrophobic pocket |
| LOO | Leave-one-out cross-validation |
| MM | Molecular mechanics |
| MM-PB | Molecular mechanics–Poisson–Boltzmann |
| Mt | Mycobacterium tuberculosis |
| MtInhA | 2-trans enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis |
| PDB | Protein Data Bank |
| PfEACP | 2-trans enoyl-acyl carrier protein reductase of Plasmodium falciparum |
| PH4 | Pharmacophore |
| QSAR | Quantitative structure–activity relationships |
| RMSD | Root-mean square deviation |
| SAR | Structure–activity relationships |
| TB | Tuberculosis |
| TS | Training set |
| VS | Validation set |
Author Contributions
K.C.K. performed the complexation study, PH4 pharmacophore generation, interaction energy analysis, the PH4-based VL searching, and the analogues evaluation under the supervision of E.M. E.M. wrote the manuscript with assistance and intellectual input from V.F. and S.M. L.C.O. established the first preliminary complexation model in order to confirm the feasibility of this work. M.K. and R.K.N.G. performed the VL generation and focusing. All the authors approved the final manuscript and had complete access to the study data.
Funding
This research was funded by Slovak Research and Development Agency (grants number APVV-15-111 and APVV-17-0239) as well as Granting Agency of Slovak Ministry of Education and Slovak Academy of Sciences (grant number VEGA 1/0228/17).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.UN General Assembly High-Level Meeting on the Fight against Tuberculosis, New York, 26.09.2018. [(accessed on 13 August 2019)]; Available online: https://www.who.int/news-room/events/un-general-assembly-high-level-meeting-on-en ding-tb.
- 2.Road map towards the implementation of the United Nations Millennium Declaration. [(accessed on 13 August 2019)]; Available online: https://www.undp.org/content/dam/undp/library/MDG/english/MDG%20Country%20Reports/MDG%20Reporting%20Guidelines/MDG%20Roadmap%20from%202001.pdf.
- 3.Global Tuberculosis Report 2018. World Health Organization, Geneva. License: CC BY-NC-SA 3.0 IGO. [(accessed on 7 August 2019)];2018 Available online: https://www.who.int/tb/publications/global_report/en/
- 4.Dye C., Maher D., Weil D., Espinal M., Raviglione M. Targets for global tuberculosis control. Int. J. Tuberc. Lung D. 2006;10:460–462. [PubMed] [Google Scholar]
- 5.Shirude P.S., Madhavapeddi P., Naik M., Murugan K., Shinde V., Nandishaiah R., Bhat J., Kumar A., Hameed S., Holdgate G., et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013;56:8533–8542. doi: 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- 6.Soutter H.H., Centrella P., Clark M.A., Cuozzo J.W., Dumelin C.E., Guiee M.-A., Habeshian S., Keefe A.D., Kennedy K.M., Sigel E.A., et al. Discovery of cofactor-specific, bactericidal Mycobacterium tuberculosis InhA inhibitors using DNA-encoded library technology. Proc. Natl. Acad. Sci. USA. 2016;113:E7880–E7889. doi: 10.1073/pnas.1610978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rozman K., Sosic I., Fernandez R., Young R.J., Mendoza A., Gobec S., Encinas L. A new ‘golden age’ for the antitubercular target InhA. Drug Discov. Today. 2017;22:492–502. doi: 10.1016/j.drudis.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 8.de Ávila M.B., Bitencourt-Ferreira G., Azevedo W. Structural basis for inhibition of enoyl-acyl carrier protein reductase (InhA) from Mycobacterium tuberculosis. Curr. Med. Chem. 2018;26 doi: 10.2174/0929867326666181203125229. [DOI] [PubMed] [Google Scholar]
- 9.Freundlich J.S., Wang F., Vilchèze C., Gulten G., Langley R., Schiehser G.A., Jacobus D.P., Jacobs W.R., Sacchettini J.C. Triclosan derivatives: Towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. Chem. Med. Chem. 2009;4:241–248. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Am Ende C.W., Knudson S.E., Liu N., Childs J., Sullivan T.J., Boyne M., Xu H., Gegina Y., Knudson D.L., Johnson F., et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorg. Med. Chem. Lett. 2008;18:3029–3033. doi: 10.1016/j.bmcl.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luckner S.R., Liu N., Am Ende C.W., Tonge P.J., Kisker C. A slow, tight binding inhibitor of inha, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 2010;285:14330–14337. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He X., Alian A., Stroud R., Ortiz de Montellano P.R. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006;49:6308–6323. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He X., Alian A., Ortiz de Montellano P.R. Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides. Bioorg. Med. Chem. 2007;15:6649–6658. doi: 10.1016/j.bmc.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Encinas L., O’Keefe H., Neu M., Remuinán M.J., Patel A.M., Guardia A., Davie C.P., Pérez-Macías N., Yang H., Convery M.A., et al. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014;57:1276–1288. doi: 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- 15.Wall M.D., Oshin M., Chung G.A., Parkhouse T., Gore A., Herreros E., Cox B., Duncan K., Evans B., Everett M., et al. Evaluation of N-(PheMe)-4-[5-(PheMe)-4,5,6,7-tetrahydro-1H-imidazo [4,5-c]pyridin-4-yl]benzamide inhibitors of Mycobacterium tuberculosis growth. Bioorg. Med. Chem. Lett. 2007;17:2740–2744. doi: 10.1016/j.bmcl.2007.02.078. [DOI] [PubMed] [Google Scholar]
- 16.Owono Owono L.C., Ntie-Kang F., Keita M., Megnassan E., Frecer V., Miertus S. Virtually designed triclosan-based inhibitors of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis and of Plasmodium falciparum. Mol. Inform. 2015;34:292–307. doi: 10.1002/minf.201400141. [DOI] [PubMed] [Google Scholar]
- 17.Kouassi A.F., Kone M., Keita M., Esmel A., Megnassan E., N’Guessan Y.T., Frecer V., Miertus S. Computer-aided design of orally bioavailable pyrrolidine carboxamide inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with favorable pharmacokinetic profiles. Int. J. Mol. Sci. 2015;16:29744–29771. doi: 10.3390/ijms161226196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Punkvang A., Kamsri P., Kumkong A., Kunasa K., Saparpakorn P., Hannongbua S., Wolschann P., Pungpo P. In: The Structural Requirement of Direct InhA Inhibitors for High Potency against M. Tuberculosis Based on Computer Aided Molecular Design, Science against Microbial Pathogens: Communicating Current Research and Technological Advances. Mendez-Vilas A., editor. Formatex Research Center; Badajos, Spain: 2011. pp. 160–168. (Microbiology Book Series No. 3). [Google Scholar]
- 19.Putz M.V., Duda-Seiman C., Duda-Seiman D., Putz A.M., Alexandrescu I., Mernea M., Avram S. Chemical structure-biological activity models for pharmacophores’ 3D-interactions. Int. J. Mol. Sci. 2016;17:1087. doi: 10.3390/ijms17071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putz M.V., Duda N.A., Isvoran A. Double variational binding—(SMILES) conformational analysis by docking mechanisms for anti-HIV pyrimidine ligands. Int. J. Mol. Sci. 2015;16:19553–19601. doi: 10.3390/ijms160819553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putz M.V., Duda N.A. Determining chemical reactivity driving biological activity from smiles transformations: the bonding mechanism of anti-HIV pyrimidines. Molecules. 2013;18:9061–9116. doi: 10.3390/molecules18089061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartkoorn R.C., Sala C., Neres J., Pojer F., Magnet S., Mukherjee R., Uplekar S., Boy-Rottger S., Altmann K.H., Cole S.T. Towards a new tuberculosis drug: Pyridomycin—nature’s isoniazid. EMBO Mol. Med. 2012;4:1032–1042. doi: 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guardia A., Gulten G., Fernandez R., Gomez J., Wang F., Convery M., Blanco D., Martinez M., Herran E.P., Alonzo M., et al. N-Benzyl-4-((heteroaryl)methyl)benzamides: A new class of direct NADH-dependent 2-trans enoyl–acyl carrier protein reductase (InhA) inhibitors with antitubercular activity. Chem. Med. Chem. 2016;11:687–701. doi: 10.1002/cmdc.201600020. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y., Goodacre R. On splitting training and validation set: A comparative study of cross-validation, bootstrap and systematic sampling for estimating the generalization performance of supervised learning. J. Anal. Test. 2018;2:249–262. doi: 10.1007/s41664-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keita M., Kumar A., Dali B., Megnassan E., Siddiqi M.I., Frecer V., Miertus S. Quantitative structure-activity relationships and design of thymine-like inhibitors of thymidine monophosphate kinase of Mycobacterium tuberculosis with favourable pharmacokinetic profiles. RSC Adv. 2014;4:55853–55866. doi: 10.1039/C4RA06917J. [DOI] [Google Scholar]
- 26.Discovery Studio Molecular Modeling and Simulation Program. Accelrys, Inc.; San Diego, CA, USA: 2009. version 2.5. [Google Scholar]
- 27.Sullivan T.J., Truglio J.J., Boyne M.E., Novichenok P., Zhang X., Stratton C., Li H.J., Kaur T., Amin A., Johnson F., et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 28.N’Guessan H., Megnassan E. In silico design of phosphonic arginine and hydroxamic acid inhibitors of Plasmodium falciparum M17 Leucyl aminopeptidase with favorable pharmacokinetic profile—2017. J. Drug Des. Med. Chem. 2017;3:98–125. doi: 10.11648/j.jddmc.20170306.13. [DOI] [Google Scholar]
- 29.Allangba K.N.P.G., Keita M., Kre N’Guessan R., Megnassan E., Frecer V., Miertus S. Virtual design of novel Plasmodium falciparum cysteine protease falcipain-2 hybrid lactone-chalcone and isatin-chalcone inhibitors probing the S2 active site pocket. J. Enz. Inhib. Med. Chem. 2018;34:547–561. doi: 10.1080/14756366.2018.1564288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Available Chemicals Directory, Version 95.1, MDL Information Systems, San Leandro, CA. [(accessed on 13 August 2019)];2003 Available online: http://cds3.dl.ac.uk/cds/cds.html.
- 31.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 32.Stec J., Vilchèze C., Lun S., Perryman A.L., Wang X., Freundlich J., Kozikowski A.P. Biological evaluation of potent triclosan-derived inhibitors of the enoyl-acyl carrier protein reductase InhA in drug-sensitive and drug-resistant strains of mycobacterium tuberculosis. Chem. Med Chem. 2014;9:2528–2537. doi: 10.1002/cmdc.201402255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.QikProp. X Schrödinger, LLC; New York, NY, USA: 2014. version 3.7, release 14. [Google Scholar]
- 34.Jorgensen W.L., Duffy E.M. Prediction of drug solubility from monte carlo simulations. Bioorg. Med. Chem. Let. 2000;10:1155–1158. doi: 10.1016/S0960-894X(00)00172-4. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen W.L., Duffy E.M. Prediction of drug solubility from structure. Adv. Drug Delivery Rev. 2002;54:355–366. doi: 10.1016/S0169-409X(02)00008-X. [DOI] [PubMed] [Google Scholar]
- 36.DrugBank: Benzoic Acid Amides. [(accessed on 07 August 2019)]; Available online: https://www.drugbank.ca/categories/DBCAT000775.
- 37.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Insight-II and Discover Molecular Modeling and Simulation Package. Accelrys; San Diego, CA, USA: 2005. version 2005. [Google Scholar]
- 39.Frecer V., Miertus S., Tossi A., Romeo D. Rational design of inhibitors for drug-resistant HIV-1 aspartic protease mutants. Drug Des. Discov. 1998;15:211–231. [PubMed] [Google Scholar]
- 40.Frecer V., Burello E., Miertus S. Combinatorial design of nonsymmetrical cyclic urea inhibitors of aspartic protease of HIV-1. Bioorg. Med. Chem. 2005;13:5492–5501. doi: 10.1016/j.bmc.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 41.Frecer V., Berti F., Benedetti F., Miertus S. Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing –Phe-Psi-Pro– core and displaying favourable ADME-related properties. J. Mol. Graph. Model. 2008;27:376–387. doi: 10.1016/j.jmgm.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Dali B., Keita M., Megnassan E., Frecer V., Miertus S. Insight into selectivity of peptidomimetic inhibitors with modified statine core for plasmepsin II of Plasmodium falciparum over human Cathepsin D. Chem. Biol. Drug Des. 2012;79:411–430. doi: 10.1111/j.1747-0285.2011.01276.x. [DOI] [PubMed] [Google Scholar]
- 43.Megnassan E., Keita M., Bieri C., Esmel A., Frecer V., Miertus S. Design of novel dihydroxynaphthoic acid inhibitors of Plasmodium Falciparum Lactate Dehydrogenase. Med. Chem. 2012;8:970–984. doi: 10.2174/157340612802084324. [DOI] [PubMed] [Google Scholar]
- 44.Owono Owono L.C., Keita M., Megnassan E., Frecer V., Miertus S. Design of thymidine analogs targeting thymidilate kinase of Mycobacterium tuberculosis. Tuberculosis Res. Treat. 2013;2013:670836. doi: 10.1155/2013/670836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frecer V., Megnassan E., Miertus S. Design and in silico screening of combinatorial library of antimalarial analogs of triclosan inhibiting Plasmodium falciparum enoyl-acyl carrier protein reductase. Eur. J. Med. Chem. 2009;44:3009–3019. doi: 10.1016/j.ejmech.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 46.Esmel A., Keita M., Megnassan E., Beguemsi T., Frecer V., Miertus S. Insight into binding mode of nitrile inhibitors of Plasmodium falciparum falcipain-3, QSAR and pharmacophore models, virtual design of new analogues with favorable pharmacokinetic profiles 2017. SDRP J. Comput. Chem. Mol. Model. 2018;2:103–124. doi: 10.25177/JCMP.2.1.5. [DOI] [Google Scholar]
- 47.De-Eknamkul W., Umehara K., Monthakantirat O., Toth R., Frecer V.K., Knapich L., Braiuca P., Hiroshi N., Miertus S. QSAR study of natural estrogen-like isoflavonoids and diphenolics from Thai medicinal plants. J. Mol. Graph. Model. 2011;29:784–794. doi: 10.1016/j.jmgm.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Maple J.R., Hwang M.-J., Stockfisch T.P., Dinur U., Waldman M., Ewig C., Hagler A. Derivation of class II force fields. I. Methodology and quantum force field for the alkyl functional group and alkane molecules. J. Comput. Chem. 1994;15:162–182. doi: 10.1002/jcc.540150207. [DOI] [Google Scholar]
- 49.Gilson M.K., Honig B. The inclusion of electrostatic hydration energies in molecular mechanics calculations. J. Comput. Aid Mol. Des. 1991;5:5–20. doi: 10.1007/BF00173467. [DOI] [PubMed] [Google Scholar]
- 50.Rocchia W., Sridharan S., Nicholls A., Alexov E., Chiabrera A., Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J. Comput. Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 51.Li H., Sutter J., Hoffmann R. In: Pharmacophore Perception, Development and Use in Drug Design. Güner O.F., editor. International University Line; La Jolla, CA, USA: 2000. pp. 171–189. [Google Scholar]